
Abstract
Objective
Myocardial ischemia/reperfusion (I/R) injury causes various severe heart diseases, including myocardial infarction. This study aimed to determine the therapeutic effect of atractylenolide I (ATR-I), which is an active ingredient isolated from Atractylodes macrocephala, on myocardial I/R injury.
Methods
Male Sprague-Dawley rats were randomly allocated to the five following groups (nine rats/group): control, I/R, and I/R + ATR-I preconditioning (10, 50, and 250 µg). The effects of ATR-I on rats with I/R injury were verified in cardiomyocytes with hypoxia/reoxygenation. Production of reactive oxygen species was determined. The proliferative ability of cardiomyocytes was detected using the bromodeoxyuridine assay. Mitochondrial membrane potential was measured using flow cytometry. Cellular apoptosis was assessed by flow cytometry and the terminal dUTP‐digoxigenin nick end labeling assay.
Results
I/R and hypoxia/reoxygenation injury increased mitochondrial dysfunction and activated caspase-3 and Bax/B cell lymphoma 2 expression in vitro and in vivo. ATR-I pretreatment dose-dependently significantly attenuated myocardial apoptosis and suppressed oxidative stress as reflected by increased mitochondrial DNA copy number and superoxide dismutase activity, and decreased reactive oxygen species and Ca2+ content.
Conclusion
ATR-I protects against I/R injury by protecting mitochondrial function and inhibiting activation of caspase-3.
Keywords
Introduction
Cardiovascular disease is the main cause of death in the global population, and its incidence rate has still been rising in recent years. 1 Acute myocardial infarction is the leading cause of death from cardiovascular disease and has the characteristics of high morbidity and mortality, which seriously threaten public health. 2 Interventional therapy for restoring coronary blood supply to rescue sudden cardiac death is the primary clinical treatment. However, myocardial ischemia/reperfusion (I/R) injury after recanalization of coronary arteries often leads to more severe myocardial injury, myocardial infarction, and abnormal heart rhythm.3,4 The pathogenesis of I/R injury includes accumulation of oxygen-free radicals, Ca2+ overload, and production of inflammatory mediators. Mitochondrial dysfunction results from excessive production of ATP and reactive oxygen species (ROS) during reperfusion, and it further stimulates oxidative damage and apoptosis of cells. 5 Additionally, increasing intracellular Ca2+ concentrations activates various apoptosis-related pathways to induce cell death. 6 Increased intracellular Ca2+ concentrations also causes formation of intracellular inflammasomes, which mediate production of inflammatory factors, such as interleukin-1β and tumor necrosis factor-α, thereby exacerbating I/R injury. 7 Therefore, determining the pathophysiological mechanism of myocardial I/R injury is important for identifying the mechanism of myocardial infarction and searching for a reasonable treatment strategy.
In recent years, sesquiterpene compounds were found to protect against I/R injury. Patchouli, which is a natural tricyclic sesquiterpene extracted from the Chinese herb Pogostemonisherba, alleviates I/R-induced brain injury. 8 Parthenolide, which is a sesquiterpene lactone isolated from the herbal medicine feverfew, attenuates I/R injury. 9 Atractylenolide I (ATR-I) is a sesquiterpene compound and an active ingredient of Atractylodes macrocephala, 10 and it has been shown to possess anti-inflammatory effects.11–13 ATR-I plays an essential role in relief of oxidative damage and pain.11,14 This evidence indicates that ATR-I might alleviate myocardial I/R injury. Although ATR-III, which is another active ingredient of A. macrocephala, participates in regulating acute myocardial infarction, 15 different bioactivity between ATR-I and ATR-III has been reported. 16 Li et al. 16 found that the inhibitory effect of ATR-I on lipopolysaccharide-induced tumor necrosis factor-α and nitric oxide production in macrophages was higher than that with ATR-III. Therefore, ATR-I may be more effective in alleviating myocardial I/R injury than ATR-III. Consequently, the role of ATR-I in I/R injury should be investigated.
In this study, we aimed to examine the protective effect of ATR-I on myocardial tissue, and investigated the underlying mechanism of action in the I/R injury model in vivo and the hypoxia/reoxygenation (H/R) model in vitro.
Materials and methods
Animal experiments
Forty-five Sprague-Dawley male rats (aged 4–6 months, weight: 220–280 g) were acquired from the Experimental Animal Center of Jingjiang People’s Hospital and randomly divided into five groups with nine rats in each group. All rats had free to access food and water and were housed under a 12-hour light/12-hour dark cycle. The I/R injury model was established using methods described in a previous study. 17 Briefly, the rats were fasted initially for 12 hours before the operation and were anesthetized with chloral hydrate (400 mg/kg). The rats were fixed in the supine position and their limbs were connected to a 12-lead electrocardiogram. The trachea was separated from the neck and a ventilator was used to keep the rats breathing smoothly (respiratory frequency was 60 times/minute and the tidal volume was 2 mL/100 g). The pericardium was cut along the mediastinum in the third intercostal space on the left side of the sternum. A suture needle was inserted 2 to 3 mm into the left atrial appendage and a 1.5-mm diameter latex tube was placed under the ligature. The ligature was tightened to induce myocardial ischemia and blocked coronary blood flow. After 30 minutes, an electrocardiogram showed ST elevation, which indicated ischemia. Subsequently, the ligature was loosened, and blood perfusion was performed for 2 hours to generate an I/R injury model. Various parameters, including the arrhythmia score, left ventricular systolic pressure, left ventricular-developed pressure, left ventricular end-diastolic pressure, left ventricular ejection fraction, and left ventricular wall thickness, were measured for all rats in each group. In this study, we attempted to minimize the number of rats used and decrease their suffering. All rats were humanely euthanized with carbon dioxide inhalation. The study was conducted according to the guidelines for the care and use of laboratory animals. All experiments were approved by the Institutional Animal Care and Use Committee of Jingjiang People’s Hospital (approval date: July 2018; approval number: SYXK (SU) 2017-0044).
Drug treatment
A total of 100 mg ATR-I (HPLC grade, Must Bio-technology, Chengdu, China) was dissolved in 1 mL of dimethylsulfoxide (Sigma, St. Louis, MO, USA). In the three drug-treated groups, the rats were intraperitoneally injected with either a low (10 µg), medium (50 µg), or high (250 µg) dose of ATR-I (I/R + ATR-I groups) for 5 days before the onset of occlusion. 18 The rats in the control group and I/R group were injected with an equal volume of dimethylsulfoxide.
Arrhythmia score
The arrhythmia score was determined as follows. A normal electrocardiogram was scored as 0 points. Sinus rhythm abnormality without significant supraventricular and ventricular arrhythmia was scored as 0.5 points. Supraventricular or ventricular premature beats were scored as 1 point. More than 30 supraventricular premature beats were scored as 2 points. Three or more consecutive premature beats with second-degree atrioventricular block were scored as 3 points. Ventricular tachycardia <30 s was scored as 4 points. One or more bursts with accumulated tachycardia >30 s was scored as 5 points. Occurrence of ventricular fibrillation was scored as 6 points. Ventricular fibrillation that lasted longer than 30 s or the rat died during observation was scored as 7 points.19–21
Staining with 2, 3, 5-triphenyltetrazolium chloride
After the rats were euthanized, the hearts of rats were collected to assess the size of infarction. The collected hearts were placed at −20°C for 10 minutes and cut into 2-mm coronal sections. The sections were then immersed into 2% 2, 3, 5-triphenyltetrazolium chloride solution for 25 minutes at 37°C, followed by fixing using 4% formaldehyde. The infarct sizes of hearts were then analyzed by ImageJ software (https://imagej.nih.gov/ij/).
Hematoxylin and eosin staining
After the rats were euthanized, myocardial tissue of rats was resected and fixed by paraformaldehyde and embedded by paraffin. The tissues were then sectioned into 4-µm slices. The slices were stained with hematoxylin and eosin. The stained slices were analyzed under a light microscope (Olympus, Tokyo, Japan).
Establishment of the H/R model in vitro
Rat cardiomyocytes were isolated from newborn Sprague-Dawley rats. Under aseptic conditions, the ventricular muscles were cut into pieces and digested into a single cell suspension using 0.1% trypsin. Cardiomyocytes were collected through centrifugation and cultured in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (Thermo Fisher Scientific) (5% CO2 at 37°C) for 3 days. To establish the H/R model, the synchronized beating cardiomyocytes were cultured in glucose and serum-free Dulbecco’s modified Eagle’s medium at a hypoxic atmosphere (1% O2, 94% N2, and 5% CO2) for 12 hours. The cells were then transferred to a standard complete medium and regular O2 atmosphere (95% air, 5% CO2) for 1 hour.
Enzyme-linked immunosorbent assay
Levels of creatine kinase-MB (CK-MB) (354798; USBiological, MA, Swampscott, USA), creatine kinase (CK) (ab187396; Abcam, Cambridge, UK), cardiac troponin I (cTnI) (ab246529; Abcam), and myoglobin (MB) (ab260068; Abcam) in myocardial tissues and cardiomyocytes with H/R were measured using corresponding enzyme-linked immunosorbent assay kits by following the protocols of the manufacturers.
Real-time reverse transcription-polymerase chain reaction
Total DNA from myocardial tissues and cardiomyocytes was extracted using the PicoPure™ DNA Extraction Kit (Thermo Fisher Scientific). Real-time reverse transcription-polymerase chain reaction conditions were as follows: 95°C for 10 minutes, 40 cycles of 95°C for 15 s, and 60°C for 1 minute. The sequences of specific micro RNA reverse transcription primers (Invitrogen, Carlsbad, CA, USA) were as follows: ND1 forward, 5′-CCC TAA AAC CCG CCA CAT CT-3′ and reverse, 5′-GAG CGA TGG TGA GAG CTA TGG T-3′; β-actin forward, 5′-AAG ACC CCA GCA CAC TTA GCC-3′ and reverse, 5′-TAG CAC AGC CTG GAT AGC AAC-3′. The experiment was conducted on the Agilent Stratagene Mx3000P real-time polymerase chain reaction instrument (Agilent Technologies, Santa Clara, CA, USA). The data were processed using the 2−△△Ct method. 22
Immunohistochemical detection of caspase-3 expression
Myocardial tissue was fixed with 10% formaldehyde and embedded in paraffin. The paraffin sections were immersed in a 0.01 mol/L (pH 6.0) citrate buffer and heated to 100°C for 5 minutes, and this was repeated twice. After cooling naturally at room temperature, the paraffin sections were washed with 0.1 mol/L phosphate-buffered saline (PBS) and blocked with sheep serum. The sections were then incubated overnight with a caspase-3 antibody (9662; CST, Boston, MA, USA). Finally, the sections were subsequently incubated with biotin-labeled antibodies. The proteins were detected at room temperature using a diaminobenzidine immunohistochemistry color development kit (Sangon Biotech, Shanghai, China).
Western blot analysis
Total protein was separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes (Millipore, Boston, MA, USA). The membranes were blocked with 5% nonfat milk and incubated with anti-B cell lymphoma 2 (Bcl-2) antibody (1:1000), anti-Bax antibody (1:1000), anti-cleaved caspase-3 antibody, anti-cleaved caspase-8 antibody (1:1000), anti-cleaved caspase-9 antibody (1:1000), anti-cleaved caspase-12 antibody (1:1000), and anti-glyceraldehyde-3-phosphate dehydrogenase antibody (1:5000) (Abcam). The membranes were then incubated with an immunoglobulin G H and L chain-specific (horseradish peroxidase) antibody (Abcam). The proteins were detected by a film scanner (Microtek, Shanghai, China), and glyceraldehyde-3-phosphate dehydrogenase was used as an internal control. The net optical density was analyzed with a gel image processing system (Image-pro Plus 6.0; Media Cybernetics, Silver Spring, MD, USA).
Determination of ROS
According to the manufacturer’s instructions, ROS production of myocardial tissue and cardiomyocytes was determined using an ROS assay kit (Qcbio S&T Co., Ltd., Shanghai, China). The fluorescence intensity of the fluorescent dye dichloro-dihydro-fluorescein diacetate was detected using a fluorescence spectrophotometer (Thermo Fisher Scientific).
Bromodeoxyuridine proliferation assay
Proliferation of cardiomyocytes was detected by the bromodeoxyuridine (BrdU) cell proliferation detection kit (Cell Signaling Technology, Boston, MA, USA) according to the manufacturer’s instructions. The ability of cell proliferation was analyzed using a fluorescence microscope (Axioplan; Carl Zeiss, Oberkochen, Germany).
Flow cytometry
Mitochondrial membrane potential (MMP, ΔΨm) was measured by flow cytometry. Trypsinized myocardial tissues were resuspended with PBS and treated with 25 µmol/L rhodamine 123 for 30 minutes. The MMP was analyzed by flow cytometry with excitation at 480 nm and emission at 530 nm.
Rat cardiomyocytes of apoptosis were also assessed using flow cytometry. The cardiomyocytes were washed with PBS and fixed in 70% ethanol (30 minutes, 4°C). The cells were then resuspended with PBS containing 50 µg/mL RNase A and 50 µg/mL propidium iodide (Roche, Shanghai, China). The samples were protected from light at 4°C for 30 minutes and then analyzed by flow cytometry with excitation at 488 nm and emission measured at 560 nm.
Terminal dUTP‐digoxigenin nick end labeling assay
Apoptosis of cardiomyocytes and myocardial tissue was determined by using the terminal dUTP‐digoxigenin nick end labeling (TUNEL) apoptosis detection kit (Abbkine Scientific Co., Ltd., Los Angeles, CA, USA) according to the manufacturer’s instructions. The proportion of cellular apoptosis was analyzed using a fluorescence microscope.
Determination of Ca2+ levels and SOD activity
Ca2+ levels were determined using the Calcium Colorimetric Assay Kit and Phosphate Colorimetric Kit (MAK022; Sigma) by following the protocols of the manufacturer. SOD activity was determined using the Superoxide Dismutase Activity Assay Kit (Colorimetric) (ab65354; Abcam) by following the protocols of the manufacturer.
Statistical analysis
All data are presented as mean ± standard deviation. One-way analysis of variance with the least significant difference post hoc test was used to assess statistical significance. Analysis was conducted using IBM SPSS version 22.0 software (IBM Corp., Armonk, NY, USA). A P value < 0.05 was considered as statistically significant.
Results
Protective effect of ATR-I on myocardial injury in rats with I/R
To investigate the effect of ATR-I on myocardial injury in I/R rats, we first established the I/R model, and the rats were pretreated with different concentrations of ATR-I (10, 50, and 250 µg). Staining by 2, 3, 5-triphenyltetrazolium chloride showed that I/R injury increased the infarction size of hearts, while ATR-I dose-dependently decreased the infarction size (all P < 0.05) (Figure 1). A histopathological examination showed that the hearts after I/R injury were seriously damaged with widespread edema, necrosis, and separation of cardiac muscle fibers. Fortunately, pretreatment with ATR-I strongly attenuated the I/R-induced myocardial injury (Figure 1b). Levels of heart injury markers (CK-MB, CK, cTnI, and MB) were significantly reduced in the ATR-I-pretreated I/R groups compared with the I/R group (all P < 0.05) (Figure 1c–f). Furthermore, in the IR + ATR-I groups, the arrhythmia score (all P < 0.05) and left ventricular end-diastolic pressure were significantly lower (P < 0.05 in the 250-µg group), and left ventricular-developed pressure (P < 0.05 in the 250-µg group), left ventricular systolic pressure (all P < 0.05), and left ventricular ejection fraction (all P < 0.05) were significantly higher compared with the I/R group (Figure 1g–l).
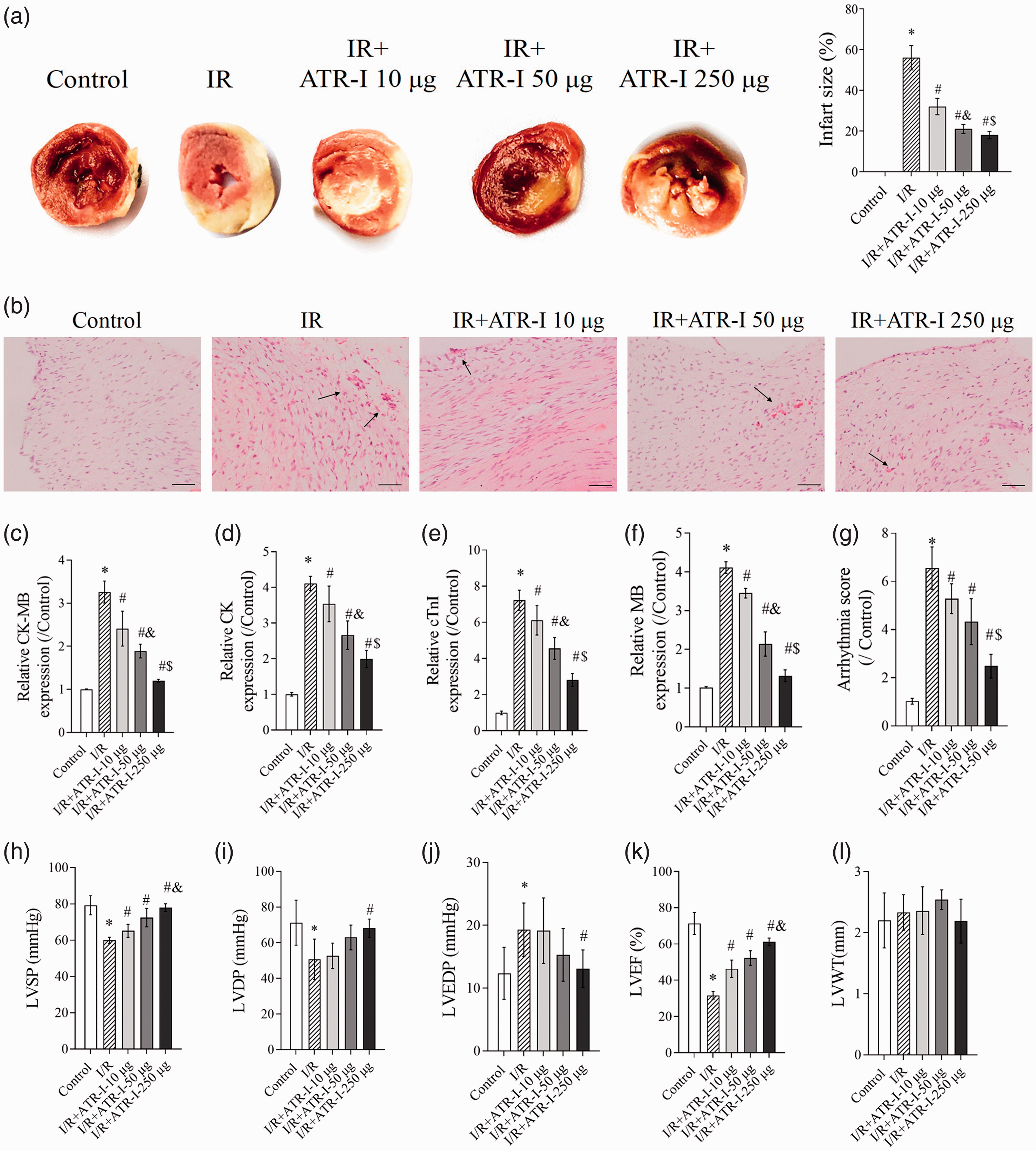
Protective effect of ATR-I on myocardial injury in rats with I/R. Male Sprague-Dawley rats were randomly allocated to the following five groups (nine rats/group): control, I/R, and I/R + ATR-I preconditioning (10, 50, and 250 µg). (a) Staining with 2, 3, 5-triphenyltetrazolium chloride was conducted to determine the infarction size of rat hearts in the different groups. (b) Representative hematoxylin and eosin-stained histological images (× 200 magnification) of myocardial tissue. Scale bars = 50 µm. (c–f) The enzyme-linked immunosorbent assay was used to determine CK-MB, CK, cTnI, and MB expression in myocardial tissue. (g) Arrhythmia score, (h) LVSP, (i) LVDP, (j) LVEDP, (k) LVEF, and (l) LVWT. *P < 0.05 vs the control group; #P < 0.05 vs the I/R group; &P < 0.05 vs the I/R + ATR-I 10-µg group; $P < 0.05 vs the I/R + ATR-I 50-µg group.
ATR-I inhibits I/R-induced apoptosis of cardiomyocytes in vivo
To investigate the effect of ATR-I on myocardial apoptosis in vivo, the apoptosis rate of myocardial tissue was detected by TUNEL assay. Additionally, expression levels of apoptosis marker proteins, such as Bcl-2, Bax, caspase-3, and cleaved caspase-3, were analyzed by immunohistochemistry or western blotting. Apoptosis of myocardial tissues was significantly induced by I/R treatment (P < 0.05 vs the control group). ATR-I dose-dependently decreased the apoptosis rate in myocardial tissue (all P < 0.05 vs the I/R group) (Figure 2a). Additionally, expression of Bax, caspase-3, cleaved caspase-3, and cleaved caspase-9 were enhanced and Bcl-2 expression was reduced in the I/R + ATR-I groups (all P < 0.05 vs the I/R group) (Figure 2b and c). Furthermore, these effects of ATR-I were dose-dependent (Figure 2b and c). However, ATR-I had no significant effects on cleaved caspase-8 and cleaved caspase-12 expression (Figure 2b and c).

ATR-I inhibits I/R-induced apoptosis of cardiomyocytes in vivo. (a) Representative photomicrographs (×200 magnification) of terminal dUTP‐digoxigenin nick end labeling staining in myocardial tissues. Scale bars = 50 µm. (b) Immunohistochemical detection of caspase-3 protein. (c) Levels of Bcl-2, Bax, cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, and cleaved caspase-12 in myocardial tissue were analyzed by western blot analysis. GAPDH was a loading control. *P < 0.05 vs the control group; #P < 0.05 vs the I/R group; &P < 0.05 vs the I/R + ATR-I 10-µg group; $P < 0.05 vs the I/R + ATR-I 50-µg group.
ATR-I protects mitochondrial function of cardiomyocytes in vivo
Several studies have suggested that mitochondrial dysfunction plays a critical role in regulating apoptosis of rat cardiomyocytes.23–25 Therefore, we further identified the function of ATR-I on mitochondrial function of myocardial tissue. ATR-I pretreatment dose-dependently reversed the I/R-induced decrease in DNA replication in mitochondria (all P < 0.05 vs the I/R group) (Figure 3a). Additionally, the decrease in MMP induced by I/R was strongly abrogated by ATR-I pretreatment (Figure 3b). Pretreatment with ATR-I resulted in significantly lower cellular ROS and Ca2+ levels, and higher SOD activity compared with the I/R group (P < 0.05 for all I/R + ATR-I groups) (Figure 3c–e). These results indicated that ATR-I protected mitochondrial function during I/R injury.

ATR-I protects mitochondrial function of cardiomyocytes in vivo. (a) The rate of DNA replication in mitochondria was detected by real-time reverse transcription-polymerase chain reaction analysis. (b) The mitochondrial membrane potential was assayed by flow cytometry. (c) ROS activity was performed using dichloro-dihydro-fluorescein diacetate. (d) Ca2+ concentrations in myocardial tissue. (e) SOD activity. *P < 0.05 vs the control group; #P < 0.05 vs the I/R group; &P < 0.05 vs the I/R + ATR-I 10-µg group; $P < 0.05 vs the I/R + ATR-I 50-µg group.
ATR-I protects cardiomyocytes against injury induced by H/R in vitro
To determine whether ATR-I protects cardiomyocytes against H/R-induced injury, injury of neonatal rat cardiomyocytes was assessed by assaying levels of CK-MB, CK, cTnI, and MB. We found that expression of CK-MB (Figure 4a), CK (Figure 4b), cTnI (Figure 4c), and MB (Figure 4d) was enhanced in H/R-induced rat cardiomyocytes (all P < 0.05 vs controls). As expected, pretreatment with ATR-I counteracted the induction effect of H/R on CK-MB, CK, cTnI, and MB expression (P < 0.05 for all H/R + ATR-I groups vs the H/R group) (Figure 4a–d).

ATR-I protects cardiomyocytes against injury induced by H/R in vitro. Rat cardiomyocytes were divided into the following five groups: control, H/R, and H/R + ATR-I preconditioning (10, 50, and 250 µg). Enzyme-linked immunosorbent assays were performed to determine the levels of CK-MB (a), CK (b), cTnI (c), and MB (d) expression in cardiomyocytes with H/R. *P < 0.05 vs the control group; #P < 0.05 vs the H/R group; &P < 0.05 vs the H/R + ATR-I 10-µg group; $P < 0.05 vs the H/R + ATR-I 50-µg group.
ATR-I reduces H/R-induced apoptosis of rat cardiomyocytes in vitro
The BrdU assay showed that H/R treatment inhibited proliferation of cardiomyocytes (P < 0.05 vs controls), which was reversed by ATR-I preconditioning (P < 0.05 for all H/R + ATR-I groups vs the H/R group) (Figure 5a). The TUNEL assay and flow cytometry showed that H/R treatment resulted in a significantly higher amount of apoptosis of cardiomyocytes compared with controls (P < 0.05) (Figure 5b and c). In contrast, ATR-I reduced apoptosis in H/R-stimulated cardiomyocytes in a dose-dependent manner (P < 0.05 for all H/R + ATR-I groups vs the H/R group). Moreover, H/R treatment enhanced expression levels of Bax, caspase-3, cleaved caspase-3, leaved caspase-8, cleaved caspase-9, and cleaved caspase-12, and reduced Bcl-2 expression levels in cardiomyocytes (all P < 0.05 vs controls) (Figure 5d). ATR-I pretreatment reduced Bax, caspase-3, cleaved caspase-3, and cleaved caspase-9 expression, and enhanced Bcl-2 expression in cardiomyocytes (P < 0.05 for all H/R + ATR-I groups vs the H/R group) (Figure 5d). However, ATR-I had no significant effects on cleaved caspase-8 and cleaved caspase-12 expression (Figure 5d).

ATR-I reduces H/R-induced apoptosis of rat cardiomyocytes in vitro. Representative photomicrographs (×200 magnification) of BrdU (a) and terminal dUTP‐digoxigenin nick end labeling (b) staining in cardiomyocytes with H/R. Scale bars = 25 µm. (c) Cellular apoptosis of cardiomyocytes with H/R was detected by flow cytometry. (d) Expression levels of Bcl-2, Bax, caspase-3, cleaved caspase-3, cleaved caspase-8, cleaved caspase-9, and cleaved caspase-12 in cardiomyocytes with H/R were analyzed by western blotting. GAPDH was a loading control *P < 0.05 vs the control group; #P < 0.05 vs the H/R group; &P < 0.05 vs the H/R + ATR-I 10-µg group; $P < 0.05 vs the H/R + ATR-I 50-µg group.
ATR-I reduces H/R-induced mitochondrial dysfunction in rat cardiomyocytes
H/R decreased DNA replication of rat cardiomyocytes (P < 0.05 vs controls) (Figure 6a), which was reversed by ATR-I pretreatment (P < 0.05 for all H/R + ATR-I groups vs the H/R group). Additionally, H/R activated cellular ROS and Ca2+ accumulation (both P < 0.05 vs controls), and ATR-I dose-dependently reduced the ROS accumulation and cellular Ca2+ concentrations (P < 0.05 for all H/R + ATR-I groups vs the H/R group) (Figure 6b and c). H/R reduced SOD activity in cardiomyocytes (P < 0.05 vs controls), which was abolished by ATR-I preconditioning (P < 0.05 for all H/R + ATR-I groups vs the H/R group) (Figure 6d).

ATR-I reduces H/R-induced mitochondrial dysfunction in rat cardiomyocytes. (a) The rate of DNA replication in mitochondria was detected by real-time polymerase chain reaction analysis. (b) ROS activity was performed using dichloro-dihydro-fluorescein diacetate. (c) Ca2+ concentrations in rat cardiomyocytes. (d) SOD activity. *P < 0.05 vs the control group; #P < 0.05 vs the H/R group; &P < 0.05 vs the H/R + ATR-I 10-µg group; $P < 0.05 vs the H/R + ATR-I 50-µg group.
Discussion
Myocardial I/R injury induces mitochondrial dysfunction, including production of ROS and accumulation of Ca2+, during reperfusion. 26 This then activates the caspase-3 dependent apoptotic pathway by releasing pro-apoptotic factors into the cytoplasm. 27 In the present study, ATR-I pretreatment reduced the size of myocardial infarction, improved pathological changes of myocardial tissues, and suppressed myocardial apoptosis via inactivation of the caspase-3 and caspase-9 signaling pathways in the I/R injury model. Additionally, ATR-I protected myocardial tissue from mitochondrial dysfunction, including mitochondrial DNA copy number, production of ROS, accumulation of Ca2+, and SOD concentrations. Furthermore, these protective effects of ATR-I in the I/R model were observed in an H/R model in vitro.
A variety of natural products, such as curcumin, quercetin, and coptisine, exert protective effects on myocardial I/R injury. Curcumin effectively protects against myocardial I/R injury by inducing expression of the reperfusion injury salvage kinase/glycogen synthase kinase-3β pathway and inhibiting activity of the p38/c-Jun NH2-terminal kinase pathway. 28 Additionally, quercetin inhibits apoptosis of myocardial I/R injury through modulating the phosphatidylinositol-3-kinase /Akt pathway. 29 Furthermore, coptisine alleviates myocardial I/R injury by inhibiting the Rho/Rho-associated protein kinase pathway. 30 Similarly, we found that ATR-I, which is an active ingredient of A. macrocephala, exerted protective effects on myocardial I/R injury by suppressing myocardial apoptosis with protection of mitochondrial function and inhibition of the caspase-3 and caspase-9 pathways. Previous research showed that ATR-I had a dual role in promoting and inhibiting apoptosis of cells. ATR-I induces human leukemia cellular apoptosis through activation of caspase-3 and caspase-9. 31 In ovarian cancer cells, ATR-I promotes cell cycle arrest in the G2/M phase and triggers cellular apoptosis by inducing the mitochondrial apoptotic pathway. 32 Furthermore, ATR-I induces apoptosis and cell cycle arrest in melanoma cells by extracellular signal‐regulated kinase/glycogen synthase kinase-3β signaling. 33 However, ATR-I attenuates 1-methyl-4-phenylpyridinium +-induced cell death by inhibiting Bax/Bcl-2 mRNA levels in SH-SY5Y cells. 34
Existing evidence suggests that mitochondrial dysfunction and mitochondrial DNA damage aggravate myocardial I/R injury through inducing inflammation, oxidative stress, and cellular apoptosis.35,36 After ischemic reperfusion, excessive production of ROS and intracellular accumulation of Ca2+ occur in myocardial tissue with I/R and neonatal rat cardiomyocytes with H/R.37,38 Consistent with these previous findings, we found that myocardial I/R injury blocked mitochondrial DNA replication and inactivated antioxidant activity. Moreover, ATR-I pretreatment dose-dependently restored mitochondrial function, including an increase in mitochondrial DNA copy number, a decrease in ROS/Ca2+ content, and activation of SOD.
Caspase-3 pathway activation plays a critical role in myocardial cellular apoptosis. 39 Bax, as a proapoptotic protein, induces permeability of the mitochondrial outer membrane, resulting in release of pro-apoptotic factors into the cytoplasm. Conversely, Bcl-2, which is an anti-apoptotic protein, reduces mitochondrial injury and cellular apoptosis. Our study showed that ATR-I abrogated the I/R-induced caspase-3 and cleaved caspase-3 expression. Additionally, ATR-I inhibited apoptosis by regulating the Bcl-2 family (Bax and Bcl-2), which is associated with mitochondrial physiology and cellular apoptosis. 40 We also found that ATR-I inhibited activation of caspase-9 in myocardial tissue and cardiomyocytes with I/R. Caspase-9 is the initiator caspase associated with the mitochondrial pathway of apoptosis. 41 Activation of caspase-9 plays a vital role in myocardial I/R injury.42–44 A study conducted by Sodhi et al. 42 reported that administration of a caspase-9 inhibitor attenuated myocardial tissue damage and apoptosis induced by I/R injury. Zhu et al. 43 found that salidroside suppressed myocardial apoptosis by inhibiting activation of caspase-9 during I/R injury. Zhao et al. 44 showed that fenofibrate alleviated rat acute myocardial I/R injury by suppressing mitochondrial apoptosis as shown by suppression of caspase-9 activation. In our study, ATR-I had no effect on activation of caspase-8 and caspase-12. However, previous studies showed that caspase-8 and caspase-12 were involved in regulation of myocardial I/R injury.45–47 In one study, miR-214 alleviated myocardial apoptosis and caspase-8 activity induced by I/R injury. 45 In another study, lipoxinA4 alleviated rat myocardial I/R injury by a mechanism related to downregulation of caspase-12. 46 Additionally, erythropoietin protects against myocardial infarction by suppressing caspase-12. 47 Caspase-8 is considered as an integral initiator and regulator of death receptor-mediated activation of apoptosis. 48 Caspase-12 is a key protein of endoplasmic reticulum stress-mediated apoptosis. 49 Therefore, based on the above-mentioned evidence, we consider that ATR-I might inhibit myocardial cellular apoptosis through the mitochondrial caspase 9 pathway. A previous study showed that ATR-I effectively induced lung carcinoma cellular apoptosis by upregulation of caspase-3, caspase-9, and Bax, and downregulation of Bcl-2 and Bcl-XL. 50 These results are inconsistent with the results in the current study. In this study, ATR-I inhibited cellular apoptosis by inhibiting caspase-3, caspase-9, and Bax, and increasing Bcl-2 expression. Similarly, a previous study showed that ATR-I attenuated 1-methyl-4-phenylpyridinium-induced cytotoxicity by suppressing caspase-3. 34 ATR-I mainly induces apoptosis in cancer cells, including cancers such as leukemia, 31 ovarian cancer, 32 and melanoma. 33 These conflicting results indicate that ATR-I has a dual role in regulating cellular apoptosis. This regulation might be caused by different pathological mechanisms or different upstream signaling regulation mechanisms of apoptotic pathways between different diseases.
There are some limitations in our study. First, the in-depth mechanism of protective effects of ATR-I on myocardial I/R injury by inactivating the caspase-3 signaling pathway was not investigated in this study. This issue will be explored in future studies. Second, the findings in our study were obtained from preclinical experiments in the rat model and cell model, which might be different from findings in human samples.
Taken together, our findings suggest that ATR-I pretreatment reduces the size of myocardial infarction, improves pathological changes of myocardial morphology, and suppresses myocardial apoptosis by inactivating the caspase-3 signaling pathway in vivo and in vitro. Furthermore, ATR-I protects myocardial tissue from mitochondrial dysfunction. ATR-I has powerful anti-apoptotic properties resulting in prevention of myocardial I/R injury and improvement of survival of cardiomyocytes.
Supplemental Material
sj-jpg-1-imr-10.1177_0300060521993315 - Supplemental material for Atractylenolide I alleviates ischemia/reperfusion injury by preserving mitochondrial function and inhibiting caspase-3 activity
Supplemental material, sj-jpg-1-imr-10.1177_0300060521993315 for Atractylenolide I alleviates ischemia/reperfusion injury by preserving mitochondrial function and inhibiting caspase-3 activity by Caiqin Sun, Xuesong Zhang, Fei Yu, Chen Liu, Fangbin Hu, Li Liu, Jing Chen and Jue Wang in Journal of International Medical Research
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.