
Abstract
Activated phosphoinositide 3-kinase delta syndrome (APDS) is a combined inborn error of immunity mainly caused by PIK3CD mutations. We herein describe a 4-year-old Chinese boy who was admitted for recurrent pneumonia and persistent hematuria and exhibited multisystem involvement and anti-neutrophil cytoplasmic antibody (ANCA) positivity. He was initially diagnosed with ANCA-associated vasculitis. However, genetic testing revealed a c.1574A>G PIK3CD mutation, resulting in a diagnosis of APDS1.
Keywords
Introduction
Activated phosphoinositide 3-kinase delta syndrome (APDS) is a recently described inborn error of immunity (IEI) due to either monoallelic gain-of-function (GOF) mutation on PIK3CD (resulting in APDS1) or biallelic loss-of-function mutation on PIK3R1 (resulting in APDS2). Patients with APDS present with highly variable manifestations such as recurrent respiratory tract infections (RTIs), bronchiectasis, herpesvirus infections, autoimmunity, non-neoplastic lymphoproliferation, and lymphoma as well as neurodevelopmental delays and growth retardation.1,2 Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitides (AAVs), which are necrotizing vasculitides that do not substantially involve the deposition of immune complexes, are also a highly variable group of diseases. The clinical presentation of AAVs is often heterogeneous, with frequent involvement of the respiratory tract, kidneys, skin, and joints. 3 The clinical manifestations of APDS and AAVs overlap, and identification is sometimes confusing. However, their recognition, correct diagnosis, and appropriate treatment are crucial because they may be life- or organ-threatening.
Increasingly more IEIs are being associated with single gene defects, and treatment depends upon the part of the immune system affected. Therefore, genetic testing is helpful and important for patients suspected to have immunodeficiency. We herein describe a child who presented with hematuria and was misdiagnosed as having AAV but was finally diagnosed with APDS1 by genetic testing.
Case presentation
On 16 December 2014, a 4-year-old Chinese boy was admitted to our hospital because of a 2-year history of persistent microscopic hematuria and repeated pneumonia. Since the age of 2 years, the patient had been hospitalized four times for pneumonia. Physical examination revealed wheezing on pulmonary auscultation, hepatomegaly, splenomegaly, otitis media, and sinusitis. Multiple enlarged lymph nodes were found in the neck, axilla, and inguinal region.
Work-up at that time revealed perinuclear ANCA positivity and immunoglobulin (Ig) G, IgM, IgA, IgG1, and IgG2 levels of 20.8 (reference range, 9–13), 2.49 (0.5–1.8), 1.98 (0.6–1.5), 24.80 (4.9–11.4), and 1.87 (1.5–6.4) g/L, respectively. Urinary microscopic examination showed 50 red cells per high-power field. Seventy percent of the urinary erythrocytes were heteromorphic. A computed tomography (CT) scan revealed pneumonia and bronchiectasis (Figure 1(a)), hepatosplenomegaly (Figure 1(b)), multiple lymph node enlargement, bilateral maxillary sinusitis, ethmoid sinusitis, and bilateral mastoiditis. A bone marrow biopsy showed granulocyte proliferation with no significant dysplasia or blast expansion. Excisional biopsy of a left inguinal lymph node showed reactive lymph node hyperplasia (Figure 2(a) and (b)). No antibodies to Epstein–Barr virus (EBV) were found in the lymph node. Bronchoscopy revealed cobblestone-like changes in the airway mucosa. In addition, the alveolar lavage fluid contained a large number of red blood cells with a high number of EBV DNA copies (2.8 × 105 copies/mL). Renal biopsy revealed mild mesangial cell proliferation with no immune deposits (Figure 2(c) and (d)).

(a) Chest computed tomography showed bronchiectasis (blue arrow). (b) Abdominal computed tomography showed hepatomegaly (blue line) and splenomegaly (red line).

(a, b) Lymph node biopsy revealed enlarged lymphoid follicle hyperplasia and lymphocytic hyperplasia in the paracortical area with no malignant clonal cells ((a) 50×, (b) 200×). (c) Light microscopy showed minimal changes in glomeruli and no necrosis or vasculitis (magnification, 200×). (d) Electron microscopy showed fusion of part of the foot processes and no immune deposits (magnification, 8000×). (e, f) Lung biopsy showed a large number of lymphocytic cells infiltrating the bronchioles, with fibrous tissue and collagen fibers hyperplasia but no necrosis or vasculitis ((e) 50×, (f) 400×).
Based on the presence of sinusitis, otitis media, recurrent pneumonia, renal involvement, multiple lymphadenopathy, ANCA positivity, and a high number of EBV-DNA copies, the patient was initially diagnosed with AAV. However, the diagnosis of AAV was not definitive because of the absence of vasculitis in the renal pathologic examination. Nevertheless, the patient began treatment with oral prednisone (initial dose of 20 mg/day). From March to July 2015, he was treated with intravenous cyclophosphamide (CTX) once every 2 weeks for a total of seven treatments (cumulative dose of 2.8 g). Thereafter, the patient was treated with oral mycophenolate mofetil (MMF) (0.25 g twice per day). Immunotherapy resulted in improvement of his hematuria and decreased his lymphadenopathy and hepatosplenomegaly. He was also started on oral trimethoprim/sulfamethoxazole prophylaxis to help prevent infections (0.24 g twice per day, subsequently reduced to once per day). Although he continued to develop intermittent RTIs, fewer infections occurred. In addition, ganciclovir was given to treat his EBV infection.
In April 2015, the patient underwent right lung biopsy, which showed peribronchiolar infiltration of lymphocytes, proliferation of fibrous tissue and collagen fibers, and alveolar dilatation; however, neither necrosis nor vasculitis was present (Figure 2(e) and (f)).
In March 2016, the patient was again found to have enlarged left cervical lymph nodes and microscopic hematuria. He underwent excisional biopsy of a left cervical lymph node. The pathologic examination revealed the same results as previously obtained, and flow cytometry identified no significant malignant clonality. Peripheral blood tests showed persistently low levels of lymphocytes, low percentages of CD19- and CD20-positive cells, decreased or reversed CD4/CD8 ratios, and persistent positivity for EBV early antigen-IgM and cytomegalovirus IgM. These findings (Table 1) indicated that the patient had immunodeficiency. He underwent one treatment session of intravenous Ig (IVIG) replacement therapy (30 g) on demand when severe infection developed.
Immune evaluation and EBV testing in peripheral blood.

EBV, Epstein–Barr virus; WBC, white blood cell; p-ANCA, perinuclear anti-neutrophil cytoplasmic antibody; c-ANCA, cytoplasmic anti-neutrophil cytoplasmic antibody; N, negative; P, positive; EA, early antigen; CA, capsid antigen; COI, cutoff index.
Beginning in April 2017, the patient’s frequency of pneumonia increased. CT scans showed aggravation of pulmonary lesions and bronchiectasis along with enlarged lymph nodes in the mediastinum and other regions. Increasing the glucocorticoid dose resulted in no improvement. Whole-exome sequencing revealed a c.1574A>G mutation in PIK3CD, which led to the final diagnosis of APDS1.
In November 2018, the patient began treatment with oral rapamycin (1 mg once per day) and regular IVIG replacement (30 g once per month). The main treatment process is shown in Figure 3. After beginning treatment with rapamycin, the patient no longer returned to our hospital for new recurrences of pneumonia; at the time of this writing, however, he was still experiencing problems with otitis media and hearing, and CT scans had shown no improvement in his bronchiectasis. In addition, his height decreased from the 20th percentile before glucocorticoid therapy to the 3rd percentile at the end of glucocorticoid therapy.

(a) Changes in peripheral blood leukocyte counts. (b) Main course of treatment.
Discussion
APDS is a heterogeneous IEI that was first reported in 2013. 4 It is caused by autosomal dominant mutations in PIK3CD (resulting in APDS1) or PIK3R1 (resulting in APDS2), increasing the activity of phosphoinositide-3-kinase delta (PI3Kδ).1,5 Hyperactivation of PI3Kδ can enhance downstream PIK/AKT/mammalian/mechanistic target of rapamycin (mTOR)/S6 kinase (S6K) signaling, leading to a series of pathological conditions. The most common clinical manifestations of APDS are RTIs, lymphoproliferation, autoimmunity, enteropathy, failure to thrive, and malignancy; hematuria is rarely seen.1,2 This is the first reported case in which the clinical presentation of APDS was consistent with AAV.
AAVs are defined as necrotizing vasculitides that predominantly affect small blood vessels such as capillaries, venules, and arterioles. They are associated with the presence of circulating autoantibodies (ANCAs) that are usually directed against myeloperoxidase or proteinase. AAVs are a family of multisystemic diseases that include microscopic polyangiitis, granulomatosis with polyangiitis, and eosinophilic granulomatosis with polyangiitis. 3
Differentiation of APDS from AAVs is sometimes difficult. Our patient presented with repeated pneumonia, bronchiectasis, otitis media, hematuria, and ANCA positivity; he was initially diagnosed with AAV and given standardized treatment with a glucocorticoid and CTX followed by MMF. The glucocorticoid and CTX effectively improved his hematuria and induced a negative ANCA titer, but he continued to develop recurrent RTIs and otitis media. In addition, his lung imaging, immunoglobulin concentrations, and lymphocyte subset abnormalities showed no improvement (Table 1). Moreover, kidney and lung pathologic examinations revealed no evidence of vasculitis and did not support the diagnosis of AAV. Finally, whole-exome sequencing revealed a heterozygous mutation in the exon region of PIK3CD, c.1574A>G (Figure 4), resulting in the substitution of glutamic acid by glycine at position 525. This finding led to the diagnosis of APDS1.
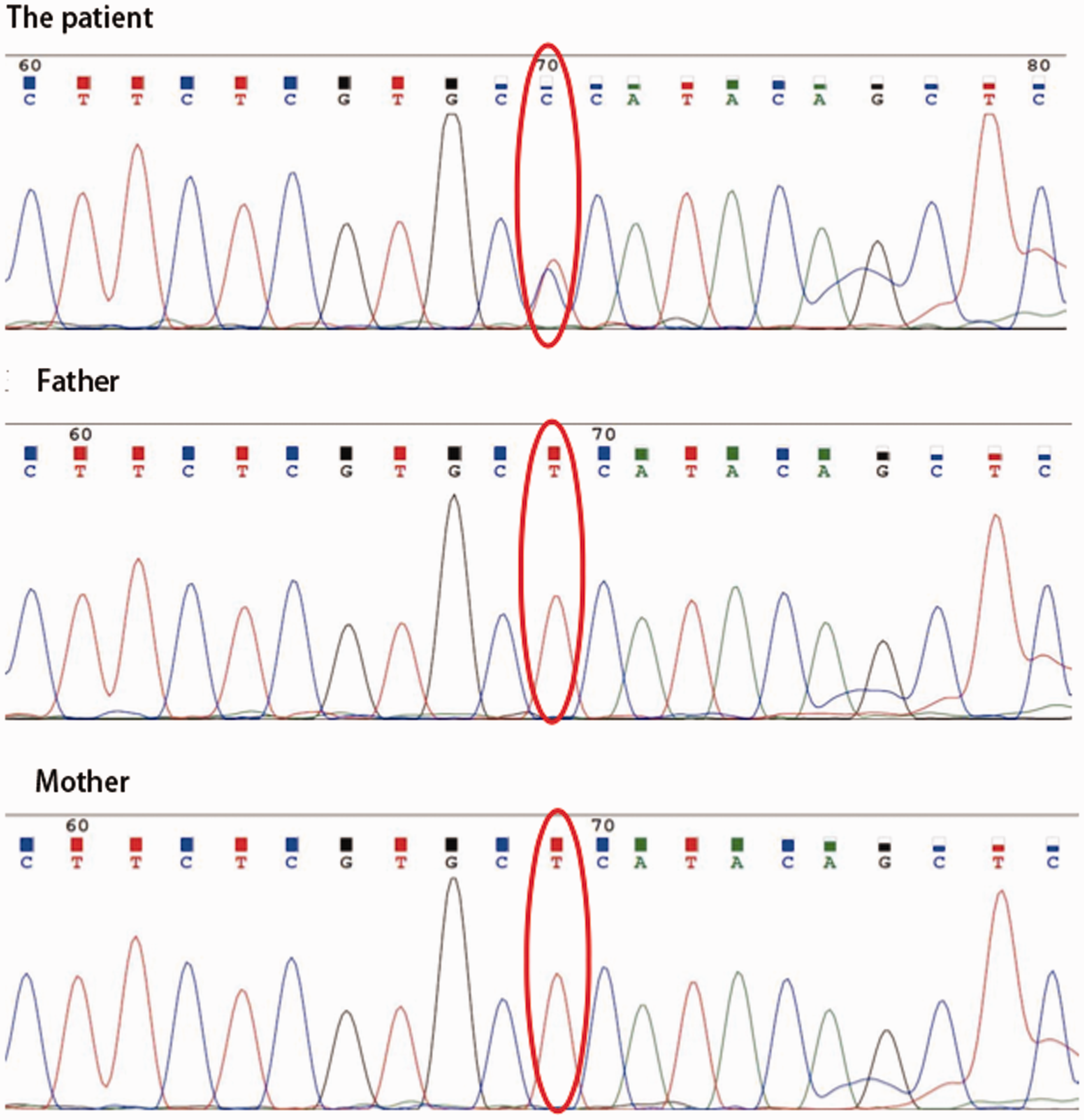
Genetic testing revealed a heterozygous mutation in the exon region of PIK3CD: c.1574A > G (adenine > guanine).
The c.1574A>G mutation has not been previously reported. However, a similar mutation c.1574A>C (E525A), has been reported in patients with APDS. This mutation results in the amino acid mutation p.Glu525Ala (glutamic acid > alanine).6,7 Functional prediction algorithms (SIFT and PolyPhen) have indicated that the novel mutation c.1574A>G is deleterious (PP3). This mutation is a newly arisen missense mutation in a child without a family history (PS2); moreover, this mutation is located in a mutational hot spot and a critical and well-established functional domain (PIK3CD) (PM1) and was absent from controls in the Exome Sequencing Project (PM2). This novel missense mutation affects the same amino acid residue as does the mutation E525A (PM5). The American College of Genetics and Genomics mutation interpretation guidelines 8 and the clinical features of our patient strongly suggest that the novel c.1574A>G mutation (E525G) is a pathogenic mutation in patients with APDS. However, because many IEIs display genetic heterogeneity, genetic pleiotropy, and incomplete clinical penetrance, we cannot rely solely on sequence data to secure a diagnosis. It is important to correlate the results of genetic testing with protein-based studies to avoid assigning a causal role to benign variants, especially in cases involving novel variants and genes. Increased PI3Kδ activity leads to aberrant activation of the AKT/mTOR/S6K pathway and is thought to be a major molecular cause of APDS.1,2,4 Elevated phosphorylation of AKT (pAKT) and S6K (pS6K) have been found in these patients and are considered valuable indicators of the pathogenic variant in APDS and the hyperactivated PIK/AKT/mTOR/S6K pathway.6,7,9 Moreover, in previous studies, patients with APDS who were treated with rapamycin or leniolisib (inhibitors of the PIK/AKT/mTOR/S6K pathway) showed decreased levels of pAKT and pS6K5,9,10 Therefore, the pAKT or pS6K levels of the patient in the present case need to be followed up to monitor the efficacy of rapamycin, which may also be a partial supplement for the functional validation of E525G.
Our patient initially developed frequent pneumonia with bronchiectasis and mucosal cobblestone-like changes evident on bronchoscopy, which was considered to indicate submucosal lymphadenopathy. Most patients with APDS develop repeated RTIs, many of which are accompanied by bronchiectasis. Historically, bronchiectasis has been thought to be secondary to repeated pulmonary infections. However, one study showed that 50% of children with APDS accompanied by bronchiectasis did not have a history of pneumonia, 2 suggesting that bronchiectasis cannot be fully explained by complications after pulmonary infections. Dysfunctional cellular and humoral immunity may lead to a sustained inflammatory response in the airway. The lung biopsy of our patient indicated obviously abnormal lymphoblastic infiltration and fibrosis (Figure 2(a) and (b)). In addition, subtracheal and extratracheal lymphoid hyperplasia may lead to airway obstruction. Collectively, the above-described abnormalities may lead to bronchiectasis in children with APDS. 11 Therefore, the possibility of immunodeficiency must be considered in children with bronchiectasis, even without a history of repeated RTIs.
PIK3CD GOF mutations overactivate the downstream PIK/AKT/mTOR/S6K signaling pathway, which is involved in regulating the biology of lymphocytes, including B cells, T cells, and natural killer (NK) cells. Consistent with previously published data,1,12,13 cell subpopulation analysis in our patient revealed decreased percentages of CD19+ or CD20+ B cells, CD4+ T cells, and CD56+ NK cells but increased percentages of CD8+ T cells (Table 1). Moreover, regardless of whether the total number of white blood cells increased or decreased, the total number of lymphocytes always remained low. A previous study revealed that PIK3CD GOF mutations may result in sustained and exaggerated activation of polyclonal CD8+ T cells; thus, many patients with APDS have increased frequencies of CD8+ T cells. However, these CD8+ T cells seem to exhibit premature immunosenescence and exhaustion,9,12 which reduces their cytotoxicity. PIK3CD GOF mutations also dysregulate the expression of key regulatory molecules and reduce the cytotoxic function of NK cells. In addition, these mutations induce B cells to express more ligands that regulate the cytotoxicity of CD8+ T cells and NK cells.12,14 Because CD8+ T cells and NK cells play a critical role in EBV control, dysfunction of these cells potentially results in reduced immunosurveillance of EBV-infected B cells and an increased incidence of EBV-associated disease. Thus, 50% of patients with APDS present with long-term EBV infections. Moreover, patients have been reported to develop B-cell lymphomas, and the risk of malignancies is likely to increase with age. 2 Although our patient underwent two lymph node biopsies that revealed no malignancies, he still needs long-term follow-up and even lymph node biopsies in the future.
Mutations in PIK3CD can affect the maturation and differentiation of B cells in the bone marrow, resulting in increased proportions of immature B cells and a reduction in mature recirculating B cells. 15 Patients have difficulties producing sufficient amounts of functional memory and effector B cells in the periphery, which impairs somatic hypermutation and class switch recombination. 16 Collectively, these defects contribute to an impaired ability of B cells to produce antibodies that neutralize and clear invading pathogens from the host. Most patients with APDS present with decreased IgG and IgA concentrations and increased IgM concentrations.1,16 Interestingly, our patient presented with elevated IgG and IgA concentrations. However, IgG subclass analysis revealed increased IgG1 and reduced IgG2 and IgG4 concentrations. Previous data have shown that while patients with APDS have normal or elevated levels of IgM, variable levels of IgG, and low or normal levels of IgA, antigen-specific antibody titers against protein- and polysaccharide-containing vaccines are consistently low. 1 IgG2 predominantly controls the antibody response to polysaccharide capsular antigens. 17 IgG4 subclass deficiency has been described in combination with IgG2 deficiency and IgA-IgG2 deficiencies. In addition, recurrent pulmonary infections and bronchiectasis have been reported in symptomatic patients. 18 Deficiencies in this IgG subclass have also been found in other patients with APDS. 19 These observations indicate that IgG2 and IgG4 subclass deficiencies, which are associated with recurrent RTIs and bronchiectasis, may be important immune features of APDS. These findings provide a theoretical basis for the use of IVIG therapy in patients with high IgG levels.
Patients with APDS display autoimmune phenotypes in addition to immunodeficiency. Almost 50% of patients with APDS exhibit autoantibodies and evidence of autoimmune-mediated organ damage, such as cytopenia (hemolytic anemia, neutropenia, and/or thrombocytopenia), colitis, glomerulonephritis, and autoantibody production. 1 Research by Lau et al. 20 demonstrated that patients with GOF mutations in PIK3CD have high levels of IgM antibodies and that PIK3CD GOF B cells in mice are activated by self-antigens to form plasmablasts that secrete high titers of germline-encoded IgM autoantibodies and hypermutating germinal center B cells. This relationship may explain why our patient exhibited ANCA positivity and a presentation similar to that of AAVs.
Delayed diagnosis of APDS is very common. Epidemiological statistics based on available data show that the median age at diagnosis of APDS is 12.0 (6.5–21.5) years, with a median diagnostic delay of 7.0 (3.4–14.0) years. 2 In our patient, for example, onset occurred at the age of 2 years, and he presented to our hospital at the age of 4 years. However, the diagnosis was not made until the age of 8 years. Unfortunately, he did not undergo genetic testing earlier, mainly because we were unaware of APDS. In fact, when our patient presented with persistently high serum IgM levels, IgG subclass deficiencies, and lymphocyte subclass deficiencies, and especially when his kidney, lung, and lymph node pathology results were not fully consistent with AAV, we suspected primary immunodeficiency disease. Therefore, thorough knowledge of the possible clinical characteristics of APDS is very important to reducing the diagnostic delay and providing more accurate treatment.
The therapeutic strategy for patients with APDS includes prophylactic antibiotics, immunosuppressive agents, IVIG replacement therapy, and hematopoietic stem cell transplantation. Our patient was treated with glucocorticoids, CTX, MMF for immunosuppression, and trimethoprim/sulfamethoxazole for antibiotic prophylaxis in the early stage. This treatment was helpful for relieving hematuria and ANCA positivity but could not prevent pneumonia, otitis media, lymphoproliferation, or bronchiectasis. Moreover, the patient’s impaired height development may have been related to long-term glucocorticoid therapy.
Rapamycin inhibits mTOR signaling downstream of activated PI3Kδ, and it has been used with good efficacy in patients with APDS. Rapamycin has also been shown to ameliorate lymphoproliferation and reduce the levels of immunologic markers in patients with APDS. 5 After the use of IVIG replacement therapy and rapamycin, our patient developed less frequent pneumonia. Regardless of whether rapamycin improved his bronchiectasis, lymphoproliferation and hearing loss in our patient still need to be monitored. However, one report indicated that a patient developed lymphoma while under rapamycin therapy; 5 therefore, the long-term adverse effects of rapamycin must be monitored. The levels of pAKT and pS6K can be used as valuable follow-up indicators to monitor the therapeutic effect of rapamycin.
Selective PI3Kδ inhibitors have recently been suggested to provide improved efficacy with fewer adverse effects. Leniolisib has been used in patients with APDS with good efficacy. 10 The in vivo effects of other PI3Kδ inhibitors, including idelalisib 21 and nemiralisib, 22 remain to be assessed. However, recent evidence has shown that treating mice with PI3Kδ inhibitors results in increased genomic instability in normal and neoplastic B cells, 23 suggesting that long-term PI3Kδ inhibitor administration could have detrimental consequences.
Hematopoietic stem cell transplantation can achieve a complete cure with no need for ongoing treatment. However, because it has high frequencies of adverse complications and engraftment failure as well as a mortality rate of nearly 20%, 24 it is generally used only in patients with APDS who have particularly complex clinical phenotypes that do not respond well to other conventional treatments.
Conclusion
APDS is a very rare IEI that has diverse clinical manifestations and lacks specific clinical features. Its presentation may be similar to that of AAVs. However, primary immunodeficiency should be considered in patients with recurrent RTIs, bronchiectasis, a high serum IgM level, a low IgG2 level, lymphoproliferation, and autoimmunities. Genetic testing is helpful for diagnosis and needs to be performed early. Our case report shows that E525G is very likely a pathogenic mutation in patients with APDS. If there is no pathological evidence to support AAVs, immunosuppressive therapy should not be started, especially in children.
Footnotes
Ethics
This study was approved by the Ethics Committee of The Children’s Hospital, Zhejiang University School of Medicine (No. 2020-IRB-053). The patient’s parent provided written informed consent.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This work was supported by the Nature Science Foundation of Zhejiang Province (LQ18H050001 and LQ19H050007).