
Abstract
Primary biliary cirrhosis (PBC)–autoimmune hepatitis (AIH) overlap syndrome is frequently associated with extrahepatic autoimmune disorders. Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired disease that is characterized by complement-mediated hemolysis due to erythrocyte membrane defects. However, autoimmune liver disease was not previously reported to be associated with PNH. A 37-year-old female patient was referred to our hospital with elevated liver enzymes and hematuria. On the basis of the symptoms and results of laboratory tests, radiographic studies, and pathologic results, she was diagnosed with PBC–AIH overlap syndrome and PNH. She was treated with a combination of ursodeoxycholic acid and prednisolone. The patient was symptom-free, with laboratory findings within near-normal range. The patient had recovered well at the 24-month follow-up evaluation. While we acknowledge that this was a single case, these findings expand our knowledge of immunological diseases that are associated with PNH and suggest an immune-mediated pathogenic pathway between PNH and PBC–AIH overlap syndrome. The combination of ursodeoxycholic acid and prednisolone can achieve therapeutic success. Routine follow-up of these patients is necessary to document disease progression.
Keywords
Introduction
Autoimmune liver diseases include autoimmune hepatitis (AIH), which is characterized by necro-inflammation, as well as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), which are characterized by progressive cholestasis. The spectrum of autoimmune liver diseases includes the overlap syndromes of PBC–AIH and PSC–AIH. PBC–AIH overlap syndrome has serology results that are consistent with PBC, and it is characterized by positive antimitochondrial antibody (AMA) and histologic findings of chronic hepatitis. 1
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired hemolytic anemia that is caused by somatic mutations in phosphatidylinositol glycan-complementation class A (PIGA) and the resulting absence of CD56, which is a key complement regulatory protein. The lack of glycosyl phosphatidylinositol-linked proteins leads to the clinical features of chronic intravascular hemolytic anemia and thromboembolism. 2 PBC–AIH overlap syndrome is frequently associated with extrahepatic autoimmune disorders; however, there has been no previously reported case associated with PNH. This is a study to report PNH in a patient with PBC–AIH overlap syndrome, suggesting immune-mediated pathogenesis as an underlying mechanism.
Case report
A 37-year-old woman was referred to our hospital for evaluation of elevated liver enzymes and hematuria. The patient provided verbal informed consent for the publication of this case and accompanying images. She did not have a history of smoking or alcohol use disorder. She did not use medications and was not exposed to toxic materials. At the time of admission, she complained of fatigue and dark urine and denied any family history of liver or hematological diseases. A physical examination subsequently revealed that she had an anemic appearance.
One month earlier, the patient had been diagnosed with PBC and a urinary tract infection at another hospital on the basis of the presence of the M2 fraction of AMA (AMA-M2) and elevated immunoglobulin (Ig) M, gamma-glutamyl transpeptidase, and alkaline phosphatase levels. Additionally, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were slightly elevated (130.7 U/L and 206.0 U/L, respectively). Abdominal ultrasound and a multi-slice computed tomography (CT) scan of her hepatobiliary area revealed that she had a fatty liver. AIH was not excluded on the basis of positive antinuclear antibodies (ANA; 1:1000) and elevated IgG and r-globulin levels. The patient was treated with 750 mg/day ursodeoxycholic acid (UDCA), which led to an improvement of the symptoms and cholestatic enzyme levels.
Initial urinalysis results revealed 2+ occult blood in the urine, 12 red blood cells (RBCs)/high-power field, and a negative urine culture. Blood test results revealed moderate anemia (RBC count, 2.47 × 1012/L and hemoglobin, 87 g/L) and liver dysfunction (AST, 130.7 U/L; ALT, 206.0 U/L; gamma-glutamyl transpeptidase, 457.7 U/L; and alkaline phosphatase, 619.1 U/L). The globulin levels were as follows: r-globulin, 37 g/L; IgG, 32.6 g/L; and IgM 7.68 g/L. Hematuria position diagnosis revealed mixed hematuria with 30% polymorphic and 70% morphologically uniform erythrocytes.
An extensive viral hepatitis workup, including polymerase chain reaction assays for hepatitis A, B, C, D, E, and G viruses, Epstein–Barr virus, and cytomegalovirus, revealed negative results. Histological examination of the percutaneous liver biopsy specimens revealed nonsuppurative destructive cholangitis and moderate interface hepatitis (Figure 1), suggesting the overlap of PBC and AIH.
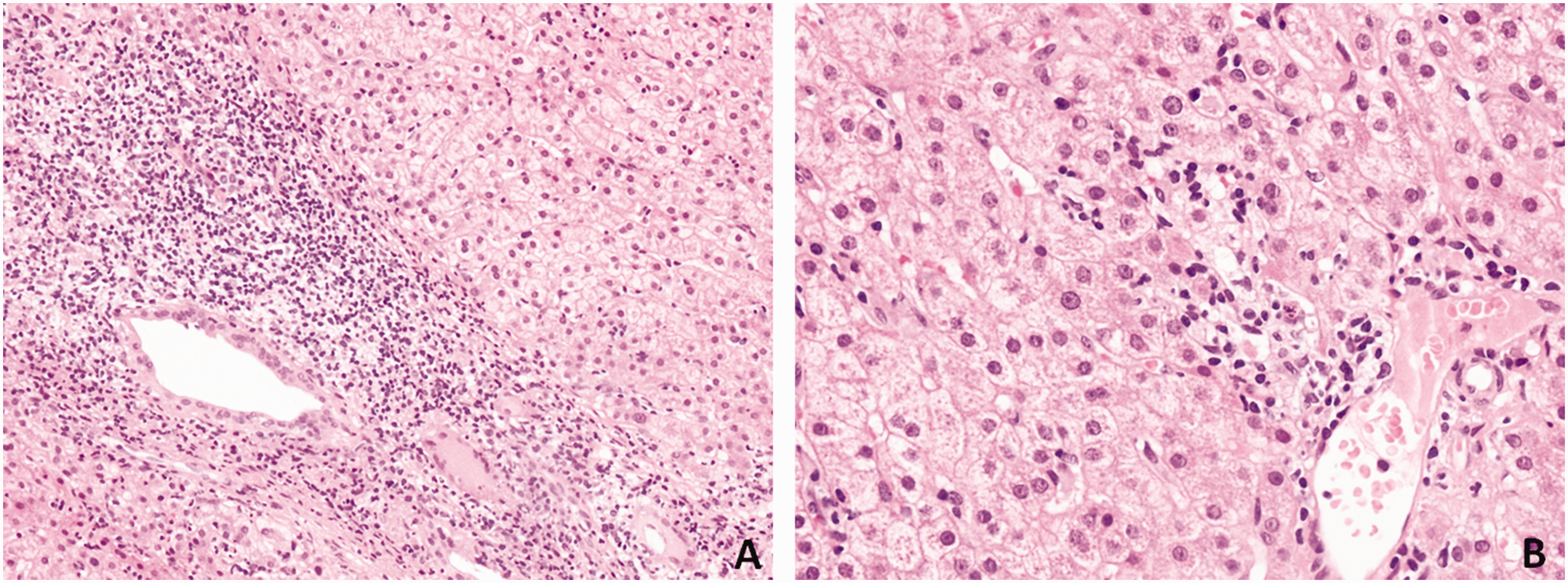
Liver biopsy specimens stained by hematoxylin and eosin show chronic nonsuppurative destructive cholangitis and moderate interface hepatitis. (A) ×200, (B) ×400.
Further investigation revealed cell-free plasma hemoglobin level of 200.5 mg/L and a reticulocyte percentage of 6.62%; the patient had negative serum acid glycerin hemolysis and anti-human globulin test results. There were no findings that suggested iron, folic acid, or vitamin B12 depletion. Bone marrow biopsy revealed hyperplastic anemia without other significant abnormalities (Figure 2). Flow cytometric analysis of the peripheral blood revealed the following: totally deficient RBCs (type III), 28.45%; partially deficient RBCs (type II), 28.22%; CD24−/fluorescent aerolysin (FLAER)− cells (PNH granulocytes), 87.36% of granulocytes; and CD14−/FLAER− cells (PNH monocytes), 89.07% of monocytes (Figure 3). These results led to the diagnosis of PNH. 2

Bone marrow biopsy revealing hyperplastic anemia.

Multiparametric flow cytometry evaluation of the patient’s peripheral blood. (a) Totally deficient (type III) and partially deficient (type II) red blood cells. (b) CD24−/FLAER− PNH granulocytes. (c) CD14−/FLAER− PNH monocytes.
On the basis of the results of the extensive workup, the patient was definitively diagnosed with PBC–AIH overlap syndrome and PNH, and she was treated with 750 mg/day UDCA and 25 mg/day oral prednisolone after she provided consent. The patient showed dramatic improvements in routine urinalysis and liver function test results over the 2 monthly follow-up examinations at the outpatient clinic after treatment was initiated (Figure 4). Prednisolone was reduced to 15 mg/day. The patient started to control her weight. Seven month later, the patient was symptom-free with a near-normal range of laboratory findings, and there were no indications of a fatty liver. However, the ANA titer results were still 1:1000. Prednisolone was discontinued, and the patient continued treatment with UDCA. At the 24-month follow-up evaluation, the patient had recovered well.

Graphic depiction of the key details of the patient’s clinical course. (a) Liver function, (b) immunoglobulin levels, (c) routine blood test results.
Discussion
PBC and AIH can occur consecutively or simultaneously in patients with PBC–AIH overlap syndrome, 3 which has been reported at frequencies ranging from approximately 10% to less than 2% in patients with AIH or PBC alone. 4 The Paris criteria, which are commonly used to define the presence of PBC with AIH features, are endorsed by the European Association for the Study of the Liver. 1
Generally, about one in three patients with an autoimmune liver disease is diagnosed with a concomitant extrahepatic autoimmune disease, which may include rheumatological, endocrine, gastrointestinal, pulmonary, or dermatological conditions. Rare cases of autoimmune liver diseases, such as cholangiocarcinoma, 5 hepatic sarcoidosis, 6 pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome, 7 and primary cutaneous amyloidosis, 8 have also been reported. The pathogenesis of these conditions includes the activation of both innate and adaptive immune responses targeting cholangiocytes as well as different extrahepatic tissues. Therefore, extrahepatic autoimmunity represents a continuous spectrum of autoimmunity involving liver and extrahepatic tissues.9–11
We could not identify any reports of patients with autoimmune liver diseases who were diagnosed with PNH. PNH is caused by an acquired defect of the erythrocyte membrane that results in complement-mediated hemolysis. 12 Liver involvement is not frequent in PNH; symptomatic liver disease is rare, whereas hepatomegaly, splenomegaly, and jaundice are the most common abnormalities. Several studies reported PNH in patients with Budd–Chiari syndrome 13 and hepatitis. 14 A high incidence of venous thrombosis has been reported almost since the first clinical description of PNH, wherein hepatic veins and portal system veins are particularly affected. Clinical examination shows an enlarged liver that is painful upon percussion. Acute ascites may occur, and the patient may develop jaundice. 15 Abnormal liver function test results in patients with PNH are not reliable predictors of the clinical course and outcome of the liver disease, whereas hepatic failure due to portal or hepatic vein thrombosis is a common cause of mortality in PNH. Anticoagulation, transjugular intrahepatic portosystemic shunts, surgical shunts, and liver transplantation may lead to transient improvement.16,17
Shaaban et al. 14 reported aplastic anemia–PNH associated with non-A–E hepatitis virus infection that resulted from an autoimmune attack directed against hematopoietic stem/progenitor cells. The abnormal immune activity in patients with PNH has been recognized. An Italian study of 11 patients showed that PNH is characterized by expansion of PIGA-defective hematopoietic cells, which is probably due to immune-mediated alterations of the bone marrow environment that selects PIGA− stem cells. 18 The presence of PNH clones in aplastic anemia usually suggests immunopathogenesis in patients. 19 The marrow aplasia due to immunological dysregulation was not found in the present patient. It is possible that PNH and PBC–AIH overlap syndrome share similar immunological characteristics.
To date, there is no clear evidence regarding the impact of concomitant extrahepatic autoimmune diseases on the natural history of autoimmune liver diseases and associated complications. However, prompt recognition of these diseases is fundamental to ensure appropriate patient referral and treatment because extrahepatic autoimmune diseases may strongly impact the quality of life in these patients. 17 The association between PNH and autoimmune liver diseases is currently unknown, and the present case may be a simple association between the two disease entities. Although it is a rare clinical presentation, the current case is an important reminder of PNH’s association with autoimmune liver diseases for physicians involved in this field.
Conclusions
In patients with autoimmune liver disease with anemia or hematuria, concurrent PNH should be considered. As illustrated in the present case, steroids in combination with UDCA therapy might achieve a good prognosis.
Footnotes
Author contributions
Conceptualization: Lin Chen, Xiaodong Shi, Wanyu Li
Funding acquisition: Wanyu Li
Resources: Wanyu Li
Visualization: Wanyu Li
Writing – original draft: Lin Chen, Xiaodong Shi, Wanyu Li, Wei Han
Writing – review and editing: Jinglan Jin, Wanyu Li, Limei Qu
Data availability statement
The datasets for this study are available from the corresponding author upon request.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This study was supported by grants from the First Hospital of Jilin University (No. JDYYJC006), Tianqing Liver Disease Research Fund (No. TQGB20180160), and the Department of Science and Technology of Jilin Province (No. 20180520116JH).