
Abstract
Background:
Switching between versions of medication products happens commonly despite challenges in achieving bioequivalence and therapeutic equivalence. Central nervous system and psychiatric drugs, especially those that are technically demanding to manufacture and have complex pharmacokinetic properties, such as long-acting injectables (LAIs), pose particular challenges to bioequivalence and safe and efficacious drug switching.
Aims:
To assess whether drugs deemed “bioequivalent” are truly interchangeable in drug switching.
Methods:
We assessed the published literature from January 2017 through June 2023 on PubMed using the MeSH terms “drugs, generic” OR “equivalency, generic” combined with terms for different psychiatric drug classes.
Results:
While most of the published studies returned in the search found that switching drug products was safe and clinically comparable, data on most drug classes other than those primarily indicated in the treatment of seizure disorder were sparse. Some studies also provided evidence that real-world outcomes such as adherence and hospitalizations may also be affected by switching. In addition, a review of bioequivalence testing guidance showed inconsistency across agencies and a lack of product-specific guidance from Health Canada, which raises questions about potential claims of bioequivalence for more complex products such as LAIs.
Conclusions:
Overall, given the difficulty in treating mental health disorders, prescribers should be cautious when switching products and formulations in a patient who has been stabilized on a drug.
Keywords
Introduction
Developing a new drug is costly and time-consuming. Research and development costs for a successful new drug are approximately $1.9–2.9 billion, and an average of 9–12 years will pass between the synthesis of a new chemical entity and approval of a new drug application (Dickson and Gagnon, 2004; DiMasi et al., 2016). For a new drug entering clinical testing, there is only a 12% chance that it will eventually be approved (DiMasi et al., 2016).
Developing novel drugs targeting the central nervous system (CNS) is particularly challenging (Brady and Insel, 2012; Procyshyn et al., 2019; Riordan and Cutler, 2012), and as few as 8% of CNS drug candidates ever become available for clinical use (Riordan and Cutler, 2012). Challenges associated with CNS drug development include a lack of knowledge of the pathophysiologic underpinnings of many CNS disorders, poor predictive quality of preclinical models, and relatively novel mechanisms of action for many CNS drugs (Riordan and Cutler, 2012). Drugs with modified-release and long-acting properties, such as long-acting injectables (LAIs), often require added technical manufacturing demands (Bauer et al., 2023; Patel et al., 2022). Notably, clinical approval success rates for small molecules (chemical compounds) are substantially lower than those for large molecules (biologics), thus contributing to much higher costs (DiMasi et al., 2016).
Given the difficulty and costs of creating new drugs targeting the CNS, it is appropriate that these novel agents undergo extensive, exacting, and time-consuming approval processes. However, generic versions of original products do not face the same approval steps and development costs (Habert et al., 2020), raising the question of whether drugs deemed “bioequivalent” are truly interchangeable.
Generic products—Pharmaceutical equivalence and bioequivalence
Original and generic versions of products are expected to exhibit comparable efficacy and tolerability, but their development and approval processes are quite different. For original products, efficacy and safety must be established through an extensive preclinical and clinical evaluation program. For generic products, which can enter the market after patent protection for the original version has expired or is invalidated by the generic product company, only evidence of pharmaceutical equivalence (same active ingredients, route of administration, and strength or concentration) and bioequivalence to the original product are required for approval; efficacy and safety data are not required (Blier et al., 2019; Habert et al., 2020), and could differ between originals and generics. For example, Rosenthal et al. (2008) reported a case series of seven patients in remission who had either worsening symptoms or relapse following a switch from branded paroxetine or citalopram to a generic version, or between different generic versions, with neither the patients’ nor their doctors’ knowledge. The authors described the challenges of managing the patient’s symptoms and the treatment decisions that led to a return to remission (for four of seven patients, this involved restarting the branded drug or switching to another branded drug).
Pharmaceutically equivalent generic products can differ in several ways from the original version and each other, which could affect efficacy and tolerability. Differences in excipients and salts conjugated to the active pharmaceutical ingredient (Borgheini, 2003) (i.e., inactive components that are typically benign substances, such as lactose, which is used in 45% of oral solid dosage forms) may cause safety and/or tolerability issues for individuals with allergies or sensitivities (Reker et al., 2019). Furthermore, tolerability issues due to a specific changed excipient may cause patients to discontinue an effective agent (Caballero et al., 2021). In addition, potentially dangerous impurities can arise in the synthesis of a drug, particularly if it is more complex and expensive to manufacture; these by-products are more difficult to formally regulate, and generic products can differ in the type and percentage of impurities (Blier, 2003; U.S. Food and Drug Administration, 2010). Physiochemical properties, such as product solubility, friability of tablets, and form of the active ingredient (e.g., the free-base preparation of desvenlafaxine) can also vary (Colvard, 2014). Variations in size, color, shape, and hardness of tablets and capsules are common (Blier, 2007; Borgheini, 2003). Differences in hardness, for instance, can affect the dissolution of a drug and, thus, its absorption from the gastrointestinal tract (Blier, 2007). To this day, batches of generic medications must be pulled from the market, most often because of failed dissolution tests (Health Canada, 2019; Sun Pharmaceutical Industries, 2023). Factors in the manufacturing process, including milling, granulation, particle size, particle shape, particle surface properties, powder flow properties, coating, tablet press, solvent extraction, and presence of moisture, may vary among products, resulting in differences in physiochemical properties (Gupta et al., 2018; Panchal et al., 2023; Procyshyn et al., 2019). Variations of these factors have the potential to influence outcomes when treating a patient with a CNS or psychiatric disorder.
Bioequivalence is established based on pharmacokinetic (PK) parameters, typically maximum concentration (Cmax) and area under the concentration–time curve (AUC) (Meyer, 2001; U.S. Food and Drug Administration, 2001). Historically, the biomedical community came to an assumption that a 20% to 25% change in relevant PK parameters would not pose a significant clinical risk (Midha and McKay, 2009). This translated into a confidence interval (CI) approach first adopted by the U.S. Food and Drug Administration (FDA) following the passage in 1984 of the Hatch-Waxman Act, which stipulated that the 90% CI for the ratio of average PK parameter values between the test and reference product should fall within a limit of 80%–125% to be “bioequivalent” (Boehm et al., 2013; Midha and McKay, 2009). The variability levels resulting from this approach could almost certainly produce inconsistencies in true bioequivalence in some drug classes and dosage forms, such as narrow therapeutic index or critical dose drugs where there is a small difference between therapeutic and toxic doses (Midha and McKay, 2009; U.S. Food and Drug Administration, 2017). As an example, patients with schizophrenia who switched to generic clozapine in a 2001 study experienced worsened symptoms and relapse, even though there were no statistically significant differences in serum concentrations of the drug (Kluznik et al., 2001). The authors explained that a difference in the rate of absorption between the formulations could explain their findings. In a similar vein, because of the nonlinear correlation between dose and dopamine D2 receptor occupancy, critical thresholds exist for dose reduction (Horowitz et al., 2021). At these points, even minor adjustments in dosing may induce substantial changes in receptor occupancy, potentially triggering a relapse.
Definitions of bioequivalence differ across countries or regulatory agencies (Table 1) (Blier et al., 2019; European Medicines Agency, 2010, 2022; Health Canada, 2018, 2023; U.S. Food and Drug Administration, 2001). For Health Canada (2018) compared with the FDA and the European Medicines Agency (EMA), the criteria are the same for AUC, but for Cmax, only the point estimate, and not the 90% CI, must fall between 80% and 125% of that of the reference drug, except for medications with a narrow therapeutic index. Reliance on only the point estimate versus the 90% CI may result in even greater variability in drug concentrations in clinical practice, particularly when considering significant individual variability in Cmax, which could translate to variability in clinical effects related to efficacy and/or safety/tolerability. Of note, these regulatory agencies all require equivalence for Cmax, but not for the time to reach Cmax (Tmax); thus, absorption rate and lag time may differ, potentially resulting in considerable variations in the onset of clinical effects relative to the reference drug (Procyshyn et al., 2019). In addition, there is even greater potential for variability of these PK parameters between different generic versions of a single reference drug (Blier et al., 2019; Meyer, 2001). In this regard, it is possible that two generic products could meet bioequivalence criteria with an original product but not with each other (Habert et al., 2020).
Comparative assessment of bioequivalence standards in Canada, the United States, and Europe (Blier et al., 2019; European Medicines Agency, 2010, 2022; Health Canada, 2018, 2023; U.S. Food and Drug Administration, 2001).

Source: Adapted with permission from Blier et al. (2019).
AUC0–t: area under the concentration–time curve from time zero (drug administration) to the last observed concentration; AUC0–∞: area under the concentration–time curve from time zero extrapolated to infinity; CI, confidence interval; Cmax: maximum concentration; FDA: Food and Drug Administration; LLOQ: lower limit of quantitation; PK: pharmacokinetics; t½: terminal elimination half-life; Tmax: time to reach maximum concentration; ɣz: terminal or elimination rate constant.
Study designs for bioequivalence testing can pose issues for therapeutic equivalence. Studies are usually conducted in small groups of healthy volunteers, which can limit generalizability to target patient populations (Blier, 2007). PK parameters, including clearance and Cmax, can differ following a single dose versus repeated doses; as such, bioequivalence established via single-dose studies may not translate to bioequivalence under steady-state conditions (Tothfalusi and Endrenyi, 2013).
Some regulatory agencies have adopted product-specific guidance to address drug products with complex or nonstandard dose forms (reviewed in the “Results” section). For example, after several studies demonstrated efficacy issues in switching from an original to a generic long-acting methylphenidate product, the FDA issued revised product-specific guidance with additional partial AUC metrics to ensure more similarity in the time–concentration PK profile (Park-Wyllie et al., 2016). The FDA also modified its requirements for approval of generic bupropion products to include assessments of both the parent compound and its active metabolites (i.e., hydroxybupropion, threohydrobupropion, and erythrohydrobupropion) following reports of clinical issues after switching to a generic version that was approved after receiving a waiver of bioequivalence testing (Kharasch et al., 2019; U.S. Food and Drug Administration, 2013; Woodcock et al., 2012). Currently, the FDA and the EMA have published guidance (European Medicines Agency, 2013; U.S. Food and Drug Administration, 2022) to assist in identifying appropriate methods for generic drug development, while Health Canada has not yet generated such documentation.
LAI products also pose unique challenges to bioequivalence testing, generic drug development, and, more importantly, achieving therapeutic equivalence. LAIs are intricate to manufacture and complicated to produce at scale, requiring highly skilled and knowledgeable personnel, as well as high capital investment (Panchal et al., 2023). LAIs have an added layer of quality requirements compared with immediate-release (IR) injectables, and aspects of the manufacturing process such as controlling particle size to create a drug with a target release profile are critical for efficacy and safety (Bauer et al., 2023). Bioequivalence studies of LAIs are challenging because they involve long duration (potentially leading to high participant dropout rates), low power, and complex dosing procedures (Gong et al., 2023). Model-integrated approaches using quantitative tools to characterize drug release, absorption, and population PK patterns have attempted to overcome some of these challenges in demonstrating bioequivalence (Gong et al., 2023; Sharan et al., 2021; Zhao et al., 2019). Loading-dose regimens present another complication for achieving therapeutic equivalence with LAIs. Current second-generation LAIs have well-defined instructions for initiating loading doses that have been tested through population PK studies and can be patent-protected independent of the product itself (Brissos et al., 2014; Lee, 2020; Riboldi et al., 2022). These loading-dose regimens, which, in some cases, negate the need for additional oral supplementation during the initiation period, play a vital role in preventing relapse during the initiation process (Brissos et al., 2014; Riboldi et al., 2022).
Switching between formulations
Despite these challenges in achieving bioequivalence and therapeutic equivalence, switching between formulations commonly occurs. It can occur at any time a prescription is filled or refilled, as well as at admission to or discharge from a hospital. Switching formulations after discharge can be particularly problematic. For instance, in a patient with bipolar disorder, a mood stabilizer initiated after a manic episode could be switched while the drug has yet to achieve its full effect (Chokhawala et al., 2023). In many jurisdictions, pharmacists are allowed to substitute between interchangeable original and generic products as part of their standard practice (unless the physician states “no substitution” on the prescription) and between different generic versions of the same product (Blier et al., 2019; Habert et al., 2020). Furthermore, pharmacists have no obligation to inform the patient or treating physician of a substitution (Rosenthal et al., 2008).
The primary driver for switching formulations is the lower cost of generic products, although switching can result in increased total healthcare costs (Blier et al., 2019). In Canada, provincial policies may encourage generic substitution to reduce provincial medication costs (Park-Wyllie et al., 2016). Of course, it would be ideal to be able to switch to less expensive generic medications if there were no negative consequences to patient outcomes. However, switching to generic medications can result in economic and quality-of-life costs, such as loss of therapeutic control and readmission, which may lead to switching back to the original product (Andermann et al., 2007; Blier et al., 2019). The risk is particularly acute for CNS and psychiatric disorders, as diagnostic and monitoring assessments to gauge therapeutic effects are not as straightforward compared with products for other medical conditions (e.g., hypertension).
A 2011 review highlighted cases of adverse consequences of switching from original to generic psychiatric products (Desmarais et al., 2011). An updated review in 2019 addressed the same topic and pointed to issues in switching itself, rather than to a generic or an original product (Blier et al., 2019). The objective of this paper is to review the updated literature on switching CNS and psychiatric medication products, with a particular focus on drugs that pose challenges for bioequivalence testing as well as therapeutic equivalence, including second- and third-generation LAIs. Given that LAI products have become more widely available and have been successfully used to treat psychosis (Riboldi et al., 2022), as well as the current drive for the development of generic LAIs and other complex drugs (Gong et al., 2023; Stern et al., 2021), it is timely to review the literature on switching CNS and psychiatric medication products.
Methods
A PubMed literature search was conducted on June 9, 2023, for articles published between January 2017 and June 2023. We used a search strategy similar to previous reviews (Blier et al., 2019; Desmarais et al., 2011). Combined MeSH terms used were “drugs, generic” OR “equivalency, generic,” with terms including “antidepressants,” “anxiolytics,” “antiepileptics,” “lithium,” “mood stabilizers,” “benzodiazepines,” “anticonvulsants,” “antipsychotics,” OR “stimulants.” Additional articles were identified from the bibliographies of relevant articles and through targeted searches of PubMed. Studies deemed by the authors to be irrelevant to this paper were excluded. Findings were summarized descriptively and considered in the context of potential clinical consequences of switching between any formulations.
Results
A total of 170 articles were identified; 110 were excluded based on title and abstract review, as they were deemed irrelevant to switching psychiatric drug formulations.
Bioequivalence challenges
Several studies highlighted how the manufacturing process for psychiatric medication products can pose challenges to achieve bioequivalence (Bauer et al., 2023; Gupta et al., 2018; Parker and Jenner, 2022; Procyshyn et al., 2019). While not included in regulatory agency bioequivalence criteria for LAIs, particle size control is essential for achieving the target drug’s release profile (Bauer et al., 2023). For example, the LAI paliperidone palmitate once monthly (PP1M) has a formulation in which particle size drives the release rate of the drug; smaller particles dissolve faster at the site of injection, affecting the PK profile of the drug (Procyshyn et al., 2019). Importantly, no dose adjustment of formulations with different particle sizes could compensate for differences in PK profiles for PP1M because changes in particle size independently affect the initial release rate, as well as Cmax and Tmax. A slower or delayed release rate caused by larger particle size could result in subtherapeutic plasma concentrations, potentially leading to higher rates of relapse and hospitalization, as well as adverse events. A version of PP1M with a smaller particle size could lead to excessively rapid delivery of the drug and adverse events including sedation, QT prolongation, and hypotension.
Manufacturing parameters other than particle size, such as the choice of salt (or no salt, e.g., the free-base form of desvenlafaxine), can also affect a drug’s release characteristics by impacting its polymorphism, solubility, stability of the formulation, and other physiochemical properties that influence its release (Gupta et al., 2018). The manufacturing process itself can play a role, as demonstrated in a study of original and generic versions of lamotrigine tablets that attributed failures in therapeutic equivalence for drugs deemed bioequivalent to “faulty” batches (Parker and Jenner, 2022).
Pharmacogenetic traits can also impact key PK parameters and drug efficacy. A bioequivalence trial of aripiprazole found significant correlations between CYP2D6 polymorphisms and aripiprazole PK parameters, highlighting the need for bioequivalence studies to account for population pharmacogenetic differences (Zhang et al., 2021). Along these lines, a bioequivalence study of risperidone, which is known to have high within-subject variability, found that controlling for genotype can allow a decreased sample size compared with what would otherwise be needed for a highly variable drug (Chen et al., 2018). Post hoc analyses of a parallel-group study of lamotrigine extended-release tablets that unexpectedly failed to achieve bioequivalence showed that the result was likely caused by uneven representation of a bimodal clearance distribution between the groups (Yang et al., 2019). The effects of polymorphisms in hepatic transferases on lamotrigine clearance were only discovered after the study was conducted. Given our incomplete knowledge of pharmacogenetics, when conducting a bioequivalence study, it may often be the case that there are clearance subpopulations unknown to the study designers. If a patient is a member of a clearance subpopulation that was not represented in a bioequivalence study, switching formulations could have unintended adverse effects.
Regulatory guidance on LAI dosage forms
LAI formulations of medications indicated for psychosis play an important role in managing the symptoms of schizophrenia. Regulatory agencies have issued drug-specific bioequivalence guidelines to guide the development of generics for LAIs and other particularly challenging drug formulations (Li et al., 2017). Supplemental Table 1 illustrates the differences in drug-specific guidance for second-generation LAIs from the EMA, FDA, and Health Canada (European Medicines Agency, 2017; U.S. Food and Drug Administration, 2014, 2016a, 2016b, 2021a, 2023). The FDA has issued product-specific guidance for aripiprazole, olanzapine pamoate, paliperidone palmitate (both PP1M and once every 3 months (PP3M)) and risperidone (intramuscular (IM) injection; no guidance has been issued for the subcutaneous injection version) (Correll et al., 2021; U.S. Food and Drug Administration, 2014, 2016a, 2016b, 2021a, 2023). The EMA has issued product-specific guidance only for paliperidone palmitate (PP1M). In Canada, second-generation LAI drugs used include aripiprazole, paliperidone palmitate (PP1M and PP3M), and risperidone IM, but Health Canada has not issued guidance on establishing bioequivalence for any injectable products.
While no generic versions of these second-generation LAIs have yet been approved and made available to patients in any of these jurisdictions, recommendations show inconsistent standards by region. For PP1M, a multidose study is required by both the FDA and EMA, but the EMA also requires a single-dose study (European Medicines Agency, 2017; U.S. Food and Drug Administration, 2021a). The addition of a single-dose study in the EMA guidelines makes it possible to detect a clinically relevant difference in absorption rate (Procyshyn et al., 2019). The FDA guideline specifies the site of injection that can be used (gluteal or deltoid), while the EMA does not (European Medicines Agency, 2017; U.S. Food and Drug Administration, 2021a). The FDA guidance also specifies the dosage strength to be used, while the EMA allows for any dosage strength if the test product has the same concentration of active substance as the reference for all the strengths. Also of note, the FDA guidance for PP3M details that quantitative methods and modeling, such as a model-integrated approach, can be used to support the demonstration of bioequivalence, which is not mentioned in the PP1M guidance (U.S. Food and Drug Administration, 2021a).
On the other hand, Health Canada has not issued guidance on LAIs, making it unclear which study design considerations and PK metrics would be used to assess a generic candidate. In 2018, Health Canada (2018) revised guidelines for multiphasic modified-release formulations, but only for oral and noninjectable formulations. They added partial AUC metrics to assess the time course of changes in the rate of drug delivery throughout the day. This revision shows a recognition of the need for specific guidelines to ensure bioequivalence for more complex products, despite a lack of current guidance for LAIs (Supplemental Table 1).
Several studies reported on the use of model-integrated evidence to demonstrate bioequivalence, showing the growing push to use these techniques. These studies have included the use of population PK modeling for the following: (1) bioequivalence tests of paliperidone; (2) simulation of the performance of partial AUC metrics in bioequivalence tests of methylphenidate drugs with different release mechanisms; (3) the use of a model for PK optimization in studies of metformin, alprazolam, and clonazepam; (4) a proposed Bayesian population model and simulated virtual crossover trial to establish dissolution specifications for oral dosage forms; (5) the development of a PK model for gabapentin; and (6) the assessment of PK models for extended-release methylphenidate products (Gajjar et al., 2022; Glerum et al., 2018; Gomeni et al., 2017; Hsieh et al., 2021; Jackson and Foehl, 2022; Nuske et al., 2021). The benefits of model-integrated approaches include cost savings, as well as reduced sample sizes and study durations while maintaining statistical confidence (Gong et al., 2023). However, modeling techniques do carry potential complications that need to be considered when assessing claims of bioequivalence. For example, when using a virtual participants approach to increase sample size, the simulation may be biased by the population PK model developed from a small population of patients. While certainly useful in optimizing dosing regimens and adjusting dosing in patient subgroups, caution should be exercised in applying model-integrated evidence to drugs used to treat mental health disorders, where there are large risks for adverse consequences if model-integrated bioequivalence does not translate to therapeutic equivalence.
PK and therapeutic outcomes of switching products
Original to generic product switching
The literature search identified 27 studies that addressed switching from original to generic products (Supplemental Table 2) (Berg et al., 2017; Bosak et al., 2019; Das et al., 2019, 2020a, 2020b, 2022; Desai et al., 2019; Elmer and Reddy, 2022; Fanella et al., 2017; Gaïes et al., 2022; Gha-Hyun and Dae, 2018; Holtkamp and Theodore, 2018; Kharasch et al., 2019, 2020; Kwan and Palmini, 2017; Lang et al., 2018; MacKrill and Petrie, 2018; Markoula et al., 2017; Oloyede et al., 2020; Olsson et al., 2019; Ratri et al., 2020; Reimers et al., 2017; Roberti et al., 2021; Trimboli et al., 2018; Van Lancker et al., 2019; Weitzel et al., 2020; Yang et al., 2021). Most of those studies (20/27) focused on antiepileptic drugs (AEDs). Generally, the studies showed that switching from an original to a generic product is safe and that the products are clinically comparable, but there is a paucity of recently published research on products other than AEDs.
Several studies detailed issues associated with switching. One such study assessed whether any AED caused more frequent switch problems or was associated with “generic brittleness” in patients (i.e., patients with a history of having problems switching to generics) (Das et al., 2020b; Markoula et al., 2017). The studies found that patients currently taking a greater number of different AEDs had significantly greater odds of having a switching problem and that taking lamotrigine IR tablets was associated with a greater probability of having switch problems compared with taking other AEDs. Another study on AEDs investigated the risk for recurrent seizures in a large cohort of German patients after switching drug formulations (Lang et al., 2018). They found a higher risk for seizure recurrence in previously seizure-free patients after switching the manufacturer of AEDs. A systematic review of switching drug products in patients with neurologic diseases found the highest-quality data for AEDs; the studies showed some evidence of increased seizure frequencies and adverse drug events, as well as impaired adherence and patient satisfaction (Weitzel et al., 2020). It is important to note that some AEDs are also indicated for other conditions (e.g., lamotrigine is indicated for bipolar disorder), so concerns should also be raised for other indications (GlaxoSmithKline, 2018). Lastly, a database study of U.S. health insurance claims found a higher rate of psychiatric hospitalizations with generics for escitalopram and sertraline, although the results may be explained by generic perception bias (Desai et al., 2019).
Generic to original product switching
The literature search identified only five studies that addressed switching from generic to original products (Supplemental Table 2) (Gaïes et al., 2022; Kharasch et al., 2020; Roberti et al., 2021; Van Lancker et al., 2019; Yang et al., 2021). These studies generally found that switching is safe and clinically comparable, although one study described potential problems associated with the switch. A retrospective study of switching generic carbamazepine formulations found a higher rate of samples with plasma concentrations within therapeutic range (69%) when patients received the original product than when patients received a generic product (44%); the percentages of samples with plasma concentrations in subtherapeutic and supratherapeutic ranges were also higher when patients received the generic (50% and 6%, respectively) than when they received the original product (31% and 0%, respectively), although none of the differences were significant (Gaïes et al., 2022).
Generic to generic product switching
The literature search identified 10 studies that addressed switching from generic to other generic product formulations (Supplemental Table 2) (Alderfer et al., 2022; Berg et al., 2017; Constantino, 2022; Holtkamp and Theodore, 2018; Kharasch et al., 2019; Lang et al., 2018; MacKrill and Petrie, 2018; Odi et al., 2021; Roberti et al., 2021; Weitzel et al., 2020). These studies generally found that switching is safe and clinically comparable, although several described potential problems. A case study found a decrease in antidepressant efficacy after a change in generic formulation (Constantino, 2022). The patient restarted her past generic formulation and showed a full return to her pre-switch mood and functioning. The systematic review of switching drug products in patients with neurologic diseases described above (see the “Original to generic product switching” section above) identified several studies that found increased seizure frequencies and adverse drug events, as well as impaired adherence and patient satisfaction, in patients switching between generic products, although other studies examined found no or few complications after switching (Weitzel et al., 2020). Also, a study on the risk for recurrent seizures in a large cohort of German patients after switching formulations of AEDs found a higher risk for seizure recurrence in previously seizure-free patients after switching the manufacturer (Lang et al., 2018).
Overall interpretation of studies from literature review on switching
Most of the published studies in this literature review found that switching drug products was safe and that the products switched were clinically comparable, although several studies identified concerns. The research on most drug classes other than AEDs is sparse. Recently published studies do not address concerns about switching to generic products that are more complex or technically demanding to manufacture or that have more complicated (i.e., modified release) PK properties. While drug formulation switching is common and there is a growing push by regulatory agencies, insurers, and even physician organizations for generic product development (American Medical Association, 2023; Lionberger, 2021; Thomas, 2020), the therapeutic outcomes of switching have not been extensively researched. It is difficult to make any generalizations about the relative risks of switching for different drug classes, as published studies are often limited to bioequivalence studies necessary for regulatory approval and few evaluate therapeutic equivalence. Problems with switching are also more easily detected when large numbers of patients are taking a product. Less frequently prescribed medications might have issues at the same percentage, but they would be less likely to be detected or reported. Minor differences in bioavailability measured in healthy young volunteers may also be amplified in a patient population.
Overall, for cases where problems were associated with switching, the cause appeared to be related to the switch itself rather than to the direction of change (i.e., original to generic, generic to generic, or generic to original), in accordance with a previous review (Blier et al., 2019).
Real-world consequences of switching
Real-world outcomes such as adherence, adverse events, and economic impact may also be affected by switching. In a study of patients in Taiwan with depression, the risk of discontinuation was higher in users of generic sertraline than in users of the original formulation, although users of generic citalopram and mirtazapine had a lower risk of discontinuation than users of the original formulations (Hsu et al., 2020). The study also found a greater risk of hospitalization for patients with depression treated with generics compared with those treated with original products. In a study of U.S. Medicare claims data on escitalopram, patients who started on an original product and switched to an alternative had a higher rate of discontinuation than those who did not switch (Li et al., 2020). However, the study found that generic escitalopram initiation and substitution provided significant savings in prescription spending.
In terms of adherence, a real-world study in Spain of pregabalin, which is used to treat neuropathic pain or generalized anxiety disorder, found greater adherence in patients who initiated with the original version than with the generic, leading to lower costs for payers (Sicras-Mainar et al., 2019). A retrospective medical records study of gabapentin for treating chronic peripheral neuropathic pain found greater adherence in patients taking the original medication than those taking generic drugs (Navarro-Artieda et al., 2018). The study also found that treatment with original gabapentin led to lower healthcare costs and greater pain relief compared with generics.
Regarding adverse events, an analysis of adverse event reports for AEDs found disproportional reporting for suicide with generics compared with original drugs (Rahman et al., 2017). A study comparing the long-term effectiveness of original dopamine D2 receptor blockers risperidone and sulpiride compared with generics found that higher daily doses were prescribed with generics than with the original drugs in clinical settings (Hsu et al., 2018). Also, original sulpiride was more effective at preventing hospitalizations than the generic formulation (Figure 1). Finally, a study of consumer cost-sharing levels for original and generic medications for major depressive disorder emphasized that cost barriers for original products may increase the odds of relapse and may result in greater impairment and additional acute care utilization (Buxbaum et al., 2018).
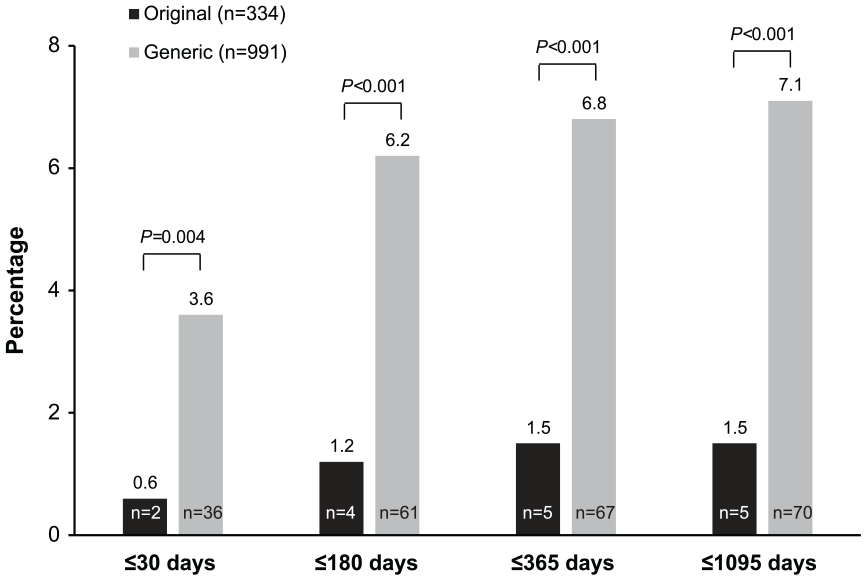
Rates of hospitalization in patients with schizophrenia treated with original and generic forms of sulpiride (Hsu et al., 2018).
Overall, switching drug formulations can result in higher costs than staying with the originally prescribed product, even if the medication costs of the new formulation are lower than the former. For example, Figure 2 illustrates the healthcare costs and drug costs for patients with major depressive disorder who switched, and who did not switch, from original drugs to generic drugs using data from a study of U.S. healthcare claims (Wu et al., 2011). Patients who switched saved on drug costs but spent more on medical costs. This depiction does not account for indirect or quality-of-life costs, which are more difficult to quantify, as even a single breakthrough seizure, for instance, can be catastrophic (LeLorier et al., 2008).
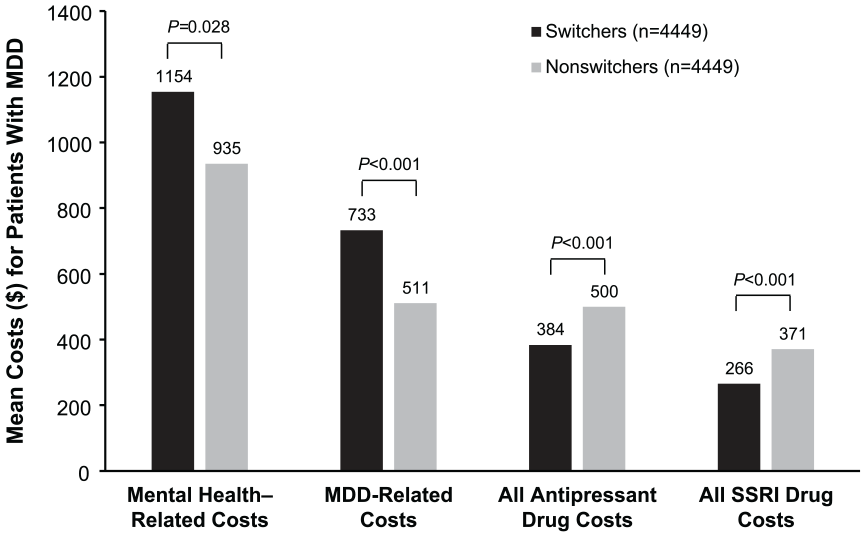
Health care costsa for patients with MDD who switched from an original to a generic SSRI versus non-switchers (Wu et al., 2011).
Discussion and conclusions
This updated review of published literature between January 2017 and June 2023 on switching CNS or psychiatric medication products shows that data generally support the safety and therapeutic equivalence of switching drug products/formulations, although some reports do highlight problems, including higher risks of seizures after switching AEDs from original to generic formulations and between generics (Lang et al., 2018; Weitzel et al., 2020). Real-world data also showed differences between original and generic products in hospitalization risk, adherence, therapeutic effects, and healthcare costs (Hsu et al., 2020; Navarro-Artieda et al., 2018; Sicras-Mainar et al., 2019).
Achieving bioequivalence and therapeutic equivalence is particularly challenging for complex drug formulations that are technically demanding to manufacture (Bauer et al., 2023; Panchal et al., 2023; Procyshyn et al., 2019). Model-integrated approaches have been developed to more efficiently conduct bioequivalence studies for complex products (Gong et al., 2023; Sharan et al., 2021; Zhao et al., 2019), although these could potentially be subject to bias (Gong et al., 2023). At the very least, these techniques must be thoroughly tested before they can be relied on for drug approval. Finally, for drugs targeting the CNS, bioequivalence studies may fundamentally be flawed given that PK measures are based on systemic exposure; it is unclear whether otherwise bioequivalent products/formulations result in equivalent CNS exposure. More advanced and rigorous bioequivalence testing may be needed for CNS and psychiatric drugs, especially for those with more complex PK properties and specialized loading-dose regimens (Brissos et al., 2014; Riboldi et al., 2022).
Importantly, drug regulatory agencies differ in basic bioequivalence standards. This means that switching products may pose greater safety and clinical risks in regions with less stringent standards. The U.S. FDA and the EMA, but not Health Canada, issue product-specific guidance to facilitate the development of generic products, such as LAIs, that require specialized considerations for bioequivalence (European Medicines Agency, 2013; Health Canada, 2018; U.S. Food and Drug Administration, 2022). Without such guidelines, it is unclear how agencies could adequately assess and confidently establish the bioequivalence of a generic candidate for complex formulations, notwithstanding therapeutic equivalence.
Based on this review, we recommend that for patients who achieve and maintain a stable response to a given CNS or psychiatric medication, switching should be approached with caution or avoided, particularly in the context of complex product formulations, as drugs deemed bioequivalent are not necessarily interchangeable or therapeutically equivalent. Healthcare providers should be aware of the possibility of switching when patients have prescriptions filled (particularly when original products have recently lost patent protection) and consider more frequent follow-up and monitoring following a switch. Pharmacists should proactively inform prescribers when a switch occurs for their patients. Overall, we call for more studies to evaluate the therapeutic effects of product switching. The drive to develop more generics (to reduce costs) needs to be balanced with more studies of the consequences of switching. Otherwise, unintentional costs can quickly outweigh potential benefits. Health agencies also need to develop specialized guidelines for evaluating the bioequivalence of drugs that have more complex PK properties. The same broad guidelines used for drugs such as blood pressure medications cannot be expected to produce reliable assessments of bioequivalence for CNS or psychiatric drugs. In addition, health agencies should employ verification task forces, as is the case in the United States (U.S. Food and Drug Administration, 2021b), to ensure that generic products are manufactured under the same strict standards as original products.
Supplemental Material
sj-docx-1-jop-10.1177_02698811241301219 – Supplemental material for Challenges for switching central nervous system and psychiatric medication products: A review of the literature
Supplemental material, sj-docx-1-jop-10.1177_02698811241301219 for Challenges for switching central nervous system and psychiatric medication products: A review of the literature by Ric M Procyshyn, Martin A Katzman, Howard C Margolese, Ofer Agid and Pierre M Blier in Journal of Psychopharmacology
Footnotes
Author contributions
All authors had access to the articles included in this review. All authors collaborated in the preparation of this manuscript, supported by a professional medical writer provided by Otsuka Pharmaceutical Development & Commercialization, Inc. All authors contributed to the critical review and revision of this manuscript and granted approval of the final manuscript for submission.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.M.P. has served as a speaker for/received consultation fees from AbbVie, Eisai, HLS Therapeutics, Janssen, Lundbeck, and Otsuka. M.A.K. has served as a speaker and/or advisor for AbbVie, Aleafia Health, Allergan, Bausch Health, Biron, Canopy, Eisai, Elvium, Lundbeck, Janssen, Lilly, Novartis, Merck, Otsuka, Pfizer, Purdue, Sante Cannabis, Sunovion, Takeda, and Tilray. He has also supported clinical trials and/or research studies for AbbVie, Aleafia Health, Biron, Canopy, Lundbeck, Janssen, Lilly, Pfizer, and Takeda. H.C.M. has received research grant support from AiFred, the Montreal General Hospital Foundation, and SyneuRx and has served as a speaker for/received consultation fees from AbbVie, Boehringer Ingelheim, HLS Therapeutics, Lundbeck, Janssen, Newron, Otsuka, and Teva. O.A. has served as a speaker and/or advisor for Allergan/AbbVie, HLS Therapeutics, Janssen-Ortho (Johnson & Johnson), Lundbeck, Mylan/Viatris, Otsuka, and Teva and has received research grants from Boehringer Ingelheim, DiaMentis, Janssen-Ortho (Johnson & Johnson), Lundbeck, Otsuka, and Neurocrine Bioscience. P.M.B. has received honoraria for lectures and/or participation on advisory boards for AbbVie, Eisai, Janssen, Lundbeck, Otsuka, Pfizer, and VilaNova, has provided expert testimony on behalf of AbbVie and Merck, and has received research grants from Allergan, Janssen, Ontario Brain Institute, Canadian Institutes for Health Research, and University of Ottawa Medical Research Fund.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This review was sponsored by Otsuka Pharmaceutical Development & Commercialization, Inc. Medical writing and editorial support were provided by Adam Fishbein, PhD, at Peloton Advantage, LLC, an OPEN Health company, and funded by Otsuka Pharmaceutical Development & Commercialization, Inc.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.