
Abstract
There is an increasing awareness of the link between chronic alcohol consumption and the development of cognitive, behavioural and functional deficits. Currently, preventative strategies are limited and require engagement in dedicated long-term rehabilitation and sobriety services, the availability of which is low. The acute alcohol withdrawal syndrome is an episode of neurochemical imbalance leading to autonomic dysregulation, increased seizure risk and cognitive disorientation. In addition to harm from symptoms of alcohol withdrawal (e.g. seizures), the underpinning neurochemical changes may also lead to cytotoxicity through various cellular mechanisms, which long-term, may translate to some of the cognitive impairments observed in Alcohol-Related Brain Damage (ARBD). Here we review some of the pharmacological and neurochemical mechanisms underpinning alcohol withdrawal. We discuss the cellular and pharmacological basis of various potential neuroprotective strategies that warrant further exploration in clinical populations with a view to preventing the development of ARBD. Such strategies, when integrated into the clinical management of acute alcohol withdrawal, may impact large populations of individuals, who currently face limited dedicated service delivery and healthcare resource.
Introduction
Alcohol withdrawal syndrome
Alcohol withdrawal syndromes (AWS) result when the balance between γ-Aminobutyric acid type A (GABAA) receptor adaptation through escalating ethanol use leads to unopposed upregulated glutamate activity (Brousse et al., 2012; Faingold et al., 2004; Hermann et al., 2012; Littleton, 1998). This leads to hyperexcitability throughout a range of central and peripheral physiological systems and the plethora of symptoms observed clinically (Becker and Mulholland, 2014). AWS may present with autonomic dysregulation, confusion, motor disturbances and complex behavioural and psychological alterations. These symptoms can range in severity from mild agitation or tachycardia, to complete disorientation, tremulousness, hypertensive crisis, delirium and seizures (Bojdani et al., 2019). Those living with significant levels of physical dependence on alcohol are more likely to experience more severe AWS (Becker and Hale, 1993; Kraemer et al., 2003). As such, in addition to factors such as co-morbidity with a mental health diagnosis and various psychosocial characteristics that are associated with relapse and continued consumption, for example availability (Bowen et al., 2022; Durazzo and Meyerhoff, 2017; Stillman and Sutcliff, 2020), consumption may also be driven by a need to avoid, and fear of, AWS (Koob et al., 2020), rather than the desire to feel pleasure associated with consumption. Furthermore, those who experience repeated episodes of AWS are more likely to undergo more severe AWS episodes and have worse long-term outcomes (Duka et al., 2004).
Management of AWS
Current guidance on the management of AWS recommends holistic assessment by a healthcare professional trained in the assessment and management of alcohol-related conditions, high-dose thiamine provision for prevention (and management) of Wernicke’s encephalopathy and symptom control with benzodiazepines (Lingford-Hughes et al., 2012; National Health and Medical Research Council, 2020; National Institute for Health and Care Excellence, 2017; Office for Health Improvement and Disparities, 2023; Wood et al., 2023). Where benzodiazepines are not suitable, or symptoms are refractory to escalating doses of benzodiazepines, anti-convulsants such as sodium valproate or carbamazepine (Eyer et al., 2011) may be employed. Furthermore, an emerging body of evidence surrounding adjuncts or alternatives to traditional management strategies is developing. For example, modulation of the γ-aminobutyric acid-B (GABAB) receptor using baclofen (Cooney et al., 2019; Liu and Wang, 2017), or the α2-adrenergic receptor through dexmedetomidine or clonidine (Aggarwal et al., 2020; Ahearn et al., 2019).
Alcohol-related brain damage
Alcohol-related brain damage (ARBD) is an umbrella term describing the long-term effects of harmful levels of alcohol consumption on cognition. The following diagnoses are often discussed as examples of pathologies that fall under the ARBD umbrella representing conditions that are directly, and indirectly associated with the long-term impact of alcohol on the brain: Wernicke’s encephalopathy, Korsakoff’s syndrome, Marchia–Fava Bignami and alcohol-related central pontine myelinolysis. However, a diagnosis of ARBD is often made clinically through repeated comprehensive assessment and engagement with patients. Absence of a prior diagnosis of an alcohol-related condition, such as Wernicke’s or Korsakoff’s syndrome for example, should not limit patient access to ARBD services. The impact of chronic alcohol consumption on cognition ranges from episodic and working memory impairments (Pitel et al., 2007; Sullivan et al., 2000), deficits in sustained and divided attention (Moriyama et al., 2002), altered sensory processing speeds, poor impulse control (Duka and Trick, 2010) and changes in behavioural and emotional regulation (Maurage et al., 2017).
The link between AWS and ARBD
Whilst the precise link between suboptimally managed or repeated cycles of withdrawal and ARBD is not yet clear, various lines of evidence suggest that an increasing number of episodes of alcohol withdrawal may be associated with more complex patterns of cognitive impairment (Duka and Stephens, 2014; Duka et al., 2003, 2004). These include impaired attentional and executive function (Loeber et al., 2010; Pitel et al., 2009), altered functional connectivity in brain regions regulating emotional perception of stimuli (O’Daly et al., 2012), dysfunction of inhibitory control (Duka and Trick, 2010) and increased emotional sensitivity (Duka, 2009); all of which are hallmarks of ARBD. As a result of these alterations in brain function and cognitive flexibility, individuals with alcohol-use disorder often face reductions in quality of life (Ugochukwu et al., 2013). Some of these are mediated by cognitive elements such as impulsivity or impaired attention (Dayal and Kaloiya, 2024), and may be mitigated by adjunctive treatments and psychosocial support (Morgan et al., 2004). Importantly, repeated episodes of alcohol withdrawal can escalate in severity through the phenomenon of kindling (Becker, 1998); whereby changes in receptor availability, sensitivity and neuronal excitability thresholds are shifted following repeated exposure to both ethanol and withdrawal episodes (Ballenger and Post, 1978; Gonzalez et al., 2001). This increased severity of withdrawal carries with it increased risks of alcohol withdrawal-related seizures, delirium tremens and mortality (Monte et al., 2010).
Various existing medications and treatment adjuncts that could be employed during the AWS management period may represent a potential opportunity to limit the development of ARBD.
Glutamate receptor modulation
The role of glutamate receptor blockade through pharmacological antagonism has demonstrated significant promise with respect to both reducing the excitotoxic burden to neuronal tissues in AWS (Glue and Nutt, 1990) but also long-term drinking behaviours and outcomes. For example, acamprosate treatment leads to improved abstinence rates at 6 months, treatment completion (Mann et al., 2004; Mason and Lehert, 2012) and favourable improvements in gamma-glutamyl transferase (Carmen et al., 2004). In addition, the role that magnesium may play by acting as a naturally occurring N-methyl-D-aspartate (NMDA) receptor blocker (Burnashev et al., 1992), its actions more generally as a neuroprotective agent (Song et al., 2024) and as a co-factor for thiamine-dependent enzymes in alcohol withdrawal and alcohol dependence has been highlighted.
Autonomic hyperarousal
The hyperglutamatergic state of AWS may also contribute to the increased autonomic symptoms observed. However, a degree of peripheral alcohol-related vagal neuropathy (Tan et al., 1984) and blunted responsiveness to adrenoreceptor modulation (Glue and Nutt, 1987; Nutt et al., 1988), also likely underpin this (Glue and Nutt, 1990). Indeed, pharmacological interventions targeting alpha-adrenergic receptors (Baumgartner and Rowen, 1987; Ip Yam et al., 1992) and pre-synaptic beta-adrenergic receptors (Bailly et al., 1992; Brewer, 1995; Glue and Nutt, 1990) for example are efficacious in mediating some of the symptoms of AWS. However, the neuroprotective effects of clonidine, an alpha-adrenergic receptor agonist, remain to be conclusively evaluated. Of note, Tiplady et al. (2005) demonstrate a lack of impairment on new memory formation in those receiving clonidine compared with temazepam despite the disproportionate impact of clonidine on alcohol-related drunkenness and sedation (Tiplady et al., 2005). In addition, neurohormonal responses associated with autonomic hyperarousal also represent potential future avenues for addressing modulating alcohol-use disorder phenotypes (Neira et al., 2023; Schreiber and Gilpin, 2018).
Thiamine
Whilst already part of most AWS treatment guidelines (Pruckner et al., 2019), the literature relating to the role that thiamine may play in neuroprotection and its potentially preventative role in the development of alcohol-related neurocognitive impairment is growing. As such the impact of thiamine provision not only during the acute phases of Wernicke’s encephalopathy and alcohol withdrawal but also long-term, on prevention of the cellular mechanisms underpinning cognitive impairment and the cognitive and functional phenotypes of individuals with Alcohol Usw Disorder (AUD) warrants review.
Neuroinflammation
Finally, the association between the gut–brain axis, neuroinflammation and proinflammatory immune responses and AUD and AWS is growing. Various lines of evidence demonstrate that alcohol withdrawal represents a period of increased brain vulnerability with respect to insult from reactive oxygen species; this is potentiated in those with pre-existing thiamine deficiency (Clergue-Duval et al., 2022a). This period of AWS-related oxidative stress is also associated with increased AWS severity and biomarkers suggestive of glial damage and astrogliosis, and neuronal and axonal damage (Clergue-Duval et al., 2022b). As a result, individuals at high risk of severe alcohol withdrawal should be identified early and an improved understanding of ways to manipulate the neuroimmune response during AWS in order to protect the brain during this at-risk period is needed.
Rationale and methods
Here, we present a review of the academic literature surrounding thiamine and magnesium supplementation, glutamate receptor blockade (particularly with acamprosate) and neuroinflammation. These domains were selected due to (1) the breadth and depth of information surrounding their utilisation in the management of AWS, (2) the likelihood of implementation – for example pre-existing licensing, guidance and relative-risk data of acamprosate, thiamine and magnesium prescribing, for example and (3) novelty and the anticipated trajectory of the role of neuroinflammatory responses and the gut–brain axis in the management of AWS, but AUD more broadly. A comprehensive search of existing scientific literature databases was performed in relation to the a-priori areas outlines above. A narrative synthesis was adopted aimed at relating the literature outcomes observed to AWS and the development of ARBD through various neurobiological or pathological themes. A consensus through literature review and expert opinion (A.L.-H. and D.N.) surrounding core themes and future research goals was developed by the research team.
Literature review
Glutamate balance
The glutamate receptor family is composed of subgroups consisting of eight metabotropic g-protein coupled receptors (Acher et al., 2023), as well as three ionotropic ones – NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) and kainite receptors (Bettler et al., 2023); the latter of which all have multiple sub-unit configurations. The glutamate receptor family plays a vital role in both the acute presentation and the long-term sequelae of AWS.
During AWS, alterations in glutamate clearance, synthesis and receptor trafficking lead to hyperfunctioning of the glutamatergic system; the effects of which are compounded by a reduction in GABAergic functioning (Abulseoud et al., 2016; Becker and Mulholland, 2014; Brousse et al., 2012). This hyperfunction underpins many of the acute symptoms observed during AWS such as tremor, tachycardia and hypertension. In addition to the impact on autonomic signalling, overstimulation of neurons via glutamate receptors can lead to initiation of apoptotic processes resulting in cell death. Gerace et al. (2019) demonstrate AMPA and metabotropic glutamate receptor-5 (mGlu5) dependent hippocampal pyramidal neuron cell death following exposure to alcohol (at concentrations comparable to those detected in the blood of those with alcohol dependence presenting to the emergency department) and subsequent withdrawal (Gerace et al., 2019). Similarly, NMDA receptor overactivation following chronic exposure to ethanol has been shown to contribute to increased cell death through apoptotic mechanisms (Chandrasekar, 2013; Hu and Ticku, 2000; Tsai and Coyle, 1998). In the long-term, these mechanisms can lead to structural brain volume loss, where repeated cycles of withdrawal-detoxification-relapse and withdrawal, exert an additive effect on withdrawal severity and brain morphometry (Duka and Stephens, 2014). Functionally, this translates to reductions in processing speed and short-term memory plus executive functioning impairments (Duka et al., 2003, 2004; Loeber et al., 2010).
Addressing the glutamate imbalance in AWS
The most common medications used to combat the neurochemical imbalance, and symptoms, of AWS are positive allosteric modulators (PAMs) of the GABAA receptor, for example diazepam and chlordiazepoxide. These medications act at sites distinct to the GABA binding domain, increasing the activity of GABAA (Campo-Soria et al., 2006; Malizia and Nutt, 1995) to enhance chloride influx and so have a net inhibitory effect on neurons. As such treatment with diazepam indirectly attenuates glutamate-mediated neurotransmission (Carton et al., 2022; Kumar et al., 2009; Schmid et al., 1999), alters hippocampal (Carton et al., 2022) and anterior cingulate cortex glutamate concentrations (Wang et al., 2021), and effectively mitigates risk of the development of alcohol withdrawal-related seizures and delirium tremens (Amato et al., 2011; Weintraub, 2017). Other medications that alter the hyperglutamatergic state in AWS include topiramate (an AMPA and kainite receptor antagonist, and GABAA receptor modulator (Mula et al., 2006)), lamotrigine (a pre-synaptic sodium channel blocker, inhibiting glutamate release) (Betchel et al., 2024)) and memantine (an activity-dependent NMDA receptor blocker) (Johnson and Kotermanski, 2006); all of which demonstrate varying efficacy (Krupitsky et al., 2007).
Glutamate receptors as a site of action for ethanol
Various lines of evidence suggest that ethanol directly interacts with the NMDA glutamate receptor leading to alterations in receptor binding parameters, translational and post-translational upregulation of receptor peptides and changes in receptor-mediated currents (Chandrasekar, 2013; Hoffman et al., 1990; Hu and Ticku, 1995). The binding of ethanol to NMDA is reported to be independent of magnesium (Wirkner et al., 1999), but dependent on both glutamate and glycine, whilst not directly binding to the same conformational site for these endogenous ligands, nor the synthetic PCP site antagonist, dizocilpine (Snell et al., 1993). Chandrasekar (2013) proposed a structural model of the NMDA receptor sub-units NR1 and NR2 pairs; with four sites of action for ethanol on the NMDA receptor, all within the third and fourth transmembrane domain interfaces (Chandrasekar, 2013). In agreement with this, the NR1 and NR2 sub-units appear to be preferentially selective to the effects of ethanol (Wirkner et al., 1999).
Behaviourally, in healthy controls, sub-anaesthetic doses of NMDA receptor antagonists such as ketamine produce symptoms comparable with ethanol intoxication including dissociation. These effects have been measured using the Clinician-Administered Dissociative States Scale, reflective of perceptual behavioural and attentional alterations and behavioural activation (Krystal et al., 1994), and by the Brief Psychiatric Rating Scale. Self-reports of ‘high’ measure using a clinician-rated visual analogue scale and, impairments in executive function and memory are also demonstrated following ketamine infusion, all of which are analogous to both the subjective symptoms and objective measures observed in ethanol intoxication (Krystal et al., 1994). This similarity of ethanol-like effects of NMDA receptor antagonists has been explored in recently detoxified alcohol-dependent individuals demonstrating that ketamine selectively increased scores on the sedative (descending limb) of the Biphasic Alcohol Effects Scale (Krystal et al., 1998). Furthermore, in those with previous experience of ethanol, cocaine and marijuana, ketamine administration was more frequently reported as ‘similar’ to the effects of ethanol, as opposed to cocaine or marijuana (Krystal et al., 1998). When compared with healthy controls, recently detoxified individuals appear to exhibit a blunted response to acute NMDA receptor blockade across various executive functioning and subjective responses domains (Krystal et al., 2003). This is consistent with the notion that chronic GABAA receptor PAMs such as barbiturates (Khanna et al., 1998), or ethanol (Rafi-Tari et al., 1996), may induce upregulation of NMDA receptor sub-units and facilitate tolerance to the effects of both ethanol and NMDA receptor antagonists.
Ethanol as a therapeutic intervention
Ethanol exhibits its effects through multiple receptor and neurotransmitter systems (Nutt et al., 2021). Behavioural- and cue-induced neurochemical changes also play a role in the psychological and physiological response to alcohol (Zoethout et al., 2011). Alongside its action at GABAA receptors, ethanol has been demonstrated to act via NMDA receptors to inhibit excitatory glutamate currents (Woodward, 1999) at clinically relevant concentrations (Möykkynen and Korpi, 2012).
Individuals living with AUD successfully manage their alcohol-withdrawal symptoms, or avoid the onset of them, through the consumption of alcohol. Various reports from the early 1900s document the effective use of alcohol to control alcohol withdrawal-related symptoms peri-operatively, or delirium tremens (Nuland, 1983; Osler, 1933; Williams, 1907). More recently, the use of ethanol to manage symptoms of alcohol withdrawal in modern clinical settings has been investigated. Whilst outcome data are mixed, likely secondary to the heterogeneity in intervention setting, clinical population and administration routes and doses, the relative safety of prescribing ethanol is promising (Gipson et al., 2016; Hansbrough et al., 1984; Wilkens et al., 1998). Moreover, oral ethanol prescribing as part of an acute NHS trust alcohol-withdrawal guidelines, led to decreased likelihood of admission and comparable safety profiles concerning the reduction in seizure documentation post-treatment, when compared with benzodiazepines (Quelch et al., 2024b). Pharmacologically, ethanol may act simultaneously at GABAA receptors within the periventricular zone of the hypothalamus as a PAM inhibiting glutamate release, and as an antagonist at NMDA receptors suppressing sympathetic outflow from the central nervous system, regulating the autonomic features of alcohol withdrawal, for example. Further evidence is emerging surrounding the role of glutamatergic projections from the paraventricular nucleus (PVN) to thalamic regions in wakefulness and arousal (Liu et al., 2022; Ren et al., 2018), which may contribute to observations of increased engagement. However, this is also likely related to ethanol-induced dopamine and opioid peptide release (Mitchell et al., 2012; Yoder et al., 2007).
Glutamate and autonomic symptoms
The PVN of the hypothalamus, alongside the medulla and brain stem, plays a crucial role in regulating autonomic tone and as such, pre-ganglionic control of blood pressure and heart rate (Coote and Spyer, 2018; Nunn et al., 2011). Neurons exiting the PVN are influenced by glutamate-mediated excitatory potentials, facilitated through collections of NMDA-NR1 and NR2 sub-unit positive neurons (Eyigor et al., 2001). Further to this, Li et al. (2006) suggest that this outflow from the PVN is under GABAA receptor mediated tonic inhibitory control (Li and Pan, 2007). Increased activation of these pathways via unopposed glutamate receptor agonism secondary to a reduction in GABAA receptor tone may therefore lead to the signs of autonomic hyperarousal (Li et al., 2006; Savić et al., 2022). Indeed, an upregulation of hypothalamic NMDA receptor sub-unit expression (Devaud and Morrow, 1999) and excitatory post-synaptic current amplitude (Marty et al., 2020) has been observed following repeated ethanol exposure in rats. Furthermore ethanol administration has been shown to lead to a decrease in glutamic acid decarboxylase (the GABA synthetic enzyme) in the hypothalamus (Seilicovich et al., 1985), and high affinity GABA binding sites, the latter of which may occur through a process mediated by clathrin-dependent internalisation (Kumar et al., 2003). Collectively these mechanisms may, in individuals who have been consuming alcohol for many years, and who are now entering withdrawal, serve to flip the homeostatic balance regulating noradrenergic tone leading to rapid rises in sympathetic nervous system activity and the resultant tachycardias, agitation, tremor and hypertensive crisis associated with alcohol withdrawal (Finn and Crabbe, 1997). Furthermore, continued consumption and repeated cycles of withdrawal may further precipitate long-term autonomic hyperarousal (Julian et al., 2020) and the kindling effect in individuals with alcohol-use disorder (Linnoila et al., 1987).
Glutamate receptor antagonism
Blockade of glutamate receptor responses during alcohol withdrawal may help not only to attenuate both the immediate autonomic responses and cytotoxic effects on neurons but also long-term reduce the development of cognitive effects through neuroprotective mechanisms.
Acamprosate is proposed to impact glutamate-mediated neurotransmission by both pre- and post-synaptic mechanisms (De Witte et al., 2005). Post-synaptically, acamprosate interacts with the NMDA receptor, acting as a partial agonist at an allosteric modulatory site at low levels of activity (Naassila et al., 1998) but also as a functional antagonist at higher levels of activity (al Qatari et al., 1998; Naassila et al., 1998), such as those occurring during alcohol-dependence (and withdrawal (al Qatari et al., 1998; Naassila et al., 1998)). Various lines of evidence suggest the main site of action for acamprosate is within the polyamine/spermidine binding site (Naassila et al., 1998; Popp and Lovinger, 2000) of the NMDA receptor. Pre-synaptically, acamprosate acts to reduce glutamate release via antagonism at mGlu5 receptors (Blednov and Harris, 2008; De Witte et al., 2005). Acamprosate is efficacious in clinical populations in terms of length of abstinence and numbers needed to treat to prevent one person from returning to any drinking (NNT = 11) (Kiritzé-Topor et al., 2004; McPheeters et al., 2023; Whitworth et al., 1996); it is generally well tolerated by patients, but should be prescribed in accordance with local guidelines, used with caution in severe hepatic failure and avoided in renal impairment if serum-creatinine greater than 120 µmol/L (National Institute for Health and Care Excellence, 2024a).
In rodents when administered prior to the onset of alcohol withdrawal, acamprosate attenuates the development of ethanol-induced withdrawal in an mGlu5-dependent manner (Blednov and Harris, 2008), and reduces handling induced convulsions during alcohol withdrawal, in mouse models (Farook et al., 2008). In related studies, acamprosate has been shown to prevent hippocampal cell necrosis during alcohol withdrawal (Mayer et al., 2002), potentially through both NMDA and mGlu5 receptor antagonist mechanisms (De Witte et al., 2005).
More recently, the neuroprotective role of acamprosate has been explored in other brain conditions such as traumatic brain injury and ischaemia. Following controlled cortical impact injury lower glutamate and zinc mediated toxicity, reactive oxygen species production, glial activation and dendritic injury was found in animals treated with acamprosate compared with vehicle (Choi et al., 2019). Acamprosate also improved outcomes in neurological severity score and the Morris water maze performance (Choi et al., 2019). Furthermore, in thread occlusion ischaemic stroke models in mice, those pre-treated with acamprosate demonstrated improved neuronal density, behavioural neurological recovery and reduced NMDA receptor-mediated excitotoxicity (Doeppner et al., 2015). In this model, acamprosate increased activation of pro-survival signalling pathways via Akt (Doeppner et al., 2015).
In agreement with pre-clinical data, reductions in anterior cingulate glutamate concentration measured with MR spectroscopy are observed in recently detoxified alcohol-dependent patients following administration of acamprosate (Khadse et al., 2019; Umhau et al., 2010). Related to this, acamprosate significantly decreased the amplitude of event-related potentials in healthy subjects (Hegerl et al., 1996). Furthermore, acamprosate attenuated withdrawal related rises in magnetoencephalography alpha-slow wave index readings (Boeijinga et al., 2004) and improved latency to Rapid Eye Movement (REM) sleep and the period of time spent in stage 3 sleep; both of which are features of deranged sleep architecture typically associated with alcohol withdrawal (Luc et al., 2006). Luc et al. (2006) and Boeijinga et al. (2004) were observed in individuals meeting DSM-IV criteria for alcohol-dependence; however, there was an absence of physical withdrawal symptoms among the cohorts. Whilst this allowed the impact of acamprosate on brain activity to be investigated without the confounder of benzodiazepines, further studies are warranted to determine not only in the influence of acamprosate both on brain activity and neurochemistry during acute physical withdrawal but also in the context of benzodiazepine co-prescribing.
The role that modulation of glutamatergic signalling through acamprosate may have in relation to longer-term cognitive processes has also been investigated. Human studies have failed to demonstrate significant improvements or detriment on working memory in healthy controls (Schneider et al., 1999) and, memory and attention measures in patients with schizophrenia and co-morbid alcohol dependence (Ralevski et al., 2011). Whereas pre-clinical evidence suggests that both ethanol consuming rats and ethanol naïve rats experience an improvement in both spatial learning in Morris Water Maze paradigms, and short-term memory tasks using social recognition tests (Mikolajczak et al., 2002; Szulc et al., 2002). Additionally, in mice chronically exposed to ethanol vapour, acamprosate demonstrated efficacy in attenuating ethanol-induced impairments in cognitive flexibility in attentional set-shifting models (Hu et al., 2015). The impact of acamprosate on cognitive flexibility and executive function has been explored in relation to goal-directed and habitual responding by Melugin et al. (2022). They demonstrated that acamprosate treatment led to improvements in cognitive outcomes through a reduction in post-synaptic NMDA-receptor mediated currents in the medial pre-frontal cortex in mice only exhibiting goal-directed ethanol consumption behaviours, not those consumed through habitual responding. This may suggest that the neuroprotective effect of acamprosate, and therefore, protective qualities it confers on alcohol-relative cognitive dysfunction, may only manifest early in the development of a dependence phenotype – prior to the neuroadaptive response in reward signalling from the ventral accumbens shell to more dorsal striatal areas observed when a switch in reward-focussed to habit-based patterns of consumption take place (Everitt and Robbins, 2005).
Acamprosate is recommended by various bodies to form a part of management plans when addressing moderate or severe drinking behaviours and reducing relapse (Lingford-Hughes et al., 2012; National Institute for Health and Care Excellence, 2011; UK Government, 2023). When incorporated into management regimens of alcohol withdrawal, acamprosate may lead to a later first relapse compared with those managed with placebo (Gual and Lehert, 2001). Furthermore, Kampman et al. (2009) demonstrate that acamprosate, when included as a part of detoxification that confers no additional harm or relapse risk compared with placebo. In addition to forming part of relapse prevention plans, based on the information above, various lines of evidence suggest that when initiated early in withdrawal (and in addition to thiamine and benzodiazepines), or ideally prior to the onset of symptoms, acamprosate may offer neuroprotective, and neurorestorative benefits (through improved sleep architecture) with respect to the development of ARBD, conferring longer-lasting protection, manifested by preservation of cognitive function.
Collectively the data we have reviewed demonstrate the impact that hyperglutamatergic tone has on neuronal function during AWS. We highlight the potential role that glutamate receptor antagonism through acamprosate for example, may play in the protection of the brain during AWS and the prevention of signs and symptoms of ARBD, long-term. In order to facilitate this translation to clinical practice, further research is required in clinical populations to investigate the long-term neurocognitive outcomes and recovery of function of patients prescribed acamprosate as part of their alcohol withdrawal management plan. Additionally, a risk-benefit exercise focussed on both the feasibility of prescribing acamprosate in the context of acute illness where renal and hepatic function may be impaired, and where capacity to consent to engage with the treatment alongside the recommended psychological intervention (National Institute for Health and Care Excellence, 2011) may be suboptimal.
Magnesium
Magnesium acts as an NMDA receptor pore blocker, reducing inward currents in a voltage sensitive manner (Paoletti and Neyton, 2007; Wang and MacDonald, 1995). The association between low magnesium concentrations and alcohol dependence has been explored in pre-clinical models (Belknap et al., 1978; Hemmingsen and Kramp, 1980) and clinical populations (Grochowski et al., 2019; Rink, 1986; Shane and Flink, 1991). Indeed, the link between alcohol consumption, low serum magnesium concentrations and neuronal damage or pathological neurobiological changes have previously been reported (Ema et al., 1998; Li et al., 2001; Shah, 2018). Patients with alcohol dependence are often magnesium depleted and an association between hypomagnesaemia and clinical outcomes including duration of most recent alcohol binge episodes, quality of life scores and impulsivity measures has also been demonstrated in alcohol-dependent groups (Ordak et al., 2017). The potential neuroprotective role that magnesium may play in NMDA receptor-mediated excitotoxicity (McDonald et al., 1990), and alcohol-related cerebrovascular damage (Ema et al., 1998), hypertension (Hsieh et al., 1992) and oxidative stress (Markiewicz-Górka et al., 2011) is increasingly recognised.
In 2013, a Cochrane review concluded that there was insufficient evidence to promote or dissuade the use of magnesium in the management of alcohol withdrawal (Sarai et al., 2013); to date a clinical consensus in relating to the provision of magnesium has not been reached. However, since publication of the Cochrane review, several reports have further explored the relationship between low serum magnesium concentrations and alcohol dependence-related outcomes. Prior et al. (2015) demonstrate that alcohol withdrawal intensity is correlated with reduction in serum magnesium concentration. Furthermore, an inverse relationship has been observed in alcohol-dependent females between serum magnesium concentration and number of heavy drinking days and pro-inflammatory serum markers (Vatsalya et al., 2020). Maguire et al. (2019a, 2019b) demonstrate that individuals presenting with alcohol withdrawal to an emergency department with serum magnesium of <0.75 mmol/L were associated with higher 1-year mortality rates. Disappointingly, at follow-up (median 126 days later), 76% still had a serum magnesium of <0.75 mmol/L and magnesium was prescribed in only 7% of these presentations (Maguire et al., 2019a, 2019b). In contrast though the majority of patients had whole blood thiamine diphosphate concentrations (indicative of thiamine reserve) within the reference ranges and thiamine was routinely prescribed. Moreover, low whole blood thiamine diphosphate was not associated with reduced 1-year survival (Maguire et al., 2019b). Maguire et al. (2019a) suggests that the paucity of magnesium supplementation observed may relate to a lack of clinician awareness and evidence surrounding hypomagnesia in AUD.
To our knowledge, two randomised controlled trials have been performed since the 2013 Cochrane review. The studies are summarised in Table 1.
Study characteristics and outcomes from two prospective trials delivering magnesium therapy as an adjunct to standard alcohol withdrawal management strategy.
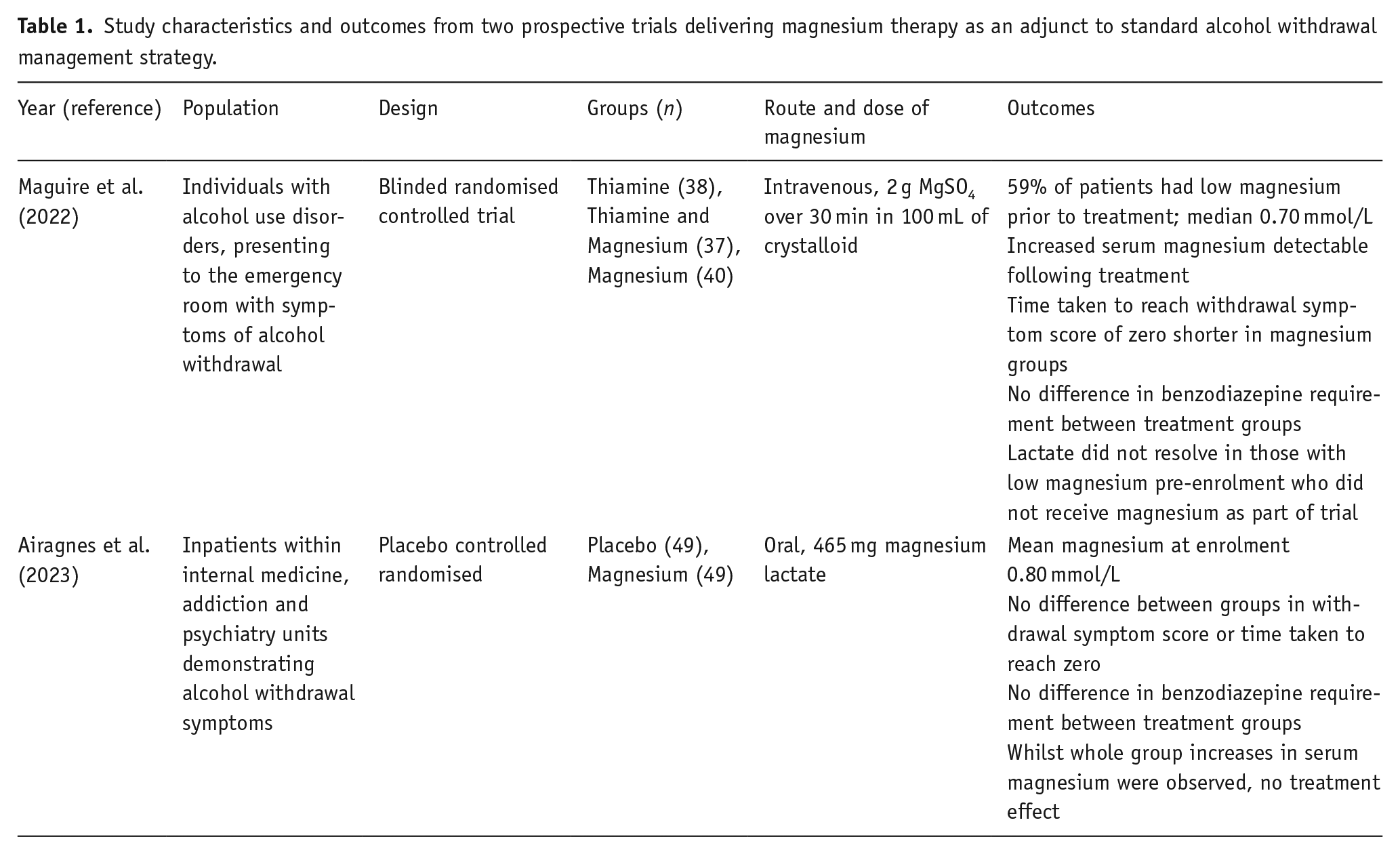
Whilst Airagnes et al. (2023) demonstrate low pre-treatment serum magnesium concentrations within their population, in agreement with various reports, magnesium supplementation had no impact on outcomes (Airagnes et al., 2023). In contrast, using a more intensive, intravenous regimen, within British National Formulary dose recommendations (National Institute for Health and Care Excellence, 2024b), a positive effect on the duration on withdrawal symptoms and resolution of raised lactate, and post-treatment serum magnesium concentrations were seen (Maguire et al., 2022). Of note is that these studies represent two distinct clinical populations, namely those presenting to acute medical services with marked alcohol-withdrawal symptoms requiring acute management, and those presenting to addiction units either in a scheduled manner or following a visit to the emergency ward. As such the impact of magnesium provision may be affected by the acuity of their symptoms and recent prior treatment with thiamine supplementation for example.
Whilst there is limited evidence from controlled trials since the seminal 2013 Cochrane Report relating to magnesium supplementation in the management of AWS, the impact that reduced serum magnesium concentrations may have on patient outcomes is clearer, as summarised in Table 2.
Impact of hypomagnesaemia on patient outcomes.
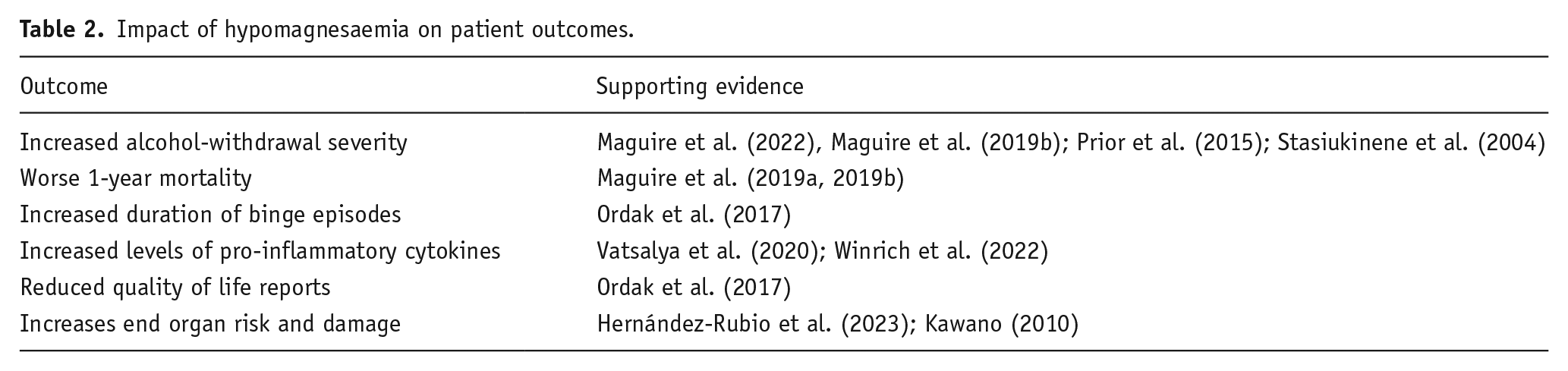
The pathophysiological role that magnesium may play in neuroprotection during the AWS period is multifaceted. Various lines of evidence suggest that magnesium supplementation may affect AWS symptom severity. The impact of this on the development of the features of ARBD requires further investigations. Increased efforts are warranted to explore the impact that administration of magnesium may confer in this population, across different management settings, using practicable and translatable dosing regimens. Both short- and long-term outcome reporting is required from these studies to inform not only the acceptability of prolonged oral magnesium, given its gastrointestinal side effects and tolerance profile for example, but also the impact of magnesium provision on alcohol-withdrawal severity scores and development of alcohol-related cognitive impairments.
Thiamine
The biochemical importance of thiamine and thiamine as a co-factor
Pyruvate dehydrogenase, branched chain α-ketoacid dehydrogenase, α-ketoglutarate dehydrogenase and transketolase are all thiamine-dependent enzymes (Tetsuka and Hashimoto, 2021). Reduced transketolase action leads to decreased ribose-5-phosphate and nicotinamide adenine dinucleotide phosphate (NADPH) production which is essential to produce fatty acids, certain steroidal molecules, nucleotides and branched amino acids. Furthermore, NADPH is required for the production of anti-oxidants such as glutathione, a crucial cellular defence against oxidative stress. Pyruvate dehydrogenase converts pyruvate to acetyl coenzyme A (acetyl-CoA) for entry into the tricarboxylic acid cycle. As such in a thiamine-deplete state, production of cellular adenosine triphosphate (ATP) is severely reduced as are the precursors for myelin production. Decreased conversion of pyruvate to acetyl CoA also leads to lactate production and deranged pH homeostasis. Finally, an accumulation of α-ketoglutarate secondary to thiamine deficiency-induced impairments in α-ketoglutarate dehydrogenase activity leads to increased glutamate concentration. This results in both direct (via NMDA receptor-mediated excitotoxicity) and indirect (via production of free radicals) neuronal cell damage and death. Genetic risk factors for the development of Wernicke’s encephalopathy, or increased sensitivity to thiamine deficiency have been described (O’Brien et al., 2022). Cumulatively, thiamine deficiency, such as that observed in alcohol-dependence, results in a broad range of metabolic perturbations that may lead to cellular starvation, neuronal damage and altered synthetic abilities. Furthermore, in response to thiamine depletion, ethanol-dependent rats show raised TNFα and IL-6 (Toledo Nunes et al., 2019), enhanced markers of oxidative stress (NO−2) and more cell apoptosis (caspase 9 activity) (Moya et al., 2022); importantly some of these markers, relate to changes in behaviour in these animals (Moya et al., 2022).
Prodromal symptoms of thiamine depletion include paraesthesia, fatigue and abdominal pain. The extremis of thiamine deficiency resulting from a marked derangement in cellular homeostasis presents as Wernicke’s encephalopathy. Whilst typically associated with alcohol-dependence, non-alcohol-related causes include severe hyperemesis gravidarum, or anorexia nervosa, and post-bowel resection syndromes. Wernicke’s encephalopathy is characterised by cerebellar ataxia, oculomotor disturbances, such as nystagmus or other ophthalmoplegias, and cognitive disturbances including altered mental state or memory impairment (Caine et al., 1997). Post-mortem examination of individuals with Wernicke’s encephalopathy demonstrate cerebellar vermis atrophy, ventricular dilation and mammillary body atrophy as common features (Harper, 1979). Interestingly, of the 51 cases reviewed by Harper, only 7 were diagnosed with Wernicke’s encephalopathy pre-morbidly (Harper, 1979; Harper et al., 1986). Cerebellar lesions, and mammillary body and thalamic lesions, have been proposed to represent the chronic and acute phases of thiamine depletion, respectively (Singleton and Martin, 2001). Imaging studies support this finding, adding that initial vasogenic and cytogenic oedema is seen in T2-FLAIR sequence MRI in acute Wernicke’s encephalopathy (Jung et al., 2012). Following, the initial oedematous stage, a process of atrophy and cerebral shrinkage is observed, with cortical involvement being associated with a poorer prognosis (Jung et al., 2012). Various studies have related mammillary body vasogenic oedema or atrophy to functional deficits during acute Wernicke’s encephalopathy (Jung et al., 2012). For example deficits of mammilo-thalamic functional connectivity in Wernicke’s encephalopathy patients is related to impairments in verbal memory test performance (Kim et al., 2009). This altered functional connectivity and the associated cognitive impairments are partially reversible following thiamine provision (Kim et al., 2010).
With the growing awareness of ARBD, the interest in alcohol-use disorders, thiamine depletion and cognitive function has increased in recent years. Alcohol dependence has consistently been shown to lead to reduction in serum thiamine concentrations which relate to negative performance of various measures of cognitive performance (Bonnet et al., 2023; Coulbault et al., 2021; Langlais, 1995; Pitel et al., 2011). Furthermore, AWS may increase the consumption of thiamine through increased catabolism, further depleting reserves, increasing the risk of the development of more severe AWS symptoms and Wernicke’s encephalopathy (Clergue-Duval et al., 2022a). Exacerbated thiamine depletion during the neurochemical imbalance and increased oxidative-stress of AWS may lead to the production of increased levels of unopposed reactive oxygen species. These may further potentiate neuronal and glial damage, and astrocytic dysfunction, leading to the development of cognitive impairments (Clergue-Duval et al., 2022a). However, importantly, the impact that effective supplementation has on recovery of cognitive function (Dingwall et al., 2022), or prevention of alcohol-related cognitive impairment has also been demonstrated (Chou et al., 2019). Of note, Listabarth et al. (2023) compared the effect of oral and intravenous thiamine supplementation regimens on cognitive performance in those receiving treatment for alcohol-withdrawal. Whilst improvements in serum thiamine were associated with better delayed recall subdomains of the Montreal Cognitive Assessment (MoCA) and the increase in thiamine pyrophosphate following treatment were correlated with improvement in memory function, and no difference was observed between the treatment routes (Listabarth et al., 2023) (Table 3).
Study characteristics from prospective trials investigating cognitive outcomes following provision of thiamine therapy as an adjunct to standard alcohol withdrawal management strategy.
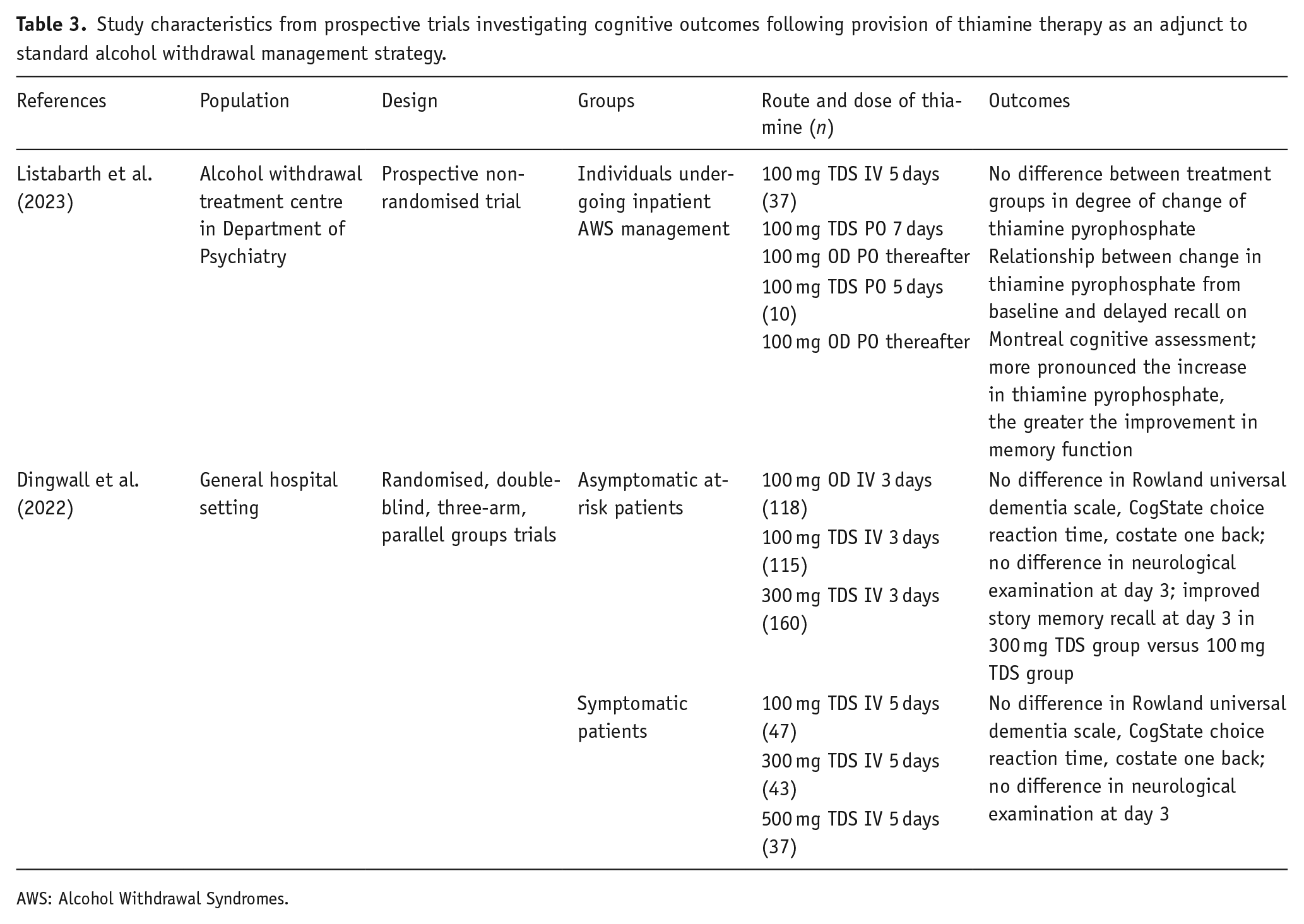
AWS: Alcohol Withdrawal Syndromes.
Whilst the protective role that thiamine plays in the prevention of Wernicke’s encephalopathy is well documented, there is less evidence surrounding the impact that thiamine has on long-term protection and cognitive recovery. A growing body of evidence supports the potentially preventative effect that thiamine may have with respect to the development of ARBD. Furthermore, an increasing level of evidence suggests that a resurgence of the importance of thiamine provision in AUD is needed. Sinclair et al. (2023) demonstrate from a UK wide audit that only 41% of patients on acute psychiatric adults wards undergoing medically assisted withdrawal had received thiamine (Sinclair et al., 2023). A lack of consistency in relation to thiamine prescribing is also observed in critical care (as few as 51% of individuals with AUD presenting with a critical illness were prescribed thiamine; of those that were 49% did not receive any within their first 12 h of admission (Pawar et al., 2022)) and acute medical settings (Brothers et al., 2023).
Neuroinflammation
A large body of literature discusses the impact of acute and chronic alcohol exposure on both the immune system and the relationship between the peripheral and central inflammatory response. Indeed, alcohol acts as a modifiable risk factor in brain vulnerability to ageing, age-related decline (Manuello et al., 2024) and the relationship between ageing, harmful levels of alcohol consumption and inflammation have been explored (Carlson et al., 2023). Ethanol exposure may induce both independent neuroinflammatory processes as well as amplify those associated with peripheral mechanisms (Anand et al., 2023; Pascual et al., 2021). Furthermore, harmful levels of alcohol consumption may lead to increased inflammatory mediators, and decreased number of available, or increased proportion of saturated, microglial cells, which can impact amyloid and tau deposition (Venkataraman et al., 2017). Lanquetin et al. (2021) demonstrate increased microglial and astrocyte staining in the cortex and hippocampus of rodents following a 50-week alcohol exposure trial. Decreased white matter tract volume was also observed (Lanquetin et al., 2021). Alongside the pre-clinical arm of this study, MRI tractography and morphometry studies in individuals with AUD demonstrate a relationship between circulating cytokines and pre-frontal, temporal and orbito-frontal cortices grey matter volumes (Lanquetin et al., 2021).
The role that the neurochemical imbalance during AWS may have on neuroinflammatory processes and the development of ARBD is increasingly recognised. Both in vitro and ex vivo studies demonstrate increases in pro-inflammatory cytokine gene expression following alcohol binge-withdrawal paradigms (Doremus-Fitzwater et al., 2014; Walter and Crews, 2017). In agreement with pre-clinical data, Yen et al. (2017) report increases in a range of interleukins, TNFα and INFγ in alcohol-dependent individuals during early withdrawal (Neuman et al., 2021; Yen et al., 2017); this increase is largely reversed by 4 weeks post-withdrawal (Yen et al., 2017). Interestingly, the change in the inflammatory cytokines during early and later-withdrawal was related to performance on the trail-making tasks (Yen et al., 2017). These fluctuations in cytokines may play an additive role in neuronal cell damage and degeneration during alcohol withdrawal (Allan and Rothwell, 2001). The gut–brain axis has also been linked to the chronic inflammatory responses observed in AUD and AWS, resulting in changes in mood, cognition and alcohol-related behavioural responses (Leclercq et al., 2017). Leclercq et al. (2012) report increased intestinal permeability, proinflammatory cytokines, measures of low mood and anxiety and slower reaction times early during AWS; all of which largely improved later during their admission (Leclercq et al., 2012).
Novel therapeutics targeting the neuroinflammatory response associated with long-term alcohol exposure warrant exploration. Those influencing the gut–brain axis, including pro-biotics such as Bifidobacterium lactobacillus and clostridium (Li et al., 2022), and butyrate (Wei et al., 2023) demonstrate promise, particularly with respect to alcohol-related cognitive impairment. However, to our knowledge, therapeutics targeting the gut–brain axis or the increased oxidative stress or expression of markers of neuronal and glial damage, specifically in AWS, have not been assessed.
Existing medications licensed for cognitive impairment
Pharmacological management of withdrawal symptoms and thiamine provision are cornerstones of effective prevention of alcohol-withdrawal seizures and delirium tremens. Avoidance of withdrawal symptoms through relapse prevention, referral to alcohol services, sobriety promotion and thiamine provision, remain the most holistic and impactful routes to reducing short- and long-term harms among those with AUD.
In addition to the avenues already discussed, various innovative approaches including outcomes from studies employing existing medications for cognitive decline warrant discussion. Trials of traditional therapies for cognitive impairment in early Alzheimer’s such as acetycholine esterase inhibitors (Angunawela and Barker, 2001; Cochrane et al., 2005; Sahin et al., 2002) in alcohol-related cognitive impairments are mixed. A larger number of participants demonstrate acceptability and feasibility of memantine in AUD (Lewis et al., 2020), with some also reporting reduced number of heavy drinking days, number of units consumed or craving (Arias et al., 2007; Krishnan-Sarin et al., 2020). In addition, memantine has been shown to improve quality of life scales and various measures of cognitive function at 12 weeks in individuals with alcohol-related dementia (Cheon et al., 2008). Finally, in a single case study, memantine improved cognitive functioning in an individual with alcohol-related cognitive impairment. Interestingly, during this trial, two relapses occurred; one where memantine was discontinued and restarted, another where memantine was continued throughout. The former was associated with a relapse of cognitive impairment, whereas the latter was not (Bonnet et al., 2014), further highlighting the potential role that AWS and NMDA receptor antagonism may play in the development of alcohol-related cognitive dysfunction.
Conclusions and future directions
We have summarised the growing body of evidence surrounding the neuroprotective and potentially prophylactic impact that changes to the management of alcohol withdrawal may have on brain neurochemistry and cognitive outcomes. These include reducing the excitotoxic burden that unopposed glutamate surges during withdrawal present through NMDA currents, via pharmacological blockade or provision of magnesium. Furthermore, we highlight the role that thiamine plays in maintaining cellular homeostasis and the increasing number of studies reporting the impact that alcohol-induced thiamine depletion may have on neuronal damage and long-term cognitive sequelae. Finally, our emerging knowledge surrounding the role of neuroinflammation in AUD and AWS, more specifically, is highlighted.
Consideration of pharmacological strategies during the AWS period that may help mitigate the development of signs and symptoms of ARBD has the potential to be hugely impactful on patients level of independence, social reintegration and well-being. Future directions in this area should include research surrounding the manipulation of the heightened reactive oxygen species burden observed during alcohol withdrawal, and how reducing this might mitigate long-term complications like cognitive impairments.
Collectively, we demonstrate that AWS represents a period of heightened vulnerability of the brain through:
exhaustion of endogenous protective factors,
excitotoxicity as a result of neuroadaptive processes during the development of alcohol dependence,
a limited understanding of the complex mechanisms such as neuro-inflammation underpinning neuronal damage during AWS that go on to impact cognition and function long-term and
a limited repertoire of evidence surrounding protective strategies that are implementable and translatable to clinical practice (Angunawela and Barker, 2001; Cochrane et al., 2005; Sahin et al., 2002).
Whilst significant efforts are being made towards improving our understanding of the role that alcohol withdrawal may have on the development of ARBD, further work is required. In order to reduce both short- and long-term morbidity and mortality associated with alcohol withdrawal, effective strategies for the prevention of alcohol withdrawal onset and prophylaxis of symptom escalation are paramount. The following questions remain to be answered, all of which may lead to significant improvements in both acute and long-term outcomes of those presenting with AWS:
Does extended periods of inpatient parenteral thiamine provision, over the days and weeks following alcohol withdrawal lead to improved recovery of cognitive function, or greater levels of protection against developing alcohol-related cognitive impairment long-term?
Whilst parenteral magnesium supplementation during the acute phase of alcohol withdrawal may offer some degree of protection, is there any merit in offering long-term oral supplementation and how acceptable would this be in the patient population being managed?
Can magnesium provision during alcohol withdrawal help mitigate some of the risks associated with the development of ARBD in the context of long-term sobriety seeking patient groups? If so, what regime is most appropriate? Could a multi-targeted combination therapy combining parenteral magnesium replacement, direct NMDA receptor blockade (perhaps through acamprosate) and prolonged parenteral thiamine protect against the development of ARBD in certain at-risk populations for example younger females?
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: D.Q., B.J., G.R.-D., S.B. have nothing to declare; within the past 5 years D.N. has advised Indivior and D&A Pharmaceutical two companies with an interest in treating AUD. A.L.-H. has received Honoraria paid into her Institutional funds for speaking and chairing engagements from Lundbeck, Lundbeck Institute, UK, Janssen-Cilag, Pfizer, Servier; received research grants or support from Lundbeck, GSK; unrestricted funds support from Alcarelle for a PhD; consulted by Silence, Sanofi-Aventis and also consulted by but received no monies from Britannia Pharmaceuticals, GLG and Dobrin. She led the British Association for Psychopharmacology ‘addiction’ guidelines who received support from Archimedes Pharma, Lundbeck, Pfizer, Link Pharmaceuticals, Schering-Plough, Reckitt Benckiser, Britannia Pharmaceuticals, Martindale and Lilly.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Darren Quelch is supported by the Sandwell and West-Birmingham NHS Trust research fellowship scheme.