
Abstract
Objective
To investigate the effects of bortezomib on human osteosarcoma cells from the HOS cell line, and the underlying associated mechanisms.
Methods
Viability of HOS cells treated with bortezomib (5–20 nM) for different time periods was measured and changes in the cell cycle were assessed. Apoptosis and autophagy in HOS cells treated with bortezomib were analysed using annexin V-fluorescein isothiocyanate assay, transmission electron microscopy and Western blotting. Surges in mitogen-activated protein kinase (MAPK) pathways including MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK1/2), ERK1/2, c-Jun N-terminal kinase (JNK) and p38 MAPK were analysed using Western blotting.
Results
Bortezomib induced growth inhibition in a time- and dose-dependent manner, and autophagy and apoptosis in a dose-dependent manner, in HOS cells. HOS cell autophagy and apoptosis in response to bortezomib, corresponded with changing levels of intracellular MAPK signalling molecules.
Conclusions
This study provided new insights into the mechanisms underlying bortezomib-induced apoptosis in human osteosarcoma HOS cells, and suggests that bortezomib could be a potent chemotherapeutic agent in the treatment of osteosarcoma.
Keywords
Introduction
Osteosarcoma is the most common primary malignant bone tumour, with two incidence peaks: one at 10–14 years and the other at >65 years of age. 1 There is a high tendency for metastatic spread in osteosarcoma and ∼20% of patients have lung metastases at initial diagnosis. 2 Osteosarcoma is highly resistant to conventional chemotherapy, indicating a need for novel therapeutic approaches that target specific cell survival pathways. 3 Knockdown of GLI family zinc finger 2 by siRNA in two- and three-dimensional cultures, has been found to decrease osteosarcoma cell proliferation and viability, and eventually induce cell death. 4 However, such targeted strategies have not been explored extensively in osteosarcoma. Tumours often possess multiple genetic and cell signalling lesions, therefore, agents that inhibit multiple cellular pathways may be therapeutically more effective than agents that only target one signalling pathway. 5
Proteasome inhibitors, including the multipathway inhibitor bortezomib, are gaining increased attention for their therapeutic potential in the treatment of cancers and inflammation. 6 Bortezomib is a dipeptidyl boronic acid compound that inhibits the 26 S proteasome (which is a large multisubunit protein complex found in eukaryote cells), and also degrades polyubiquitinated target proteinsincluding the cyclins, apoptotic regulators, and p53. 7 Bortezomib has been approved by the US Food and Drug Administration for the treatment of patients with multiple myeloma who have received at least one prior therapy. 8 The multicatalytic ubiquitin-proteasome system is known to play a key role in cell proliferation, cell-cycle control, cell death and signal transduction, 7 although the mechanisms underlying bortezomib-induced cell death remain poorly defined. The effects of bortezomib appear to be cell type-specific or context dependent, for instance, in melanoma and head and neck carcinoma cell lines, increased Noxa expression appears to be critical for apoptosis induction. Bortezomib blocks nuclear factor of κ-light-chain enhancer of activated B cells (NF-κB) activation by stabilizing the NF-κB inhibitor, IκB, and thus allowing IκB to maintain its inhibitory influence over NF-κB. 9 Bortezomib can also stabilize p53, allowing it to function as a proapoptotic transcription factor in cells that are exposed to bortezomib. 10
Programmed cell death is an intracellularly mediated and highly regulated process, comprising three different types: apoptosis (type I); autophagy (type II); necrosis (type III). Autophagic cell death involves an evolutionarily conserved membrane trafficking pathway that mediates the transport of cytosolic materials, superfluous organelles and long-lived proteins to the lysosome; this occurs in all eukaryotic cells. During autophagy, a cup-shaped structure, the preautophagosome, engulfs cytosolic components including organelles and closes, forming an autophagosome, which subsequently fuses with a lysosome; this leads to the proteolytic degradation of internal components of the autophagosome by lysosomal lytic enzymes. 11 Autophagy primarily functions as a cell survival mechanism under stress, however, persistent stress can promote extensive autophagy, leading to cell death. There is complex interplay between autophagy and apoptosis. In some instances, autophagy can serve as a cell survival pathway, inhibiting apoptosis, and in others, autophagy can promote apoptosis. 12 The relationship between bortezomib, osteosarcoma, apoptosis and autophagy is unclear, although resistance to apoptosis is known to be an important mechanism by which osteosarcoma cells escape therapeutic control.13,14
The precise mechanism by which bortezomib kills neoplastic cells has been related to inactivation of cytoprotective signalling pathways, and activation of stress-related pathways. 14 The mitogen-activated protein kinase (MAPK) pathways govern cell proliferation, differentiation, stress responses and survival. p38 is thought to act as a tumour suppressor, largely mediated by both negative regulation of cell-cycle progression and the induction of apoptosis. 13 In general, c-Jun N-terminal kinase (JNK) and p38 MAPK activation are associated with apoptotic induction, whereas MAPK kinase/extracellular signal-regulated kinases (ERK) kinase 1/2 (MEK1/2)/ERK1/2 activation is cytoprotective. 15 Studies have suggested that disturbances in MAPK pathways may be involved in regulating proteasome inhibitor-mediated lethality. 14
Osteosarcoma and Ewing sarcoma cell lines have been reported to undergo apoptosis when treated with proteasome inhibitors,16,17 however, the exact molecular mechanism by which bortezomib induces apoptosis in cancer cells, specifically in osteosarcoma cells, has not been clearly defined. The purpose of the present study was to define the relationship that might exist between MAPK activation, autophagy and apoptosis in human osteosarcoma cells exposed to bortezomib, using pharmacological approaches.
Materials and methods
Reagents and antibodies
Bortezomib (Selleck Chemicals, Houston, TX, USA) was dissolved in dimethylsulphoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA) at a stock concentration of 1 mM, and stored at −20℃. Antibodies to microtubule-associated protein 1 light chain 3-α (LC3I and LC3II) (rabbit anti-, No. NB100-2220, diluted 1 : 500) and sequestosome 1 (p62) (rabbit anti-, No. NBP1-48320, diluted 1 :500) were obtained from Novus Biologicals (Littleton, CO, USA). Antibodies to phosphorylated (p)p-MEK (rabbit anti-, No. 9121), p-ERK1/2 (rabbit anti-, No. 9102), p-JNK (rabbit anti-, No. 9251), p-p38 (rabbit anti-, No. 9211), ERK1/2 (rabbit anti-, No. 9101), MEK1/2 (rabbit anti-, No. 9122), poly (ADP-ribose) polymerase 1 (PARP) (rabbit anti-, No. 9532), B-cell chronic lymphocytic leukaemia/lymphoma 2 (BCL2)-associated X protein (BAX) (rabbit anti-, No. 5023), Bcl-2 (rabbit anti-, No. 2876), JNK (rabbit anti-, No. 9258) and p38 (rabbit anti-, No. 9212) were obtained from Cell Signaling Technology (Danvers, MA, USA). The antibody to glyceraldehyde 3-phosphate dehydrogenase (mouse anti-GAPDH) was obtained from Santa Cruz Technology (Santa Cruz, CA, USA).
Cell culture
Human osteosarcoma HOS cells (American Type Culture Collection, Manassas, VA, USA) were cultured in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (Hyclone Laboratories, Logan, UT, USA) in a humidified atmosphere of 5% CO2 and 95% air at 37℃. Exponentially growing cells were detached using 0.25% trypsin-EDTA (Gibco, Grand Island, NY, USA) at 37℃ for 2 min. For exposure to bortezomib, 1 mM stock solutions were freshly prepared before every experiment and filter sterilized using a 0.2-µm syringe filter. Bortezomib was added to the culture medium in a concentrated form and mixed gently. The cultures were incubated with bortezomib at various concentrations for various times at 37℃ as described in the following sections.
Cell morphology
The HOS cells, seeded at 5 × 105 cells/ ml into 6-well flat-bottomed microplates, were incubated for 24 h at 37℃ in culture medium (with or without bortezomib at 5, 10 and 20 nM) and examined under an Eclipse TE2000-S microscope (Nikon, Mississauga, ON, Canada) equipped with a Retiga 1300R digital charged-coupled camera (Qimaging, Burnaby, BC, Canada). Photographs were taken at 100 × magnification.
Cell viability assay
Viability of HOS cells following treatment with bortezomib was measured using the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Tokyo, Japan). HOS cells were seeded at 1 × 104 cells/well into a 96-well flat-bottomed microplate and allowed to attach at 37℃ for 12 h. Cultures were then treated with: 100 µl culture medium containing either DMSO (0.02% [v/v]), or bortezomib at 5, 10 or 20 nM, and incubated for 24 h at 37℃; 10 nM bortezomib and incubated for 0, 12, 24 or 48 h at 37℃. Following these treatments, CCK-8 (10 µl) was added to each well and incubated for 2 h at 37℃ in order to perform viable cell counts. Viable cells were counted by absorbance measurements at 450 nm using an Infinite® M200 auto microplate reader (Tecan, Grödig, Austria). All experiments were performed in triplicate in three separate experiments.
Cell-cycle analysis
In this experiment, HOS cells, seeded at 5 × 105 cells/ml into 6-well flat-bottomed microplates, were treated with DMSO (0.02%) or bortezomib (5, 10, or 20 nM) for 24 h at 37℃. HOS cells were then suspended in ice-cold 0.01 M phosphate-buffered saline (PBS, pH 7.4) and fixed in 70% ethanol at −20℃ for 16 h. Cells were stained with propidium iodide (PI), as previously described. 18 The population of nuclei in each phase of the cell cycle was determined using CellQuest™ software, version 3.3 (BD Biosciences, San Jose, CA, USA) and analysed using WinMDI software, version 2.8 (Becton Dickinson, San Jose, CA, USA).
Annexin V and PI staining
HOS cells, seeded at 5 × 105 cells/ml into 6-well flat-bottomed microplates, were treated with DMSO (0.02%) or bortezomib (5, 10, or 20 nM) for 24 h at 37℃. Apoptosis was assessed by observing the translocation of phosphatidyl serine to the cell membrane surface, detected using annexin V-fluorescein isothiocyanate (FITC)/PI apoptosis detection kit (KeyGEN BioTECH Co. Ltd., Nanjing, China), as previously described. 19 Apoptotic cells were detected using a FACSCalibur™ flow cytometer (BD Biosciences) and data were analysed using CellQuest™ software, version 3.3 (BD Biosciences). Quadrants were positioned on annexin V/PI plots to distinguish living cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−) and late apoptotic/secondary necrotic cells (annexin V+/PI+).
Transmission electron microscopy
Changes in cell ultrastructures caused by bortezomib treatment were visualized using transmission electron microscopy (TEM). Autophagy was evaluated by examining autophagosome formation. HOS cells, seeded at 5 × 105 cells/ml into a 6-well flat-bottomed microplate, were treated with DMSO (0.02%) or bortezomib (20 nM) for 24 h at 37℃. Bortezomib-treated and untreated HOS cells were fixed with a solution containing 2.5% glutaraldehyde plus 2% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.3) for 1 h at room temperature. Following fixation, samples were postfixed in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer (pH 7.3) for 1 h at room temperature. Cells were then dehydrated in a graded series of ethanol, substituted with propylene oxide, then embedded in epoxy resin (Oken, Tokyo, Japan). Ultrathin sections were observed under TEM (JEOL JEM-1200EX; JEOL, Japan) at 100 kV. Images were digitally acquired from a randomly selected pool of 10–15 fields under each condition.
Protein extraction and Western blots
To investigate whether MAPKs play a role in apoptosis and autophagy, HOS cells, seeded at 5 × 105 cells/ml into 6-well flat-bottomed microplates, were treated with DMSO (0.02%) or bortezomib (5, 10, or 20 nM) for 24 h at 37℃. Collected cells were washed with 0.01 M cold PBS (pH 7.4). Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton™ X-100, 1 mM phenylmethylsulphonyl fluoride and protease inhibitor cocktail set I (Calbiochem, La Jolla, CA, USA). Protein content was quantified using a Bicinchoninic Acid Protein Assay Kit (Biocolor Biotech, Shanghai, China) according to the manufacturer’s instructions. Total protein extracts (80 µg) were separated by 10% or 15% sodium dodecyl sulphate polyacrylamide–gel electrophoresis using XCell SureLock® Mini-Cells (Bio-Rad, Hercules, CA, USA) and transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 5% nonfat milk in Tris-buffered saline Tween-20 buffer (10 mM Tris pH 7.5, 150 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature, then incubated overnight at 4℃ with various primary antibodies (p-MEK; p-ERK1/2; p-JNK; p-p38; ERK1/2; MEK1/2; PARP; BAX; BCL-2; JNK; p38 – all diluted 1 : 500 – and GAPDH (as the loading control), diluted 1 : 2 000 in blocking buffer(pH 7.3). Membranes were then washed for 3 min with Tris-buffered saline Tween-20 buffer (10 mM Tris pH 7.5, 150 mM NaCl, and 0.1% Tween 20) five times, and incubated with appropriate horseradish peroxidase-labelled secondary antibodies (goat antimouse [diluted 1 : 4 000; Santa Cruz]; goat antirabbit [diluted 1 : 3 000; Santa Cruz]) for 1.5 h at room temperature. Membranes were then washed for 3 min with Tris-buffered saline Tween-20 buffer (10 mM Tris pH 7.5, 150 mM NaCl, and 0.1% Tween 20) five times. Western blot membranes were visualized using an enhanced chemiluminescence kit (ECL Western Blotting Substrate, 250 ml kit; Pierce Chemical, Rockford, IL, USA) and exposed to X-radiography film (Kodak, Japan).
Statistical analyses
Data were presented as mean ± SD. Statistical analyses were performed using the SPSS® software package, version 13.0 (SPSS Inc., Chicago, IL, USA) for Windows®. Statistically significant between-group differences were determined using a two-tailed Student's t-test for comparison between the means. Differences were considered statistically significant when P < 0.05.
Results
Morphological examination of HOS cells showed that incubation with bortezomib (10 and 20 nM) for 24 h caused many cells to round up and start to detach from the polystyrene surface. In the 5 nM bortezomib and DMSO (0.02%) control groups, most of the HOS cells remained well attached (Figure 1A–D).
Representative photomicrographs showing morphology of human osteosarcoma (HOS) cells following culture for 24 h at 37℃ with (A) 0.02% dimethylsulphoxide (control); (B) 5 nM bortezomib; (C) 10 nM bortezomib; (D) 20 nM bortezomib.
Using the CCK-8 assay, HOS cells treated with increasing concentrations of bortezomib (5 nM–20 nM) showed a concentration-dependent decrease in cell growth (Figure 2A). Treatment with 10 or 20 nM bortezomib resulted in significantly reduced cell growth compared with control and 5 nM bortezomib treatment (P < 0.05). Treatment with bortezomib (10 nM) for 0, 12, 24 and 48 h revealed a time-dependent decrease in cell growth that was significantly decreased in the 24 and 48 h groups compared with controls (P < 0.05; Figure 2B).
Human osteosarcoma (HOS) cell viability, measured using Cell Counting Kit-8, following incubation with: (A) 0.02% dimethylsulphoxide (control) or bortezomib (5, 10 or 20 nM) for 24 h at 37℃; (B) 10 nM bortezomib for 0, 12, 24 or 48 h at 37℃; n = 6 wells per treatment for all CCK-8 assays. Data presented as mean ± SD. *P < 0.05, compared with control, two-tailed Student's t-test.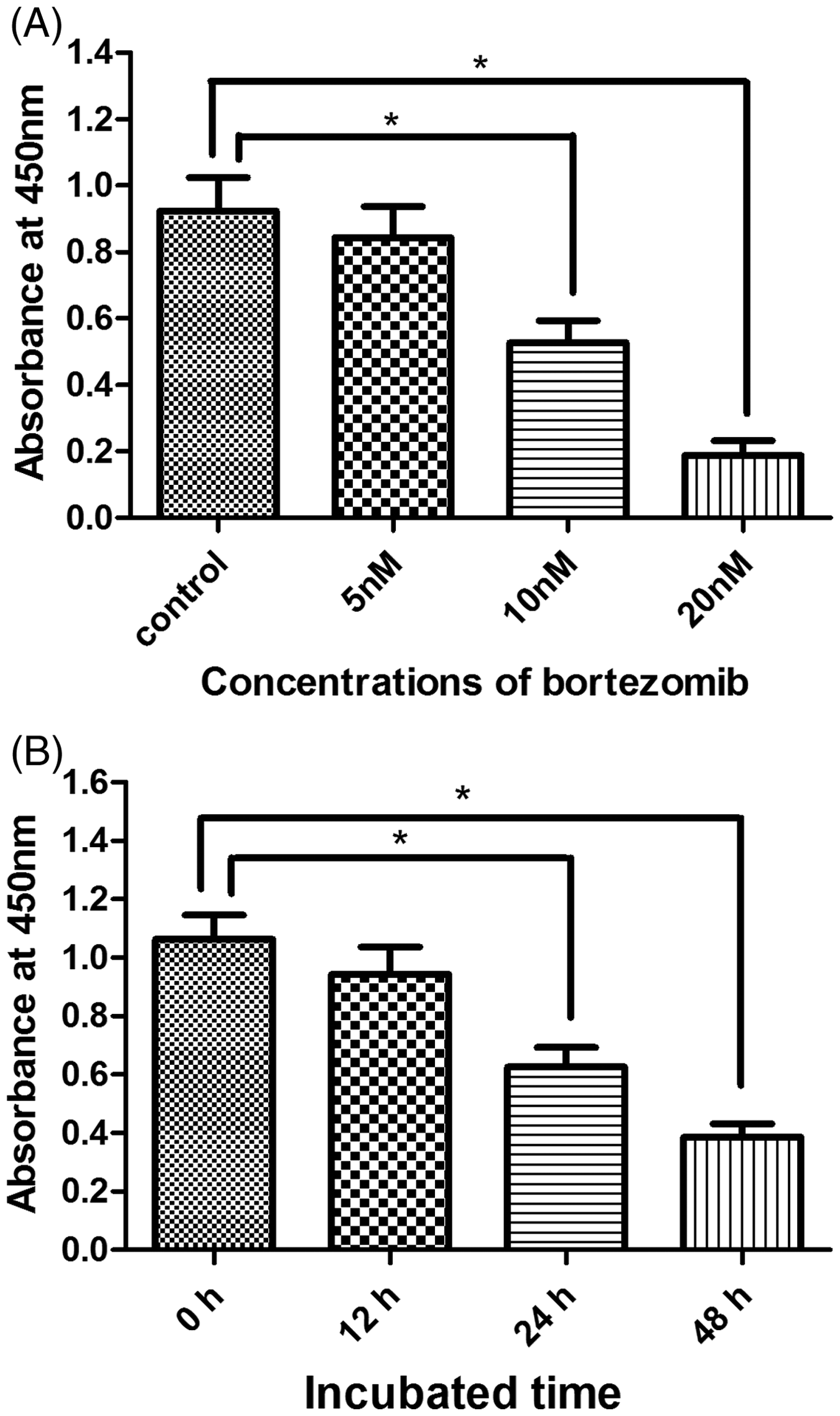
Cell morphology experiments indicated no significant increase in Gap 2 (G2)/Mitosis (M) cell-cycle arrest in HOS cells treated with bortezomib (5 nM) compared with controls (Figures 3A and 3B). As bortezomib concentration increased, the percentage of HOS cells in the G2/M phase significantly increased in cells treated with 10 and 20 nM compared with controls (P < 0.05; Figures 3C–E).
Representative flow cytometry profiles of DNA content in human osteosarcoma (HOS) cells stained with propidium iodide, showing cell-cycle arrest at Gap (G)2/Mitosis (M) phase following treatment with: (A) 0.02% dimethylsulphoxide (control); (B) 5 nM bortezomib; (C) 10 nM bortezomib; (D) 20 nM bortezomib, each for 24 h at 37℃; (E) Percentage of HOS cells in G2/M phase following treatment with bortezomib (0, 5, 10 or 20 nM) for 24 h at 37℃. Data presented as mean ± SD of three independent experiments. *P < 0.05, compared with control, two-tailed Student's t-test.
Representative flow cytometry results revealed the fraction of annexin V+ HOS cells was 9.00% following DMSO treatment and 11.06, 21.25 and 41.87% following treatment with bortezomib at 5, 10 and 20 nM, respectively. The fraction of annexin V+/PI+ HOS cells was 6.26% following DMSO treatment and 7.99, 16.34 and 37.14% following treatment with bortezomib at 5, 10 and 20 nM, respectively (Figure 4A–D). The mean results from three separate experiments showed that <12% of cells were apoptotic when incubated with DMSO and bortezomib (5 nM), whereas 22%–60% of cells were apoptotic following exposure to higher concentrations of bortezomib (10 and 20 nM; Figure 4E), detected by annexin-V/PI staining.
Representative flow cytometry scatter plots showing apoptosis of human osteosarcoma (HOS) cells stained with annexin V-fluorescein isothiocyanate (FITC)/propidium iodide following treatment with (A) 0.02% dimethylsulphoxide (control); (B) 5 nM bortezomib; (C) 10 nM bortezomib; (D) 20 nM bortezomib, each for 24 h at 37℃. Numbers at the corners represent the percentage of cells found in each quadrant; (E) Percentage of annexin V-stained HOS cells following treatment with bortezomib (0, 5, 10, or 20 nM; mean ± SD values, 11.55 ± 2.341, 13.10 ± 1.866, 26.85 ± 4.915 and 52.55 ± 9.521, respectively). *P < 0.05, compared with control, two-tailed Student's t-test.
Western blot analysis demonstrated cleavage of PARP, reduction of Bcl-2, and activation of BAX in HOS cells treated with bortezomib (5, 10, and 20 nM; Figure 5).
Representative Western blots showing cleavage of poly (ADP-ribose) polymerase 1 (PARP), reduction in B-cell chronic lymphocytic leukaemia/lymphoma (Bcl-2) levels, and activation of Bcl-2-associated X protein (BAX) in human osteosarcoma HOS cells treated with bortezomib (0, 5, 10 or 20 nM) for 24 h at 37℃. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. Values at the bottom of each lane represent relative densities compared with GAPDH control bands. Data represent three independent experiments. Relative densities of cleaved PARP, Bcl-2 and BAX in cells treated with 5, 10, or 20 nM bortezomib were significantly different from controls (P < 0.05, two-tailed Student's t-test).
Experiments undertaken using TEM, and Western blotting of LC311 and degradation of p62, showed that bortezomib induced autophagy in HOS cells compared with controls. TEM examination revealed that treatment of HOS cells with 20 nM of bortezomib for 24 h resulted in the formation of autophagosomes (Figure 6). Western blot analysis showed increased conversion of LC3I to LC3II with increased concentrations of bortezomib, accompanied by a progressive decrease in p62 levels (Figure 7).
Representative transmission electron micrographs showing human osteosarcoma HOS cells treated (A) without or (B) with bortezomib (20 nM) for 24 h at 37℃. Arrows indicate intracellular autophagosomes. Following treatment, cells were fixed in 2.5% glutaraldehyde and postfixed in 1% osmium tetroxide. Ultramicrotome sections were poststained and imaged on a JEOL JEM-1200EX transmission electron microscope at 100 kV. Representative Western blots showing increased conversion of microtubule-associated protein 1 light chain 3-α (LC3)I to LC3II and reduced levels of sequestosome 1 (p62) in human osteosarcoma HOS cells treated with 0.02% dimethylsulphoxide (0) or bortezomib (5, 10 or 20 nM) for 24 h at 37℃. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. Values at the bottom of each lane represent relative densities compared with GAPDH control bands. Data represent three independent experiments. Relative densities of LC3 II and p62 in cells treated with 5, 10, or 20 nM bortezomib were significantly different from controls (P < 0.05, two-tailed Student's t-test).

Following bortezomib treatment, no significant changes in MEK, ERK, p38 and JNK levels were observed. However, levels of phosphorylated forms of MEK, and ERK decreased, whereas levels of phosphorylated p38 and JNK proteins increased, in a bortezomib concentration-dependent manner (Figure 8).
Representative Western blots showing levels of phosphorylated (p)-mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (p-MEK), MEK, p-ERK, ERK, p–p38, p38, p-c-Jun N-terminal kinase ([p]-JNK) and JNK, in human osteosarcoma (HOS) cells treated with 0.02% dimethylsulphoxide or bortezomib (20 nM) for 24 h at 37℃. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control. Values at the bottom of each lane represent relative densities compared with GAPDH control bands. Data represent three independent experiments. Relative densities of p-MEK, p-ERK, p-p38 and p-JNK in cells treated with 20 nM bortezomib were significantly different from controls (P < 0.05, two-tailed Student's t-test).
Discussion
The present study demonstrated that bortezomib was capable of inducing both apoptosis and autophagy via MAPK pathways, in a human osteosarcoma cell line, by decreasing p-MEK/ERK and increasing p-JNK/p38, in a concentration-dependent manner. These results suggest that bortezomib could be a potentially important chemotherapeutic drug for the treatment of osteosarcoma.
Limb salvage and multiagent chemotherapy, including drugs that cause apoptosis, are the standard treatments for patients with osteosarcoma. The present study showed that nanomole ranges of the proteasome inhibitor bortezomib could inhibit cell proliferation and induce both apoptosis and autophagy in the human osteosarcoma HOS cell line, in a time- and concentration-dependent manner. First, it was shown that treatment of HOS cells with increasing concentrations of bortezomib led to cell death-related changes in morphology and a concentration-dependent decrease in cell growth. Secondly, treatment with bortezomib resulted in a significantly larger proportion of annexin V-stained cells, and induced cell-cycle arrest in a larger fraction of HOS cells at G2/M phases, suggesting that cell-cycle arrest at this phase is one of the mechanisms of botezomib-induced cell growth inhibition. These results are consistent with those of other investigations into cancer cells including tumour necrosis factor receptor superfamily, member 8 (CD30)-positive anaplastic large cell lymphoma, 16 and cell lines derived from Ewing’s sarcoma family of tumours 17 and human colon cancer cells. 20
In addition to inducing cell-cycle arrest at G2/M phases, the present study showed concentration-dependent inhibition of ERK phosphorylation by botezomib in HOS cells, suggesting that bortezomib may inhibit HOS cell proliferation through inhibition of ERK phosphorylation. These results indicate that bortezomib-induced growth inhibition in osteosarcoma cells is mediated, at least in part, via inhibition of MAPK pathways. Other investigators demonstrated that bortezomib induced G2/M arrest through intracellular reactive oxygen species-inducible ataxia telangiectasia mutated phosphorylation in human colon cancer cells. 20
Bortezomib was shown to induce HOS cell apoptosis in the present study by the significantly increased proportion of annexin V-stained cells in response to increasing concentrations of bortezomib, increased cleavage of PARP, reduced levels of Bcl-2 and increased levels of BAX. PARP is an important regulator of apoptosis, and increased cleavage of PARP is an indicator of apoptosis. 21 The antiapoptotic protein, Bcl-2, and the proapoptotic protein, BAX, are members of the Bcl-2 protein family, with opposite roles in regulation of apoptosis. The increased levels of Bax and decreased levels of Bcl-2 observed in the present study provided further evidence for bortezomib-induced apoptosis in HOS cells. In addition to altered levels of Bcl-2 family proteins, increased levels of JNK and p38 were observed in bortezomib-treated HOS cells, suggesting that bortezomib induced apoptosis through activation of JNK and p38 MAPK pathways. The contribution of elevated JNK and p38 activity to bortezomib-induced apoptosis was also supported by the observation of a concomitant increase in annexin V staining. The present findings suggest that, in addition to altering the ratio between the antiapoptotic (Bcl-2) and proapoptotic (BAX) proteins, JNK/p38 activation plays an important role in mediating bortezomib-induced apoptosis in HOS cells. Bortezomib-induced apoptosis has been reported in several types of cancer cells,16,17,20,22 and proteasome inhibition has been found to induce runt-related transcription factor 2 (RUNX2) and RUNX2-dependent BAX expression, triggering osteosarcoma cell apoptosis. 23 The present study supports the theory that bortezomib modulates MAPK pathways to induce apoptosis.
The present study revealed that bortezomib could induce autophagy, which is a tightly regulated lysosome-dependent catabolic pathway, important in the regulation of cancer development and progression, and in determining the response of tumour cells to anticancer therapy. 24 Bortezomib-induced autophagy was demonstrated in HOS cells by the dose-dependent conversion of LC3I to LC3II, the reduction of p62 and the maturation of autophagosomes. LC3 is a major constituent of the autophagosome. During autophagy, the cytoplasmic form (LC3I) is processed and recruited to the autophagosomes, where LC3II is generated by site-specific proteolysis and lipidation near to the C-terminus. The hallmark of autophagic activation is the formation of cellular autophagosome punctae containing LC3II, while autophagic activity is measured biochemically as the amount of LC3II that accumulates in the absence or presence of lysosomal activity. Thus, the conversion of LC3I to LC3II is indicative of autophagic activity. 25 Protein p62 is an autophagic adapter, and a receptor for cargo destined to be degraded by autophagy, including ubiquitinated protein aggregates destined for clearance; it is able to bind ubiquitin and also LC3, thereby targeting the autophagosome and facilitating clearance of ubiquitinated proteins. 26 Autophagy generally plays dual roles in cellular death or survival, either inducing type II programmed cell death, or preventing the accumulation of damaged proteins and organelles during stress. 27 The results of the present study suggest that autophagic cell death may contribute, at least in part, to bortezomib-induced apoptosis. Manipulation of autophagy may be useful in preventing cancer development, by limiting tumour progression and increasing the efficacy of cancer treatments.
The present study supports the theory that bortezomib induces apoptosis and autophagy via modulating MAPK pathways. Apoptosis and autophagy may be interconnected under some circumstances, and their interactions may manifest themselves in several ways. Autophagy is thought to be a primary cell response to apoptosis induction. 28 When the autophagic capacities are overloaded, mitochondrial proapoptotic factors activate cell death. 28 Depending on the cell type and stimulus, autophagy may be a necessary preceding step in switching on apoptosis.23,29
There are substantially overlapping signalling networks between autophagy and apoptosis, including various kinase pathways, such as MAPKs. MAPK families play a vital role in complex cellular processes including proliferation, differentiation, development, transformation and apoptosis. The activation of MAPKs is induced by various molecules, from MEKKs to MEKs, through a multistep process. MEKs phosphorylate ERK1 and ERK2 respectively, which increases their enzymatic activities. Activated ERKs translocate to the nucleus and transactivate transcription factors, altering gene expression to promote growth, differentiation or mitosis. 30 The JNK signal transduction pathway is implicated in multiple physiological processes, and JNK activation was an important event in apoptosis in the present study. p38 MAPK appears to play a major role in apoptosis, and can be activated by cellular stress including ultraviolet irradiation, heat shock, high osmotic stress, lipopolysaccharide, protein synthesis inhibitors and pro-inflammatory cytokines. 31 Activated p38 kinase can cause mitotic arrest in somatic cell cycles at the spindle assembly checkpoint. 30 It is likely that MAPK pathways could be central in the regulation of both apoptosis and autophagy, and that bortezomib may induce both apoptosis and autophagy via modulation of MAPK pathways.
The results of the present study suggest there is crosstalk between bortezomib-induced autophagy and apoptosis in HOS cells. According to our findings, MAPK activities were affected by bortezomib (MEK/ERK activity was inhibited, whereas JNK and p38 kinase activity was stimulated). Bortezomib resulted in cytotoxicity in Waldenstrom macroglobulinaemia cells, mediated through ERK signalling pathways, which were found to be critical for survival of the Waldenstrom macroglobulinaemia cells. 32 The present study suggested that inhibition of the MEK/ERK MAPK pathway by bortezomib represents an important mechanism in cytotoxic activity. JNK and p38 MAPK family members function in a cell context-specific and cell type-specific manner to integrate signals that affect proliferation, differentiation, survival and migration. 13 The present results have demonstrated that activation of JNK and p38 and concurrent inhibition of MEK/ERK are critical for induction of apoptosis in osteosarcoma cells. Activation of either JNK or p38 MAPK signalling pathways, or both, may be necessary but not sufficient for apoptosis under all conditions. 33 Proteasome inhibitors are multipathway inhibitors, therefore the present findings may represent a small part of a large bortezomib-induced signalling network in osteosarcoma cells.
In conclusion, the present study demonstrated that the differential effects of bortezomib on the MEK/ERK and JNK-p38 MAPK pathways led to significant cytotoxic effects in osteosarcoma cells. The inhibitory effects of bortezomib appeared to rely, in part, on inhibition of the MEK/ERK pathway and augmentation of the JNK/p38 MAPK pathway, which may be important therapeutic targets in osteosarcoma cells. Proteasome inhibition may affect a wide range of cellular mechanisms, however. Regardless of which pathways are involved, suppression of tumour growth and induction of autophagy and apoptosis by bortezomib in osteosarcoma cells represent promising and potentially important findings. Based on the present observations, a pre- or postoperative bortezomib regimen may be an effective complement to current chemotherapies for osteosarcoma, and perhaps other types of cancer.
Footnotes
Declaration of conflicting interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by grants from The National Natural Sciences Foundation of China (No. 81172543 and 81001193).