
Abstract
Background:
Binge-eating disorder (BED) is a common psychiatric condition with adverse psychological and metabolic consequences. Lisdexamfetamine (LDX) is the only approved BED drug treatment. New drugs to treat BED are urgently needed.
Methods:
A comprehensive review of published psychopathological, pharmacological and clinical findings.
Results:
The evidence supports the hypothesis that BED is an impulse control disorder with similarities to ADHD, including responsiveness to catecholaminergic drugs, for example LDX and dasotraline. The target product profile (TPP) of the ideal BED drug combines treating the psychopathological drivers of the disorder with an independent weight-loss effect. Drugs with proven efficacy in BED have a common pharmacology; they potentiate central noradrenergic and dopaminergic neurotransmission. Because of the overlap between pharmacotherapy in attention deficit hyperactivity disorder (ADHD) and BED, drug-candidates from diverse pharmacological classes, which have already failed in ADHD would also be predicted to fail if tested in BED. The failure in BED trials of drugs with diverse pharmacological mechanisms indicates many possible avenues for drug discovery can probably be discounted.
Conclusions:
(1) The efficacy of drugs for BED is dependent on reducing its core psychopathologies of impulsivity, compulsivity and perseveration and by increasing cognitive control of eating. (2) The analysis revealed a large number of pharmacological mechanisms are unlikely to be productive in the search for effective new BED drugs. (3) The most promising areas for new treatments for BED are drugs, which augment noradrenergic and dopaminergic neurotransmission and/or those which are effective in ADHD.
Keywords
Introduction
Binge-eating disorder (BED) is a psychiatric disorder characterised by loss of control leading to frequent, compulsive episodes of excessive eating (binges). BED differs from anorexia nervosa (AN) or bulimia nervosa (BN) because it is not associated with the regular use of inappropriate compensatory behaviour (e.g. purging, fasting and excessive exercise). As discussed in the review, BED is a major causal factor in obesity and is an independent risk factor for a wide range of metabolic, physical and other psychiatric disorders. Lisdexamfetamine (LDX) (Vyvanse®) has been approved to treat BED in the USA and a limited number of other countries. However, with only one medication available in some countries and none in others, new drugs are urgently needed to provide physicians with prescribing choices when treating this disorder.
In view of the extensive overlap between BED and obesity, we compare the diagnostic criteria for these two disorders, discuss their neurobiology and pathology, and the pharmacology of drugs used to treat them. The following objectives are addressed:
➢ Differentiate the neurobiology of BED and obesity.
➢ Define the pharmacology of current drugs that are effective in BED.
➢ Define the pharmacological target profile of the ideal BED drug.
➢ Evaluate drug-candidates for BED in non-clinical and clinical development.
➢ Propose research avenues for developing improved drugs.
In a complementary article (Heal and Gosden, 2021), we reviewed results from clinical trials in BED including not only anti-obesity drugs but also drugs for attention deficit hyperactivity disorder (ADHD), depression, epilepsy and substance use disorders to re-evaluate the evidence for efficacy. For that reason, we have only briefly summarised the findings here (Table 1).
Summary of the evidence from clinical trials in treating binge-eating disorder.
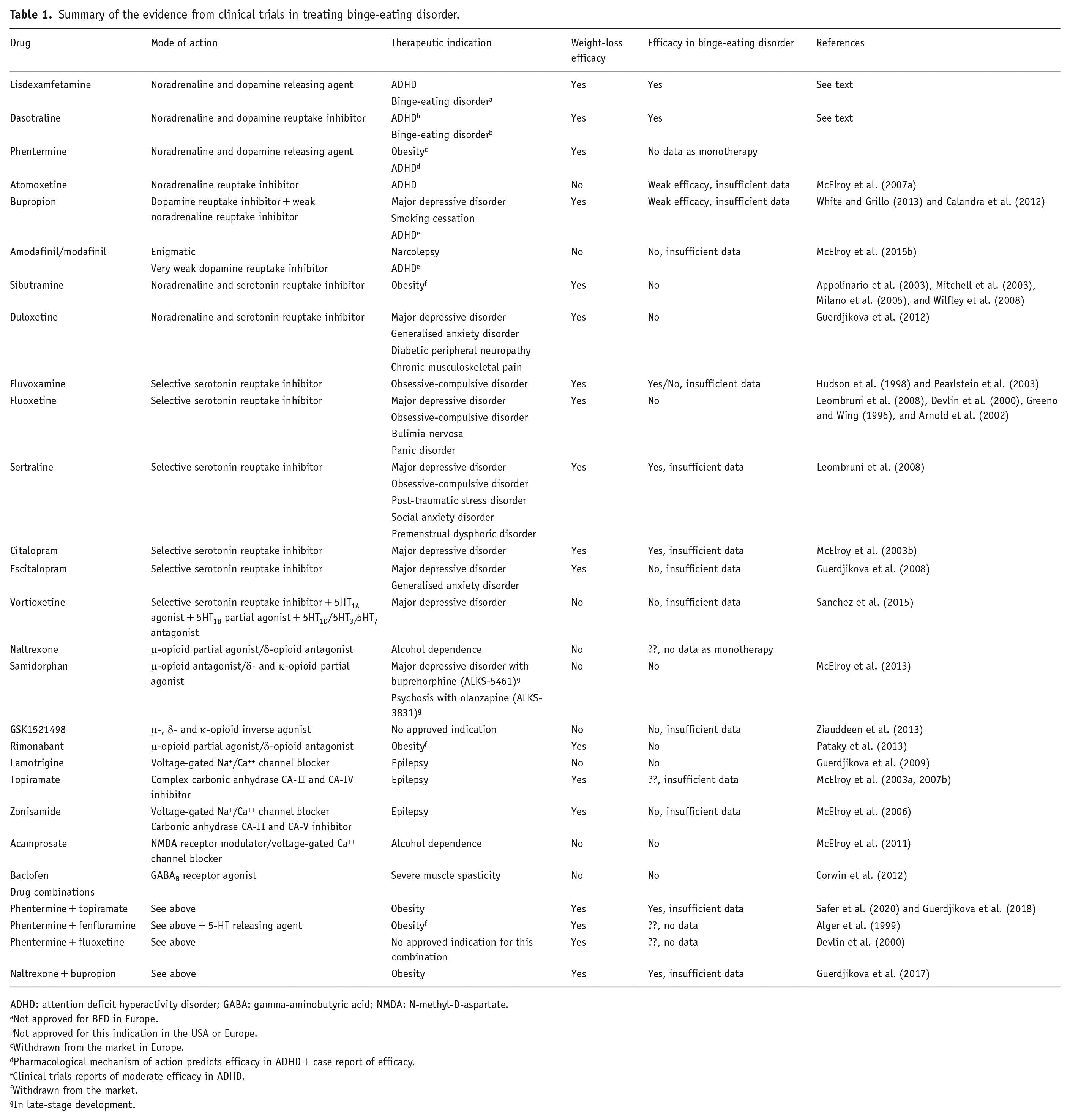
ADHD: attention deficit hyperactivity disorder; GABA: gamma-aminobutyric acid; NMDA: N-methyl-D-aspartate.
Not approved for BED in Europe.
Not approved for this indication in the USA or Europe.
Withdrawn from the market in Europe.
Pharmacological mechanism of action predicts efficacy in ADHD + case report of efficacy.
Clinical trials reports of moderate efficacy in ADHD.
Withdrawn from the market.
In late-stage development.
In this review, we discuss similarities in the psychopathology of ADHD and BED and the pharmacology of drugs that have proved to be effective in treating both disorders. An analysis of the successes and failures of drug trials BED and ADHD will be used to provide insights into pharmacological mechanisms relevant to BED and its treatment.
Clinical characteristics of binge-eating disorder versus obesity
BED was first recognised as a discrete eating disorder in the American Psychiatric Association (APA): Diagnostic and Statistical Manual of Mental Disorders, Edition 5 (APA, 2013: DSM-V); its symptoms are defined under five criteria:
Eating much more rapidly than normal. Eating until feeling uncomfortably full. Eating large amounts of food when not hungry. Eating alone because of embarrassment over how much is eaten. Feeling disgusted, depressed or very guilty after overeating.
The severity rating of BED is defined in APA: DSM-V as ranging from Mild (1–3 episodes/week) to Extreme (⩾14 episodes/week).
It is important to emphasise that none of the BED diagnostic criteria refer to weight, the metabolic sequelae of obesity, or its risk factors. The inference is the effectiveness of BED treatments is based exclusively on enabling the patient to regain self-control, reduce the impulsive, compulsive and perseverative drive to binge-eat and decrease the frequency and severity of BE episodes.
BED is the most common eating disorder with a lifetime prevalence rate in the young >1% compared with 0.3% for AN and ~1% for BN (Cossrow et al., 2016; Hoek and van Hoeken, 2003). A more recent meta-analysis estimated the lifetime prevalence of BED at 2.22% compared with 0.21% and 0.81% for AN and BN, respectively (Qian et al., 2013). An analysis of BED and BN rates across 14 countries, which included those with different average income levels, produced similar lifetime prevalence rates of 1.9% and 1.0%, respectively, with no difference due to income classification (Kessler et al., 2013). BED is slightly more common in females than males (Hay et al., 2015; Hudson et al., 2007; Kessler et al., 2013). With ~40% comorbidity between the two disorders, BED is strongly associated with obesity (Fairburn et al., 2000; Goldschmidt et al., 2011; Hudson et al., 2007; Kessler et al., 2013) and BED in adolescence predicts the development of obesity with an odds ratio (OR) = 3.58 (Micali et al., 2015). Nonetheless, a significant proportion (17%–30%) of BED sufferers have normal body weights (body mass index (BMI) 18.0–25 kg/m2) (Fairburn et al., 2000; Goldschmidt et al., 2011; Kessler et al., 2013), and according to Kessler et al. (2013) and Hudson et al. (2007), the majority (~60%) are in the normal weight/overweight categories (BMI 18.5–29.9 kg/m2). Evidence that BED is a causal factor in extreme obesity comes from the Longitudinal Assessment of Bariatric Surgery-2 (LABS-2) before/after surgery (Mitchell et al., 2015). Of 2266 severely obese subjects, 15.7% subjects satisfied the criteria for BED, 17.7% for night eating syndrome, but only 2% for BN (Mitchell et al., 2015).
BED has a strong association and comorbidity with other psychiatric conditions. Obese BED subjects have greater concerns about their appearance and body weight and exhibit greater body dissatisfaction than those without BED (Lynch et al., 2008; Sonneville et al., 2012). There is also an association between BED and anxiety or substance use disorders (Wonderlich et al., 2009). Mitchell et al. (2015) reported that taking medication for psychiatric or emotional problems, having symptoms of alcohol use disorder, lower self-esteem and greater depressive symptoms were among the factors that independently increased the odds of BED. In addition to psychiatric disorders, BED is independently associated with an increased risk of physical comorbidities including back/neck pain, chronic headaches and other types of chronic pain, as well as the cardiometabolic diseases like type 2 diabetes and hypertension, but not heart attacks or strokes (Kessler et al., 2013).
The primary endpoint in BED trials is a reduction in the frequency of binge episodes. Although most patients in these trials are obese because of the severity of their BED (Citrome et al., 2019; McElroy et al., 2015a, 2016a; Navia et al., 2017), subjects are enrolled exclusively on a confirmed BED diagnosis generally with BMI inclusion criteria of ⩾18 to ⩽45. Consistent with the focus on BE episode frequency, decreased appetite and weight loss are treated as part of the safety and tolerability assessment, not as indices of clinical benefit (Citrome et al., 2019; McElroy et al., 2015a, 2016a; Navia et al., 2017).
In contrast to BED, the diagnosis and treatment of obesity focuses on weight, adiposity and a reduction of risk factors for cardiometabolic disease and cancer. The primary efficacy endpoint for new anti-obesity drugs is a decrease in body weight (absolute and/or categorical analyses), and an important secondary measure is a reduction in waist circumference or waist/hip ratio (Center for Drug Evaluation and Research (CDER)/Food and Drug Administration (FDA), 2007; Committee for Medicinal Products for Human Use (CHMP)/European Medicines Agency (EMA), 2014). The key assumption is a reduction in weight and visceral adiposity translate into a decreased risk of developing type 2 diabetes, and suffering strokes, heart attacks and some cancers, while simultaneously, directly treating comorbidities like osteoarthritis of the knee and sleep apnoea (Albaugh et al., 2021; Center for Drug Evaluation and Research (CDER)/Food and Drug Administration (FDA), 2007; Chao et al., 2020; Colman, 2012; Committee for Medicinal Products for Human Use (CHMP)/European Medicines Agency (EMA), 2014; Hemmingsson, 2011; Malik et al., 2021; Vincent et al., 2012; Yee et al., 2007). The objective of the therapeutic intervention is to reduce the disease burden and ultimately to increase patients’ life expectancy and quality of life. Since none of the approved drugs alters metabolic rate, weight reduction is solely driven by reduced food consumption that is decreased appetite or increased satiety. What is absent from these primary and secondary outcome measures are items to explore effects on abnormal eating patterns or eating disorders, for example BED, night-time eating or BN. The reason is simple. It is because anti-obesity drugs were not designed or developed with a view to producing weight loss in patients with eating disorders by treating the underpinning psychopathology of their conditions.
Psychopathology of binge-eating disorder
The diagnostic criterion ‘a sense of lack of control of eating during the episode’ implies that BED is a classical impulse control disorder. It also illustrates the compulsive nature of the behaviour. The criteria of ‘eating alone because of being embarrassed by how much one is eating’ and ‘feeling disgusted with oneself, depressed, or very guilty after overeating’ are indices of BED’s emotional impact. Binges are defined discrete hyperphagic episodes within a short time-frame. The classification of BED as ranging from ‘mild: 1–3 episodes/week’ to ‘extreme: ⩾14 episodes/week’ demonstrates it is a frequent and recurrent behaviour, which can justifiably be described as perseverative.
Psychiatric risk factors for BED include conduct problems, negative affect, anxiety, impulse control and substance abuse disorders and perfectionism (Hilbert et al., 2011, 2014; Hudson et al., 2007; Kessler et al., 2013; McCuen-Wurst et al., 2018). Moreover, there is an emerging body of clinical evidence to show that a loss of impulse control in BED is a causal factor in bingeing on palatable foods (Colles et al., 2008; Galanti et al., 2007; Nasser et al., 2004; Schag et al., 2013; Svaldi et al., 2014; Wu et al., 2013). McElroy et al. (2016b) investigated different facets of impulsiveness in BED patients and found they exhibited deficits in motor and non-planning impulsiveness, but not attentional impulsiveness. Intolerance of delayed reward and enhanced delay discounting are established indices of impulsive choice and enhanced delay-discounting is exhibited in BED (Davis et al., 2010; Mole et al., 2015; Stojek et al., 2014). Mole et al. (2015) studied delay discounting in obese subjects with/without BED and showed both groups exhibited greater delay discounting, that is increased cognitive impulsivity, compared with normal, healthy volunteers. Stojek et al. (2014) showed dysregulated eating behaviours were associated with enhanced delay discounting. Moreover, increased intolerance of delayed reward predicted higher levels of dietary restraint and weight and shape concerns (Stojek et al., 2014). Negative urgency in delay discounting (increased delay discounting in negative mood states) predicted BE as well as concerns about body weight and body shape (Stojek et al., 2014).
Neural circuits and neurotransmitter systems involved in BED have been investigated by functional magnetic resonance imaging (fMRI) and positron emission tomography (PET). However, the findings need to be viewed cautiously because several of the studies conflate BED with obesity, (Aviram-Friedman et al., 2018; Wang et al., 2011) or BN (Fischer et al., 2017) rather than specifically targeting BED by using weight- or BMI-matched non-BED subjects as controls.
Schienle et al. (2009) performed fMRI on female subjects who were (i) overweight with BED, (ii) overweight healthy controls, (iii) normal-weight healthy controls or (iv) normal-weight BN patients. Following an overnight fast, participants’ brain activation in response to visual exposure to high-caloric food, unpleasant and neutral images were measured. Although all groups experienced food pictures as very pleasant with increased activation in orbitofrontal cortex (OFC), anterior cingulate cortex (ACC) and insula, BED subjects reported enhanced reward sensitivity and stronger medial OFC responses than all other groups. In contrast, BN subjects displayed greater arousal, ACC activation and insula activation than the other groups. Additional evidence of abnormalities in reward signalling and executive cognitive control come from the finding that patients with BED and BN exhibited aberrant functional connectivity in the dorsal ACC within the salience network, as well as in the medial prefrontal cortex (PFC) within the default mode network compared with BMI-matched control groups (Stopyra et al., 2019). Interestingly, the functional connectivity within each network differed between the BED and BN groups (Stopyra et al., 2019).
A series of fMRI studies have investigated neural connectivity and functioning during executive control and reward processing in BED patients (Balodis et al., 2013a, 2013b, 2014). Relative to BMI-matched obese and lean controls, obese BED subjects were hypoactive in brain areas involved in self-regulation and impulse control with diminished activity in ventromedial PFC (vPFC), inferior frontal gyrus (IFG), and insula during Stroop test performance. Dietary restraint scores were negatively correlated with right IFG and vPFC activation in BED subjects, but not in either control group (Balodis et al., 2013a). In a monetary win/loss paradigm, obese subjects with BED exhibited diminished bilateral ventral striatal activity during anticipatory reward/loss processing relative to BMI-matched obese subjects, but not lean controls (Balodis et al., 2013b). The relatively diminished fronto-striatal activity occurred in both anticipatory and outcome phases and during win/loss conditions indicating a generalised pattern of diminished fronto-striatal processing of rewards and losses in BED (Balodis et al., 2013b). These derangements were evidently core to BED because they persisted in treatment-resistant patients, but not in successfully treated BED subjects (Balodis et al., 2014).
Research into CNS neurotransmitter systems involved in altered connectivity and neural function in BED has focused on the dopaminergic and endogenous opioid systems because of their pivotal role in reward, motivation and cognitive control. Eating disorders have been linked with dopaminergic dysregulation in the CNS (Geiger et al., 2009; Johnson and Kenny, 2010; Pothos et al., 1995). Endogenous opioids that are important in motivational aspects of feeding are dysregulated in BED and BN (Bencherif et al., 2005; Davis et al., 2009; Nathan and Bullmore, 2009). The implicated brain areas are striatum, including ventral striatum and nucleus accumbens (ABC) which play a pivotal role in motivation, emotional responding and reward processing (Balleine, 2007; Delgado, 2007; Valbrun and Zvonarev, 2020). PFC mediates attention, cognitive function and decision-making (Arnsten, 2001, 2011; Robbins and Arnsten, 2009) and is anatomically linked to striatum via the striato–cortical pathway (Arnsten, 2001), and hypothalamus, which integrates central and peripheral signals to regulate ingestive behaviour and thermogenesis (Clapham, 2012; Harrold et al., 2012; Parker and Bloom, 2012). 5-Hydroxytryptamine (5-HT; serotonin) function has recently been identified as being dysregulated in BED with increased serotonin reuptake transporter (SERT) binding in the parieto–occipital cortical regions in BED subjects, with parallel decreases in ABC, inferior temporal gyrus and lateral OFC (Majuri et al., 2017). Although brain imaging techniques have provided a wealth of information on the neuronal systems and transmitters involved in CNS disorders, there are important technical limitations to imaging research. Some of the findings discussed are based on small numbers of subjects (e.g. Balodis et al., 2013a, 2013b; Schienle et al., 2009). Furthermore, noradrenergic systems are involved in the mode of action of LDX and dasotraline and potentially also in the psychopathology of BED; however, as there are no good noradrenergic PET tools, its potential contribution is often overlooked.
Viewing the evidence overall, BED subjects show enhanced responsiveness to palatable food cues, with reduced ability to integrate and compute the positive and negative outcomes that inevitably result from their bingeing sessions. BED subjects have substantial decrements in their ventral striatal reward pathways and diminished ability to recruit fronto-cortical impulse-control circuits to implement dietary restraint. The findings also reveal that the pattern of dysregulated reward processing in limbic brain structures, combined with decreased saliency, impulse control and cognitive decision-making in cortical regions is unique to BED and not shared with other eating disorders or obesity.
The Yale–Brown Obsessive–Compulsive Scale (YBOCS), which was introduced by Goodman et al. (1989a, 1989b), has been modified for BED (YBOCS-BE) by including additional items related to impulsivity, behavioural restraint, and distress (Deal et al., 2015). The efficacy of LDX in BED made it possible to determine the validity of the YBOCS-BE scale by a post hoc assessment of the association between LDX-induced decreases of BE frequency and changes in the YBOCS-BE scores (Citrome et al., 2018; McElroy et al., 2016b; Yee et al., 2019). The results showed YBOCS-BE is a valid scale for assessing the obsessive, compulsive and impulsive features of BED (Citrome et al., 2018; McElroy et al., 2016b; Yee et al., 2019). Although the argument is somewhat circular because of LDX’s ability to reduce impulsivity and increase cognitive control in ADHD, nonetheless it supports the hypothesis that efficacy in BED is dependent on treating its core obsessive, compulsive and impulsive behaviours.
Most mental health risk factors for BED, including conduct problems, negative affect, anxiety and impulse control and substance abuse disorders (Hilbert et al., 2011, 2014; Hudson et al., 2007; Kessler et al., 2013; McCuen-Wurst et al., 2018), are prevalent and commonly comorbid with ADHD (De Alwis et al., 2014; Eme, 2013; Ishii et al., 2003; Pliszka, 1998). Impulsivity is a core symptom in ADHD and attention deficit disorder (ADD) and manifests itself as a loss of control causing the subject to engage in risk-taking behaviours despite an awareness of the adverse consequences that may follow, for example alcohol, nicotine and illicit drug use, dangerous driving, criminal activities, etc. Impulsive choice as revealed by enhanced delay discounting is exhibited by subjects with ADHD (Anokhin et al., 2011; Jackson and Mackillop, 2016; Mostert et al., 2015; Shiels et al., 2009). Furthermore, there is often a perseverative component to these actions in ADHD, and for drug and alcohol abuse, an increasingly compulsive component as the disorder progresses. It is, therefore, unsurprising that the prevalence of substance use disorders, their rate of development and severity are several-fold higher in people with ADHD than the general population (Chen et al., 2018; Ilbegi et al., 2018; Molina et al., 2018; Polyzoi et al., 2018; Romo et al., 2018). ADHD is also associated with higher rates of eating disorders and behavioural addictions (gambling, compulsive buying disorder and Internet addiction) (Romo et al., 2018) and anxiety and depression are frequently comorbid with ADHD (Chen et al., 2018; Polyzoi et al., 2018). This synopsis reveals not only substantial overlap between the psychopathology of BED and ADHD but also a clear association between these two disorders. Furthermore, LDX and dasotraline, which have clinically proven efficacy in treating BED (Citrome et al., 2019; McElroy et al., 2015a, 2016a; Navia et al., 2017), are either approved to treat ADHD, that is LDX, or have shown clinically significant efficacy in ADHD in randomised, placebo-controlled trials, that is dasotraline (Findling et al., 2019; Koblan et al., 2015; Wigal et al., 2020).
Target product profile of the ideal binge-eating disorder drug
Although BED is often a causal factor in obesity the neurobiological drivers of excessive food consumption are very different, and therefore, the pharmacological characteristics of the ‘ideal’; drug to treat BED will be unique to this psychiatric disorder. We propose the following target product profile (TPP) for the ideal BED drug.
The ‘ideal’ drug should:
➢ Prevent the incidence of uncontrolled BE episodes.
➢ Reduce impulsive, compulsive and perseverative symptoms of BED.
➢ Increase cognitive restraint over food consumption.
➢ Restore healthy eating patterns.
➢ Reduce bodyweight to help overweight/obese subjects achieve a healthy BMI.
➢ The weight-loss effect should not be so powerful that it causes large decreases in BMI in normal weight subjects with BED.
➢ Reduce food intake by increasing satiety not by suppressing appetite thereby disrupting normal meal patterns.
➢ It should be safe when used long term.
The ‘ideal’ drug should not:
➢ Produce pharmacological tolerance that would result in dose-escalation.
➢ Cause psychological or physical dependence.
➢ Be subject to human abuse.
➢ Be a controlled drug.
The neural circuits that fail to adequately regulate food consumption in obesity are different from those responsible for compulsive and perseverative bingeing in BED. Therefore, it logically follows that the pharmacological mechanisms of drugs, which are effective in obesity may not work in BED and vice versa. Furthermore, the psychopathology and neurobiology of BED is unique and distinct other eating disorders, implying that pharmacological mechanisms that are effective in BED may not be effective in AN and BN and vice versa.
An independent weight-loss effect has been included in the TPP of the ideal BED drug. As discussed later in the review, the actions of LDX and dasotraline include an independent effect to reduce food intake by decreasing appetite or increasing satiety. If patients in the LDX and dasotraline clinical trials are representative of those seeking treatment, their BMI values show that the majority are obese or severely obese (Citrome et al., 2019; Grilo et al., 2020; McElroy et al., 2015a, 2016a, 2020); they require clinically meaningful weight loss as part of their treatment. Evidence shows that the degree of achieved weight loss increases along with initial BMI (Aftab et al., 2014; De Pergola et al., 2020; Dhurandhar et al., 2019), suggesting that this independent weight-loss effect would be more in patients who are severely obese than those patients in the overweight and normal weight ranges.
In the following sections, we review the pharmacology of drugs and drug-candidates that have been clinically evaluated in BED to elucidate the mechanisms, which offer promise for new medications and those which are unlikely to be effective.
Lisdexamfetamine and dasotraline
LDX is the only approved drug to treat BED. Although dasotraline was in pre-registration for adult BED in the USA, its development was recently discontinued (Sunovion Press Release, 2020). The FDA had already issued a Complete Response Letter for the New Drug Application (NDA) declining to approve dasotraline for the treatment of ADHD without further clinical trials to establish its efficacy and safety in August 2018 (Sunovion Press Release, 2018). When Sunovion announced it was discontinuing development of dasotraline in ADHD and BED, it stated that that further clinical studies would be needed to support a regulatory approval for dasotraline in both indications (Sunovion Press Release, 2020). On this basis, the company took the decision not to invest further in developing this drug.
These drugs share some key features. Both drugs have catecholaminergic mechanisms. LDX is a d-amphetamine prodrug comprising d-amphetamine covalently bonded to L-lysine. The active moiety, d-amphetamine, is a close analogue of the catecholamine neurotransmitters, dopamine and noradrenaline (norepinephrine), and by mimicking their chemical structures, it serves as a competitive substrate for the dopamine and noradrenaline reuptake transporters (DAT and NET, respectively) and the vesicular monoamine transporter-2 (VMAT-2) (see review by Heal et al., 2013a). d-Amphetamine is translocated into presynaptic terminals by these ATP-driven carrier systems and where it displaces dopamine and noradrenaline from the cytosolic (newly synthesised) and vesicular storage pools. They are expelled into the synaptic cleft by reversal of DAT and NET’s direction of transport (‘reverse transport’) (Heal et al., 2013a). The rapid surge of synaptic dopamine and noradrenaline produced by d-amphetamine (Géranton et al., 2003; Heal et al., 2009, 2013a; Rowley et al., 2012, 2014; Wortley et al., 1999) underpin its efficacy in ADHD. Potentiating mesolimbic dopaminergic neurotransmission is partly responsible for its efficacy, and at supratherapeutic doses, make d-amphetamine a stimulant substance of abuse (Heal et al., 2009, 2013a; Rowley et al., 2012, 2014). LDX is highly unusual because it is metabolised by a rate-limited enzymatic hydrolysis in red blood cells (Pennick, 2010; Sharman and Pennick, 2014). The pharmacokinetics (PK) of LDX profoundly influence its pharmacological actions resulting in more gradual and sustained increases in dopamine and noradrenaline concentrations in the PFC and striatum compared with immediate-release d-amphetamine (IR-d-amphetamine) (Figure 1) (Hutson et al., 2014; Rowley et al., 2012, 2014). LDX has a much longer duration of action than IR-d-amphetamine and is considerably less stimulant in animals (Ermer et al., 2016; Hutson et al., 2014; Rowley et al., 2012, 2014) and humans (Jasinski and Krishnan, 2009a, 2009b). Unlike IR-d-amphetamine, its potency cannot be increased by switching from the oral to intravenous or intranasal routes (Heal et al., 2013a; Hutson et al., 2014). In a rat BED model (Figure 2, (Binge-eating rat)) (Vickers et al., 2015), LDX and its active metabolite, d-amphetamine, dose-dependently reduced chocolate BE (Figure 3). They also preferentially suppressed the consumption of chocolate (the highly palatable food that elicited the BE) compared with normal chow. Because rats consume ~40% of their daily kJ intake in these binges, they also markedly reduce 24 h food consumption (Figure 3). LDX and IR-d-amphetamine also reduce food intake and bodyweight in a rat dietary-induced obesity (DIO) model (Figure 2, (DIO rat)) (Dickinson et al., 2001; Heal and Jagger, 2005). The large initial reduction of food intake produces steep weight loss that is followed by sustained weight loss (Figure 4). Based on these findings LDX was predicted to have a dual action in BED (i) to suppress BE and (ii) to reduce overall food consumption to induce weight loss.
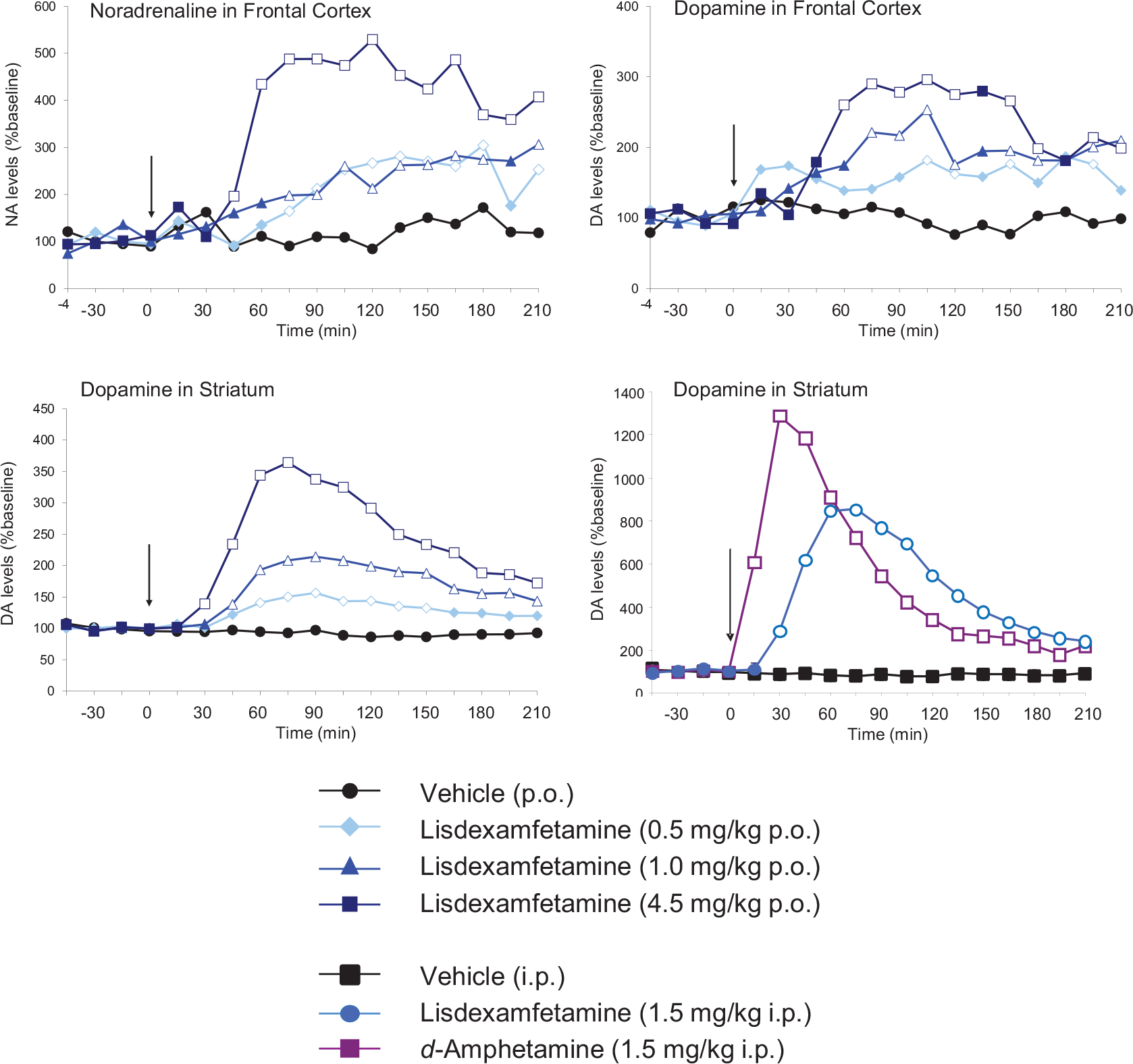
Effects of lisdexamfetamine (LDX) on catecholaminergic neurotransmission in the frontal cortex and striatum. Extracellular concentrations of dopamine and noradrenaline were investigated by intracerebral microdialysis in freely moving rats. Results are adjusted means; n = 5–8 rats/group. To demonstrate pharmacological equivalence, the doses of LDX dimesylate and d-amphetamine hemi-sulphate are expressed in terms of d-amphetamine free base. The vertical arrow indicates time of drug administration. Microdialysate samples were collected at 15 min intervals and concentrations of dopamine and noradrenaline were measured by high-performance liquid chromatography (HPLC) with electrochemical detection. Data were analysed by analysis of covariance (ANCOVA) followed by the multiple t-test (d-amphetamine) and Williams’ test (LDX).
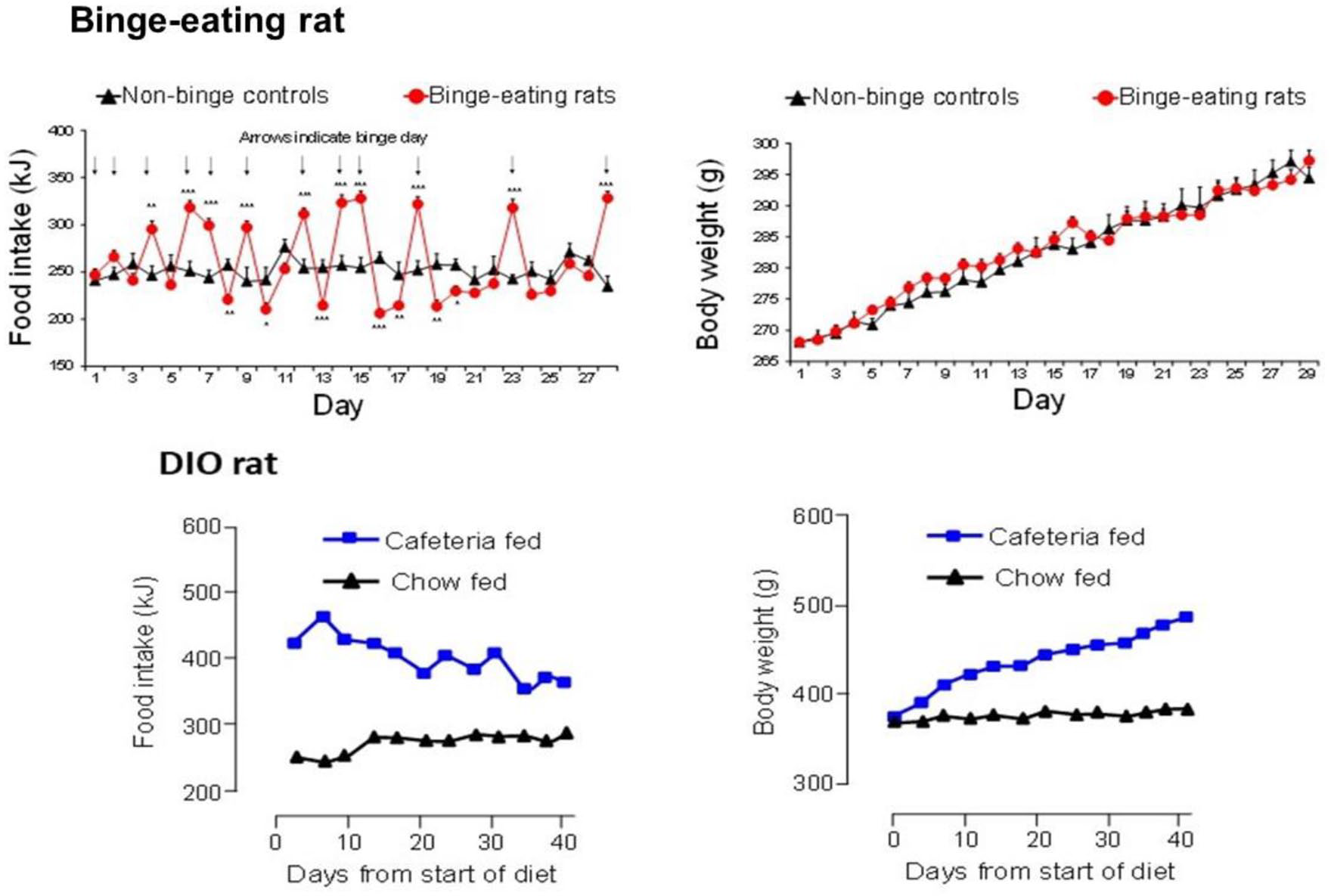
Comparison of the daily patterns of food intake and weight gain in binge eating (BE) and dietary-induced obese (DIO) female rats. BE is established in freely fed rats by giving them unpredictable, intermittent, 2 h access to powdered chocolate. Opportunities for chocolate binges are shown by the arrows in the top left panel. Rats develop a characteristic saw-tooth pattern of daily food intake with hyperphagia on chocolate binge days followed by voluntary restriction of food intake on non-binge days. Full details of the rat BE model are reported in Vickers et al. (2015). This highly abnormal pattern of BE induces impulsive and compulsive behaviours (Heal et al., 2016; Vickers et al., 2017), but not an obese phenotype (top right panel). The DIO rats are given ad libitum access to powdered chocolate as well as high-fat chow and ground salted peanuts. These rats show consistent hyperphagia over time (bottom left panel) and develop a profoundly obese phenotype (bottom right panel), which reaches a weight plateau after ~12 weeks on the diet. Full details of the DIO rat are reported in Dickinson et al. (2001).
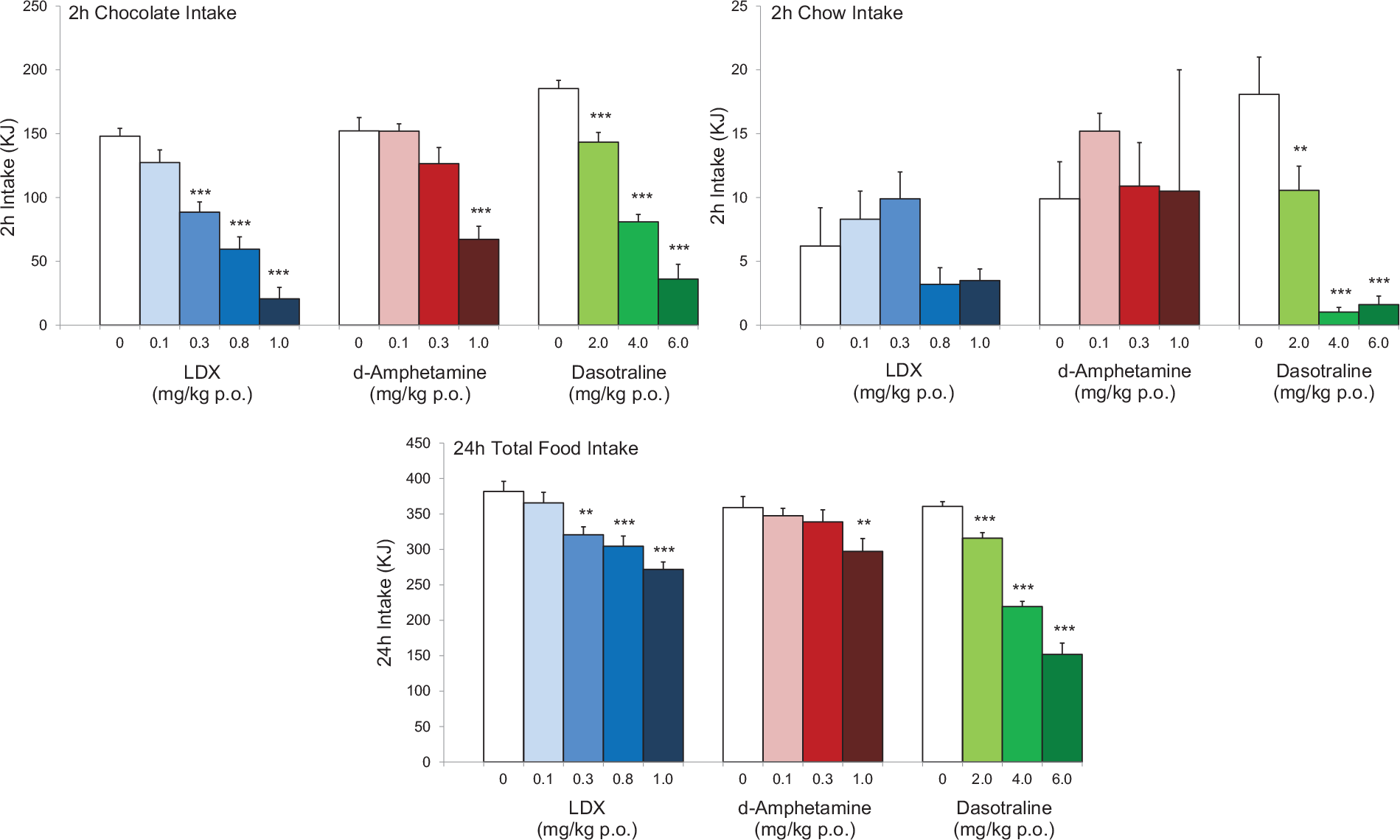
Comparison of the effects lisdexamfetamine (LDX), its active metabolite, d-amphetamine and dasotraline on binge-eating (BE) behaviour in rats. The figures show the effects of LDX, d-amphetamine and dasotraline on the consumption of chocolate and chow (normal diet) in a 2 h BE session and the overall food consumption in the 24-h period including the BE session. To demonstrate pharmacological equivalence, the doses of LDX dimesylate and d-amphetamine hemisulphate are expressed in terms of d-amphetamine-free base. The data for LDX and d-amphetamine were abstracted from Vickers et al. (2015) and the data for dasotraline from Heal et al. (2018). The results show that all of the compounds produce marked reductions in chocolate bingeing and 24 h food intake predicting that they will reduce BE and produce weight loss.
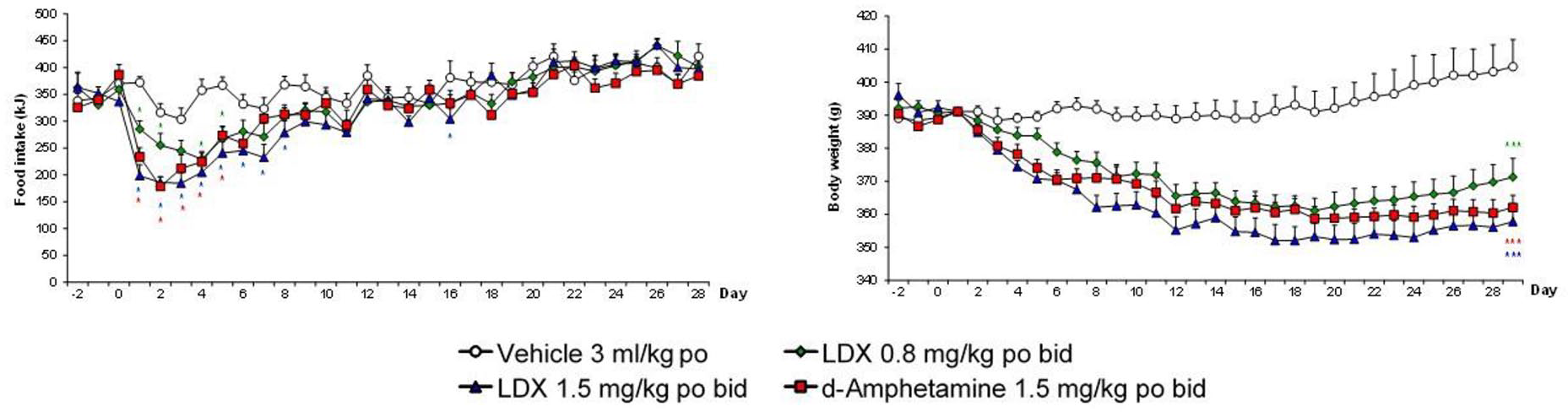
Effects of lisdexamfetamine (LDX) and its active metabolite, d-amphetamine, on daily food intake and bodyweight of dietary-induced obese (DIO) female rats. To demonstrate pharmacological equivalence, the doses of lisdexamfetamine dimesylate (LDX) and d-amphetamine hemisulphate are expressed in terms of d-amphetamine free base. Daily food consumption results were analysed by analysis of covariance (ANCOVA) with average baseline intake as covariate. Bodyweight data were analysed by ANCOVA with body weight on day 1 as covariate.
LDX significantly reduced the number of BE days/week relative to placebo in randomised, controlled trials and increased the percentage of subjects who were in remission (McElroy et al., 2015a, 2016a). Beneficial effects of LDX on BED psychopathology included significant decreases in the YBOCS-BE obsessional and compulsive scales, and at the highest dose, significant reductions in the Barratt Impulsiveness Scale, version 11 (BIS-11) self-reported questionnaire scores for non-planning and motor impulsivity (McElroy et al., 2016b). Although the lowest 30 mg/day dose of LDX did not significantly reduce the number of BE days/week, it produced substantial weight loss (McElroy et al., 2016a). Since placebo-treated patients experienced no weight loss, the results indicate that LDX has a dual mode of action, that is, it reduces food intake by suppressing appetite or enhancing satiety and is efficacious in BE at higher doses, that is, 50 and 70 mg/day. In summary, there is consensus between the findings from the non-clinical models and clinical trials demonstrating that the non-clinical models have excellent predictive validity for discovering new BED drug-candidates.
Lisdexamfetamine is approved by the FDA for the treatment of moderate to severe BED in adults. As a d-amphetamine prodrug, LDX is a schedule two controlled drug (C-II) and carries a ‘boxed warning’ in its Product Label stating that CNS stimulants (amphetamines and methylphenidate-containing products) have a high potential for abuse and dependence. It instructs prescribers to assess the risk of abuse prior to prescribing and monitor for signs of abuse and dependence while on therapy. The most common adverse events (AEs) associated with the use of LDX (incidence ⩾5% and at a rate at least twice placebo) were dry mouth, insomnia, decreased appetite, increased heart rate, constipation, feeling jittery and anxiety.
Dasotraline [(1R,4S)-4-(3,4-dichlorophenyl)-1,2,3,4-tetra-hydronaphthalen-1-amine] is a potent catecholamine reuptake inhibitor (DAT: IC50 = 3 nM and NET: IC50 = 4 nM) with weaker effects on the 5-HT transporter (SERT: IC50 = 15 nM) (Koblan et al., 2016). Dasotraline is slowly absorbed after oral administration in humans with a tmax of 10–12 h and a very long t1/2 (terminal elimination half-life) of 47–77 h (Chen et al., 2016; Hopkins et al., 2016; Koblan et al., 2015). It takes 2 weeks of daily dosing to reach steady state plasma concentrations (Chen et al., 2016; Koblan et al., 2015). Microdialysis measurements of ACB dopamine efflux were consistent with human PK (Heal et al., 2017; Rowley et al., 2017), that is small, dose-dependent increases that were slow in onset and sustained for many hours (Figure 5). Dasotraline is clearly different from the stimulants, d-amphetamine and methylphenidate, which produce rapid, large short-lasting increases in dopamine efflux (Figure 5). There is an important difference between the mechanism of releasing agents and reuptake inhibitors. The former are transporter substrates that expel neuronal monoamines by firing-independent reverse transport, whereas the latter are transporter blockers, which potentiate and prolong synaptic monoamines after firing-dependent exocytosis (Heal et al., 2013a). Tetrodotoxin blocks neuronal firing and it abolished dasotraline’s ability to increase synaptic monoamine concentrations, thereby demonstrating that dasotraline is a reuptake inhibitor not a monoamine releaser (Heal et al., 2017). In our rat BED model (Figure 2 (Binge-eating rat)) (Vickers et al., 2015), dasotraline dose-dependently suppressed chocolate bingeing with a much smaller effect on the consumption of normal chow (Figure 3) (Heal et al., 2018) predicting it would be effective in treating BED in humans. Dasotraline’s ability to independently evoke weight loss cannot be predicted without information on its effect in animal models of obesity.
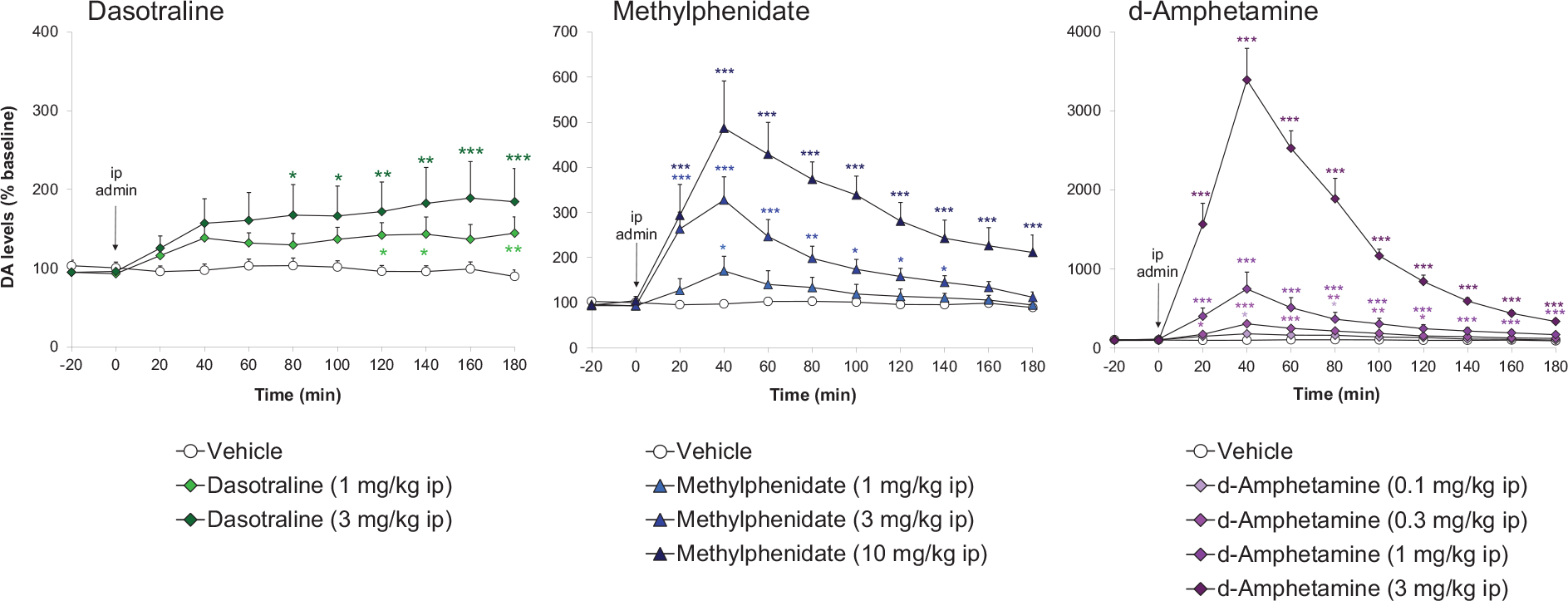
Comparison dasotraline, d-amphetamine and methylphenidate on extracellular dopamine concentrations in rat nucleus accumbens. Dopamine (DA) concentrations in the dialysates were quantified by high-performance liquid chromatography (HPLC) with electrochemical detection. Results were back-transformed, adjusted mean ± SEM (n = 6–9 rats/dose group). Drug doses are reported as free base and the time of administration is indicated by the vertical arrow. Data were log-transformed and analysed by the analysis of covariance (ANCOVA) with log(baseline) as covariate followed by the Williams’ test.
The efficacy of dasotraline in treating BED has been demonstrated in randomised, controlled trials (Citrome et al., 2019; Navia et al., 2017, 2018; Tsai et al., 2019). Dasotraline significantly reduced the psychopathological symptoms of BED with large falls in the YBOCS-BE obsession and compulsion scores (Navia et al., 2018). Although impulsivity scores were not reported, dasotraline-treated subjects showed a marked and significant increase in the dietary restraint score on the Eating Disorder Examination Questionnaire Brief Version (EDE-Q7) scale (Navia et al., 2018). Dasotraline (4 and 6 mg/day) produced significant reductions in YBOCS-BE total score from week 2 to week 12 (Tsai et al., 2019). Like LDX, dasotraline clearly reduces appetite and/or increases satiety because subjects experienced 4.78 kg decrease in bodyweight, whereas placebo controls experienced 0.4 kg weight gain despite a reduction of 3.76 BE days/week (Navia et al., 2017). A post hoc analysis of completers found dasotraline-induced weight loss increased with the patients’ BMI (Citrome et al., 2019). However, there was also evidence that some normal weight subjects experienced substantial weight decreases (Citrome et al., 2019).
Although LDX is approved and dasotraline discontinued, it is nonetheless worthwhile to assess their similarities and differences. Both drugs have a catecholaminergic mode of action, they increase noradrenergic and dopaminergic neurotransmission in the frontal cortex (FC), increase cognitive control and reduce impulsivity in ADHD. Both drugs also stimulate the mesolimbic dopaminergic reward pathway. The differences are (i) mechanism of action, that is, d-amphetamine is a catecholamine releasing agent while dasotraline is a potent, competitive catecholamine reuptake blocker with a very slow off-rate, and (ii) their PK profiles, that is, LDX has ⩽14-h duration of action while dasotraline produces continuous NET and DAT blockade. Consistent with their similar pharmacological mechanisms, LDX and dasotraline exhibit similar efficacy in reducing BE frequency and its underpinning psychopathology (Citrome et al., 2019; McElroy et al., 2015a, 2016a, 2016b; Navia et al., 2017, 2018; Tsai et al., 2019).
Placebo responses in these clinical trials revealed that substantially reducing binge frequency did not result in weight loss (Citrome et al., 2019; McElroy et al., 2015a, 2016a; Navia et al., 2017). Therefore, reduced calorie intake from fewer BE episodes was compensated by the subjects increasing their meal sizes or snacking. If treating BED does not materially improve obesity or its metabolic comorbidities, drugs need to have an independent weight-loss effect to be of maximum benefit to patients with BED who are overweight or obese.
Pharmacological approaches to treat binge-eating disorder: Insights from clinical trials with older drugs
There have been many drug trials in BED employing anti-obesity drugs and those approved for psychiatric or neurological disorders, which have been reported to cause weight loss. In a complementary article (Heal and Gosden, 2021), we have reviewed the results from these trials to dissect out which drugs are effective in treating the core psychopathology of BED as opposed to simply reducing weight loss. The clinical trial results are summarised in Table 1. In this review, we focus on the successes and failures to provide insights into pharmacological mechanisms relevant to BED and its treatment and particularly focus on the possible link between efficacy in treating ADHD and BED.
Monoaminergic drugs
Phentermine is a β-phenylethylamine, catecholamine releasing agent that differs from d-amphetamine by a single methyl group on the side chain of the molecule making it an obvious choice to evaluate in BED. Phentermine is a powerful noradrenaline and dopamine releasing agent (Rothman et al., 2001), administration of phentermine produces substantial increases in dopamine efflux in the ACB at anorectic doses and marked locomotor activation (Rowley et al., 2000). It is a stimulant reinforcer in animals (Stafford et al., 2001) and humans (Brauer et al., 1996; Carter et al., 2018). In addition to its approved obesity indication, phentermine has been reported to be efficacious in ADHD (Rothman, 1996). Although the evidence predicts phentermine would be effective in treating BED and its underlying psychopathology, no reports on phentermine as monotherapy exist; only those describing its use combined with topiramate (Qsymia®), fenfluramine, fluoxetine or topiramate (Table 1). The evidence from these drug combination trials indicates phentermine reduced the frequency and severity of BED and its underlying psychopathology (reductions in the YBOCS-BE and BIS scores and sub-scale scores). As discussed below, the evidence from monotherapy trials with the selective serotonin (5-HT) reuptake inhibitors (SSRIs) and topiramate predict that although these drugs produce weight loss, efficacy against BED psychopathology is probably mediated by phentermine’s catecholaminergic pharmacology.
Sibutramine (Meridia®, Reductil®), which has been withdrawn as an anti-obesity drug, is a potent noradrenaline and 5-HT reuptake inhibitor (Buckett et al., 1988; Heal and Cheetham, 1997; Luscombe et al., 1990) with minimal dopamine reuptake inhibition (Heal et al., 1992; Luscombe et al., 1990; Rowley et al., 2000). Noradrenaline and 5-HT reuptake inhibition operate synergistically to reduce food intake (Jackson et al., 1997a) via activation of α1-adrenergic and 5-HT2C receptors (Jackson et al., 1997b). Like ADHD drugs, sibutramine increases synaptic noradrenaline concentrations in the FC (Wortley et al., 1999). Because synaptic dopamine levels in FC are regulated by NET rather than DAT (Heal et al., 2009), it is predicted that sibutramine would also increase synaptic dopamine concentrations, c.f. atomoxetine (Bymaster et al., 2002). Unlike the stimulants used to treat ADHD, sibutramine does not influence mesolimbic dopaminergic reward mechanisms (Heal et al., 1992; Rowley et al., 2000). Sibutramine has been evaluated in four placebo-controlled trials in BED (Appolinario et al., 2003; Milano et al., 2005; Mitchell et al., 2003; Wilfley et al., 2008) where it consistently produced weight loss but had minimal effects on BED psychopathology (Table 1).
Duloxetine (Cymbalta®) is a noradrenaline+5-HT reuptake inhibitor antidepressant with a more balanced effect on noradrenaline and 5-HT reuptake (Guerdjikova et al., 2012) than sibutramine, which is more potent on noradrenaline. Duloxetine increases synaptic dopamine and noradrenaline concentrations in rat FC (Kihara and Ikeda, 1995; Umehara et al., 2013), but the effect is small, and duloxetine is minimally effective in treating ADHD (Park et al., 2014). In clinical trials, dasotraline produced a small weight loss, but it was ineffective in BED (Table 1).
Armodafinil (Nuvigil®) is the active, R(-)-enantiomer of modafinil (Provigil®). Armodafinil is approved to treat narcolepsy and other conditions involving excessive sleepiness. Although the parent racemate, modafinil, is stimulant, its pharmacology is enigmatic, and to date, no definitive mode of action has been demonstrated (Heal et al., 2009). Modafinil is a weak dopamine reuptake inhibitor (Madras et al., 2006; Volkow et al., 2009). However, with extremely low potency, that is Ki/IC50 values in the micromolar range (Cao et al., 2016; Karabacak et al., 2015; Minzenberg and Carter, 2008), it is debatable whether this mechanism is clinically irrelevant (Heal et al., 2009). Modafinil produces small increases in synaptic dopamine and noradrenaline concentrations in FC (Rowley et al., 2014), which is surprising because it has no NET blocking effect (Cao et al., 2016). Because of its micromolar DAT affinity, modafinil produces very small increases of extracellular dopamine in the striatum (Rowley et al., 2014). Consistent with its ability to increase noradrenergic and dopaminergic neurotransmission in the FC, modafinil is effective in ADHD (Wigal et al., 2006), but its efficacy is slower in onset and lower than the stimulants (Cortese et al., 2018; Wigal et al., 2006). The poor quality of the results from a single, small trial where armodafinil was administered to obese subjects with BED precludes a realistic assessment of its potential efficacy (Table 1).
Bupropion is approved for major depression (Wellbutrin®), as an aid to smoking cessation (Zyban®), and in combination with naltrexone for obesity (Contrave®). Bupropion is a weak DAT inhibitor (Hyttel, 1982; Richelson and Pfenning, 1984). Bupropion and its major metabolite have only micromolar affinities for NET (Ascher et al., 1995; Hyttel, 1982; Richelson and Pfenning, 1984). Nonetheless, NET inhibition appears to contribute to its pharmacological effects. Bupropion increases extracellular dopamine concentrations in PFC in microdialysis experiments (Li et al., 2002; Zocchi et al., 2003). It has been evaluated in ADHD, but the evidence is mixed with some trials showing efficacy (Casat et al., 1987; Wilens et al., 2001, 2005) and others not (Daviss et al., 2001). What is evident is the efficacy of bupropion in ADHD is substantially lower than the stimulants (Heal et al., 2012). Bupropion has been evaluated in BED where it produced weight loss, especially when administered in combination with naloxone (Contrave®), but at best bupropion is only marginally effective in treating BED psychopathology (Table 1).
SSRIs were initially developed as antidepressants but are now used in a broad range of psychiatric disorders. The selectivity of 5-HT versus noradrenaline reuptake inhibition varies from moderate (10–20-fold), for example fluoxetine, though selective (~100-fold), for example fluvoxamine and sertraline, to totally selective (>100-fold), for example citalopram (Bolden-Watson and Richelson, 1993; Richelson and Pfenning, 1984). Hypothalamic 5-HT plays an important role in the regulation of food intake (Halford et al., 2007) and various SSRIs have been shown to reduce hunger and/or decrease food intake in animals and humans (Greeno and Wing, 1996; Grignaschi et al., 1992; Halford et al., 2007; Ward et al., 1999). In part, these observations prompted the evaluation of several SSRIs as treatments for obesity. Although they produced short-term weight loss (Levine et al., 1989; Ward et al., 1999), tolerance developed (Abell et al., 1986; Goldstein et al., 1995; Ward et al., 1999) making them unsuitable as obesity treatments. Serotonergic drugs have never been thought to be effective in ADHD, which is a catecholaminergic disorder. This view is supported by paroxetine’s lack of efficacy in a randomised, double-blind, placebo-controlled trial in adults with ADHD (Weiss et al., 2006). The results from trials with SSRIs in BED are contradictory but viewing the data overall points to the conclusion that they are ineffective in treating BED psychopathology and are only efficacious in situations where their mild anti-obesity effect reduces daily food intake (Table 1). This view is supported by recent results with the antidepressant drug, vortioxetine (Trintellix®) showing no efficacy or weight loss in BED patients (Sanchez et al., 2015) (Table 1). Vortioxetine (Trintellix®) is a SSRI with 5-HT1A agonist, 5-HT1B partial agonist and 5-HT1D, 5-HT3 and 5-HT7 receptor antagonist properties.
Atomoxetine (Strattera®) is a highly selective, nanomolar potency, noradrenaline reuptake inhibitor (Bolden-Watson and Richelson, 1993; Wong et al., 1982) that is approved to treat ADHD in children, adolescents and adults. Atomoxetine is a classical monoamine reuptake inhibitor, which lacks the ability to evoke firing-dependent (c.f. methylphenidate) or firing-independent catecholamine release (c.f. d-amphetamine) (Heal et al., 2009, 2012, 2013a). Consequently, its potentiating effects on synaptic dopamine and noradrenaline concentrations in the FC are moderate and gradual in onset (Bymaster et al., 2002). As a non-stimulant, atomoxetine does not increase the synaptic concentration of dopamine in the mesolimbic reward system (Bymaster et al., 2002) further differentiating it from stimulant ADHD drugs. These differences in pharmacology and pharmacodynamics give atomoxetine a slow and gradual upward efficacy trajectory in ADHD that is very different from the rapid effects of the stimulants (Heal et al., 2009, 2012, 2013a). Atomoxetine has been evaluated in one, small trial in BED where it produced modest improvements in psychopathology with clinically insignificant weight loss (Table 1).
Antiepileptic drugs
Epilepsy is strongly associated with increased risk of cognitive and behavioural disorders including ADHD (Dunn and Kronenberger, 2005; Dunn et al., 2003; Hamoda et al., 2009; Pellock, 2004; Schubert, 2005). The reason for this association is unclear though it may be due to underlying neurodevelopmental vulnerability, the effects of chronic seizures and subclinical epileptiform activity (Hamoda et al., 2009). When assessing antiepileptic drugs as BED treatment, it is important to consider that some anticonvulsants, for example lamotrigine, produce relatively few cognitive adverse effects, whereas others, for example topiramate, produce substantial cognitive impairment (Schubert, 2005).
Lamotrigine (Lamictal®) is an approved anticonvulsant and it is also used in bipolar disorder. Lamotrigine inhibits voltage-sensitive, sodium channels to prevent the high-frequency repetitive burst-firing that occurs during seizure spread (Rogawski and Löscher, 2004). It also inhibits high voltage-activated calcium (N- and P/Q-type) channels but does not affect low voltage-activated T-type calcium channels (Rogawski and Löscher, 2004). Although lamotrigine does not directly alter excitatory or inhibitory synaptic responses, its effect on action potentials translates into reduced transmitter output at synapses, particularly on glutamate release (Rogawski and Löscher, 2004). No randomised, placebo-controlled trials of lamotrigine in ADHD have been conducted but results from open-label trials in children and adults suggest it is weakly efficacious (Han et al., 2017; Öncü et al., 2014). The limited clinical trial data suggest that lamotrigine has no effect on BED psychopathology and no anti-obesity effect to deliver minimal efficacy (Table 1).
Topiramate, which is a derivative of the naturally occurring monosaccharide D-fructose, is approved to treat epilepsy (Topamax®) and combined with phentermine for obesity (Qsymia®). Topiramate has complex pharmacology with multiple actions. Like lamotrigine, topiramate inhibits voltage-sensitive sodium and high voltage-activated calcium (N- and P/Q-type) channels (Langtry et al., 1997; Rogawski and Löscher, 2004; Shank et al., 2000; White, 1999). Topiramate also enhances gamma-aminobutyric acid (GABA)A-evoked Cl− ion currents (Langtry et al., 1997; Shank et al., 2000; White, 1999), selectively inhibits kainate receptors, and to a lesser extent, AMPA receptors (Langtry et al., 1997; Rogawski and Löscher, 2004; Shank et al., 2000; White, 1999). Topiramate is also an inhibitor of carbonic anhydrase enzymes, CA-II and CA-IV (Langtry et al., 1997; Shank et al., 2000). Although this last mechanism does not contribute to its anticonvulsant effect, it is probably involved the drug’s action to reduce food intake and body weight. Since topiramate’s side effects include difficulty with attention, concentration and memory, speech problems and psychomotor slowing (Langtry et al., 1997), many of which are similar to the cognitive deficits in ADHD, topiramate is unsuitable as an ADHD treatment. Topiramate decreased the frequency and severity of BED with significant decreases in the BED psychopathology and substantial weight loss (Table 1). However, a careful review of the findings indicates topiramate’s effects on impulsivity and compulsivity probably result from a generalised increase in inhibitory neurotransmission rather than an improvement in cognitive control. Moreover, topiramate’s unacceptable AE profile at these dose levels led to all development in obesity and type-2 diabetes being halted. For this reason, topiramate is unsuitable as monotherapy in BED.
Zonisamide (Zonegran®) is a synthetic 1,2-benzisoxazole derivative (1,2-benzisoxazole-3-methanesulfonarnide) approved to treat various types of epileptic seizures. Zonisamide inhibits voltage-sensitive sodium, high voltage-activated (N- and P/Q-type) and low voltage, fast-acting, T-type calcium channels, which are pivotal in inhibiting repetitive neuronal firing and seizure spread (Leppik, 1999; Masuda et al., 1998). Zonisamide also inhibits carbonic anhydrase II and V (De Simone et al., 2005; Shank et al., 2008), which plays no role in its anti-seizure activity (Masuda et al., 1994), but is probably important in zonisamide’s weight-loss effect. Zonisamide reduced antipsychotic-induced weight gain in animals and humans (Ghanizadeh et al., 2013; Wallingford et al., 2008) and produced sustained weight loss in obese subjects (Gadde et al., 2012; Nguyen et al., 2013). In BED, zonisamide produced weight loss but failed to reduce BE frequency or BED psychopathology (Table 1). Zonisamide’s side effects were very burdensome. The findings are similar to those observed when topiramate, another carbonic anhydrase inhibitor, was evaluated in BED.
Miscellaneous CNS drugs
Rimonabant (Acomplia®) is a cannabinoid CB1 receptor antagonist/inverse agonist (Pertwee, 2005; Shire et al., 1996, 1999) anti-obesity drug that was withdrawn in 2007. In obese subjects with BED, rimonabant reduced weight but had no effect on BE frequency or BED psychopathology (Table 1).
Acamprosate (Campral®) is approved for alcohol dependence. It has a relatively simple chemical structure, but complex pharmacology. Recent research indicates it attenuates glutamatergic neurotransmission by acting as a co-agonist at the spermidine-sensitive modulatory site on the N-methyl-D-aspartate (NMDA) receptor (Mann et al., 2008; Mason and Heyser, 2010; Zornoza et al., 2003). Acamprosate also changes NMDA receptor subunit composition, is a high voltage-gated Ca++ channel blocker, and it enhances taurine release (Mann et al., 2008; Mason and Heyser, 2010; Zornoza et al., 2003). Acamprosate had no effect on BE frequency or BED psychopathology in obese subjects with this eating disorder (Table 1).
Naltrexone is a nanomolar affinity, partial agonist/antagonist at μ- and δ-opioid receptors (<20% intrinsic efficacy) and κ-opioid receptor partial agonist (~40% intrinsic efficacy) (Wentland et al., 2009). Naltrexone is approved for alcohol dependence (Vivitrol®) and combined with bupropion for obesity (Contrave®). Clinical trials of naltrexone as a weight loss agent have been conducted based on the hypothesis the endogenous opioid system is dysregulated in obesity but the drug was found to be ineffective as monotherapy (Atkinson et al., 1985; Mitchell et al., 1987). There are no trials in BED of naltrexone as monotherapy. As discussed above, naltrexone+bupropion may be moderately effective in treating BED (Table 1). However, the failure of other opioid receptor inhibitors in BED, that is samidorphan and GSK1521498, argues that this pharmacological mechanism delivers no additional benefit, that is, efficacy of the combination deriving exclusively from the catecholamine reuptake inhibitor, bupropion, leaving naltrexone to contribute nothing but AEs.
Baclofen (Lioresal®, Gablofen®) is a GABAB receptor agonist (Costantino et al., 2001; Misgeld et al., 1995) muscle relaxant that is approved to treat severe spasticity. In a small clinical trial in obese subjects with BED, baclofen failed to reduce BED psychopathology or produce weight loss (Table 1). This outcome is similar to clinical findings with other drugs that increase CNS inhibitory tone, for example topiramate, zonisamide and lamotrigine, or those that are effective in treating substance use disorders, for example naltrexone and nalmefene.
Pharmacological approaches to treat binge-eating disorder: Insights from clinical trials with novel drug-candidates
Samidorphan (ALKS-33) is in clinical development in combination with buprenorphine for major depression (ALKS-5461) and with olanzapine as an antipsychotic with reduced potential for weight gain (ALKS-3831). Samidorphan is a highly potent, μ-opioid receptor antagonist and a nanomolar potency δ- and κ-opioid receptor partial agonist (intrinsic efficacy ~35%) (Wentland et al., 2009). In severely obese, BED subjects, samidorphan did not reduce the frequency or severity of BE episodes, reduce the core psychopathology or decrease weight (Table 1). Adverse events produced by samidorphan were prominent and onerous leading to a large number of discontinuations (McElroy et al., 2013).
GSK1521498 is an inverse agonist of μ-, δ- and κ-opioid receptors (Ignar et al., 2011). It has nanomolar affinity for the μ-receptor with 10–20-fold selectivity over the δ- and κ-receptor subtypes (Ignar et al., 2011). In subjects with BED, GSK1521498 reduced the hedonic effect of palatable foods and decreased calorie intake in test meals, but it did not reduce subjects’ BE scores or produce weight loss (Table 1).
Overall, the results predict that opioid receptor antagonists are unlikely to be effective in BED.
Current knowledge about the pharmacology of binge-eating disorder
In our review of drug trials in BED, we found the quality of the data was often inadequate and the evidence to support the claimed efficacy of drugs from many pharmacological classes did not stand up to scrutiny (Heal and Gosden, 2021).
With a focus on pharmacological mechanisms, the evidence shows that catecholaminergic enhancing drugs are effective in BED. Thus, LDX and dasotraline that have proven efficacy in BED increase noradrenergic and dopaminergic neurotransmission in PFC through catecholamine release (LDX) or noradrenaline reuptake inhibition (dasotraline). This is combined with increased dopaminergic neurotransmission in ventral striatum and ACB through dopamine release (LDX) or reuptake inhibition (dasotraline). Furthermore, many pharmacological mechanisms can be discounted as being effective in BED including SSRIs, SNRIs, opioid antagonists, CB1 antagonists, calcium channel blockers, carbonic anhydrase inhibitors, NMDA, GABAB agonists and GABAA receptor modulators. To this list, we would add drugs which target hypothalamic systems regulating food intake.
Although BED is a causal factor in obesity, its neurobiology and neuropharmacology are almost totally distinct from those mediating obesity; in short, excessive food consumption is the only common factor in these two disorders. BED’s psychopathology is underpinned by a loss of behavioural and cognitive control resulting in repetitive, episodic consumption of excessive quantities of food. The disorder’s episodic nature explains why a significant proportion of BED sufferers are in the normal/overweight range. The brain areas implicated in BED’s psychopathology are higher cognitive centres of FC and PFC, and the mesolimbic reward system. It is why BED is responsive to catecholaminergic stimulants, which increase cognitive control but is not responsive to drugs that decrease food intake at the hypothalamic level, for example SNRIs, SSRIs, carbonic anhydrase inhibitor anti-epileptics or CB1 antagonists.
BED is an eating disorder with a psychopathology, neurophysiology and neuroanatomy that is distinct from BN. This has been shown by fMRI studies and by the observation that although the SSRIs are moderately effective in BN, they are ineffective in BED. Correspondingly, there is no evidence to show that the catecholaminergic stimulants, which are effective in BED, are of benefit in BN.
Evidence from animal and human studies indicates that dopaminergic and opioid dysregulation contributes to BED’s psychopathology. Analogies to neurobiological abnormalities in substance use disorders and similarities between the behavioural patterns of BE and drug abuse led to the ‘food addiction’ hypothesis and BED as an addiction disorder. ‘Food addiction’ is a highly controversial concept with proponents for (Avena et al., 2011; Corsica and Pelchat, 2010; Gearhardt et al., 2011) and against (Cassin and von Ranson, 2007; Kirschenbaum and Krawczyk, 2018; Ziauddeen and Fletcher, 2013) its existence. An in-depth critique of the hypothesis or taking sides in the debate is beyond the scope of this review. However, we take this opportunity to provide some evidence for consideration. Deficits in reward processing are present in ADHD (Carmona et al., 2009; Plichta et al., 2009; Scheres et al., 2007; Tomasi and Volkow, 2012) and are correlated with increased intolerance of delayed reward leading to increased impulsive responding (Carmona et al., 2009, 2012; Costa-Dias et al., 2013; Scheres et al., 2010; Ströhle et al., 2008). Although ADHD subjects have deficits in reward processing, are intolerant of delayed gratification, and at greater risk of developing a substance use disorders (Charach et al., 2011; Erskine et al., 2016; Faraone and Wilens, 2007; Wilens, 2004), it does not infer that ADHD is an addiction disorder. Similar logic also applies to BED’s psychopathology. Looking at the issue from the perspective of effective treatments points to the same conclusion. Three drugs, which are approved to treat substance use disorders, that is bupropion, acamprosate and naltrexone, have been evaluated as treatments for BED and none has shown clear evidence of efficacy. On the other hand, LDX, which failed to show benefit as a treatment for cocaine dependence (Mooney et al., 2015), reduces impulsivity in ADHD and BED and is an effective medication for treating both disorders. Based on the neurobiological and pharmacological evidence and the findings from a systematic review of drug trials in BED, we conclude that BED is an impulse control disorder; this conclusion has also been proposed by other researchers (Kessler et al., 2016; Reinblatt, 2015; Ural et al., 2017). Recently, Kaisari et al. (2018) have provided a valuable contribution to the hypothesis that there is a shared psychopathology between BED and ADHD by showing there were significant correlations between disinhibited eating and both the hyperactive/impulsive and the attentional symptoms of ADHD. One cautionary note is these associations were based on symptoms from self-reported questionnaires and not on cohorts of subjects who had received a definitive clinical diagnosis of ADHD with or without BED.
Research directions into new drugs to treat binge-eating disorder
The TPP of the ideal BED drug has been described earlier in this review. To achieve the TPP requires two separate strands of preclinical research. The first is to demonstrate the test compound selectively or preferentially reduces bingeing on palatable food and is effective against the psychopathology underpinning the disorder, that is, impulsivity, compulsivity and perseveration. The second is to demonstrate that test compound independently reduces food intake and produces sustained weight loss (see Figures 3 and 4). In our experience, the unpredictable, intermittent access to palatable food model described by Corwin (2004) is a robust test with excellent predictive validity for clinical efficacy (Vickers et al., 2015). The model’s strengths are the rats develop a syndrome that mirrors the core psychopathology of BED in terms of increased impulsivity/intolerance of delayed reward (Vickers et al., 2017), compulsive and perseverative bingeing behaviour when aware of the adverse consequences (Heal et al., 2016) and dysregulated neurochemistry in the brain’s reward systems (Davis et al., 2009; DiFeliceantonio et al., 2012; Heal et al., 2017; Tarazi et al., 2015). Since many BED subjects are in the normal weight/overweight range, substantial calorie restriction is not relevant to developing a BED phenotype. Depriving the rats of access to binge food, for example ground chocolate, is stressful enough to induce BED without resorting to other stressors, for example immobilisation or foot shocks. On the other hand, allowing rats regular or unlimited access to palatable food produces hyperphagia, but as shown by the examples of sibutramine, baclofen and naltrexone, efficacy in these models (Berner et al., 2009; Buda-Levin et al., 2005; Corwin and Wojniki, 2009; Popik et al., 2011) is not replicated in clinical trials (Table 1). In contrast, baclofen and sibutramine had no selective effect on BE in the unpredictable, intermittent access model and performed as non-selective inhibitors of food consumption (Vickers et al., 2015); these results were reflected by baclofen’s and sibutramine’s lack of efficacy in BED trials (Table 1).
We have evaluated many drugs and test compounds in the rat BE model (Heal et al., 2018; Hurley et al., 2020; Vickers et al., 2015). Our current view is efficacy in this model is an essential support for clinical evaluation, but it needs supplementing with efficacy in models of BED’s psychopathology, that is compulsive/perseverative responding (Heal et al., 2016) and impulsivity (Vickers et al., 2017). This view is supported by the finding that when LDX was repeatedly administered to BED rats, it maintained its ability to decrease chocolate bingeing, but it had no effect on food intake on non-binge days (Hurley et al., 2020). Therefore, to achieve the TPP criteria, the drug-candidate demonstrating weight loss and weight-loss maintenance in a rat DIO model is essential.
If BED is an impulse control disorder that is treatable with drugs that reduce impulsivity and increase cognitive control, then compounds which increase catecholaminergic drive in PFC and dopaminergic drive in ventral limbic system are key mediators of efficacy. The most efficacious ADHD drugs are d-amphetamine (and as the active moiety in LDX) (catecholamine releasing agent), methylphenidate (stimulant catecholamine reuptake inhibitor) and dasotraline (long-acting catecholamine reuptake inhibitor). We have previously reviewed various drug-candidates with novel mechanisms under evaluation in ADHD (Heal et al., 2012). Many of these drug-candidates directly or indirectly enhance catecholaminergic signalling in the PFC or increase cognitive control through other mechanisms. Table 2 provides an update on ADHD clinical trials. If efficacy in ADHD and BED are linked, the catecholamine reuptake inhibitor, centanafadine (EB1020), is strongly predicted to be efficacious in BED and the SNRI, viloxazine (SPN-812), may also be effective. The findings also predict that triple reuptake inhibitors, α4/β2 nicotinic agonists (partial or full), glutamate and AMPA modulators, or histamine H3 antagonists are unlikely to prove successful in search for novel BED drugs. However, it should be emphasised that these theoretical predictions need to be tested in clinical trials.
New molecular targets to treat binge-eating disorder – update on drug-candidates in development for ADHD.
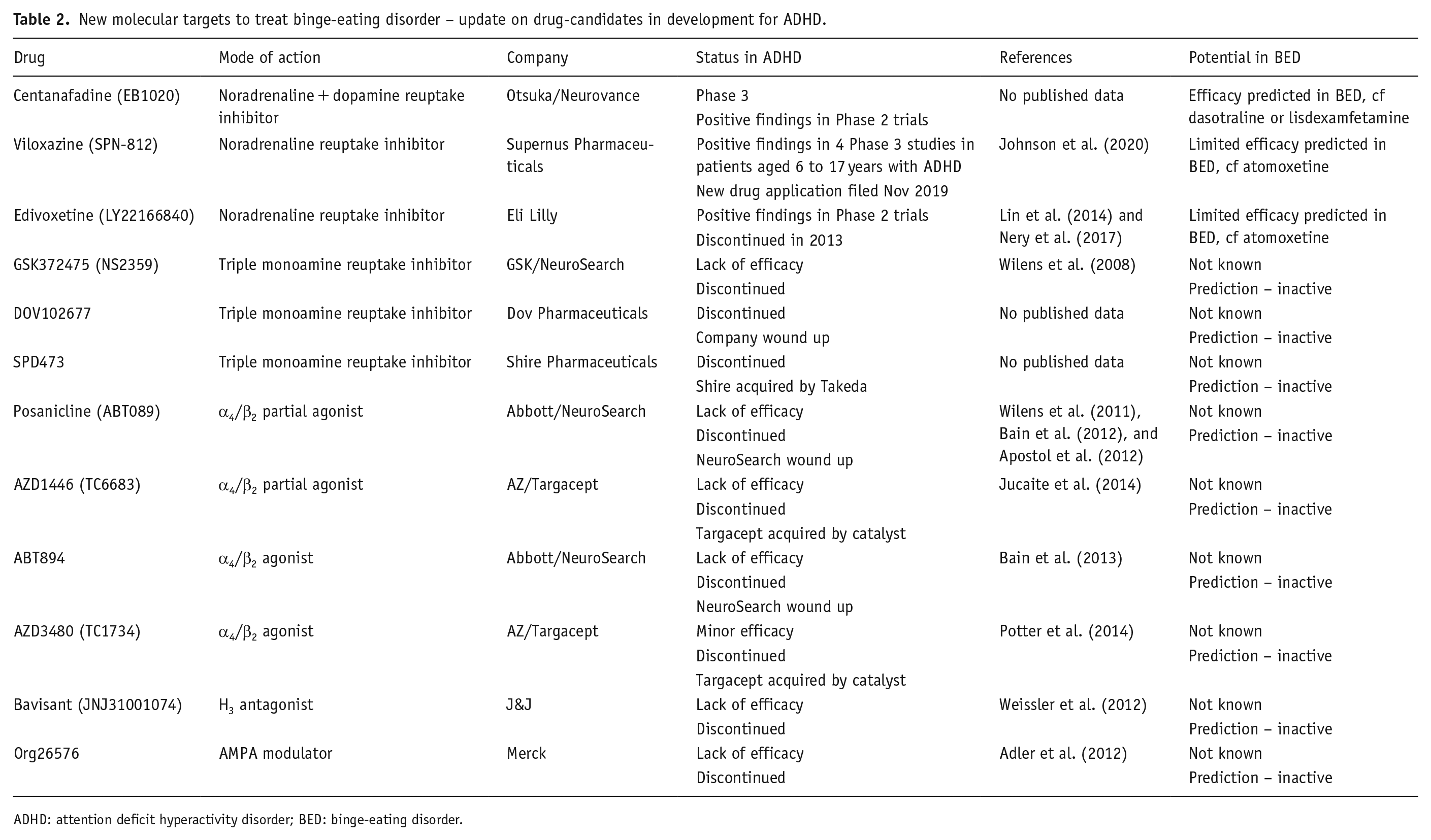
ADHD: attention deficit hyperactivity disorder; BED: binge-eating disorder.
In the last 5 years, relatively few drug-candidates in BED have emerged in the research phase. As discussed below, several compounds that are claimed to prevent BE have been evaluated in normal rats exhibiting a normal hyperphagic response to highly palatable foods. These models are not relevant; testing in a rodent BED phenotype is essential.
The most promising among them is the trace amine-associated receptor-1 (TAAR-1) antagonist, RO5256390, which has been shown to reduce the operant responding of normal rats for palatable food without affecting chow intake (Ferragud et al., 2017). Selective inhibition of palatable food consumption sparing normal chow is encouraging. However, the rat model based on predictable, daily access to palatable food lacks the stress component required to generate a BED phenotype. Validation of RO5256390 as a promising treatment needs to explore its efficacy in a BED phenotype and in BED rats in models of impulsivity, for example delay discounting (Vickers et al., 2017), and compulsivity/perseveration, for example modified CAR model (Heal et al., 2016). A similar conclusion applies to cannabidiol which reduced sucrose self-administration in normal rats and mice (Bi et al., 2020). With no BED phenotype, the result demonstrates only an effect to decrease hyperphagia, not efficacy against the psychopathology of BED.
Cifani et al. (2020) have recently reported that sigma-1 antagonists show potential as drugs to treat BED. This result requires further replication because the fasting/stress BED model is claimed to have predictive validity partly based on the differential actions of sibutramine and topiramate (Cifani et al., 2009). As discussed above, although topiramate is reported to be efficacious in BED trials, the validity of the findings is doubtful, and the authors have not studied clinically proven drugs, that is LDX or dasotraline, in this model. Further caution about the predictive validity of the model comes from Hicks et al. (2020), who reported that prazosin reduced BE. This α1-adrenoceptor antagonist had no effect on BE in our model that had been validated with LDX and dasotraline (Vickers et al., 2015). These authors have also predicted efficacy in BED for salidroside (active principle in Rhodiola rosea extract) (Cifani et al., 2010), Hypericum perforatum extract (Di Bonaventura et al., 2012a), orexin antagonists (Piccoli et al., 2012), adenosine A2A antagonists (Di Bonaventura et al., 2012b) and oleoylethanolamide (Romano et al., 2020). Since the translational and predictive validity of this BED model is unproven, the effects demonstrate only an ability to decrease stress-induced palatable food intake, not necessarily efficacy in BED.
LY2940094, a nociceptin receptor antagonist, is claimed to be a potential BED treatment (Statnick et al., 2016). The only evidence is decreased fasting-induced feeding, reductions in palatable food intake in normal rats, and effects on food intake and weight gain in DIO rats. As discussed above, these findings demonstrate the potential of LY2940094 as an anti-obesity drug, and clinical trials have shown anti-obesity drugs are ineffective in BED (Table 1).
The 5-HT2C agonist, lorcaserin, has shown efficacy in various substance use disorder models. Lorcaserin and the 5-HT2A antagonist/inverse agonist, pimavanserin, were evaluated in a rat model of bingeing (Price et al., 2018). Both drugs produced small reductions in palatable food intake, but not binge-frequency. Their effects on normal chow intake were not determined so a selective effect on bingeing cannot be supported. Lorcaserin and another highly selective 5-HT2C agonist, CP-809101, decreased deprivation-induced feeding in rats (Higgins et al., 2016). In models of impulsivity, they decreased motor impulsivity in the 5-choice serial reaction time test (5-CSRT), but critically did not increase tolerance of delayed reward in delay discounting. The findings suggest lorcaserin-induced reductions of food consumption are due to its anti-obesity properties and 5-HT2C agonists will be ineffective in addressing BED’s psychopathology. This conclusion is also consistent with our hypothesis that BED is not an addiction disorder. Lorcaserin was withdrawn from sale in February 2020. The question whether 5-HT2C agonists are effective in BED can only be definitively answered by testing in an adequately controlled and powered clinical trial. However, as the weight-loss efficacy of lorcaserin was marginal in clinical terms and several other 5-HT2C agonists have been discontinued in development, it is unclear whether such a trial will ever be conducted.
Piracetam, an anticonvulsant with enigmatic pharmacology, is claimed to have promise in BED (Hussain and Krishnamurthy, 2018). In a starvation (no food for 48 h)/refeeding rat model of BED, piracetam and LDX (reference comparator) decreased bingeing on cookies. They also reduced chow intake, suggesting the effects were anorectic rather than selective for BE. What was most concerning was the oral doses studied: piracetam was administered at 200 mg/kg, and LDX at 100 mg/kg. As shown in Figure 3, LDX is orally active in BE models at doses between 0.3 and 1.0 mg/kg.
Very recently, Hurley et al. (2020) presented preliminary results on the serotonergic hallucinogen, psilocybin, which had been evaluated in our rat BED model. A single injection of psilocybin (1, 3 or 10 mg/kg ip) decreased chocolate bingeing 1 h after administration. The effect of the highest dose persisted for 24 h but was not maintained at 5 days. In contrast, daily administration of LDX (0.8 mg/kg po) consistently suppressed BE with no effect on food consumption on non-binge days. Since psilocybin is a drug that is intended to be administered infrequently in the clinic, the results suggest that it will not be useful as a treatment for BED.
Conclusions
In the last decade, we have learned that BED is a distinct eating disorder with a unique psycho- and neuropathology. The relatively high prevalence of BED coupled with the clinical evaluation of LDX and latterly dasotraline, which both demonstrated unequivocal efficacy in reducing the psychopathology of this disorder, have generated renewed interest in developing new drugs for this indication. Moreover, the availability of clinically effective drugs as well as those which failed to show efficacy in BED trials provides sufficient positive and negative controls to facilitate the development of animal models with translational validity for clinical outcomes.
One of the complexities is bingeing in BED is a manifestation of the psychopathology of the disorder. It is not due to a failure of the hypothalamic regulation of food intake. This point is clearly revealed by the failure of a large number of clinically effective anti-obesity drugs to show efficacy in BED in patient trials. Another is the discovery that in many instances remission in BED is not accompanied by weight loss in patients. If patients in clinical trials are representative of those seeking treatment, mean BMI values suggest that the majority are obese or severely obese. These patients require weight loss, which is the rationale for suggesting the TPP for a novel BED drug should include an independent effect to reduce food intake by decreasing appetite or increasing satiety.
Having reviewed historical clinical trials with drugs for diverse therapeutic indications and widely differing pharmacological mechanisms, we were surprised by the limited number that would meet current efficacy criteria (Heal and Gosden, 2021; summarised in Table 1).
Although there is a need for new drugs to support healthcare professionals in treating BED, the scope for drug targets appears to be restricted to compounds that markedly augment catecholaminergic neurotransmission. This conclusion is based on the diverse pharmacological mechanisms of drugs, which have already failed in BED trials together with those which we have tentatively predicted would fail because they have not proven to be effective in ADHD.
On a more positive note, behavioural phenotyping of compounds in animals identified TAAR-1 ligands as potential treatments for schizophrenia, a disorder that historically was only treatable with dopamine receptor antagonists. As described above, RO5256390 shows some promise as a potential BED treatment. We have animal models of BED including its core psychopathology with good predictive validity for clinical efficacy. Testing compounds in these validated animal models could, therefore, open a path to discovering new drugs which act through non-catecholaminergic mechanisms.
Footnotes
Author Note
David J Heal is now affiliated to Department of Pharmacy and Pharmacology, University of Bath, Bath, UK.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: David J Heal and Sharon L Smith are shareholders and employees of DevelRx Ltd. DevelRx provides consultancy support to the pharmaceutical industry.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.