
Abstract
Guillain–Barré syndrome (GBS) and Miller Fisher syndrome (MFS) are acute immune-mediated peripheral neuropathies. In addition to their classic presentations, a variety of other signs and symptoms have been reported; however, headache appears to be relatively uncommon. We describe a 53-year-old woman who presented with acute bulbar palsy as the first symptom of overlapping MFS/GBS accompanied by severe headache. The first important clinical impairment of the patient was acute bulbar palsy along with prominent headache, without limb weakness. Although her initial diagnosis was acute bulbar palsy plus, she subsequently developed lower limb diffuse weakness, and her final clinical diagnosis was overlapping MFS/GBS. Anti-ganglioside antibodies were positive for anti-GQ1b and anti-GT1a immunoglobulin G. The patient received intravenous immunoglobulin on day 2 of admission. Early identification of these overlapping syndromes is important for the management of patients, to avoid respiratory failure or severe weakness with axonal degeneration. We therefore remind clinicians of the importance of further examination in patients with headache and acute bulbar palsy of unknown origin.
Keywords
Introduction
Guillain–Barré syndrome (GBS) and Miller Fisher syndrome (MFS) are considered to form a continuous clinical spectrum. 1 Pain is an important symptom of GBS, and although moderate and severe back pain is frequently noted, headache appears to be rare. In a large prospective study of pain in GBS, Moulin et al. 2 cited a headache incidence of just 2%. Furthermore, in a case series of 27 patients with MFS, 22% of the patients had painful experiences during the acute phase of MFS; of these, only three had unilateral periorbital pain. 3 Headache onset is variable; it can exist prior to, concurrent with, or following the onset of weakness. 4 Herein, we report a woman who presented with acute bulbar palsy as the first symptom of overlapping MFS/GBS accompanied by severe headache.
Case presentation
A 53-year-old right-handed woman presented with acute onset of mild numbness in the perioral region and distal limbs. Within 24 hours, she successively developed dysarthria, dysphagia, and diplopia. Distention pain was also present around both orbits and throughout the brain. Her headache was constant; she described it as a severe bilateral throbbing headache and deep orbital pain associated with nausea. There was no associated photophobia or vomiting. The pain would wake her up at night, and non-steroidal anti-inflammatory drugs did not provide significant relief. She did not have limb weakness or bowel or bladder difficulties. She had experienced a runny nose and sore throat, suggestive of an upper respiratory tract infection, 7 days before the neurological symptoms began. Her past medical history constituted of hypertension only, for which she did not take any medicine. She had no prior history of headaches and her family history was unremarkable.
Examination on admission revealed normal mental status, difficulty swallowing, bilateral lateral rectus palsies, bilateral finger-to-nose dysmetria, and areflexia, without fever. Her pupils were symmetrical and of a normal size, and reacted briskly to light. Her initial motor examinations were normal. There were no signs indicating autonomic or sphincter dysfunction. Over 2 days of hospitalization, her dysarthria worsened, and she became unable to swallow fluids. Her gag reflex was absent. A nasogastric tube was inserted. The patient developed bilateral ptosis and lower limb weakness, with a strength of 4/5 on the Medical Research Council scale. Facial nerves were normal upon examination. She had no weakness of the neck or upper limb muscles and no shortness of breath or difficulty breathing.
Computed tomography imaging of the head was performed; the results were unremarkable. A head magnetic resonance imaging showed no signs of ischemia or vascular disease (Figure 1). Nerve conduction studies on day 2 showed a reduced amplitude of sensory nerve action potentials and delayed nerve conduction velocity in the bilateral median nerves (Table 1). However, motor conduction study results were otherwise normal. The frequency and latency of F-waves in the upper and lower limbs were within the normal range. A repetitive nerve stimulation study did not reveal neuromuscular junction disorder. An intraocular pressure of 14 mmHg was measured because of the presence of eye pain in the patient. A repeat electrophysiology on day 82 revealed improved sensory nerve action potential amplitudes.

Brain magnetic resonance imaging on day 2 of the illness. No obvious abnormalities were observed in axial T2-weighted fluid-attenuated inversion recovery images at the cerebral hemisphere (a) and brainstem (b) levels.
Motor and sensory nerve conduction studies.
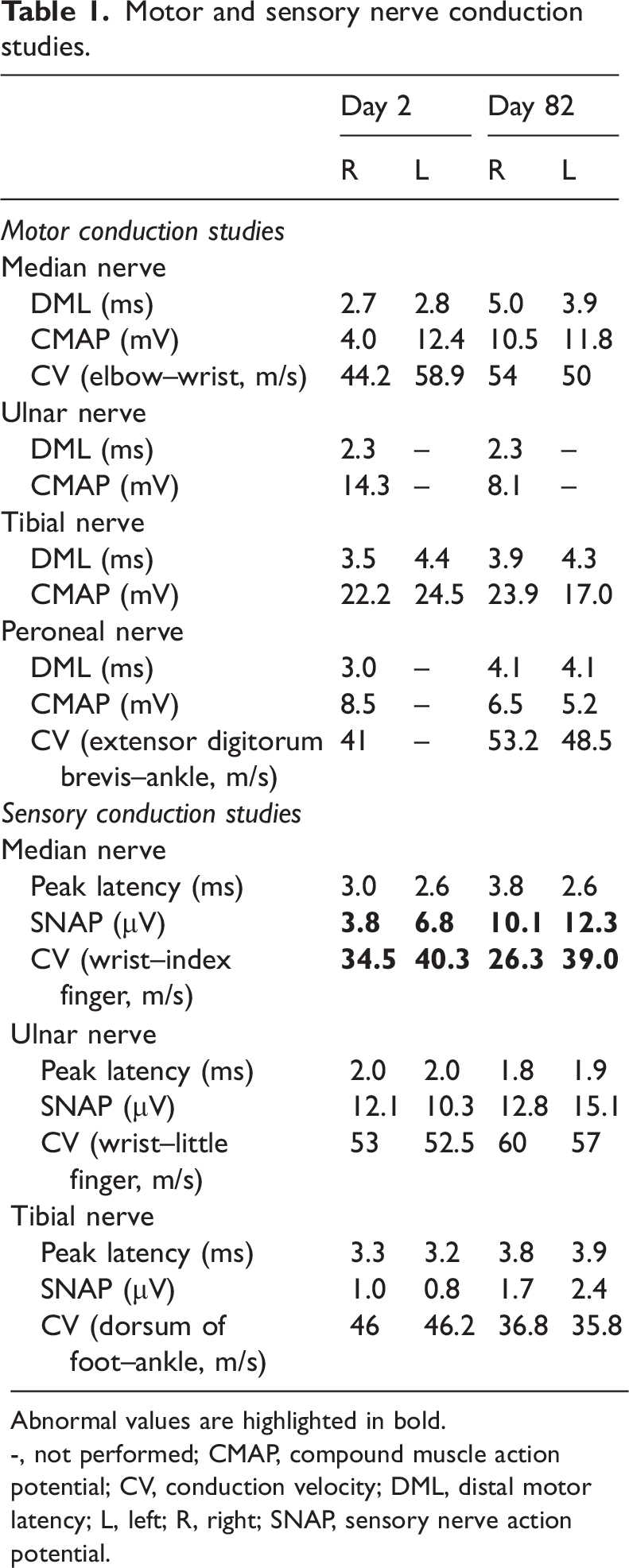
Abnormal values are highlighted in bold.
-, not performed; CMAP, compound muscle action potential; CV, conduction velocity; DML, distal motor latency; L, left; R, right; SNAP, sensory nerve action potential.
The results of laboratory studies, including a complete blood count, liver enzymes, folic acid, urea, creatinine, vitamin B12, and coagulation tests, were within normal ranges. A lumbar puncture was performed on day 2 of the illness, and showed a normal opening pressure of 180 mmH2O, a normal glucose concentration, and no pleocytosis or albuminocytological dissociation. Serum immunoglobulin G (IgG) antibodies to gangliosides were measured on admission using enzyme-linked immunosorbent assay; the patient was positive for anti-GQ1b and anti-GT1a IgG antibodies.
Early in the disease course, the patient’s most prominent and initial symptom was acute bulbar palsy, along with acute ophthalmoplegia diplopia, ataxia, areflexia, and prominent headache and eye pain, without limb weakness. The positive anti-GQ1b and anti-GT1a antibodies and the normal magnetic resonance imaging were initially in keeping with a diagnosis of acute bulbar palsy plus syndrome. However, because our patient subsequently developed diffuse lower limb weakness, the final clinical diagnosis was overlapping MFS/GBS.
After obtaining informed consent, we started treatment with intravenous immunoglobulin (IVIg) for 5 days (0.4 g/kg/day) on day 4 of the illness. On day 4 of treatment, the patient’s bulbar palsy, lower limb weakness, and diplopia started to improve. However, she still had persistent eye and head pain (unaccompanied by nausea), although its severity was reduced. The pain gradually improved under the control of simple analgesics. The patient’s dysphagia and lower limb weakness were almost completely recovered at discharge, on day 13 of the illness, and her nasogastric tube was removed; however, she still had mild bilateral lateral rectus palsies and bilateral ptosis. At this point, the pain in her eyes and head was completely relieved. Her diplopia completely disappeared after 3 months. The reporting of this study conforms to CARE guidelines (for CAse REports). 5
Discussion
Recently, new diagnostic criteria—based on an inclusive set of clinical features—were proposed for the diagnosis of GBS and its variants using a simple, yet all-inclusive, classification system. 1 Most cases of GBS can be included into this classification system. However, some GBS cases present with acute bulbar palsy overlapping with other neurological symptoms, such as cranial nerve paralysis (especially ophthalmoplegia) and ataxia, but with an absence of neck or limb weakness; these cases do not satisfy the diagnostic criteria for any subtype of GBS, MFS, or their variants. In such a situation, Kim et al. 6 proposed the concept of acute bulbar palsy plus syndrome, which can be considered a new regional variant of GBS. In the current case, the initial important clinical impairment was acute bulbar palsy along with acute ophthalmoplegia, diplopia, ataxia, and areflexia, without limb weakness. With combined anti-GQ1b and anti-GT1a positivity, a diagnosis of acute bulbar palsy plus syndrome was first made. However, because our patient subsequently developed diffuse lower limb weakness, her final clinical diagnosis was overlapping MFS/GBS. 7 In terms of clinical significance, our case indicates the importance of including GBS as a differential diagnosis for acute bulbar weakness. Our case indicates that acute bulbar palsy can be described as a transitional symptom that may evolve into generalized GBS. In addition, pharyngeal palsy may be the characteristic first step in the development of MFS/GBS but not pure MFS. Notably, our patient also experienced severe headache and eye pain in the initial stage of illness. Unilateral severe headache with extraocular involvement is a common manifestation of ophthalmoplegic migraine, and unilateral retro-orbital pain with ophthalmoplegia is the core symptom of Tolosa–Hunt syndrome. Our case indicates the importance of considering GBS as a differential diagnosis for patients with headache and bulbar paralysis of unknown origin; these are important events that should be recognized by clinicians.
Anti-ganglioside antibodies play a key role in the pathophysiology of GBS and MFS. Anti-ganglioside antibodies are often closely associated with clinical phenotypes and specific symptoms.8,9 For example, the pattern of neuropathy can be partially explained by the anatomical distribution of gangliosides. Previous studies have reported that GQ1b is densely localized in the paranodal regions of cranial nerves innervating the extraocular muscles, a subpopulation of large neurons in dorsal root ganglia, and muscle spindles in limbs.10,11 Therefore, anti-GQ1b IgG antibodies may account for ophthalmoplegia and ataxia in patients with MFS through their specific binding in these regions. Furthermore, serum IgG antibodies to the ganglioside GQ1b are a possible sensitive and specific serum marker for MFS.12,13 Anti-GT1a antibodies have been detected in a relatively high percentage of patients with GBS with bulbar palsy.14–16 Moreover, it has been reported that a monospecific anti-GT1a antibody that does not cross-react with GQ1b is essential for the development of bulbar palsy in patients with GBS. 17 This finding may be attributed to the abundant GT1a gangliosides in glossopharyngeal and vagal nerves. 18 Serum anti-GT1a IgG antibodies are therefore suggested to be associated with bulbar palsy. However, given that IgG anti-GQ1b antibodies have often been identified as cross-reactive with GT1a, the role of anti-GT1a IgG antibodies remains to be elucidated. Here, we have reported a case of overlapping MFS/GBS with positivity for both anti-GQ1b and anti-GT1a IgG. Our case initially had a variety of clinical manifestations: bulbar palsy, ophthalmoplegia, diplopia, and areflexia. The coexistence of anti-GQ1b and anti-GT1a IgG may explain this range of clinical features, at least in part.
In keeping with the ganglioside complex theory, the examination of anti-ganglioside complex antibodies to investigate antibodies against various ganglioside complexes containing GQ1b or GT1a may better explain frequent overlaps or transitional syndromes. Previous studies have reported that MFS most frequently overlaps with GBS regional variants of pharyngeal–cervical–brachial weakness. 19 With these variants, initial pharyngeal palsy is usually followed by weakness that extends downward to the neck and brachia and finally reaches the lower limbs. Notably, our patient developed lower limb weakness after progression, without weakness in the upper limbs. This finding is very rare and may have been caused by our early and timely use of IVIg, thus avoiding severe flaccid paralysis of the limbs and respiratory function deterioration. Physicians should therefore pay close attention to pharyngeal palsy and identify any overlaps as early as possible.
To our knowledge, very few cases of MFS/GBS with severe headache and acute bulbar paralysis as the initial symptoms have been previously reported. Most clinical information about the characteristics of headache in typical GBS and MFS comes from case reports.20,21 A case series recruited 38 patients with MFS and evaluated symptoms or signs beyond the classic triad. Six patients (16%) had headache with moderate severity. The authors reported that the location of headaches was variable, including periorbital, temporal, or the whole area, in pure MFS. 22 An association between headache and GBS has also been described in the literature. Headache in GBS appears to occur in four general clinical settings: posterior reversible encephalopathy syndrome (PRES), increased cerebrospinal fluid (CSF) pressure and papilledema, MFS, and in the rare scenario where aseptic meningitis occurs after IVIg use.
After the initial description of PRES in 1996, many case reports were published of PRES complicating the acute phase of GBS. However, the mechanism underlying PRES and GBS co-occurrence remains unknown. A leading explanation is that hypertension caused by autonomic dysfunction can exceed the limits of cerebral blood vessels in regulating blood flow, leading to vasogenic edema. 23 Chen et al. 24 reviewed the clinical features of all 12 previously reported patients with GBS and PRES co-occurrence. Almost all cases occurred in women over the age of 55 years, raising the possibility of increased sensitivity to dysautonomia in this patient group. Although the study included cases where headache was a prominent clinical feature, our patient with headache had no evidence of PRES on brain magnetic resonance imaging. Elevated intracranial pressure as a result of increased CSF protein concentration, thus slowing reabsorption in arachnoid granulations, may also be considered a potential cause of headache. 23 However, we did not observe elevated CSF protein levels or increased CSF pressure in our patient. Additionally, she had no papilledema, vomiting, or diminished visual acuity. Aseptic meningitis associated with IVIg therapy is a rare phenomenon that occurs within 48 hours of therapy initiation. 25 Its main clinical manifestations include headache, nausea, vomiting, neck stiffness, and photophobia. However, given that our patient had headaches before receiving IVIg therapy, this is not a reasonable explanation in our case.
The mechanism of headache in patients with MFS is uncertain; however, a potential explanation involves antibody-mediated effects on the trigeminovascular pain pathway. Friedman and Potts 26 reported one adult patient with confirmed MFS who presented with severe and persistent headache. The authors hypothesized that this headache might be explained by the presence of autoantibodies against GD3 and GD1b, which tend to be highly expressed in all cranial nerves and sensory cervical roots. Although we tested for anti-GD3 and anti-GD1b antibodies in our case, they were negative. However, Kaida et al. 27 also revealed that although GQ1b and GT1a gangliosides are densely distributed throughout the paranodal myelin of cranial nerves innervating extraocular muscles, they are also present in other cranial nerves and dorsal root ganglia. Thus, in the present case, demyelination of the cervical and cranial sensory nerves by anti-ganglioside antibodies may have activated the trigeminovascular pain pathway, thus causing headache. However, this remains mere speculation; repeated CSF studies, measurements of opening pressure, and integral intracranial imaging will be helpful to further investigate this concept. Future studies are also needed that focus on evaluating the antibody composition of patients with headache in conjunction with MFS, GBS, or overlapping MFS/GBS.
In conclusion, the present case indicates that GBS and MFS can show atypical clinical manifestations, such as bulbar paralysis and/or headache, before classic manifestations. Comprehensive examinations, such as the detection of anti-ganglioside antibodies, should be performed in patients with headaches and bulbar paralysis of unknown origin.
Footnotes
Author contributions
MC and LH made the clinical and electrophysiological diagnosis. MC and ZLZ were involved in the direct management of the patient. MC and LH drafted the first manuscript and ZLZ reviewed all manuscript versions. All authors read and approved the final manuscript.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethics statement
The study was approved by the Institutional Ethical Committee of Tianjin Huanhu Hospital (approval number 2021-054). The patient’s legal guardian provided written informed consent for the treatment. The patient’s details were de-identified, and verbal and written informed consent was obtained from a legally authorized representative for the publication of this case report and any accompanying images.
Funding
This work was funded by Tianjin Key Medical Discipline (Specialty) Construction Project (No. TJYXZDXK-052B).