
Abstract
Background
Genomic tests may improve upon clinical risk estimation with traditional prognostic factors. We aimed to explore how evidence on the prognostic strength of a genomic signature (clinical validity) can contribute to individualized decision making on starting chemotherapy for women with breast cancer (clinical utility).
Methods
The MINDACT trial was a randomized trial that enrolled 6693 women with early-stage breast cancer. A 70-gene signature (Mammaprint) was used to estimate genomic risk, and clinical risk was estimated by a dichotomized version of the Adjuvant!Online risk calculator. Women with discordant risk results were randomized to the use of chemotherapy. We simulated the full risk distribution of these women and estimated individual benefit, assuming a constant relative effect of chemotherapy.
Results
The trial showed a prognostic effect of the genomic signature (adjusted hazard ratio 2.4). A decision-analytic modeling approach identified far fewer women as candidates for genetic testing (4% rather than 50%) and fewer benefiting from chemotherapy (3% rather than 27%) as compared with the MINDACT trial report. The selection of women benefitting from genetic testing and chemotherapy depended strongly on the required benefit from treatment and the assumed therapeutic effect of chemotherapy.
Conclusions
A high-quality pragmatic trial was insufficient to directly inform clinical practice on the utility of a genomic test for individual women. The indication for genomic testing may be far more limited than suggested by the MINDACT trial.
New biomarkers and genomic tests hold substantial promise in improving upon clinical risk estimation with traditional prognostic factors. 1 Various genomic profiles are under development, in which genomic testing of tumors improves upon risk prediction with clinical and pathological characteristics in women with breast cancer. 2 Better risk prediction should lead to more targeted use of treatments, that is, that treatment is offered to those for whom the benefits outweigh the harms, such as the burden and costs of tests and treatments.
Ideally, we find predictive markers that relate to the mechanism of action of a treatment. There are at least 2 such success stories in breast cancer. Hormone receptors were discovered about 30 y ago and are key to guiding hormone therapies, which include tamoxifen and aromatase inhibitors. 3 The second success is the discovery of human epidermal growth factor receptor 2 (HER2), a protein that appears on the surface of some breast cancer cells. For HER2-positive women, trastuzumab (Herceptin) is an effective treatment, a this drug directly targets the HER2 receptor. 4 There is an intensive search for “druggable mutations,” with some success in other cancers (e.g., melanoma and lung cancer). 5
Prognostic markers are defined as markers related to the outcome of a disease irrespective of the treatment. Far more examples can be found for prognostic factors than predictive factors such as HER2. Examples in oncology include the extent of disease (e.g., TNM or other staging systems) and functional status (e.g., indicated by comorbidity). 6 Genomic tests, based on the combination of multiple genetic variants, may also be primarily prognostic rather than predictive tests.7,8 One such test is a 70-gene signature for women with breast cancer, the Mammaprint, which has been approved for the claim to have a prognostic effect and not for a predictive effect. 9 The Mammaprint was studied recently in a large randomized controlled trial: Microarray in Node-Negative and 1 to 3 Positive Lymph Node Disease May Avoid Chemotherapy (MINDACT). 10 The study goal was to help patients and clinicians avoid chemotherapy, which is the standard for these patients, by adding the genomic signature to the clinical evaluation of women. The MINDACT trial concluded that foregoing chemotherapy led to only a minor reduction in survival without distant metastasis among women with early-stage breast cancer who were at high clinical risk and low genomic risk. These women hence might not require chemotherapy. The study had sufficient statistical power to estimate the prognostic effect of the genomic signature but was too small to reliably estimate the effect of chemotherapy. 11 Moreover, clinical risk was categorized as either high or low. Contemporary risk calculators, such as Adjuvant!Online and PREDICT, provide continuous risk predictions.12,13
We aimed to explore how evidence on the prognostic strength of a gene signature can contribute to individualized decision making on chemotherapy for women with early breast cancer. We hereto supplement the MINDACT analysis with a more refined decision-analytic approach, focused on estimating benefit for individual women. We find that the indication for testing and treatment needs to be restricted radically given plausible estimates of the decision threshold and the therapeutic effect of chemotherapy.
Methods
Design of the MINDACT Trial
We based our analyses on data reported from a large randomized controlled trial (MINDACT). 10 Genomic risk was evaluated in 6693 women using the Mammaprint, which had been validated before as a prognostic factor in several studies. 14 . The genomic risk was categorized as low or high (“G-low” or “G-high”), excluding those in which the MammaPrint was not feasible, because of too few tumor cells, an inadequate/absent sample, or other reasons.
Clinical risk was evaluated using a modified version of Adjuvant!Online (version 8.0 with HER2 status). Adjuvant!Online has been promoted as a web tool for assessing the risk of breast cancer mortality for individual women. Prognostic factors in Adjuvant!Online include patient information (age, menopausal status, comorbidity) and tumor characteristics (tumor size, number of positive axillary nodes, hormone receptor status). 12
Prognostic factors from Adjuvant!Online were categorized and linked to a binary classification as “low” or “high” clinical risk. Women at low clinical and low genomic risk did not receive chemotherapy (C-low/G-low), whereas those at high clinical and high genomic risk were recommended chemotherapy (C-high/G-high). Women with discordant risk results (i.e., C-low/G-high, or C-high/G-low) were randomly allocated to chemotherapy or no chemotherapy (discordant pair design). A secondary analysis assessed the effectiveness of chemotherapy, although the study was admittedly underpowered to reliably quantify a small treatment effect. 10 The primary research question in MINDACT focused on the C-high/G-low patients, to test whether the lower boundary of the 95% confidence interval for the 5-y distant metastasis–free survival (5y DMFS) was 92% or higher among the C-high/G-low patients who did not receive chemotherapy. The accompanying editorial specifies the underlying reasoning: if the 5y DMFS is already high, the benefit from chemotherapy cannot be that large that it is worth the burden of treatment. 11 This reasoning is in line with classical decision theory on treatment choice. 15
Design of the Decision Analysis
We set out to refine the indication for genomic testing and chemotherapy based on absolute benefit from adjuvant chemotherapy for individual women. 16 We hereto performed the following steps:
We started with estimating the continuous distributions of clinical and clinical plus genomic risks to closely match the observed risks with binary categorizations in the MINDACT trial.
We based the relative effect of chemotherapy on previous randomized trials for individualized estimates of benefit of treatment in terms of absolute 5y DMFS improvement.
We estimated the fractions of women who would be reclassified as benefiting from chemotherapy or not given a threshold for minimum required benefit. 16
We then estimated the impact on fraction tested, receipt of chemotherapy, and prognosis and summarized the benefits of targeted treatment according to genomic risk in a summary measure for net benefit (NB). 17
Sensitivity analyses were performed to assess key drivers of the robustness of our findings.
1. Risk distributions
We created a hypothetical MINDACT cohort 1000 times the size of the trial to mitigate the role of chance (n = 6,693,000 women, see R code in the Supplementary Appendix). We generated a random log-normal variable to simulate the distribution of clinical risk (defined as 1 – 5y DMFS), motivated by the fact that most risk distributions from clinical prediction models follow such a distribution when considered at the appropriate scale. 18 We added the genomic risk result as a correlated random binary variable, such that the higher frequency of women with high genomic risk among those at high clinical risk was closely approximated (Supplementary Table S1). Finally, a binary variable for chemotherapy was created to simulate randomization. We calibrated these risk distributions with the 2.41 relative risk of the genomic test result (details in the Supplementary Material).
2. Benefit of chemotherapy
In MINDACT, the pooled relative risk was 0.88 for chemotherapy, or a relative risk reduction of 12%, combining the results from the 2 discordant groups with inverse variance weighting of the log(HR).
In a base-case analysis, we assumed a relative effect for chemotherapy of 0.8, in line with effects in recent meta-analyses. 19 In sensitivity analyses, we considered relative effects of 0.9 and 0.5, corresponding to pessimistic and highly optimistic perspectives on the effect of chemotherapy, respectively. Individual benefit was the difference in calculated 5y DMFS risk with and without chemotherapy. Absolute benefit was estimated for each of the simulated patients. The median 5-y risks for simulated, MINDACT-like, patients were 3.2% and 4.0% with and without chemotherapy, respectively, or a median absolute benefit of 0.8% (Figure 1).
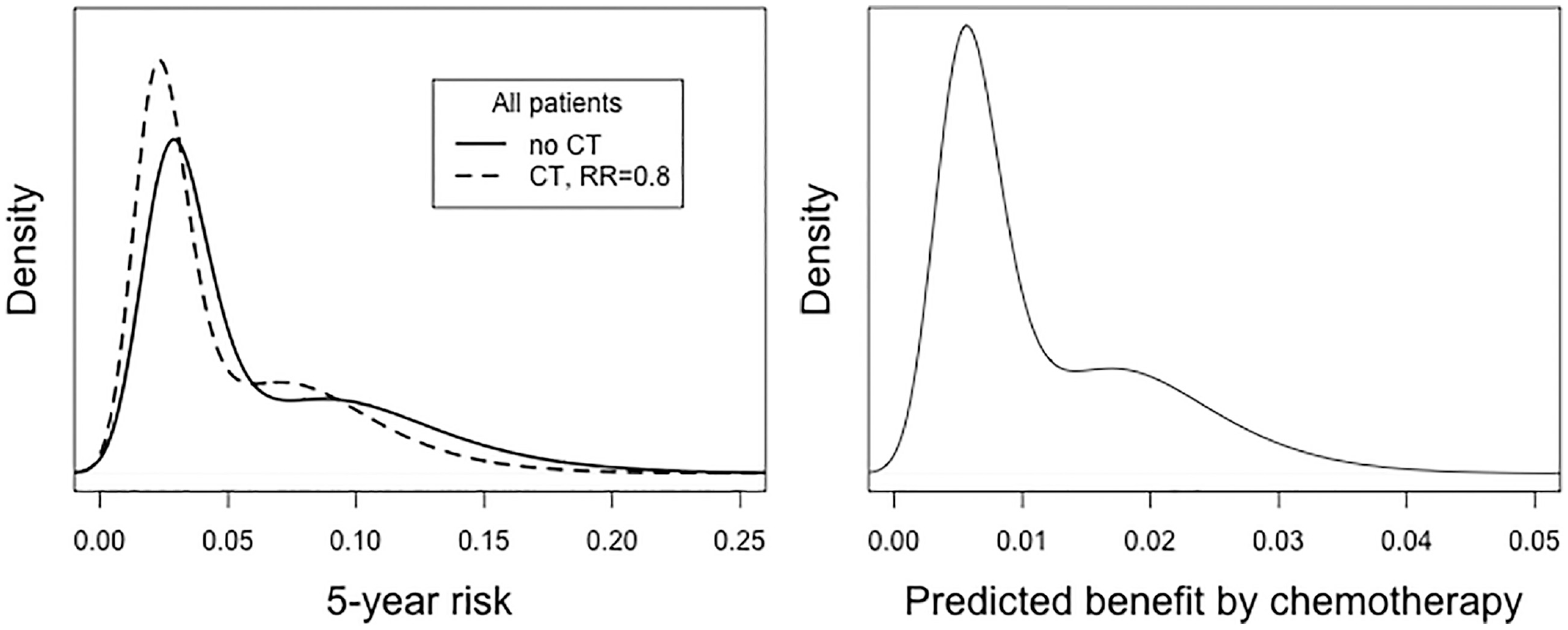
Simulated risk distribution of women enrolled in the MINDACT trial based on clinical and genomic risk, calibrated to MINDACT 5-y distant and metastasis free survival (DMFS), with and without chemotherapy (CT), assuming a relative risk reduction by CT of 20% (hazard ratio = 0.8). The median risks were 3.2% and 4.0% with and without chemotherapy, respectively (left panel), or a median benefit of 0.8% (right panel).
3. Reclassificaton by genomic risk
We followed clinical consensus in defining the threshold for the minimum benefit of chemotherapy as 3%. 20 Empirical studies eliciting preferences suggested a wide range of required benefits (medians 0.1%, 2%, 5%, and 7%).21,22 This 3% threshold was also included in a American Society of Clinical Oncology guideline. 23 Here, the relative risk reduction by chemotherapy was assumed to be about 30%. With a baseline risk of 10%, a benefit of about 3% balances the harms of chemotherapy (2%–3% assumed).
4. NB of treatment
The decision-making strategies differed in the fraction tested, the fraction receiving chemotherapy, and the overall prognosis. To summarize strategies, we follow the decision-analytic principle of defining benefits and harms. 17 The increase in 5y DMFS was the benefit of testing, and the burden of chemotherapy is the harm. The decision threshold defines the relative weight w of the burden of chemotherapy to the gain in 5y DMFS. A 3% threshold implies that women are willing to receive up to 1/w = 1/0.03 = 33 courses of chemotherapy to prevent one 5-y event. The NB is defined as:
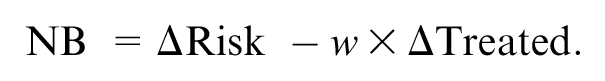
Here,
Sensitivity analyses
Key parameters were varied for NB of testing by the Mammaprint and targeted treatment by chemotherapy. Plausible ranges were considered as indicated above.
Results
Key Results from the MINDACT Trial
The 5y DMFS was estimated at 94.7% (95% confidence interval, 92.5%–96.2%) in the C-high/G-low women who did not receive chemotherapy and were in the primary test population. 10 Based on this result, the criterion for noninferiority was met, because the lower boundary of the 95% confidence interval exceeded 92%. In the C-high/G-low women who were randomized and in the intention-to-treat population, the 5y DMFS with chemotherapy was 95.9% (95% confidence interval [CI], 94.0 to 97.2) versus 94.4% (95% CI, 92.3 to 95.9) without chemotherapy. Hence, those with chemotherapy had a 1.5 percentage point higher 5y DMFS than those who did not receive chemotherapy. The adjusted hazard ratio for DMFS with chemotherapy versus no chemotherapy in this group was 0.78 (95% CI, 0.50 to 1.21; P = 0.27). 10
There was no evidence in favor of directing chemotherapy on the basis of genomic risk among randomized patients in the C-low/G-high group. The 5y DMFS was 95.8% (95% CI, 92.9 to 97.6) among those allocated to chemotherapy and 95.0% (95% CI, 91.8 to 97.0%) among those allocated to no chemotherapy. The adjusted hazard ratio for DMFS with chemotherapy versus no chemotherapy in this group was 1.17 (95% CI, 0.59 to 2.28; P = 0.66).
The hazard ratio for the genomic test was 2.41 (1.79–3.26) in a Cox proportional hazards model, adjusting for clinical and pathological factors and treatment (Supplementary Table S10. 10 This is the key piece of evidence for the prognostic effect of the test (G-high v. G-low, Table 1).
Results for the Discordant-Pairs Design as Applied in the MINDACT Trial versus Decision-Analytic Modeling of Individual Benefit
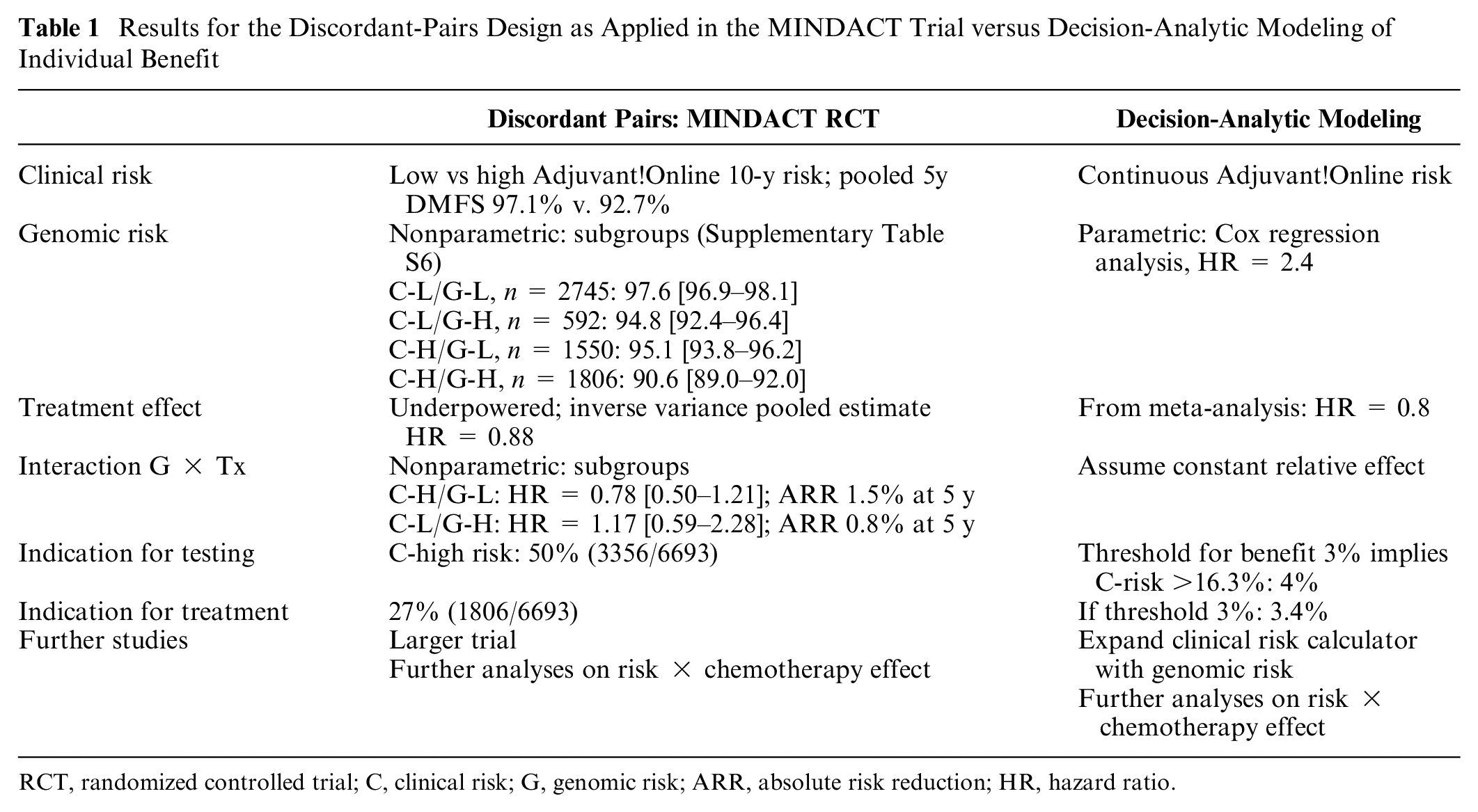
RCT, randomized controlled trial; C, clinical risk; G, genomic risk; ARR, absolute risk reduction; HR, hazard ratio.
Interpretation of the MINDACT Trial
The MINDACT investigators concluded that chemotherapy could be avoided in C-high/G-low women, at a cost of a slightly lower 5y DMFS. Women defined as C-high should be tested for genomic risk (3356/6693, 50%). Among these, 1550 had a low genomic risk (23% of the total group) and would not need chemotherapy, whereas 1806 (27% of the total group) had a high genomic risk and would be candidates for adjuvant chemotherapy. 10
Results from Decision Analysis
Individual benefit was the difference in calculated 5y DMFS risk with and without chemotherapy (Figure 1). In line with the MINDACT findings, the predicted benefit from chemotherapy was limited for C-low women, irrespective of the genomic risk (Figure 2). The benefit distribution among those in the C-high/G-low group varied widely, with a median of 0.8% (2.5% and 97.5% percentiles, p2.5 – p97.5: 0.7%–1.4%, Figure 2). The widest benefit distribution was among those in the C-high/G-high group: median 2.2% (p2.5–p97.5: 1.6%–3.8%). Remarkably, 87% of the women in this high-risk group were expected to have less than 3% benefit. Overall, only 3.3% of the women were expected to have more than 3% benefit from chemotherapy considering their risk estimate from the combination of clinical plus genomic information. Thus, the MINDACT strategy would suggest chemotherapy for far too many women using a 3% threshold for the required benefit and assuming a 0.8 relative effect of chemotherapy.
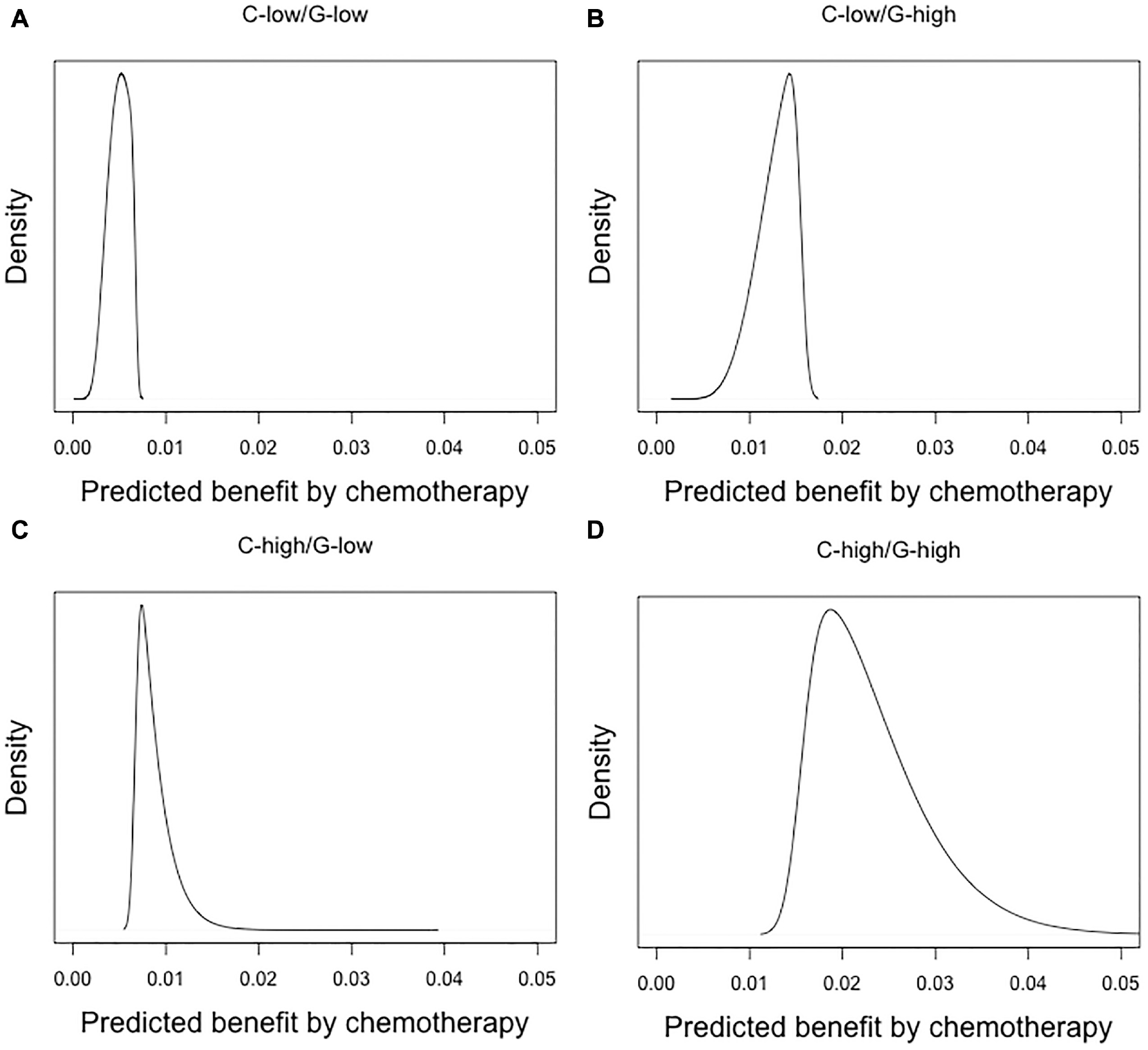
Distribution of predicted absolute benefit in terms of 5-y risk of mortality or distant metastases for women enrolled in MINDACT based on a relative risk reduction by chemotherapy of 20% (hazard ratio = 0.8). Subgroups were defined by combinations of clinical risk (C-low or C-high) and genomic risk (G-low or Ghigh). The median benefits were 0.4%, 1.1%, 0.8%, and 1.9%, respectively.
With clinical risk at a continuous scale, we should administer chemotherapy if the predicted risk without chemotherapy exceeded 16%. Above this risk, the absolute benefit would exceed 3%, assuming a relative effect of chemotherapy of 0.8. Only 2.2% of women were in this category. With the genomic test, 0.3% would be reclassified to less than 3% expected benefit. On the other hand, the test would reclassify 1.3% of women from lower than 3% benefit according to clinical risk to a higher than 3% benefit according to clinical plus genomic risk estimation (Figure 3). Thus, the genomic risk would change decision making in 1.6%, leading to chemotherapy for 3.3% rather than 2.2% based on clinical risk alone.
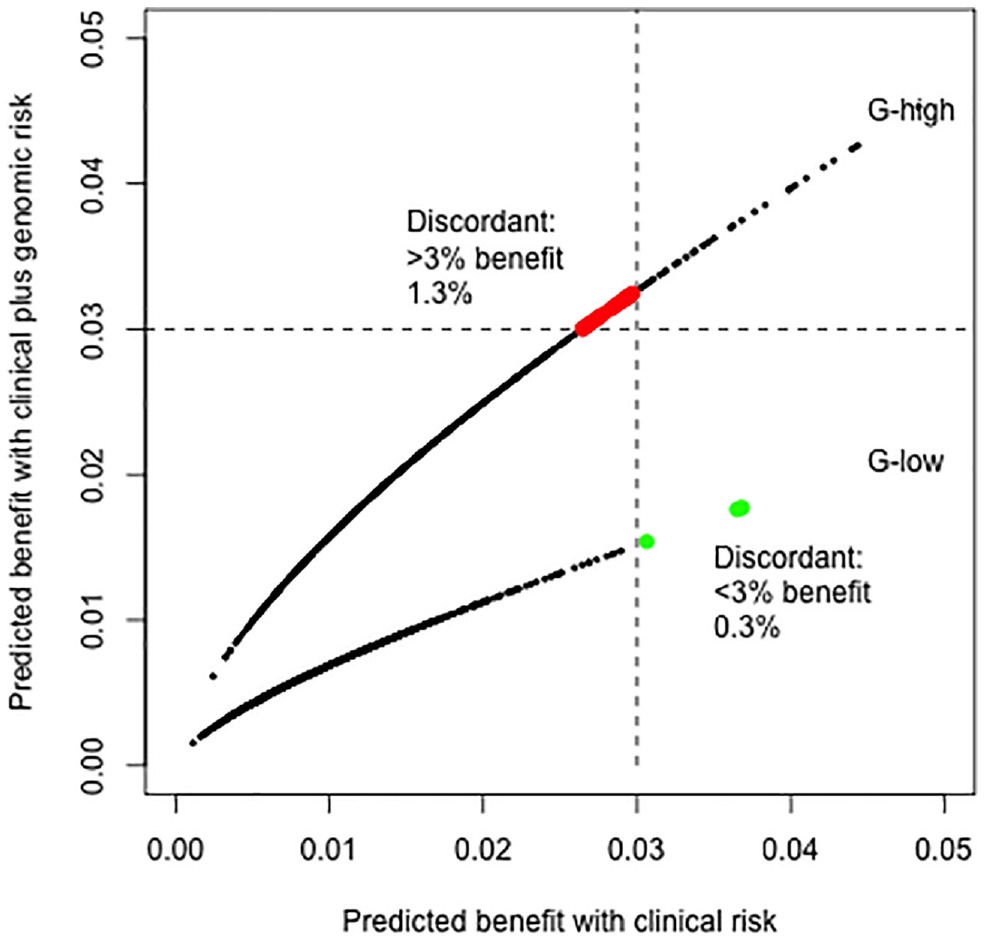
Reclassification by genomic risk. If a 3% threshold (indicated with dotted lines) is used to decide on the use of chemotherapy, 4% of women should be tested for genomic risk, with 1.3% receiving chemotherapy, whereas they would not if decision making were based on clinical risk alone (red dots). In contrast, 0.3% would not receive chemotherapy, although while they would if decision making were based on clinical risk alone (green dots). Thus, the genomic risk would change decision making in 1.6% of the patients. The dots are for a random sample of 2000 hypothetical patients.
Impact on Fraction Tested, Receipt of Chemotherapy, and Prognosis
Genomic testing was not indicated for those with predicted benefits less than 2.6% according to clinical risk, because a positive genomic test result (“G-high”) would not lead to an indication for chemotherapy (Figure 3). Most women fell into this group (96%). Virtually no women would have such high clinical risk that chemotherapy would also be indicated with a G-low result. Thus, 4.0% of the women would be candidates for genomic testing.
Overall, decision making based on clinical risk would lead to an indication for chemotherapy in only 2.2% (as discussed above), with a 5-y risk of 5.89%. Decision making based on clinical plus genomic risk would lead to an indication for chemotherapy in 3.3% and a 5-y risk of 5.86%. Thus, in 1000 women, chemotherapy would be given to 11 more women (+1.1%), leading to 0.3 more with 5y DMFS (+0.03%). The simpler strategy based on the MINDACT dichotomization of clinical and genomic risk would lead to more testing (for 50% C-high women), more chemotherapy (for 27% in the C-high/G-low group), and a lower 5-y risk (5.36% overall; Table 2).
Fraction Tested, Receiving Chemotherapy, overall 5-y risk of death or Distant Metastasis, and Net Benefit of treatment in a simulated MINDACT population a
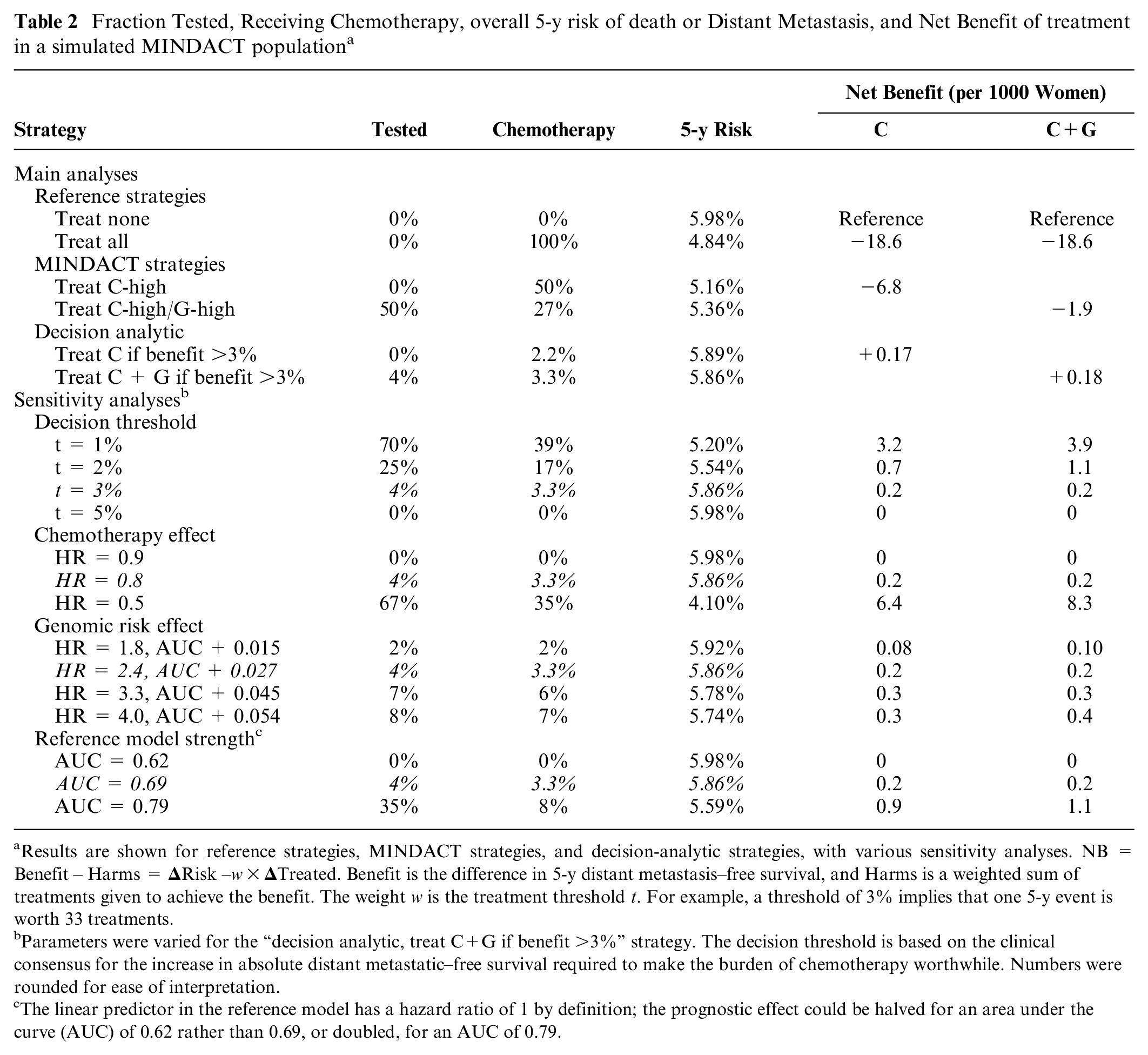
Results are shown for reference strategies, MINDACT strategies, and decision-analytic strategies, with various sensitivity analyses. NB = Benefit – Harms =
Parameters were varied for the “decision analytic, treat C+G if benefit >3%” strategy. The decision threshold is based on the clinical consensus for the increase in absolute distant metastatic–free survival required to make the burden of chemotherapy worthwhile. Numbers were rounded for ease of interpretation.
The linear predictor in the reference model has a hazard ratio of 1 by definition; the prognostic effect could be halved for an area under the curve (AUC) of 0.62 rather than 0.69, or doubled, for an AUC of 0.79.
NB of Treatment
The NB of “treat none” is 0 (no reduction in risk, no treatment) and serves as a reference to other strategies. The NB of “treat all” is negative, since the expected overall risk reduction is below the 3% threshold. Treating only those in the C-high/G-high category implies 270 women receive chemotherapy for 6.2 per 1000 more surviving without distant metastasis at 5 y (5.98%–5.36% = 0.62%). The NB = 6.2 – 3% × 270 = −1.9 per 1000. Treating the C-high category was also overall harmful (NB = −6.8). The decision-analytic strategies based on clinical risk and clinical plus genomic risk had small positive NBs (+0.17 and +0.18 per 1000 respectively; Table 2). In other words, 1 extra woman would survive at 5 y without distant metastasis per 100,000 for the same number of chemotherapy administrations, if decision making were based on the clinical plus genomic risk rather than clinical risk alone.
Sensitivity Analyses
If absolute 5y DMFS required to make the burden of chemotherapy acceptable were 1% rather than the 3%, the indication for testing and targeted chemotherapy would be much larger (70% and 39%, respectively), leading to lower 5-y risk (5.20% rather than 5.86%). Such a reduction in decision threshold could arise if chemotherapy were better tolerated and significantly cheaper than currently. The associated marginal NB of genomic testing would increase markedly if chemotherapy had such a lower threshold level for DMFS, increasing from 0.01 per 1000 population (0.18 v. 0.17) to 0.70 (3.9 v. 3.2; Table 2).
In contrast, a 5% or higher threshold would imply that none of the patients should be tested nor receive chemotherapy. The MINDACT strategy would have a positive NB for thresholds below 2.3%. At a 1% threshold, the NB would be +0.3 net DMFS event prevented per 1000, as compared with decision making based on categorized clinical risk (NB 3.5 v. 3.2).
If chemotherapy would approximately halve the 5-y risks (HR = 0.5), this would have a major impact as compared with assuming HR = 0.8. Among 1000 patients, 667 would have an indication for testing, 350 would be treated, reducing the 5-y risk to 4.10%, for an NB of +83 (compared with +64 per 1000 for decision making on clinical risk alone). The combination of assuming an HR of 0.5 and a threshold for benefit between 1% and 3% would lead to a positive NB (Figure 4). Conversely, a modest treatment effect such as HR = 0.9 would make testing and treating moot (Table 2) unless a low threshold such as 1% was acceptable (Figure 4).
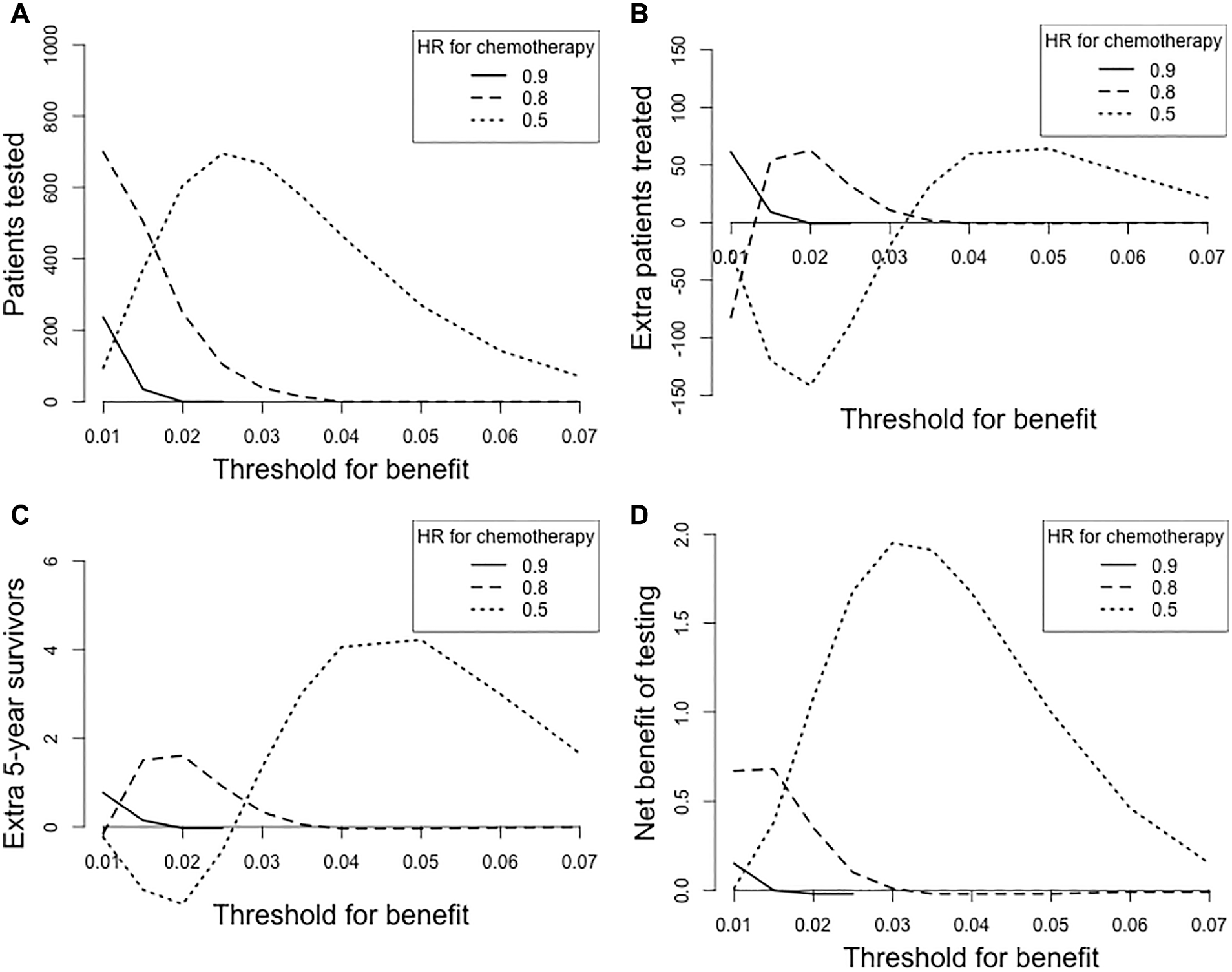
Sensitivity analyses for the “decision analytic, treat C+G if benefit >3%” strategy in Table 2. The threshold was varied between 1% and 7%, with a hazard ratio (HR) for chemotherapy of 0.9, 0.8, or 0.5 (relative risk reductions of approximately 10%, 20%, and 50%, respectively). All numbers are scaled per 1000 women with early breast cancer. For example, for a threshold of 3%, nearly no patients are tested or treated differently with an HR for chemotherapy of 0.8. With an HR for chemotherapy of 0.5, we should test 667 patients (with clinical risk between 1.6% and 5.5%), which would lead to treatment of 20 fewer patients (350 rather than 370), an improvement in 5y DMFS (95.8% to 95.9%, +1.3 per 1000), and a net benefit of 1.9 patients with DMFS (1.3% to 3% × 20 = 1.3 + 0.6 = 1.9 per 1000).
Assuming a larger prognostic effect of the genomic profile had minor impact. A stronger prognostic strength of the clinical risk model would imply somewhat more clinical value of the genomic test, with more testing, more treatment, and better survival (Table 2).
Discussion
This study evaluated the clinical utility of a genomic profile that is prognostic in women with early-stage breast cancer. The MINDACT trial confirmed the clinical validity of the genomic profile and suggested a simple approach for the clinical implementation: test all women who are at high clinical risk and treat only those with high genomic risk. 10 Unfortunately, this trial was underpowered to reliably answer the question about the impact of changing the indication for treatment directly. Moreover, the definition of “high risk” was arbitrary. Our more detailed decision-analytic modeling approach allowed for a more individualized assessment of the indication for testing, following basic decision-analytic principles.16,24 We found that far fewer women would be candidates for genetic testing and far fewer women would be selected for chemotherapy compared with the trial recommendation, given the current evidence for the effectiveness of chemotherapy and the preferences of women for such treatment. In fact, following the MINDACT strategy was harmful according to our model (Table 2). The proposed MINDACT strategy had an unfavorable balance of benefits in terms of improved 5-y disease and DMFS and harms in terms of the burden of chemotherapy. Among women in the high clinical and high genomic risk group, 87% did not meet the 3.0% absolute risk improvement in DMFS to justify use of chemotherapy. If we would add the genomic test to the risk estimation in an optimal way, the NB was close to null according to our decision analysis (1 extra net survivor at 5 y without distant metastasis per 100,000).
Limitations
The input for our analyses was from 3 sources, all of which have their limitations.
Clinical context: The design of the MINDACT trial specified the criterion of a 5y DMFS >92%. This definition of a threshold for absolute risk is uncommon but could be translated to a minimum required benefit of approximately 1.6% if the relative risk of chemotherapy were 0.8. Such a threshold of 1.6% defines the balance between the harms and benefits of treatment. This threshold is in contrast to the minimum benefit of 3% in 10-y survival set at the St Gallen convention, 20 which may translate to a 3% threshold for benefit in 5y DMFS. Preferences may vary widely according to various studies, up to a median required benefit of 7% in 10-y survival in one study. 22 The decision threshold is a key consideration in defining the indication for testing and candidates for chemotherapy (Figure 4).
Trials on treatment effect: The sample sizes in the discordant groups of the MINDACT trial were too small for reliable estimation of the treatment effect. The summary estimate for the 2 groups in MINDACT was 0.88, not far from estimates in meta-analyses of randomized controlled trials. 19 Ideally, the treatment effect would be based on the MINDACT trial, considering the specifics of the treatment regimens, potential historic changes in this therapeutic area, and application in relatively low-risk women. We note that the relative effect of chemotherapy was small in the recently completed TAILORx trial. 25 The overall effect was 1/1.08 = 0.93 in an intermediate-risk group according to the OncotypeDx 21-gene classifier.
MINDACT trial: the MINDACT trial reported on the 5-y risks in 4 distinct groups of patients, based on combinations of clinical risk (high v. low) and genomic risk (high v. low). 10 The relative risk of the genomic profile was studied with multivariable regression analysis (HR = 2.4). We could calibrate the relative risk of the clinical risk to the observed risks in the low and high groups with high accuracy. We hence approximated the continuous risk distribution from Adjuvant!Online, mitigating the categorization used in the MINDACT trial. Ideally, we would have had access to the individual patient data from the trial. Such access was sought and unfortunately denied despite repeated requests.
Sensitivity Analyses
We recognize that the decision threshold will differ between patients, which is why we varied it in sensitivity analyses. In addition, future chemotherapy might be better tolerated than currently available regimens. Moreover, the treatment effect may not be constant across risk groups, although this assumption may be reasonable in many medical domains. 18 In breast cancer, there is substantial interest in how the relative effect of treatment might differ by clinical profile. We might hypothesize that chemotherapy confers a constant absolute risk of harm to all women, irrespective of risk, which would imply a nonconstant relative effect. Our study illustrates the well-known phenomenon that the absolute benefit varies widely between patients, even if we assume that the relative effect is constant.18,26 Our study also confirmed that the magnitude of the treatment effect was the key factor in clinical utility for patients and far more important than refining the indication for such therapy with a genomic profile with a relatively small prognostic effect. Remarkably, the impact of uncertainty of the prognostic effect of the genomic profile on the clinical utility was rather limited (Table 2). Having a better reference model would have more impact by increasing the number of women with risks close to the decision threshold and hence indicating genomic testing for more. Small improvements can easily be imagined for the current version of Adjuvant!Online, for example, including the rather cheap Ki-67 marker, 27 as included in the PREDICT model. 13 The PREDICT model is presented as an easy-to-use web-based decision support tool (https://breast.predict.nhs.uk/).
Further Research
We recognize that the clinical utility of a genomic test can be assessed in more detail according to analyses that focus on quality-adjusted life-years. 24 Utility estimates for the short-term and long-term burden of chemotherapy are then required, as well as estimates of short-term and long-term survival. In our modeling approach, we tried to stay close to the empirical data from a major study and used the threshold concept to define NB as the summary measure for clinical utility. NB combines the benefit of better DMFS versus the burden of chemotherapy in a single number. If we would perform a full cost-effectiveness analysis, we might also consider direct and indirect medical costs. The costs of the genomic test are nontrivial (more than 2500 Euro in Europe) but may be offset by reduced administration of chemotherapy when considered at a group level. 28
An immediate improvement over the current MINDACT strategy is to extend current risk calculators with the MammaPrint as a prognostic factor (Table 1). Indeed, the PREDICT model is regularly updated, and work is under way to include the Mammaprint for refined decision making (https://www.health-holland.com/project/2019/improved-tools-for-breast-cancer-treatment-decisions). Given a clinical profile, the likelihood of a positive MammaPrint result should be estimated, and for both a negative and positive result the impact of treatment should be calculated. Such information will allow for shared decision making on ordering the test.29,30
In the Netherlands, recent clinical guidelines specify that ordering of the Mammaprint test needs to be discussed. Starting from a simple categorization as “you are at high clinical risk, and this test might reclassify you as low risk such that you don’t need chemotherapy” is an oversimplification. The shared decision-making process should focus on the required benefit of chemotherapy. This requires elicitation of a woman’s preferences at the individual level. The question then is whether this personal decision threshold is likely exceeded while considering the clinical risk of the individual woman and the revised risk estimate if the MammaPrint would be negative or positive. A threshold of about 3% implies that, for many women, chemotherapy will not be a viable treatment option and that there will be no value in testing. The benefit of chemotherapy will often be too low, even with a positive MammaPrint. Hence, the test often does not need to be ordered.
Further research is also desired on the effect of chemotherapy, both if assumed constant at the relative risk scale as in the current analysis or if assumed to be risk or subgroup specific. 18 Large-scale randomized trials are required for such estimates, such as the recently completed TAILORx study. 25 An alternative design compared with MINDACT might randomize patients based on a range of expected benefit and allow for evaluation of statistical interaction (risk × treatment). A fuzzy regression discontinuity design could also be considered.31,32 This design was followed in the TAILORx trial: low-risk women did not receive chemotherapy, whereas high-risk women did, and intermediate-risk women were randomized. 25
Conclusions
A pragmatic trial, such as MINDACT, may provide high-quality evidence on the clinical validity of a genomic test. Estimates of the clinical utility depend on the required benefit of treatment. The estimated treatment effect may often be impossible to estimate reliably from a trial with randomization in relatively small discordant groups with few events. Our study illustrates that detailed risk estimation at a continuous rather than dichotomized scale and further decision-analytic modeling is essential to support optimal clinical implementation of trial results evaluating genomic markers.
Supplemental Material
sj-docx-1-mdm-10.1177_0272989X21991173 – Supplemental material for Personalized Decision Making on Genomic Testing in Early Breast Cancer: Expanding the MINDACT Trial with Decision-Analytic Modeling
Supplemental material, sj-docx-1-mdm-10.1177_0272989X21991173 for Personalized Decision Making on Genomic Testing in Early Breast Cancer: Expanding the MINDACT Trial with Decision-Analytic Modeling by Ewout W. Steyerberg, Liesbeth C. de Wreede, David van Klaveren and Patrick M. M. Bossuyt in Medical Decision Making
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.