
Abstract
Objective
To evaluate the performance of a DNA methylation-based digital droplet polymerase chain reaction (ddPCR) assay to detect aberrant DNA methylation in cell-free DNA (cfDNA) and to determine its application in the detection of hepatocellular carcinoma (HCC).
Methods
The present study recruited patients with liver-related diseases and healthy control subjects. Blood samples were used for the extraction of cfDNA, which was then bisulfite converted and the extent of DNA methylation quantified using a ddPCR platform.
Results
A total of 97 patients with HCC, 80 healthy control subjects and 46 patients with chronic hepatitis B/C virus infection were enrolled in the study. The level of cfDNA in the HCC group was significantly higher than that in the healthy control group. For the detection of HCC, based on a cut-off value of 15.7% for the cfDNA methylation ratio, the sensitivity and specificity were 78.57% and 89.38%, respectively. The diagnostic accuracy was 85.27%, the positive predictive value was 81.91% and the negative predictive value was 87.20%. The positive likelihood ratio of 15.7% in HCC diagnosis was 7.40, while the negative likelihood ratio was 0.24.
Conclusions
A sensitive methylation-based assay might serve as a liquid biopsy test for diagnosing HCC.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common tumour type and the third most common cause of cancer-related deaths worldwide according to the World Health Organization report in 2020. 1 In China, HCC is the third most common cause of cancer-related deaths in male and female patients. 2 The majority of patients are diagnosed and treated at the late stages of HCC, which leads to poor prognoses. Although abdominal ultrasonography and serum alpha-fetoprotein (AFP) measurement is widely accepted, the sensitivity and specificity of existing tumour biomarker tests are relatively low when screening for HCC.3,4 In recent years, several protein biomarkers, gene mutations and epigenetic modifications have been analysed for the molecular diagnosis of HCC.5,6 As a non-invasive liquid biopsy analyte, circulating cell-free DNA (cfDNA) has gained considerable attention for the diagnosis of various cancer diseases.7,8 Circulating nucleic acid is derived from apoptotic cells, and typically, cfDNA is lower in healthy individuals. 9 cfDNA, released from tumour cells, is termed circulating tumour DNA (ctDNA). 10 It exhibits primary tumoral heterogeneity, which is a potential biomarker for cancer diagnosis, mostly detectable methylated ctDNA at the beginning of tumorigenesis.10,11 Specifically, the detection of ctDNA, genetic material from cancer cells naturally found in the bloodstream, has also been utilized for cancer detection and disease monitoring. 12 As a new biomarker, obtained by analysing cancer-associated gene mutations, 13 aberrant DNA methylation, 14 copy number variation, 15 microsatellite instability, 16 and other epigenetic alternations in ctDNA, bioinformative ctDNA in peripheral circulation implies dynamic changes in the tumour in cancer patients. These alterations not only indicate potential responses to treatment but also predict the prognosis of patients in many cancers.17–19
DNA methylation is a crucial regulator of epigenetic modification in cell physiology. 20 It has been found to be associated with the occurrence and development of tumours by silencing tumour suppressor gene promoter regions and activating oncogene expression. 21 Owing to the tissue specificity, the DNA methylation signature is considered as the fingerprint of the tumour, with characteristics of primary tumorigenesis. 22 These modifications in a single cell precede tumour formation, which could help identify tumorigenesis and manage cancer patients. 23 Recent studies showed that the ctDNA methylation pattern occurring early in carcinogenesis is a novel robust biomarker that improves the clinical utilization of liquid biopsy in cancer diagnosis.24,25 It has been demonstrated that specific DNA methylation patterns are reliable for early diagnosis, surveillance and prognosis of HCC. 26 Currently, digital droplet polymerase chain reaction (ddPCR) is utilized for the quantitation of DNA mutations. However, the DNA methylation pattern in the circulating DNA of HCC remains unclear and a limited number of methylations are arranged on the ddPCR platform.27–30 In contrast, although many methylated genes have been shown to play a major role in HCC, 6 a specific indicator has not been confirmed in HCC diagnosis.
The epigenetic alternation plays a significant role in a variety of disease processes, including cancer and other common diseases. 14 DNA methylation regulates the expression of tumour suppressor genes. 31 An increased number of methylated tumour suppressor genes are detected at the beginning of tumorigenesis events, which makes monitoring cancer based on DNA methylation patterns possible. 32 Currently, there are several strategies for the testing and validation of DNA methylation biomarkers. The majority of these approaches are based on quantitative real-time polymerase chain reaction (qPCR) and bisulfite genomic sequencing (BSP). The qPCR-based technique for methylation analysis identifies the methylation status by relative calibration curve analysis based on that generated by a reference for the standard.33–35 BSP is used to read uracil from cytosine directly after bisulfite conversion, rendering it as an important method in cfDNA analysis. 36 It provides robust and comprehensive genetic information on cancer monitoring. 37 Sensitive technologies, such as digital PCR, 38 droplet digital PCR (ddPCR), 39 an BEAMing, 40 are required for ctDNA analysis. These PCR-based and NGS analytical methodologies are complicated, whereas ddPCR can be used to evaluate the methylation status by directly counting the number of methylated and unmethylated copies. 28
This current study validated the potential of a non-invasive DNA methylation test to detect the presence of cancer and provide quantitative data using plasma samples via the ddPCR platform.
Patients and methods
Patient samples
The present study recruited consecutive patients with liver cancer and liver-related chronic diseases and healthy control subjects in the Department of Clinical Laboratory Medicine, Xijing Hospital, Air Force Medical University, Xi'an, Shaanxi Province, China, between March 2018 and May 2019. The eligibility criteria for HCC patients were as follows: (i) age >18 years; (ii) without other malignant tumours; (iii) with liver function test results; (iv) with pathological or imaging evidence of HCC or non-HCC or diagnosis of HCC based on the diagnostic criteria from the ‘Evidence-based practice guidelines for the standardized pathological diagnosis guidelines of primary liver cancer in 2015’. 41 Patients that presented with HCC of Barcelona-Clinic Liver Cancer (BCLC) stage A–D were defined in this study. The HCC stage was categorized by the criteria of the BCLC staging system. 42 Patients with chronic hepatitis were confirmed by clinicians. A group of healthy volunteers was enrolled as negative controls in addition to those without liver diseases and their serum AFP level was <7 ng/ml. Healthy individuals were local residents that came to Xijing Hospital for routine physical screening. All participants were selected for routine physical body examination, liver-related function testing and imagining screening. Patients with chronic hepatitis with an abnormal AFP level and long-term hepatitis virus infection, as well as HCC patients with BCLC A and B, without clinical liver-related symptoms, were enrolled for the evaluation of diagnostic efficiency.
The study was approved by the Ethics Committee, the First Affiliated Hospital (Xijing Hospital) of the Air Force Military Medical University (no. KY20182078-C-1). All study participants provided written informed consent.
Sample preparation
A 5-ml sample of peripheral whole blood was collected from each participant by venipuncture. The blood samples were stored in K2-ethylenediaminetetra-acetic acid tubes, followed by centrifugation at 2000
Quantification of cfDNA by qPCR
The amount of cfDNA in the plasma samples was quantified using qPCR. The initial cfDNA sample template was used at a dilution of 1:100. A volume of 7.5 μl reaction buffer was mixed with 2.5 μl diluted plasma sample and amplified in a 96-well optical reaction plate on the CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA). The reaction buffer contained: 2.8 µl 5× HemoKlenTaq Reaction Buffer (New England Biolabs, Ipswich, MA, USA); 0.2 µl Deoxynucleotide (dNTP) Solution Mix (New England Biolabs); 0.1 µl SYBR™Green I Nucleic Acid Gel Stain (Thermo Fisher Scientific, Waltham, MA, USA); 0.4 µl primers (Guangzhou Youze Biological Pharmaceutical Technology Company Ltd., Guangzhou, China) and 4.0 µl DNase-free H2O. The PCR protocol was as follows: preliminary denaturation at 95°C for 3 min, followed by 35 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and elongation at 65°C for 5 s, followed by a final elongation step at 95°C for 10 min. Standard samples were prepared using a Qubit™ dsDNA HS Assay Kit (Thermo Fisher Scientific), using 1 ng/ml cfDNA as the initial concentration and five serial dilutions for the standard curve. The relative cfDNA concentration of the sample was further calculated by Cq value, slopes and y-intercepts derived from the calibration curves of qPCR. The primers for the qPCR reaction were selected and designed as described previously. 43
cfDNA extraction and measurement
The cfDNA was extracted from the plasma using the EliteHealth cfDNA extraction kit (EliteHealth, Guangzhou Youze, China) following the manufacturer’s instructions. Briefly, 1.2–1.5 ml plasma was mixed with 0.2 ml proteinase K. Then, 1.6 ml of buffer ACL was added and the reaction incubated at 60°C for 30 min, followed by the addition of 3.2 ml of buffer ACB and incubation on ice for 5 min. Subsequently, the mixture was filtered through the column. The bound cfDNA was consecutively washed with 600 µl of buffer DCW1, 600 µl of buffer DCW2 and 600 µl of ethanol, with centrifugation at 13000
Bisulfite conversion
The extracted cfDNA was treated with bisulfite using a cfDNA methylation kit (EliteHealth, Guangzhou Youze, China) according to the manufacturer’s instructions. Briefly, 130 μl of bisulfite reagent was mixed with 19 μl of purified cfDNA and incubated at 98°C for 8 min, 54°C for 60 min and then at 4°C for 20 h. Then, 600 μl M-binding buffer was mixed with a hybridized solution, followed by centrifugation at 12000
ddPCR analysis
All ddPCR analyses were performed using the QX100 Droplet Digital PCR System according to the manufacturer’s instructions (Bio-Rad). Bisulfide cfDNA subsequently determined the methylation ratio using the markers provided by the Guangzhou Youze Biological Pharmaceutical Technology Company Ltd. (Guangzhou, China). The assay was designed to measure the methylation status at target sites within specific genes of cfDNA extracted from blood samples by using a ddPCR platform. These target sites are hypermethylated in specific cancers. 19 The target region was amplified by the primer pair, cg23612220-Forward: 5′-GTAATGGTGGTAGAGGAAT, cg23612220-Reverse: 5′-AAAACTAAACTAAACTCTACAAAAA; fluorescent probe for methylated allele detection, cg23612220-M 5'/6-FAM/TGTGAAATTTTCGTTTGTATAATTTTTGG/BHQ1/-3'; probe for unmethylated allele detection, cg23612220-NM5'/HEX/TGTGAAATTTTTGTTTGTATAATTTTTGGG/BHQ1/-3'. An equivalent of 10 ng bisulfite cfDNA was utilized for ddPCR, which amplified the target region under the following conditions: 95°C for 10 min, followed by 45 cycles at 94°C for 30 s and 54°C for 60 s, 98°C for 10 min, and maintained at 12°C. Subsequently, the droplet plate was read using the QuantaSoft™ Analysis Pro 1.0.596 software (Bio-Rad) and the data analysed. The test results were reported as a quantitative methylation percentage, which indicated the methylation status of the target sites. For each sample, ddPCR analytic data were presented as copies in a 20 μl reaction system. The total copies were calculated as the methylation copies plus non-methylation copies. The cfDNA methylation ratio was calculated as the methylation copies/(methylation copies +unmethylation copies).
Statistical analyses
All statistical analyses were performed using IBM SPSS Statistics for Windows, Version 22.0 (IBM Corp., Armonk, NY, USA). The correlation analysis of relative and absolute cfDNA quantitation methods between qPCR and DNA fluorometric quantitation was undertaken using Spearman’s correlation coefficient. Mann–Whitney U-test was used to compare the HCC patients with or without liver-related clinical symptoms. Student’s t-test was used to compare the mean value differences of cfDNA amount between the tumour, chronic hepatitis and healthy control groups. The methylation ratio of each sample was calculated. Kruskal–Wallis test and post-hoc Dunn test were performed to assess the variations in cfDNA amount, methylation copies and methylation rates in the BCLC subgroups. Youden index was generated from the area under the receiver operating characteristic (ROC) curve (AUC) in order to determine the optimal cut-off value of cfDNA methylation patterns. The figures were generated using GraphPad Prism for Windows 8.0.1 (GraphPad Software Inc., San Diego, CA, USA). A P-value < 0.05 was considered statistically significant. Venn diagrams were generated using Venny 2.1 (Oliveros, J.C. (2007-2015) Venny) An interactive tool for comparing lists with Venn's diagrams. 44
Results
Optimized qPCR was performed to quantify the cfDNA concentration at the beginning of the study. Of the 534 participants enrolled for the relative cfDNA quantitation, 97 were HCC patients, 46 were patients with chronic hepatitis B/C virus infection and 80 were healthy control subjects, with well-integrated clinical information and reliable statistics for further methylation analysis (Figure 1). The remaining 311 participants were excluded from the study as they met the exclusion criteria. The cfDNA quantification results showed optimized qPCR as assessed by Qubit™ (Figure 2a; r2 = 0.8389, P < 0.0001). The Cq values generated from the relative qPCR test represented the cfDNA amount in the study population (Figure 2b).
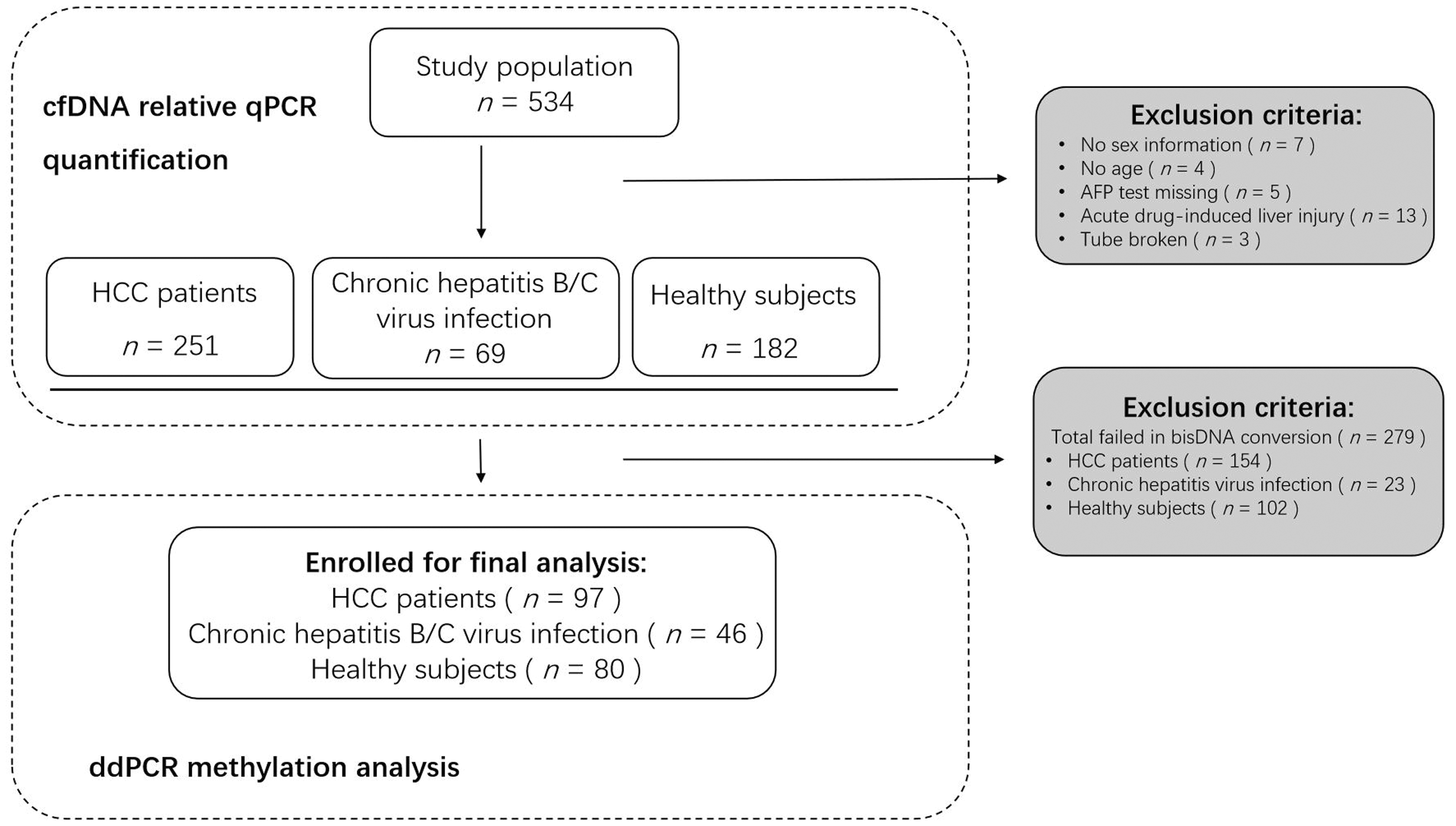
Workflow showing sample selection and inclusion in a study designed to determine the potential of a non-invasive DNA methylation test to detect the presence of cancer and provide quantitative data using plasma samples via the ddPCR platform. HCC, hepatocellular carcinoma; AFP, alpha-fetoprotein; cfDNA, cell-free DNA; qPCR, quantitative real-time polymerase chain reaction; ddPCR, digital droplet polymerase chain reaction.

Correlation analysis of cell-free DNA (cfDNA) quantification. (a) Relative (optimized quantitative real-time polymerase chain reaction [qPCR]) and absolute (Qubit™) calculation; (b) Relative cfDNA quantification and its Cq value in healthy individualized by optimized qPCR.
The mean ± SD plasma cfDNA level in the healthy control group was 14.22 ± 8.09 ng/ml compared with 23.46 ± 16.18 ng/ml in the HCC group and 13.78 ± 10.70 ng/ml in the group with chronic hepatitis B/C virus infection (Figure 3). The plasma cfDNA level in the HCC group was significantly higher than that in the group with chronic hepatitis B/C virus infection and the healthy control subjects (P < 0.0001 for both comparisons). There was no significant difference in the cfDNA level between the healthy control subjects and the group with chronic hepatitis B/C virus infection.

Cell-free DNA (cfDNA) analysis in all study participants (n = 502). Data presented as mean ± SD. Between-group comparisons undertaken using Student’s t-test. The central black horizontal line in each group is the median; the upper extremity of the box is the 25th percentile and the lower extremity of the box is the 75th percentile; the error bars represent minimum and maximum outliers; and the circles above and below the error bars represent extreme outliers. Control, healthy subjects; CH, patients with chronic hepatitis B/C virus infection; HCC, patients with hepatocellular carcinoma.
Demographic, clinical and cfDNA data of the 223 study participants are presented in Table 1. The cfDNA methylation ratio reflected the percentage of methylated alleles of CpG. The HCC subgroup analysis in which patients were stratified according to the BCLC staging system classification demonstrated significantly higher cfDNA levels and methylation ratio in patients with clinical symptoms and a heavy tumour burden (the symptomatic groups BCLC C and BCLC D) compared with the asymptomatic groups (groups BCLC A and BCLC B) (P < 0.05 for both comparisons) (Table 2). When considering the methylation patterns, the asymptomatic patients (groups BCLC A and BCLC B) carried a low number of cfDNA fragments and presented with a low methylation status (Figure 4a). Both the methylated copies (Figure 4b) and methylation ratio (Figure 4c) in HCC patients with BCLC C and BCLC D were significantly higher than in those patients with BCLC A and BCLC B (P < 0.001 for all comparisons). The methylation ratio was significantly lower in the healthy control subjects compared with each of the four subgroups of HCC patients (P < 0.05 for all comparisons) (Figure 4c). Methylated copies (Figure 4b) and total methylated copies (Figure 4d) were significantly higher in patients with late-stage BCLC (C and D) compared with the healthy control subjects (P < 0.05 for all comparisons).
Demographic and clinical characteristics of all study participants (n = 223).

Data represented as median (interquartile range, IQR).
cfDNA, cell-free DNA; AFP, alpha-fetoprotein.
Comparison of demographic and clinical characteristics in patients with hepatocellular carcinoma (HCC) stratified according to the presence or absence of clinical symptoms.
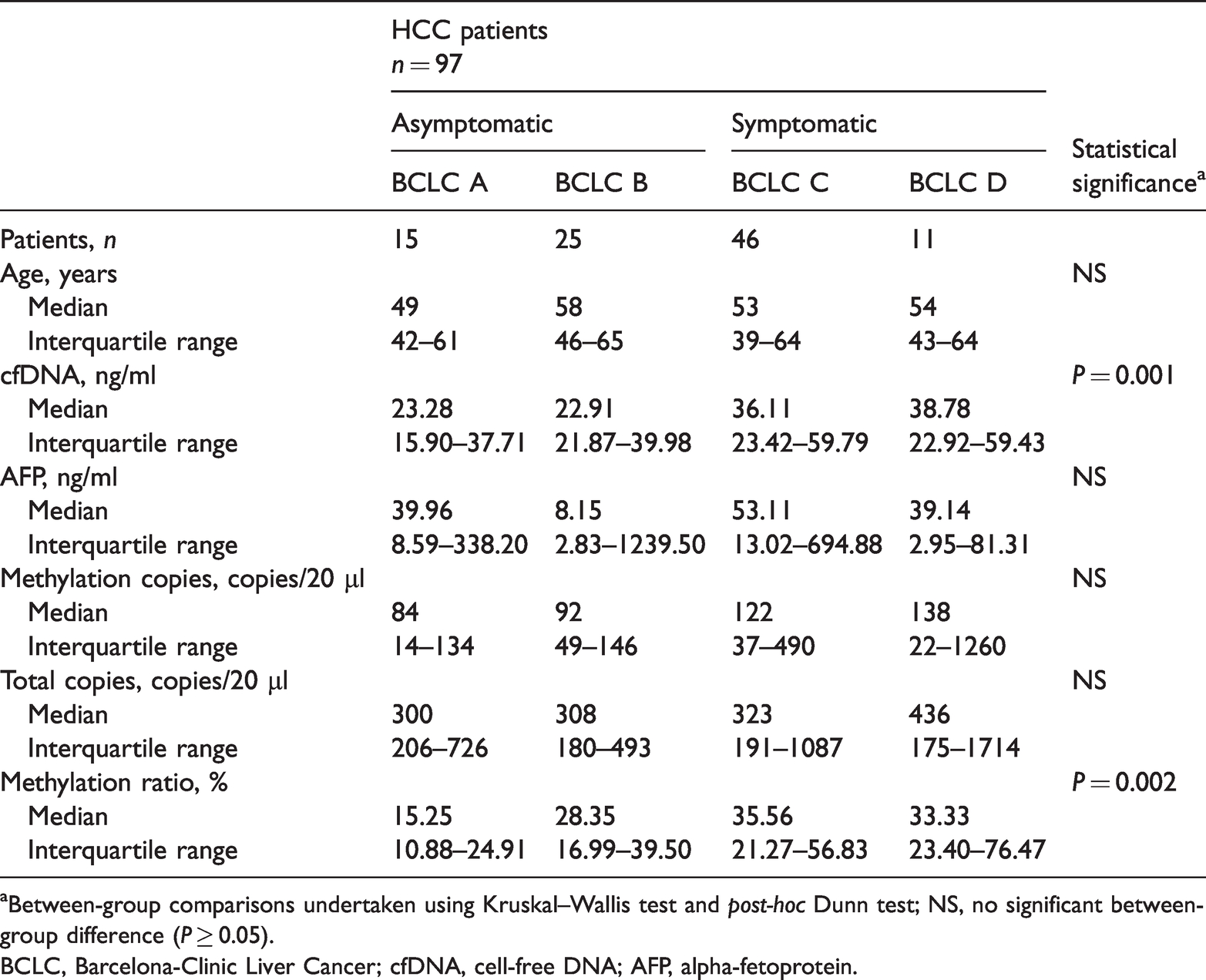
aBetween-group comparisons undertaken using Kruskal–Wallis test and post-hoc Dunn test; NS, no significant between-group difference (P ≥ 0.05).
BCLC, Barcelona-Clinic Liver Cancer; cfDNA, cell-free DNA; AFP, alpha-fetoprotein.

DNA methylation parameters in healthy control subjects (n = 80) and hepatocellular carcinoma (HCC) groups (n = 97; subgroups: BCLC A, BCLC B, BCLC C, BCLC D). (a) Comparison of cell-free DNA (cfDNA) concentration; (b) comparison of methylated copies; (c) comparison of methylation ratio; (d) comparison of total copies. Data presented as median ± interquartile range. Between-group comparisons undertaken using Kruskal–Wallis test and post-hoc Dunn test. BCLC, Barcelona-Clinic Liver Cancer.
Patients with (i) chronic hepatitis B/C virus infection as well as an abnormal AFP level and (ii) asymptotic HCC patients were enrolled in an AUC analysis. The efficiency of relevant tumour markers for HCC diagnosis was assessed (Figure 5). The ROC curve analysis determined the AUCs as follows: 0.786 (95% confidence interval [CI] 0.686, 0.886) for the AFP level and 0.809 (95% CI 0.718, 0.900) for the methylation ratio to differentiate HCC from non-cancer participants. The optimal cut-off value of the methylation ratio was 15.7%, which was obtained using the ROC curve analysis (AUC of 0.809 (95% CI 0.718, 0.900) for HCC diagnosis. A series of laboratory diagnostic indexes were then analysed using the cut-off value of 15.7% for the methylation ratio (Table 3). Based on the cut-off value of 15.7% in HCC screening, the sensitivity and specificity of HCC detection were 78.57% and 89.38%, respectively. The diagnostic accuracy was 85.27%, the positive predictive value was 81.91% and the negative predictive value was 87.20%. The positive likelihood ratio of 15.7% in HCC diagnosis was 7.40, while the negative likelihood ratio was 0.24. The combination of AFP, cfDNA and methylation ratio can distinguish asymptomatic HCC in the population with an AUC of 0.958 (95% CI 0.919, 0.996) (Figure 5). By combining AFP and methylation ratio, 13 new cases were confirmed to have HCC (Figure 6a). When screening the population, 12 individuals with a positive AFP level (range, 7–20 ng/ml; reference range, 0–7 ng/ml), but with a methylation ratio < 15.7%, were later confirmed by ultrasound examination without HCC tumours (Figure 6b).

Receiver operating characteristic curve analysis of the relative markers in a diagnosis of hepatocellular carcinoma. cfDNA, cell-free DNA; AFP, alpha-fetoprotein.
Use of cell-free DNA methylation ratio for predicting hepatocellular carcinoma.

*Study individuals selected from: (i) healthy control group (n = 80); (ii) patients with chronic hepatitis B/C virus infection (n = 46); (iii) patients with hepatocarcinoma (n = 97); (iv) new chronic hepatitis out-patient cases (n = 35).
PPV, positive predictive value; NPV, negative predictive value; +LR, positive likelihood ratio; –LR, negative likelihood ratio.
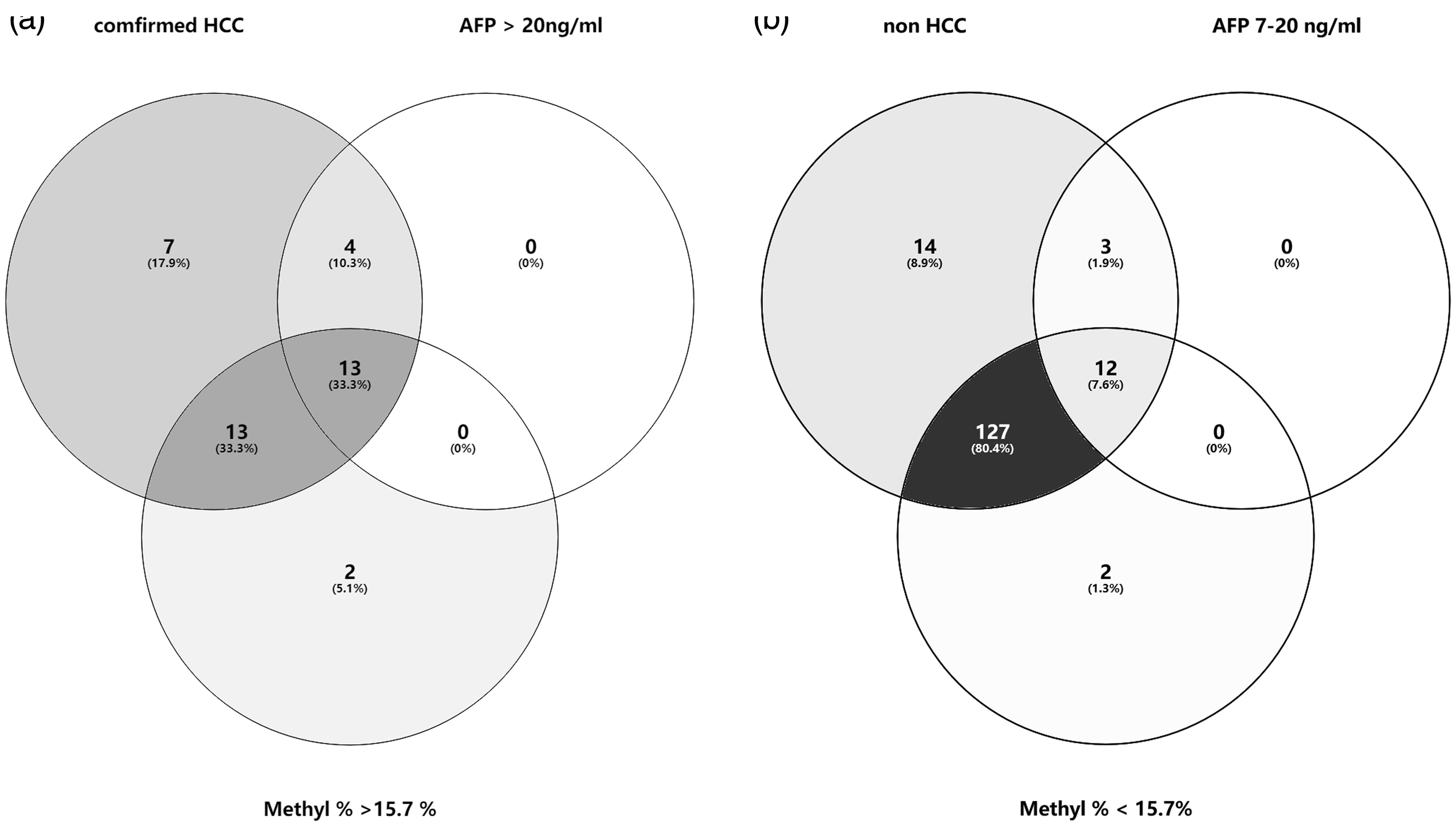
Venn diagrams representing the diagnostic ability of various clinical markers in hepatocellular carcinoma (HCC) diagnosis. (a) Combined with a methylation ratio ≥ 15.7% in screening asymptomatic individuals. (b) Combined with a methylation ratio < 15.7% in screening individuals suspected of having HCC. AFP, alpha-fetoprotein; M%, methylation ratio.
Discussion
This current study quantified cfDNA variations under different clinical conditions. Subsequently, the study validated the dynamic changes in the peripheral DNA methylation ratio that corresponded with HCC progression. Next, the diagnostic value of the methylation ratio for HCC diagnosis was investigated. This current study confirmed the potential application of tissue-specific methylation markers, combined with AFP and cfDNA analysis, in the investigation of susceptible HCC patients differentiated from the asymptomatic population. The differential methylation status between HCC patients, patients with chronic hepatitis B/C virus infection and healthy control subjects was investigated using a cut-off value of 15.7% for the methylation ratio combined with the AFP level and cfDNA level. This yielded a sensitivity of 78.57%, a specificity of 89.38% and a diagnostic accuracy of 85.27%. It was possible to distinguish symptomatic individuals from the population. Moreover, this current study demonstrated that cfDNA methylation analysis was a reliable and robust method for the early diagnosis of HCC. The DNA methylation patterns were highly correlated with the pathological subgroups based on the BCLC classification system. This current study also demonstrated that the ddPCR method efficiently detects trace amounts of DNA methylation. This relatively inexpensive approach is generalizable and could be easily used for personalized medicine, which would potentially improve health management.
The current study used the BCLC classification system, instead of the AJCC/TNM stage, to define the status of the HCC patients. The reason is because BCLC shows a strong ability to classify and predict prognosis, especially in high-risk populations in which it can identify early liver cancer patients for diagnosis and treatment. 45 It was interesting to explore if the methylation ratio had the same power as the BCLC system to predict prognosis. However, the results of this current study were not as expected due to lost follow-up data. It has been reported that using a combination of several methylation patterns would increase prognostic ability. 26 This study focused on the diagnostic performance of the explored DNA methylation markers. In cancer patients, increased cfDNA levels indicate pathological progression.46,47 According to the BCLC classification system, patients with stages C and D are defined as having tumours that have started to spread via the blood vessels or there has been extrahepatic spread.48,49 The current study demonstrated higher DNA methylation ratios and cfDNA levels in patients with advanced (BCLC C) and late (BCLC D) stages of HCC.
Alpha-fetoprotein is a conventional biomarker that presents as an increased level in pregnant women and in patients with acute liver inflammatory diseases. 50 The American College of Radiology Appropriateness Criteria® Chronic Liver Disease guidelines do not recommend screening populations for AFP levels because it is not associated with a statistically significant improvement in HCC detection in the USA.3,4,51 An AFP level > 20 ng/ml provides a sensitivity of 65% and a specificity of 94% for HCC screening. 4 A previous study analysed a HCC index, which included age, cfDNA and AFP level; and it demonstrated a sensitivity of 87% and a specificity of 100% for the diagnosis of HCC. 49 The current study demonstrated an AUC for AFP in asymptomatic population screening of 0.786; and this was improved to 0.958 by combining AFP with the DNA methylation ratio and cfDNA level.
In conclusion, these current findings emphasize the potential utility of a DNA methylation-based ddPCR platform for the minimally invasive, blood-based early detection of cancer, including HCC. The combined detection of the methylation of multiple genes might improve the diagnostic efficiency. 26 This current study also confirmed that ddPCR technology is a robust methodology that can be used in a clinical laboratory and that the analysis of peripheral DNA methylation using the ddPCR platform can be easily implemented in a clinical setting. In addition, it showed great potential for the diagnostic, prognostic and evaluation of therapeutic efficacy during individualized cancer management.
Footnotes
Authors' contributions
Juan Wang contributed to the data analysis and drafted the manuscript. Liu Yang, Jiayun Liu and Yueyun Ma helped to critically revise the manuscript. Yanjun Diao contributed to acquisition and analysis of data. Jinjie Li and Rui Li participated in the interpretation and analysis of data. Lianghong Zheng contributed to the design and critically revised the manuscript. Kang Zhang contributed to the conception and critically revised the manuscript. Xiaoke Hao contributed to the conception, design and critically revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We gratefully acknowledge all of the participants in Youze Biological Pharmaceutical Technology Company Ltd. for their technical assistance. We thank Dr Dhruvajyoti Roy from the Laboratory for Advanced Medicine (West Lafayette, IN, USA) for his lively, helpful discussions.
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This research was funded by a grant from the Key Research and Development Plan of Shaanxi Province, China (no. 2019SF-092).