
Abstract
Objective
MicroRNA (miR)-22-3p is expressed in atherosclerosis (AS), but its function and regulatory mechanisms remain unclear. Therefore, the effects of miR-22-3p in AS were assessed in this study.
Methods
MiR-22-3p expression was assessed in AS, and miR-22-3p target genes were predicted using sequencing transcriptomics. The effect of miR-22-3p agomir on atherosclerotic lesions in an AS mouse model were determined by Oil red O, Masson’s, and sirius red staining, and by anti-smooth muscle actin and macrophage antigen-3 immunostaining. Gene expression in AS was evaluated by western blot and immunofluorescence.
Results
MiR-22-3p was expressed in AS and control samples (32.5% and 33.9% levels, respectively, relative to total miRNA among six highly expressed miRNAs). In the mouse model of AS, miR-22-3p agomir significantly reduced lipid deposition, proliferation of aortic collagen fibres, and macrophage content. Additionally, inducible nitric oxide synthase, interleukin-6, and tumour necrosis factor-α levels were significantly reduced, and levels of arginase 1 and CD206 were significantly enhanced. MiR-22-3p was found to target janus kinase 1(JAK1), and significantly inhibited the activation of NLR family pyrin domain containing 3 (NLRP3) and JAK1 in mice.
Conclusions
MiR-22-3p appears to reduce the inflammatory response in AS, which might be achieved by inducing the M2 macrophage phenotype and suppressing NLRP3 activation via JAK1.
Introduction
Atherosclerosis (AS) is the leading cause of death and disease worldwide, 1 with the main AS lesions comprising smooth muscle cell (SMC) abnormalities, fibrous stromal hyperplasia, and macrophage infiltration. 2 Current preventive and therapeutic modalities mainly consist of surgery and pharmacological interventions, and the most commonplace diagnostic technique applied in clinical settings is arteriography. 3 However, most clinical treatments are focused on the thrombus caused by AS rather than the atherosclerotic lesion. Diagnostic methods also present disadvantages of short retention times, weak signals, potential side effects, and the inability to predict early lesions. 4 Therefore, exploring the underlying complex molecular mechanism and developing novel and effective therapeutic targets to prevent AS is essential.
MicroRNAs (miRNAs) are small non-coding RNAs of 20–25 nucleotides in length that play an essential role in post-transcriptional regulation of gene expression, 5 and participate in the pathogenesis of many human diseases, particularly cardiovascular diseases. 6 A substantial body of evidence implicates miRNAs in cardiovascular disease development, and their aberrant expression or genetic deletion is linked to abnormal cardiac cell differentiation.7–10 In addition, research has indicated that the progress of AS may be correlated with miRNAs due to their significance in driving the dysregulation that affects endothelial cells, SMCs and leukocytes. 11 For example, miR-99a-5p, miR-24, miR-145, miR-221, and miR-222 are involved in regulating vascular SMC proliferation, migration, and apoptosis.12–15 MiR-22-3p (also known as miR-22) is shown to be capable of regulating various diseases, 16 and miR-22-3p expression has been observed in AS. 17 More importantly, miR-22-3p has been shown to be expressed in all AS-associated cell types. 18 In addition, miR-22-3p might affect the regulation of inflammatory cytokines, and has been shown to inhibit monosodium urate-induced gouty inflammation, via the NLR family pyrin domain containing 3 (NLRP3) inflammasome.16,19 However, research on the potential mechanism underlying the role of miR-22-3p in AS is still lacking. We hypothesize that miR-22-3p may participate in the process of AS by regulating NLRP3. Thus, the aim of the present study was to investigate the effects and mechanisms of miR-22-3p in AS.
Materials and methods
Approach to open data
The following datasets were downloaded from the Gene Expression Omnibus (GEO) database: GSE137582, comprising 3 wild-type low-density lipoprotein receptor (LDLR) mice (controls) and 3 high-fat-diet fed homozygous mutated LDLR (ldlr W483Stop knockin) mice as the AS samples (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137582); and GSE137580, comprising 3 samples of oxidative low-density lipoprotein (LDL)-treated human aortic endothelial cells [HAEC] as the AS model, and 3 samples of untreated HAECs as controls (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137580). Quantile standardized preprocessing and analysis of differentially expressed genes (DEGs) was conducted using the R language package. Statistically significant DEGs were obtained using |logFC| ≥ 1 and P < 0.05 in this study. The cluster analysis heatmap for the DEGs was produced using the R language package ‘heatmap’. In addition, the upstream miRNAs of miR-22-3p were predicted by the following databases: TargetScan, 20 GSE137580, 21 and miRDB. 22
Enrichment analyses of DEGs
The DEGs in AS and normal control samples were analysed using the Gene Ontology (GO), Kyoto Encyclopaedia of Genes and Genomes (KEGG) enrichment pathway using the GSE137582 and GSE137580 datasets. David's online tool (https://david.ncifcrf.gov) was applied to obtain the GO terms and KEGG biological process based on DEGs. The interaction between DEGs was processed by STRING databases. The ggplot2 software package, version 3.4.2 (https://cran.r-project.org/) was used to depict the GO and KEGG pathway enrichments and Gene Set Enrichment Analysis (GSEA) methods were used to calculate DEG enrichments.
Animal models
Ten apolipoprotein E (ApoE)-deficient mice (ApoE–/–), aged 6–8 weeks (Skbex Biotechnology Co., Ltd, Henan, China), were randomly divided into two groups: miRNA-22-3p negative control (NC) group, and miR-22-3p agomir group. The miR-22-3p agomir (an artificially synthesized miR-22-3p mimic; Shanghai GenePharma, Shanghai, China) or miRNA-22-3p NC (Shanghai GenePharma) was dissolved in 0.2 mL normal saline and injected into the mice at a dose of 5 nM via the tail vein every 3 days for 2 consecutive weeks. All animals were housed at a temperature of 22–25 °C and relative humidity of 60% under a 12 h light: 12 h dark cycle throughout all the experiments. In a specific pathogen-free environment for 12 weeks, animals were given a continuous supply of drinking water and a high-fat diet including 21% fat and 0.25% cholesterol (Beijing Keao Xieli Feed Company Ltd., Beijing, China). The animal model experiments were approved by the Ethics Committee on Animal Experimentation of the Fourth Affiliated Hospital of Hebei Medical University (No. IACUC-2020017; approval date, 27 March 2020).
Oil red O, Masson’s and sirius red staining
After euthanasia by cervical dislocation, the mouse aorta was separated from the left ventricle, then the aorta was cut longitudinally and pinned open. Whole aorta specimens were immediately frozen for cryosectioning (and stored at –80 °C). The frozen, 6-µm thick sections were then immersed in 60% isopropanol alcohol (for 10 min), stained with 60% Oil red O solution (Sigma-Aldrich, Taufkirchen, Germany) for 3–4 h, and washed 6 to 7 times with 60% isopropanol. The stained aortic tissues were photographed under a light microscope to calculate the percentage of lipid striation in the aorta area.
Aortic roots were also prepared for Masson’s staining and Sirius red staining. Tissue samples were fixed in a 10% formalin solution, dehydrated through an ethanol gradient, embedded in paraffin, and sectioned into slices measuring 4 μm in thickness. Mouse aortic tissue sections were then deparaffinized and subjected to the following staining procedures. For Masson’s staining, Wiegerts’ iron haematoxylin solution (Beyotime, Shanghai, China) was applied for a 5-min incubation to stain cell nuclei. After three rinses with sterile deionized water, the sections were stained with 0.7% Masson-Ponceau-acid fuchsin solution (Beyotime) for 10 min. Subsequently, the slices were washed with 2% glacial acetic acid and differentiated with phosphomolybdic acid for 4 min, followed by direct staining with a 2% aniline blue dye solution (Beyotime). After ethanol dehydration, xylene clearing, and mounting with neutral resins, images of the stained sections were captured using a light microscope. Subsequent to deparaffinization, tissue sections underwent Sirius Red staining (Beyotime) according to the manufacturer’s instructions, and the samples were observed and captured using a light microscope.
Immunohistochemistry
Mice were anesthetized with 4% isoflurane and euthanized by cervical dislocation. The aortic root was then dissected and immersed in formalin, then embedded in paraffin wax and sectioned into 4-mm thick slices. Tissue sections were dewaxed in xylene and soaked in Bouin’s solution (Sigma-Aldrich) overnight at room temperature. Sections were then rinsed with water for 3–5 min, stained with haematoxylin for 2–3 min, and differentiated with 1% hydrochloric acid. The sections were again rinsed with distilled water for 5 min, stained with magenta for 10 min, and rinsed again with distilled water. Subsequently, the sections were dehydrated with 95% ethanol, anhydrous ethanol, and sealed with xylene, respectively. Dewaxed tissue sections also underwent immunostaining. Sections were initially incubated with 3% goat serum for 30 min at room temperature to block nonspecific staining, then incubated with rabbit anti-α-smooth muscle actin (SMA) primary antibody (1: 200 dilution; AF1032; Affinity Biosciences, Changzhou, China), and rabbit anti-macrophage antigen-3 (Mac-3) primary antibody (1: 200 dilution; DF6719; Affinity Biosciences) overnight at 4 °C to label macrophages. Sections were then rinsed three times (5 min each) with 0.1% phosphate buffered saline-Tween 20 (PBST) and then incubated with goat anti-rabbit horse-radish peroxidase-conjugated polyclonal secondary antibody (1: 500 dilution; S0001; Affinity Biosciences) at room temperature for 30 min. The immunoreactivity was visualized using 0.05% diaminobenzidine, then observed at 400 × magnification under an Olympus light microscope (CX41, Olympus, Tokyo, Japan) and evaluated with Adobe Photoshop, version 2020, image analysis software (Adobe.com).
Immunofluorescence
Tissue samples were fixed in a 10% formalin solution, dehydrated through an ethanol gradient, embedded in paraffin, and sectioned into 4-μm thick slices. Subsequent to deparaffinization, all slides were incubated with 3% goat serum for 30 min at room temperature to prevent non-specific staining. The following primary antibodies: anti-cluster of differentiation (CD)68 (1:1200 dilution; DF7518; Affinity Biosciences); anti-inducible nitric oxide synthase (iNOS) (1:1000 dilution: Ab178945; Abcam, Cambridge, UK); CD206 (1: 1000 dilution; ET1702-04; Huabio Inc, Hangzhou, China), and anti-arginase-1 (1:1000 dilution; DF6657; Affinity Biosciences) were diluted in tris-buffered saline (TBS) and incubated with the tissue sections overnight at 4 °C. After rinsing, tissues were incubated with the secondary antibody (1: 2000 dilution; Affinity Biosciences) for 1 h at room temperature. All glass slides were washed 3 times with TBS for 2–3 min each time, then stained with 4′,6-diamidino-2-phenylindole (DAPI). The slides were then washed in acidified water, dehydrated, and sealed with neutral gel. Fluorescent images were analysed and quantified using ImageJ software, version 4.3.67.
Luciferase reporter assays
Fragments of the 3′-UTR of the janus kinase 1 (JAK1) mRNA sequences containing the binding sites for putative (wild-type) or mutated miR-22-3p were amplified and cloned into luciferase reporter constructs (RiboBio, Guangzhou, China). These constructs and miR-22-3p agomir or NC were subsequently cotransfected into human embryonic kidney (HEK 293) cells using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. After transfection for 48 h, the cells were treated with dual-luciferase reporter assay reagent (Promega, Madison, WI, USA) and luciferase activity was measured, according to the manufacturer’s instructions.
Western blotting
Total mouse aortic protein was extracted with radioimmunoprecipitation assay buffer (Beyotime Biotechnology, Shanghai, China) containing 1% phenylmethanesulfonyl fluoride (Beyotime Biotechnology). Following lysis, samples were centrifuged, the supernatant was collected, and protein levels were assessed via a BCA assay kit (Beyotime), following the manufacturer’s instructions. Protein samples (30 µg each) were then loaded into a 12% polyacrylamide gel for electrophoresis, and the separated proteins were transferred to a polyvinylidene difluoride membrane. Membranes were blocked with 3% bovine serum albumin in tris-buffered saline-Tween (TBST; 150 mM NaCl, 10 mM Tris-HCl pH 8.0 and 0.1% (v/v) Tween 20). The membranes were incubated at 4 °C overnight with the following primary antibodies (all from Life Technologies): JAK1 (1: 800 dilution); phosphorylated (p)-JAK1 (1: 800 dilution); NLRP3 (1: 1000 dilution); iNOS (1: 1000 dilution); interleukin (IL)-6 (1: 1000 dilution); tumour necrosis factor (TNF)-α (1: 1000 dilution); arginase-1 (Arg-1) (1: 1200 dilution); and CD206 (1: 1200 dilution), with β-actin (1: 1000) as the loading control. Membranes were then washed three times with TBST and incubated with a secondary antibody (Affinity Biosciences) coupled with labelled chemiluminescence or fluorescent molecule, and band intensities were visualised and analysed using the Gel-Pro-Analyzer software system, version 4.1 (Media Cybernetics Inc., Bethesda, MD, USA).
Statistical analysis
All data are presented as mean ± SD, and were statistically analysed using GraphPad Prism 6.0 software (www.Graphpad.com). Between-group differences were assessed using Student’s t-test, and P < 0.05 was considered to be statistically significant.
Results
Expression of miR-22-3p in atherosclerosis
MiR-22-3p expression in atherosclerosis was initially assessed based on the GEO database (GSE137582). By searching the database, the samples used in the GSE137582 dataset were miRNA expression profiles of a normal control sample (ascending aorta tissue from mouse 133 with wild-type low-density lipoprotein receptor [LDLR]) and an AS case (ascending aorta tissue from mouse 179 with homozygous mutated LDLR). The GSE137582 expression profiling platform was mouse GPL21265 and Agilent-070155 mouse miRNA Microarray (miRBase release 21.0, miRNA ID version), comprising 1881 miRNAs. The ratio of each miRNA expression level to total miRNA in GSE137582 was calculated, then the relative expression of the top six highly expressed miRNAs (including MiR-22-3p) was plotted in a pie chart (Figure 1). Of the six top miRNAs, miR-22-3p expression showed the highest relative level (32.5% in AS samples and 33.9% in control samples).

Relative expression of microRNAs from the GSE137582 dataset, known to be highly expressed in atherosclerosis (AS), including miR-22-3p. Relative expression of the six most highly expressed miRNAs showed miR-22-3p to be the highest in AS (Case) and controls (Ctrl).
Atherosclerosis lesion development is inhibited in vivo by MiR-22-3p agomir
To evaluate the impact of miR-22-3p agomir, an miR-22-3p mimic, on atherosclerotic lesion progression, Oil red O staining of frozen aortic arches was performed to visualize lipid-rich atherosclerotic plaques. As shown on the representative oil red O-stained images, miR-22-3p agomir significantly reduced lipid deposition versus miR-22-3p NC (P < 0.01; Figure 2a). For further analysis of the atherosclerotic lesion area, collagen content was assessed by Masson’s trichrome staining and sirius red staining, macrophage content by Mac-3 macrophage marker staining, and smooth muscle cell area by α-SMA staining. MiR-22-3p agomir mice were found to exhibit a significant reduction in aortic collagen fibre proliferation (Figure 2b and 2c; all P < 0.05), and macrophage content (Figure 2d; P < 0.01) compared with miR-22-3p NC mice. Additionally, the lesions of miR-22-3p agomir mice showed a trend towards fewer SMCs than miR-22-3p NC mice, but there was no statistically significant between-group difference (Figure 2e). These in vivo data suggest that miR-22-3p suppressed atherogenesis.
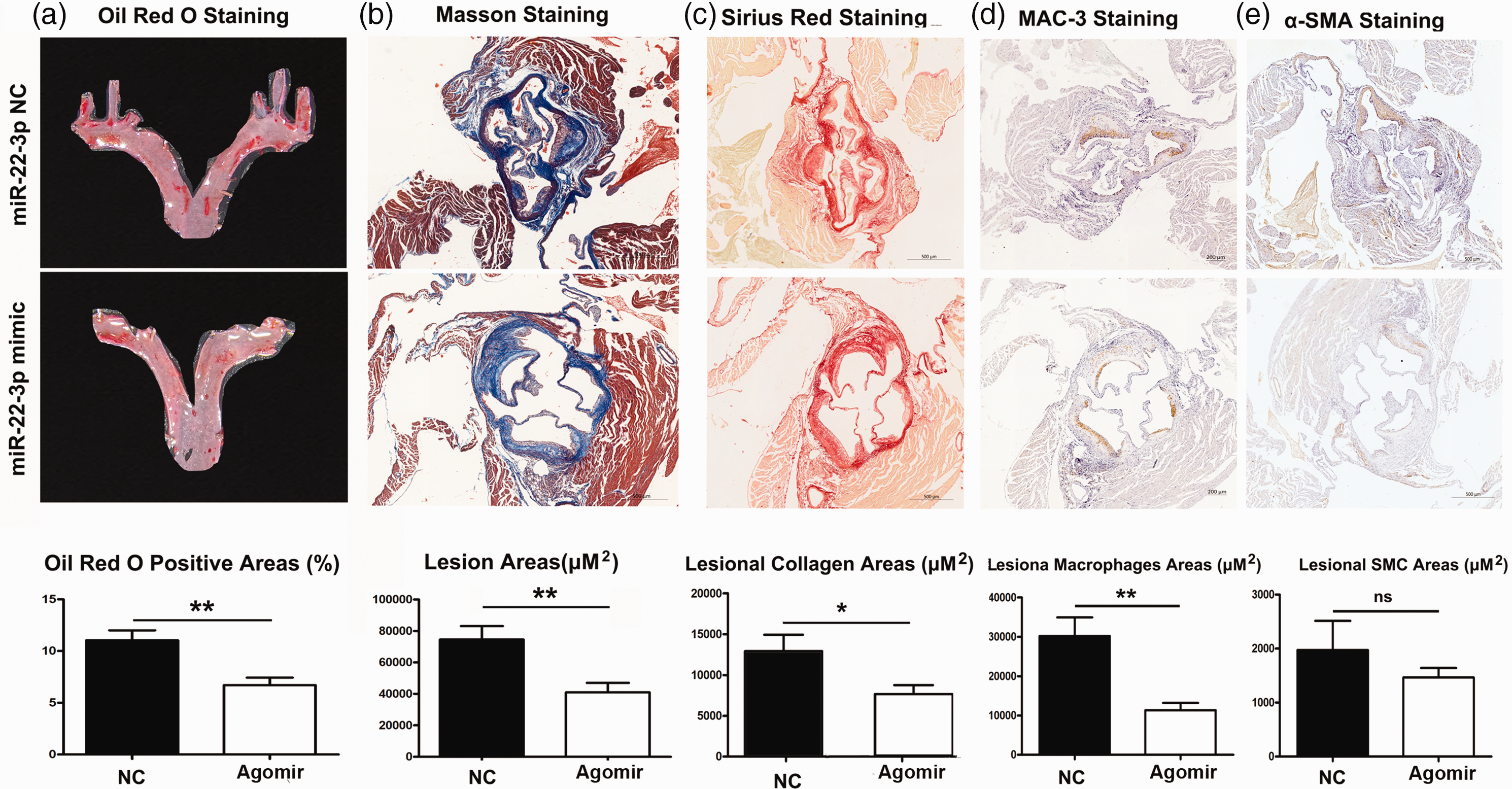
MiR-22-3p suppresses atherosclerotic lesion development in ApoE–/– mice fed a high fat diet: (a) oil red O staining and quantitative analysis of aortic sections from MiR-22-3p agomir or negative control (NC) mice; (b) Masson’s staining and quantitative analysis of aortic sections from MiR-22-3p agomir or NC mice (original magnification, × 40; bar = 500 μm); (c) sirius red staining and quantitative analysis of aortic sections from MiR-22-3p agomir or NC mice (original magnification, × 40; bar = 500 μm); (d) macrophage antigen-3 (Mac-3) immunostaining and quantitative analysis of aortic sections from MiR-22-3p agomir or NC mice (original magnification, × 40; bar = 200 μm); and (e) quantitative analysis of smooth muscle cells by α-smooth muscle actin immunostaining of aortic sections from MiR-22-3p agomir or NC mice (original magnification, × 40; bar = 500 μm). (*P < 0.05; **P < 0.01; or ns, no statistically significant difference versus NC).
MiR-22-3p agomir inhibits inflammatory infiltration and induces macrophage conversion to the M2 phenotype
To investigate the effect of miR-22-3p agomir on inflammation, the macrophage marker CD68, M1-type marker iNOS, M2-type marker arginase-1, and CD206 were detected by immunofluorescence in aortic tissues from MiR-22-3p agomir mice versus MiR-22-3p NC mice. The CD68-positive staining exposed that miR-22-3p agomir decreased macrophage infiltration in the atherosclerotic plaque, and iNOS was significantly decreased (P < 0.01, miR-22-3p agomir versus miR-22-3p NC; Figure 3a). Meanwhile, Arg-1 and CD206 were significantly increased (P < 0.01, miR-22-3p agomir versus miR-22-3p NC; Figure 3b). In addition, the M1-markers (iNOS, IL-6, and TNF-α) and M2-markers (Arg1 and CD206) in aorta tissues were analysed by western blot to examine the polarization of macrophages. Protein levels of iNOS, IL-6, and TNF-α were significantly reduced by miR-22-3p agomir, whereas Arg1 and CD206 were significantly enhanced (all P < 0.05 versus miR-22-3p NC; Figure 3c). Thus, these findings suggest that miR-22-3p agomir repressed inflammatory infiltration and induced macrophage M2 phenotype in the areas of atherosclerotic plaque lesions.

MiR-22-3p induces macrophages into the M2 phenotype in ApoE–/– mice fed a high fat diet: (a) immunofluorescence staining (original magnification, × 40, bar = 50 μm) and quantitative analysis of macrophage accumulation and macrophage polarization by detection of (a) the macrophage marker cluster of differentiation (CD)68 and M1 marker inducible nitric oxide synthase (iNOS), and (b) the M2 markers CD206 and arginase (Arg)-1, in aortic sections from MiR-22-3p agomir or normal control (NC) mice; and (c) western blot and quantitative analysis of relative protein levels of the M1 markers (iNOS, interleukin [IL]-6, and tumour necrosis factor [TNF]-α) and M2 markers (Arg1 and CD206) in aortic sections from MiR-22-3p agomir or NC mice. (*P < 0.05; **P < 0.01).
MiR-22-3p targeted JAK1 and suppressed the activation of NLRP3 and JAK1
To predict the target genes of miR-22-3p, three miRNA databases were searched, including TargetScan, 20 GSE137580, 21 and miRDB. 22 Nine common elements were identified among the databases (Figure 4a). Further database sequence analysis revealed targeted binding regions between miR-22-3p and JAK1 (Figure 4b), suggesting that miR-22-3p might target JAK1. Next, the 3’-UTR sequences of JAK1 were found to match the ‘seed sequence’ of miR-22-3p, indicating that miR-22-3p targeted JAK1 genes (Figure 4c). The dual-luciferase reporter assay further verified JAK1 as a target gene of miR-22-3p: Compared with cells treated with the NC group, the luciferase activity of JAK1 3′-UTR-wt was decreased in cells following miR-22-3p agomir transfection (P < 0.01), but no difference was found in the luciferase activity of JAK1 3′-UTR-mut (Figure 4d). To assess the inflammatory signalling pathway in AS, microarray data were analysed from the six samples in the GSE137582 dataset, 23 which contained arteriosclerosis and normal control samples. DEGs were shown in heat maps, and among the AS samples, 125 DEGs were down-regulated, whereas 148 DEGs were up-regulated (Figure 4e). Simultaneous analysis of another dataset, GSE137580, 24 was applied to validate 268 DEGs, in which 103 DEGs were down-regulated and 165 DEGs were up-regulated (Figure 4f). Next, GO term pathway analysis was performed, based on the DEGs from GSE137580, to identify the critical pathways that affect AS. The top 31 up-regulated pathways were found to include inflammatory response, cell proliferation, cellular response to DNA damage stimulus, and mitotic metaphase plate congression (Figure 4g). Biological process terms were collected and the KEGG pathway terms in GO analysis. JAK1 was identified as a top DEG with a high fold change in AS, according to the data from GSE137580 (Figure 4h). The partial results of gene-enriched GO items in KEGG pathways were also depicted (Figure 4i), and GSEA enrichment data showed that most of the genes related to the inflammatory response signalling pathway in AS were functionally enriched in JAK1 expression (Figure 4j). Finally, the western blot results further showed that the expression of NLRP3 and phosphorylated-JAK1 were significantly decreased in miR-22-3p agomir mice versus controls (P < 0.01; Figure 4k), suggesting that miR-22-3p inhibited the activation of NLRP3 and JAK1.
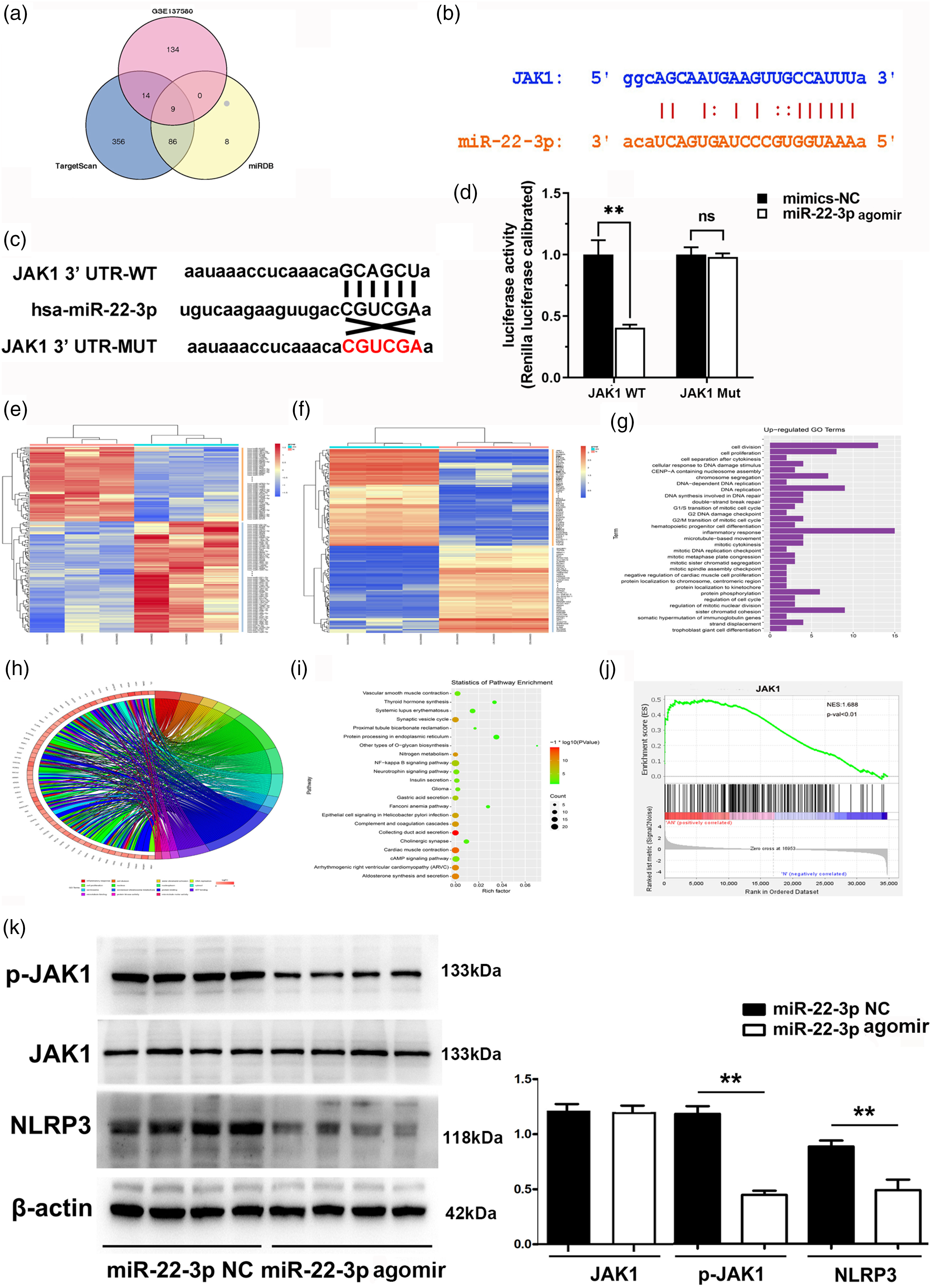
MiR-22-3p might suppresses NLR family pyrin domain containing 3 (NLRP3) activation via direct targeting of janus kinase 1 (JAK1): (a) Upstream miRNAs of miR-22-3p were predicted by TargetScan, GSE137580 and miRDB databases; (b) database sequence analysis revealed that miR-22-3p bound to the 3′-UTR of JAK1; (c) the 3′ UTR of JAK1 was predicted to contain a complementary region of miR-22-3p seed Continued.sequences, and mutation was generated in the 3′UTR of JAK1; (d) luciferase reporter plasmids harbouring the wild type (WT) or mutant (Mut) 3′-UTR of JAK1 were co-transfected with negative control (NC) agomir or miR-22-3p agomir into HEK 293 cells, and revealed that miR-22-3p targeted JAK1; (e) heatmap showing down-regulated and up-regulated differentially expressed genes (DEGs) in three atherosclerosis (AS) and three normal control (NC) samples from the GSE137582 mouse dataset; (f) heatmap showing down-regulated and up-regulated DEGs in three AS and three NC samples from GSE137580 human aortic endothelial cell dataset; (g) The top 31 up-regulated Gene Ontology (GO) pathways based on DEGs identified in the GSE137580 dataset; (h) STRING analysis was used to obtain gene-enriched items of GO biological processes; (i) partial results of Kyoto Encyclopaedia of Genes and Genomes pathways; (j) Gene Set Enrichment Analysis enrichment in AS was performed; and (k) miR-22-3p was shown to affect the expression of NLRP3 and JAK1 by quantitative analysis of western blots. (**P < 0.01, miR-22-3p agomir versus miR-22-3p NC).
Discussion
Atherosclerosis is recognized as a complex chronic inflammatory disease caused by continuous dyslipidaemia, which will evolve into fibrous plaques in conduit arteries leading to rupture of some plaques, causing thrombosis or stenosis. 25 Although the specific mechanisms remain unclear, miRNAs have been shown to play an important part in AS progression. 26 For example, miR-126, miR-155, miR-206, and miR-223 prevent unfavourable lipid metabolism and reduce inflammation via many signalling pathways; other miRs, such as miR-let-7g, miR-143, and miR-34a, modulate endothelial cell senescence by regulating related protein; also, miR-10a, miR-31, and miR-17-3p regulate inflammation by modulating the expression of adhesion molecules. 27 Most recently, miR-22-3p has been reported to inhibit human aortic SMC proliferation and migration, affecting vascular pathologies. 17 However, the mechanism of miR-22-3p has not been fully explored in AS. Therefore, the effect of miR-22-3p in AS, and its internal control mechanism, were explored in the present study.
In the current study, initial bioinformatics analysis of the GSE137582 dataset was used to screen the expression of miRNA in AS. The result showed that miR-22-3p was the highest expressed miRNA in AS samples of this study compared with other miRNAs known to be highly expressed in blood vessels, particularly miR-133 and miR-155,28,29 which is consistent with other research results. 30 Next, the role of miR-22-3p in the development of AS was further explored by constructing a mouse model of AS using ApoE–/–mice fed a high fat diet. Lipid and collagen accumulation in the arterial wall is one of the causes of AS. 25 Thus, the effect of miR-22-3p on lipids in arteries were investigated by Oil red O staining, and collagen was investigated by Masson’s staining and sirius red staining in the present study. The arterial tissues from miR-22-3p agomir mice showed significantly reduced lipid and collagen accumulation. In addition, macrophages and SMCs are the main cell types involved in AS, with evidence to indicate that macrophage infiltration and SMC proliferation are crucial players in accelerating AS progression.31,32 The macrophage-specific antigen Mac-3 was assessed to measure the effect of miR-22-3p agomir on macrophage content, and revealed that aortic tissue from miR-22-3p agomir mice exhibited significantly reduced macrophage accumulation within the lesion. In addition, α-SMA immunostaining showed that miR-22-3p agomir mice exhibited reduced areas of SMCs in arteries. The above results suggest that miR-22-3p may effectively inhibit the development of AS. Consistent with the present investigation, previously published studies have shown that miR-145 and miR-342-5p mitigate AS by modulating vascular SMC phenotype and protecting endothelial cells, respectively.33,34
Macrophage polarization regulates inflammation and clears the infection, which plays a key role in development of the inflammatory response in AS. 35 In recent years, studies have shown macrophage polarization into two phenotypes, M1 and M2, with the M1-type being associated with pro-inflammatory responses, but the M2-type weakening the inflammatory response. 36 Recently, miR-22-3p expression was found to be up-regulated in bone marrow stromal cells cocultured with M2 macrophages, suggesting that the expression of Mir-22-3p was associated with M2 macrophages. 37 The mechanism of miR-22-3p on inflammatory responses in AS were further investigated using immunofluorescence and western blots to detect the expression of macrophage polarizing cytokines at the protein level. Expression of the M1 markers iNOS, IL-6, and TNF-α was significantly reduced by miR-22-3p agomir compared with the NC group, whereas expression of the M2 markers Arg1 and CD206 was significantly enhanced by miR-22-3p agomir. These assays revealed that miR-22-3p inhibited the inflammatory response and alleviated the progression of AS by promoting the polarization of M2 macrophages in AS. This is consistent with another study, which found that miR-22-3p enhances M2 polarization of macrophages and suppresses inflammation via up-regulation of interferon regulatory factor 5. 19
Phosphorylated JAK1 is the best-characterized immunologic molecule that increases the production of inflammatory markers and mediates inflammatory response activities, 38 with inflammation being one of the important cellular responses mediated by the JAK1 signalling pathway. 39 Using the GEO, miRDB and TargetScan databases, miR-22-3p and JAK1 were discovered to have targeted binding regions, implying that JAK1 may be a target gene for miR-22-3p. Luciferase reporter assays confirmed that JAK1 is indeed a target of miR-22-3p. Analysis of microarray data from the GSE137582 and GSE137580 datasets, to further explore the JAK1-regulated signalling pathway in AS, showed that inflammatory response was an important enrichment pathway in AS, and most genes related to the inflammatory response signalling pathway in AS were functionally enriched in JAK1 expression. These results confirmed that JAK1 is related to the inflammatory response pathway in AS and demonstrated that miR-22-3p inhibits inflammation by targeting JAK1. Inflammasomes are intracellular multimolecular complexes that mediate the inflammatory reaction, and the NLRP3 inflammasome is an important cytoplasmic dot pattern recognition receptor involved in promoting the activation of inflammatory vesicles. 40 In the present study, the expression of NLRP3 was found to be significantly decreased in miR-22-3p agomir mice, suggesting that MiR-22-3p suppresses NLRP3 activation. JAK1 is one of the target genes shown to inhibit activation of the NLPR3 inflammasome. 41 The administration of a JAK1 inhibitor has been shown to inhibit expression of the NLRP3 inflammasome in the inflammatory response, which is similar to the present study results showing that expression of NLPR3 and p-JAK1 was inhibited in miR-22-3p agomir mice, suggesting that miR-22-3p might regulate the activation of NLPR3 via JAK1 to affect the inflammatory reaction. In addition, MiR-9 suppressed downstream NLRP3 inflammasome activation through its target gene JAK1 in AS, 41 which might help explain the interaction between JAK1, NLRP3, and miRNAs. In the present study, miR-22-3p was not found to affect JAK1 expression at the protein level (despite it being a direct target). Thus, the specific mechanisms by which miR-22-3p inhibits the activation of NLRP3 by JAK1-regulated signalling in AS requires further investigation.
Some limitations of the present study remain to be further explored. First, JAK1 is one of the numerous target genes of miR-22-3p, and other genes regulated by miR-22-3p may also be involved in the progression of AS. Thus, the effect of JAK1 and other gene targets of miR-22-3p are worth exploring. Secondly, since only the highly-expressed miRNAs were analysed when screening the targeted miRNAs associated with atherosclerosis using GEO datasets, some miRNAs with low expression levels but also important in atherosclerosis may have been neglected. Therefore, the role of other miRNAs in AS also warrants further investigation. Thirdly, despite the present data demonstrating that miR-22-3p promoted the polarization of M2 macrophages in AS, namely, the significantly increased protein levels of M2-markers, the role of JAK1 in M2 polarization, and the mechanistic role of NLRP3 in this context, remain unclear.
Conclusion
In conclusion, the present study results support the role of miR-22-3p in reducing the inflammatory response in AS, which might be via induction of the M2 macrophage phenotype and suppression of NLRP3 activation via JAK1. This study might generate novel insight into the mechanism of AS and strengthen the novel microRNA-based gene therapy strategies in atherosclerosis.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231197071 - Supplemental material for MicroRNA-22-3p alleviates atherosclerosis by mediating macrophage M2 polarization as well as inhibiting NLRP3 activation
Supplemental material, sj-pdf-1-imr-10.1177_03000605231197071 for MicroRNA-22-3p alleviates atherosclerosis by mediating macrophage M2 polarization as well as inhibiting NLRP3 activation by Xiaoyan Bian, Haoyang Peng, Yin Wang, Hongjiang Guo and Gaofeng Shi in Journal of International Medical Research
Footnotes
Author contributions
XY and GF wrote the manuscript, and designed and performed the experiments. GF conducted the research. HY performed the statistical analyses and organized the data. HJ created the figures. XY and GF supervised the research design and revised the manuscript. All authors have viewed and approved the final manuscript.
Data availability
The datasets analysed during the current study are available from the corresponding author upon reasonable request.
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.