
Abstract
Objective
To identify key genes in hepatitis C virus (HCV)-induced cirrhosis and to predict effective drugs for its treatment.
Methods
Three datasets were used to screen for differentially expressed genes (DEGs) and differentially methylated genes (DMGs) in HCV-induced cirrhosis. DEGs were subjected to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses using the clusterProfiler R package. Their respective protein–protein interaction (PPI) networks were constructed using Cytoscape. Cross analysis of DEGs and DMGs was performed to identify the genetic landscape of HCV-induced cirrhosis, and five genes were validated by receiver operating characteristic curve analysis. Molecular autodocking between ISG15 and natural products was performed using AutoDock Tool 1.5.6.
Results
A total of 357 DEGs and 8,830 DMGs were identified. DEG functional analysis identified several pathways involved in the pathogenesis of HCV-induced cirrhosis. Cross analysis of DEGs and DMGs identified 212 genes, and PPI network analysis identified 25 hub genes. Finally, five genes including ISG15 were identified and confirmed in dataset GSE36411. Artesunate and betulinic acid were shown to have a strong binding affinity to ISG15.
Conclusion
Our study provides novel insights into the mechanisms of HCV-induced cirrhosis which could lead to the identification of new therapeutics.
Keywords
Introduction
Cirrhosis is a chronic liver disease characterized by esophageal varices, ascites, and high portal pressure following the formation of fibrous septae and nodules; it ultimately leads to liver structure collapse. 1 Cirrhosis usually occurs after liver injury induced by viral infections, alcoholic abuse, immune dysfunction, and biliary disease. 2 Among these etiologies, hepatitis C virus (HCV) infection plays an important role. Although the hepatitis B virus epidemic is becoming controlled in China, 3 it remains a challenge to prevent hepatitis C from progressing to cirrhosis, especially when it is caused by HCV genotype 1. 4 Therefore, it is necessary to better understand the underlying molecular features of HCV-mediated cirrhosis for enhanced and adequate treatment.
DNA methylation is a basic biological process that plays an important role in cellular processes and organ function. 5 DNA hypermethylation is thought to lead to the downregulation or inhibition of gene expression, 6 but this hypothesis has been challenged. 7 Liver fibrosis is usually accompanied by changes in methylation in the entire genome or within particular genes.8–10 However, the mechanisms by which genes are regulated by DNA methylation in cirrhosis remain unclear.
Bioinformatics is an emerging and rapidly developing field that relies on sequencing and information technologies to analyze big data. 11 The present study investigated the combined gene expression and DNA methylation profiles of HCV-mediated cirrhosis to provide novel insights into underlying molecular mechanisms and the relationship between DNA methylation and gene expression. Two gene expression profiles (GSE6764 and GSE14323) and one methylation profile (GSE60753) obtained from the Gene Expression Omnibus (GEO) database were used to identify differentially expressed genes (DEGs) and differentially methylated CpG sites and genes, respectively. Potential associations between the DEGs was investigated by constructing protein–protein interaction (PPI) networks, and the potential pathological role of key genes was further validated with an independent cohort (GSE36411).
Materials and methods
Access of GSE datasets
The terms “HCV” and “cirrhosis” or “liver fibrosis” were used to retrieve valuable datasets from the GEO database with the following inclusion criteria: 1) including at least two groups (control and HCV-mediated cirrhosis), and 2) a sample size >20. Two expression datasets (GSE6764 and GSE14323) were included in the study (Table 1). Ethical approval was not applicable for the present study because no patients were recruited and no animal work was carried out.
Dataset used for analysis.

Identification of DEGs
R packages (www.r-project.org) limma and preprocessCore were used to perform background correction and quantile normalization, respectively. Each probe name was converted into gene symbols according to GPL570 and GPL96 annotation files. The limma R package was further used to identify DEGs between HCV-mediated cirrhosis and control samples. A false discovery rate < 0.05, adjusted p-value < 0.05, and |log2fold change| >1 were considered cutoff values for DEG screening.
Functional enrichment analysis of DEGs
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analyses were performed of the identified DEGs using the clusterProfiler R package. Key pathways or processes with p < 0.05 and a gene number ≥5 were recognized.
Methylation analysis of cirrhosis
The GEO dataset GSE60753 containing DNA methylation profiles of the liver was analyzed using the ChAMP R package. After quality control, two samples (GSM1487212 and GSM1487117) were discarded (Table 1). Thus, 34 control samples and 39 samples from patients with HCV-mediated cirrhosis were included in the analysis.
Differentially methylated probes (DMPs) were defined as cytosine–phosphate–guanine (CpG) sites with |deltaBeta| ≥0.1 and adjusted p < 0.05. Genes displaying at least one DMP were identified as differentially methylated genes (DMGs).
Cross-analysis of DEGs and DMGs
To explore the effect of DNA methylation on gene expression in HCV-mediated cirrhosis, all DEGs with methylation changes were identified and the regions of methylation in these genes were analyzed.
Construction of PPI network
All DEGs were submitted to the STRING database (http://string-db.org) prior to being filtered into the defined PPI network and modified using the Cytoscape platform (v3.6.0). Then, the maximal clique centrality method (MCC) in the plugin cytoHubba was used to identify hub genes, with a score >1 × 1011 considered significant.
Receiver operator characteristic (ROC) curve analysis of methylated hub genes
The whole methylation status of 25 hub genes was analyzed based on previously identified DMPs. The expression of genes with a DNA methylation change was evaluated in the GSE14323 dataset by ROC curve analysis, with p < 0.05 considered statistically significant and the genes considered relevant to HCV-mediated cirrhosis.
Validation of key genes
To further validate the study findings, GEO dataset GSE36411 (containing information on 21 HCV-mediated cirrhosis and 21 control samples) was used to confirm the expression of hub genes. Expression comparisons were performed by a Student’s t-test in GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA), with p < 0.05 considered statistically significant.
Molecular docking
To explore compounds that might target key genes, we downloaded the structure of 31 small molecules targeting liver cirrhosis in pdb format 12 from PubChem (https://pubchem.ncbi.nlm.nih.gov/), as well as the crystal structure of ISG15 (1Z2M) from RCSB PDB (https://www1.rcsb.org/). Molecular docking was performed using Auto Dock Tools 1.5.6 and a rigid docking protocol that used a genetic algorithm to generate binding poses of the protein–ligand complexes. The first binding energy of each complex was recorded.
Results
Identification of DEGs in HCV-cirrhosis
A total of 856 and 273 genes were found to be upregulated and downregulated, respectively, in HCV-mediated cirrhosis samples compared with controls in the GSE6764 dataset, in addition to 737 upregulated and 10 downregulated genes in the GSE14323 dataset (Figures 1 and 2). In total, 357 genes (350 upregulated and 7 downregulated) that were differentially expressed in HCV-mediated cirrhosis samples compared with controls in both datasets were identified as DEGs (Table 2).

Heatmap of DEGs in HCV-mediated cirrhosis. All DEGs identified in (a) GSE6764 and (b) GSE14323 datasets are shown.

Volcano plots and Venn diagrams of DEGs. Each point in the volcano map represents a gene. Red, blue, and gray represent upregulated, downregulated, and non-significantly changed genes, respectively in (a) GSE6764 and (b) GSE14323 datasets.
Differentially expressed genes identified in the two datasets.

Functional enrichment analysis of DEGs
To explore the function of the identified DEGs, GO and KEGG analyses were performed. The top eight significant GO and KEGG pathways are shown in Figure 3. Overall, extracellular matrix structural constituent, extracellular matrix organization, collagen-containing extracellular matrix, and focal adhesion were the most significant functions or pathways identified pertaining to molecular function, biological processes, and cellular component, as well as KEGG terms. Response to virus was also identified as a significant biological process, and influenza A as a significant pathway in KEGG analysis.

Top eight significantly enriched GO terms and KEGG pathways of DEGs in HCV-mediated cirrhosis. (a) Molecular function. (b) Biological process. (c) Cellular component. (d) KEGG pathways. x axis shows the frequency of each term; y axis indicates GO terms or KEGG pathways; colors represent adjusted p-values.
Identification of DMGs in HCV-mediated cirrhosis
A total of 25,125 CpG sites were identified as DMPs and 8830 genes identified as DMGs. Of these, 3831 genes had only one DMP, whereas the others had at least two DMPs.
Cross analysis of DMGs and DEGs identified 212 genes (Figure 4 and Supplementary Table 1), of which 131 genes were upregulated and hypermethylated, 77 were upregulated and hypomethylated, two were downregulated and hypermethylated, and two were downregulated and hypomethylated in HCV-mediated cirrhosis samples compared with controls. Almost half of the DMPs were located in the gene body (12,777), whereas only 4365 CpG methylation events occurred.

DMGs in the GSE60753 dataset. Heatmap of the top 212 DMGs and DEGs.
PPI network construction and hub genes identification
To better understand which of the shared DEGs were most likely to be key genes in the development of HCV-mediated cirrhosis, a PPI network of 357 DEGs was constructed (Supplementary Figure 1). Fifty-four genes were excluded from this network while 303 were included. A total of 303 nodes and 1932 edges were displayed. According to the MCC method in CytoHubba, 25 genes (OASL, OAS1, OAS2, MX1, OAS3, ISG15, IFIT1, TRIM22, MX2, IFI35, IFI6, STAT1, IFIH1, IFI44L, IFI44, BST2, IFI27, CXCL10, HERC6, RTP4, HERC5, DDX60, ISG20, IFI16, and SP110) were deemed hub genes, suggesting that they are likely to be essential for fibrogenesis in patients with HCV (Figure 5).
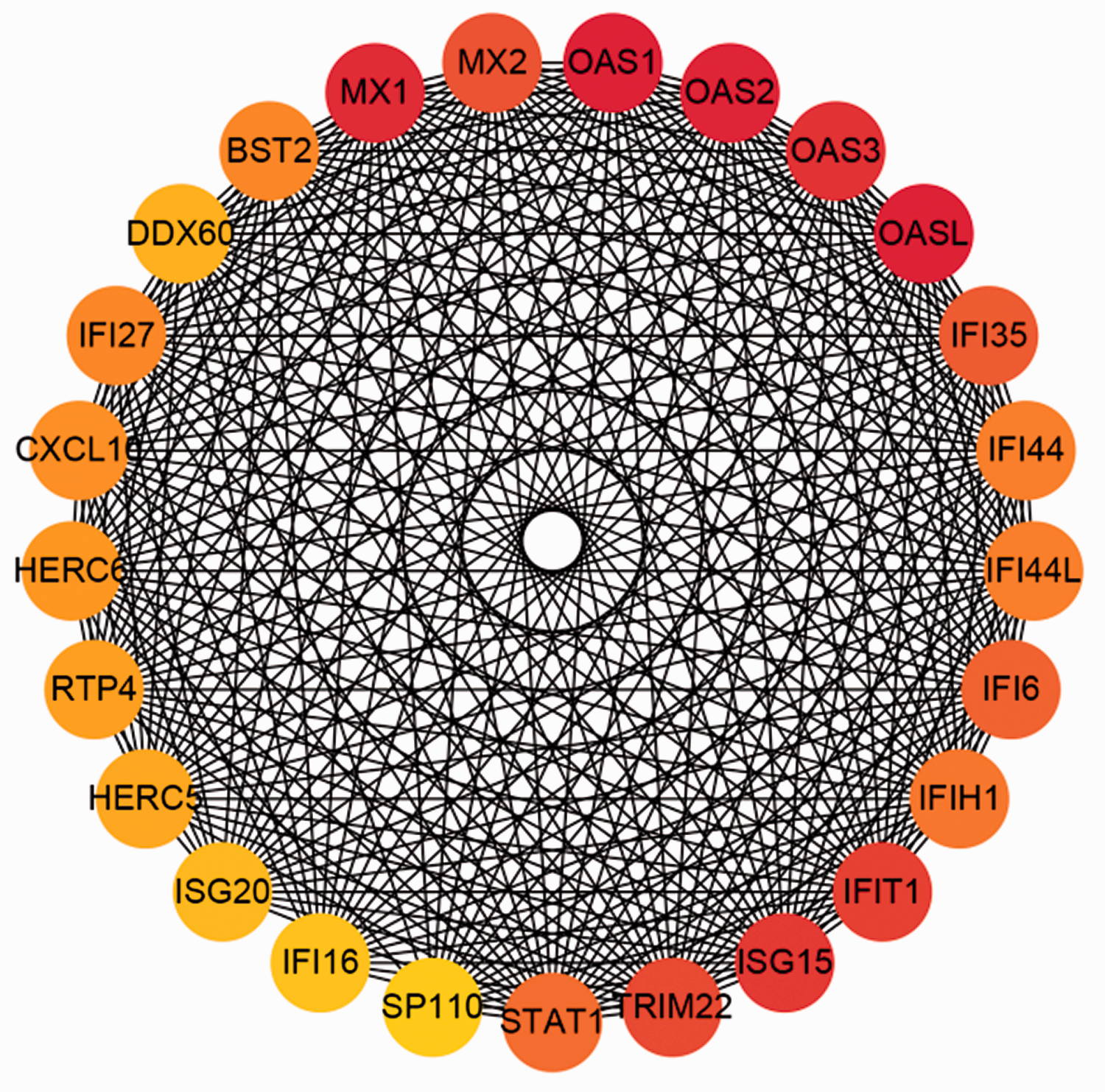
Network of the top 25 hub genes identified in HCV-mediated cirrhosis. A redder color represents a higher score in Cytoscape based on the MCC method.
Methylation status of the hub genes
Next, changes in the methylation status among the 25 hub genes were analyzed. Overall, 11 genes (OASL, OAS3, IFI35, IFI6, IFIH1, IFI44, CXCL10, HERC6, RTP4, HERC5, and SP110) displayed no significant methylation changes, whereas the other 14 hub genes were hypermethylated, showing significant changes in at least one CpG site (Supplementary Table 2).
ROC curve analysis
The performance of the 14 differentially methylated hub genes as diagnostic biomarkers was examined by ROC curve analyses. ISG15 (area under the curve [AUC] = 0.9987 [0.9945–1.000, p < 0.0001]), TRIM22 (AUC = 0.9974 [0.9906–1.000, p < 0.0001]), IFI44L (AUC = 0.9961 [0.9868–1.000, p < 0.0001]), IFI27 (AUC = 1 [1.000–1.000, p < 0.0001]), and IFI16 (AUC = 0.9936 [0.9794–1.000, p < 0.0001]) were identified as potential markers (Figure 6).

ROC curves of ISG15 (a), TRIM22 (b), IFI44L (c), IFI27 (d), and IFI16 (e) for an HCV-mediated cirrhosis diagnosis.
Validation of hub genes
The expression of ISG15, TRIM22, IFI44L, IFI27, and IFI16 was next determined in GSE36411, and that of ISG15, TRIM22, FI27, and IFI16 shown to be significantly higher in patients with HCV-mediated cirrhosis compared with controls (p < 0.001; Figure 7).

Validation of ISG15, TRIM22, IFI44L, IFI27, and IFI16 differential expression in the GSE36411 dataset. **p < 0.01, ***p < 0.001 using a Student’s t-test comparison.
Binding results
Compounds with the highest binding affinities for ISG15 were artesunate (−6.27 kcal/mol) and betulinic acid (−6.23 kcal/mol) (Supplementary Figure 2). Detailed information including numbers of hydrogen bonds formed between ISG15 and small molecules is listed in Table 3.
Binding energy of 31 small molecule drugs showing affinity to ISG15.

Discussion
Treatment approaches to cirrhosis are limited, in part because disease progression is not fully understood. In the present study, cross analysis of HCV-mediated cirrhosis genetic and epigenetic profiles was performed with the aim of improving our understanding of HCV-mediated cirrhosis.
In total, 357 DEGs were identified, including the fibrosis initiation factor transforming growth factor (TGF)-β, PDGFA, and PDGFD, which are critical to the activation of hepatic stellate cells (HSCs).13,14 Moreover, the expression of collagen deposition-related genes, including COL1A1, COL1A2, COL4A1, COL4A2, and COL4A3, was increased in HCV-mediated cirrhosis samples. Other important genes, such as STAT1, and the CXCL family, which is associated with cirrhosis, also showed increased expression.
GO enrichment analysis of DEGs demonstrated that their molecular function was mainly enriched in extracellular matrix structural constituent, glycosaminoglycan binding, and receptor ligand activity, while their biological function was mainly enriched in extracellular matrix organization, extracellular structure organization, response to virus, defense response to virus, and the regulation of cell–cell adhesion. Moreover, cellular components were mainly enriched in collagen-containing extracellular matrix, endoplasmic reticulum lumen, and the external side of the plasma membrane. This suggests that extracellular matrix deposition is a basic pathological change in cirrhosis. The response to virus was also enriched in identified DEGs, and previous research found that the response to HCV causes chronic inflammation, which is a vital activator of myofibroblast transdifferentiation. 15 HCV infection not only destroys hepatocytes, but also induces the expression of TGF-β family members. 16 Therefore, antivirals are the principal therapeutic approach for HCV-mediated cirrhosis. 17 A previous study found that cirrhosis is reversible when its etiological factors are abolished, 18 further indicating the importance of TGF-β in cirrhosis.
KEGG analysis of DEGs in this study revealed that the main pathways involved in HCV-mediated cirrhosis are focal adhesion, influenza A, the extracellular matrix–receptor interaction, protein digestion, and absorption. Focal adhesion kinase, a member of the tyrosine kinase superfamily, was previously found to regulate the activation of HSCs and liver fibrosis. 19 Phosphoinositide 3-kinase (PI3K) is a member of the focal adhesion pathway, and the PI3K/AKT/mammalian target of rapamycin pathway induces autophagy during HSC activation, thereby leading to fibrosis. 20 Furthermore, identified DEGs within the advanced glycation endproducts (AGE)/receptor for AGE signaling pathway, including TGF-β1, BCL2, STAT1, and MMP2, were also associated with cirrhosis. 21 Moreover, VEGFC was found to be beneficial for cirrhotic rats with hepatic encephalopathy, 22 but the detailed mechanisms require further investigations.
DNA methylation is a key event in cellular and molecular biological processes. Although past studies have documented reduced methylation levels in CCL4-induced liver fibrosis, 10 controversial findings regarding the hypermethylation of particular genes that are upregulated in fibrosis models have been reported. 23 For example, longer CCL4 exposure was shown not to significantly alter the DNA methylation status. 24 In turn, the methylation of promoters of several genes, such as P14, P15, P73, and MGMT, was increased in liver disease including cirrhosis. 25
The diversity of the DNA methylation status in cirrhosis is undeniable. Indeed, gene expression might not negatively correlate with methylation levels as they could increase the binding affinity of corresponding transcription factors. 26 Although methylation at CpG islands of transcription start sites (TSSs) represses gene expression, its effect on non-CpG island TSSs is unknown. 27 Our present results indicate that not all DEGs were hyper- or hypomethylated. Of the 25 hub genes, 11 showed no methylation changes and 6 were hypermethylated at only one CpG site, whereas the others were hypermethylated at more than one site. Thus, it is difficult to judge the impact of DNA methylation on the expression of genes that are methylated at different sites/levels. More detailed studies are needed to clarify the underlying mechanisms.
Of the 25 hub genes identified in this study, including OAS, MX, IFI, and HERC family members, OAS1 and MX1 polymorphisms were previously found to be associated with the severity of liver disease in patients co-infected with the human immunodeficiency virus and HCV.28,29 Moreover, MX1 is upregulated in activated HSCs. 30 Thus, it is reasonable to speculate that MX1 promotes HSC activation and is related to the prognosis of HCV-mediated cirrhosis, although this hypothesis should be verified. Moreover, while OAS1 and IFI44 expression was increased in patients with systemic sclerosis-related interstitial lung disease, their function in cirrhosis has not been addressed. Of the five identified biomarkers, IFI27 and ISG15 were previously shown to be significantly increased in patients with non-sustained virological response (SVR) compared with patients with SVR; 31 in contrast, the functions of TRIM22, IFI44L, and IFI16 require further investigation. Although OAS1 and MX1 could be epigenetic biomarkers of HCV-mediated cirrhosis, their function should be further investigated.
The observed binding energy of natural compounds artesunate and betulinic acid for ISG15 indicates their promising anti-fibrotic effects. Notably, both have shown anti-fibrotic effects in rat models of liver fibrosis.32,33 Thus, we speculate that ISG15 is a promising therapeutic target for HCV-induced liver fibrosis.
Conclusion
Herein, DEGs in cirrhosis were identified, along with their related pathways which include focal adhesion, influenza A, and protein digestion and absorption. Furthermore, the effect of DNA methylation on gene expression in cirrhosis was explored, but this warrants further detailed assessment. Last, OAS1 and MX1 were identified as potential targets of HCV-mediated cirrhosis.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605221074525 - Supplemental material for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis
Supplemental material, sj-pdf-1-imr-10.1177_03000605221074525 for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis by Junxiong Cheng, Zhiwei Chen, Guoqing Zuo and Wenfu Cao in Journal of International Medical Research
Supplemental Material
sj-pdf-2-imr-10.1177_03000605221074525 - Supplemental material for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis
Supplemental material, sj-pdf-2-imr-10.1177_03000605221074525 for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis by Junxiong Cheng, Zhiwei Chen, Guoqing Zuo and Wenfu Cao in Journal of International Medical Research
Supplemental Material
sj-pdf-3-imr-10.1177_03000605221074525 - Supplemental material for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis
Supplemental material, sj-pdf-3-imr-10.1177_03000605221074525 for Integrated analysis of differentially expressed genes, differentially methylated genes, and natural compounds in hepatitis C virus-induced cirrhosis by Junxiong Cheng, Zhiwei Chen, Guoqing Zuo and Wenfu Cao in Journal of International Medical Research
Footnotes
Author contributions
Wenfu Cao and Guoqing Zuo conceived and designed the study. Junxiong Cheng and Zhiwei Chen performed the data analysis. Junxiong Cheng wrote the manuscript. All authors were responsible for reviewing data. All authors read and approved the final manuscript.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the National Natural Science Foundation of China (Grant No: 81573860).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.