
Abstract
Major depressive disorder (MDD) is a prevalent and disabling psychiatric disease with rates of non-responsiveness to antidepressants ranging from 30–50%. Historically, the monoamine depletion hypothesis has dominated the view on the pathophysiology of depression. However, the lack of responsiveness to antidepressants and treatment resistance suggests that additional mechanisms might play a role. Evidence has shown that a subgroup of depressive patients may have an underlying immune deregulation that could explain the lack of therapeutic benefit from antidepressants. Stimuli like inflammation and infection can trigger the activation of microglia to release pro-inflammatory cytokines, acting on two main pathways: (1) activation of the hypothalamic–pituitary adrenal axis, generating an imbalance in the serotonergic and noradrenergic circuits; (2) increased activity of the enzyme indoleamine-2,3-dioxygenase, resulting in depletion of serotonin levels and the production of quinolinic acid. If this hypothesis is proven true, the subgroup of MDD patients with increased levels of pro-inflammatory cytokines, mainly IL-6, TNF-α and IL-1β, might benefit from an anti-inflammatory intervention. Here, we discuss the pre-clinical and clinical studies that have provided support for treatment with non-steroidal anti-inflammatory drugs in depressed patients with inflammatory comorbidities or an elevated immune profile, as well as evidences for anti-inflammatory properties of standard antidepressants.
Keywords
Introduction
Major depression disorder (MDD) is an important public health issue (Dowlati et al., 2010; Miller et al., 2009), predicted to be the second leading cause of disability by the year of 2020 behind only ischemic heart disease (Rush et al., 2004). MDD is the most commonly diagnosed psychiatric disorder in adults over 60 years of age (Dowlati et al., 2010). The Diagnostic and Statistical Manual of Mental Disorders (DSM-V) describes that for the diagnosis of MDD, five or more symptoms have to be present during a 2-week period and represent a change from previous functioning; at least one of the symptoms should be either: (i) depressive mood or (ii) loss of interest or pleasure for the major part of the day. The other symptoms that may be present are significant weight loss or weight gain, insomnia or hypersomnia, fatigue or loss of energy, diminished ability to concentrate or indecisiveness, recurrent thoughts of death and suicidal ideation or attempt (American Psychiatric Association, 2013). Not only the high incidence of MDD and the disability associated with the disease, but also the high rate of inadequate treatment of the disorder remains a serious concern (Kessler et al., 2003). It is estimated that 30–50% of the patients do not respond to treatment with antidepressants (Bschor et al., 2012) due to either lack of efficacy or intolerable side effects (Rush et al., 2006). Another possible reason for the ineffectual treatment of MDD has been the incomplete understanding of the nature of depression (Friedman, 2014). The high rate of treatment resistance, together with the high suicide risk in unresponsive patients and the overwhelming economic costs to society constitute the basis of the search for new therapeutic agents (Brunello et al., 2006), aiming to improve the quality of life or even cure these patients. Remission, that is, (virtual) absence of symptoms should be the objective of MDD treatment, since it is related to better functioning and a better prognosis than a response without remission (Rush et al., 2006; Trivedi et al., 2006).
Even though information concerning the epidemiology, symptoms and complications of mood disorders are well documented, the etiology and pathophysiology of depression are not completely elucidated (Rosenblat et al., 2014). The monoamine depletion hypothesis has historically dominated the view on the pathophysiology of depression. It suggests that an imbalance, mainly in serotonergic and noradrenergic neurotransmission is the core of the pathophysiology of depression (Massart et al., 2012; Prins et al., 2011). However, the lack of responsiveness to conventional treatment with antidepressants and high rates of treatment resistance suggests that additional mechanisms might play a role in depression. Over the last 20 years, psychiatric research has provided support for the hypothesis that inflammatory processes and brain–immune interactions are involved in the pathogenesis of MDD and may contribute to the serotonergic and noradrenergic dysfunction (Song and Wang, 2011). Stimuli like inflammation, chronic stress and infection can trigger the activation of microglia, the brain’s immune cells, to release pro-inflammatory cytokines that can act on two pathways that may lead to MDD and neurodegeneration, such as: (1) activation of the hypothalamic–pituitary adrenal axis, generating an imbalance in the serotonergic and noradrenergic circuits; (2) increased activity of the enzyme indoleamine-2,3-deoxygenase (IDO), resulting mainly in depletion of serotonin. Considering that MDD is a very complex and heterogeneous disorder, it is possible that immune deregulation is not present in all depressed patients, but only in specific sub-populations (Vogelzangs et al., 2012). Evidence also shows that lack of therapeutic benefit of antidepressants might be associated with persistent immunological impairment (Carvalho et al., 2013).
In this review, we aim to discuss the potential role of anti-inflammatory treatment in MDD. We first address the most relevant immunological mechanisms by which increased levels of pro-inflammatory cytokines may lead to MDD, highlighting the hypothalamic–pituitary–adrenal (HPA) axis hyperactivation and the indoleamine-2,3-dioxygenase (IDO) pathway. Next, we summarize the most recent studies concerning monotherapy with non-steroidal anti-inflammatory drugs (NSAIDs) in MDD patients, discuss the anti-inflammatory effects of standard antidepressant drugs and augmentative strategies with NSAIDs.
The hypothesis of immunological involvement in the pathophysiology of major depressive disorder (MDD)
An exhaustive discussion on all the possible immunological pathways that might play a role in the pathophysiology of depression is out of the scope of this article. Before focusing on the possible anti-inflammatory treatment for depression, however, we would like to review key points and molecular markers that are most relevant for the anti-inflammatory therapeutic strategies further discussed.
Association between pro-inflammatory cytokines and depression: the role of microglia
The hypothesis of a causal relationship between pro-inflammatory cytokines and depression was first described by Smith et al. in 1991, in the macrophage theory of depression. The theory was based on observations that cytokines produced by macrophages, when given to healthy volunteers, induced symptoms of depression and had brain effects that included the activation of the HPA axis (Dantzer et al., 2011; Smith, 1991). Afterwards, Maes et al. corroborated the theory by collecting biochemical evidence for the immunological activation in depressed patients (Maes et al., 1995, 1997). In response to infection or inflammatory conditions, peripherally produced cytokines can act on the brain and cause behavioral symptoms (Dantzer et al., 2008), such as malaise, prostration, fatigue, numbness and anorexia (Frick et al., 2013). The main elucidated pathways to which pro-inflammatory cytokines can reach the brain include: (1) cytokine passage through leaky regions in the blood–brain barrier (BBB); (2) active transport via saturable transport molecules; (3) activation of endothelial cells and other cell types (including perivascular macrophages) lining the cerebral vasculature (which in turn produce cytokines and other inflammatory mediators); (4) binding to cytokine receptors associated with peripheral afferent nerve fibers (e.g. vagus nerve), delivering cytokine signals to relevant brain regions including the nucleus of the solitary tract and hypothalamus (Dantzer et al., 2008; Miller et al., 2009). The nuclear factor NF-κB has been identified as an essential mediator at the blood–brain interface that communicates peripheral inflammatory signals to the central nervous system (CNS). Production of inflammatory cytokines can also be induced directly within the brain, via stress or other processes (e.g. vascular insults in late life depression) (Miller et al., 2009; Nadjar et al., 2005).
In the CNS, microglia cells are the main cellular regulators of the innate immune response to both physiological and pathological conditions (Czeh et al., 2011). They transform from an immunesurveillant into an activated state in response to pathogens and to synaptic and neuronal injury in several neurological disorders. During their activation, microglia change from a ramified to a hyper-ramified (Pace et al., 2006; Rawdin et al., 2013; Zunszain et al., 2011) phenotype and subsequently adopt an amoeboid morphology, a mechanism which has been suggested to help microglia to invade lesions (Raivich, 2005). This activation can be acute or chronic, depending on the type of stimulus (inflammation, stress, infection, neuronal injury) and its duration (Czeh et al., 2011). Thus, activation of microglia in stress might be different from microglial activation during inflammation or infection (Sugama et al., 2009). When chronically activated, microglia can produce a wide variety of neurotoxins such as pro-inflammatory cytokines, free radicals, nitric oxide, chemokines, proteinases and eicosanoids (Venneti et al., 2013) that may cause neuronal dysfunction and aggravate underlying pathologies (Venneti et al., 2006). As such, activated microglia can be a triggering factor for mood disorders (Rosenblat et al., 2014). Activated microglia have already been found in the brain of stress-induced animal models of depression (Wohleb et al., 2011, 2012), however the data that would confirm the presence of activated microglia in humans are still limited (Kreisel et al., 2014). Evidence for neuroinflammation in MDD could be obtained non-invasively by positron emission tomography (PET) using radioligands that bind to the translocator protein (TSPO), a receptor that is upregulated in the mitochondria of activated microglia cells (Doorduin et al., 2008). Recently, the presence of neuroinflammation in depressed patients during a major depressive episode was demonstrated using PET with the TSPO radioligand [18F]FEPPA (Setiawan et al., 2015). The study was conducted on 20 patients in a major depressive episode secondary to MDD that were medication free for at least 6 weeks, and 20 healthy controls. A significant increase in the uptake of the tracer was found in the prefrontal cortex, anterior cingulated cortex and insula, indicating the presence of activated microglia in these brain regions. Moreover, PET tracer uptake (microglia activation) was correlated with the Hamilton Depression Rating Scale (HDRS) score (Hamilton, 1960) in the anterior cingulated cortex (Setiawan et al., 2015). Hannestad et al. (2013) also conducted a study to evaluate the presence of neuroinflammation in patients with mild-to-moderate depression using [11C]PBR28, another TSPO ligand. No difference between patients and controls was found in this study (Hannestad et al., 2013). This could be due to the small sample size (n = 10) and the fact that patients with signs of peripheral immune activation (as defined by elevated high-sensitive C-reactive protein, hsCRP) were excluded. Further studies with PET imaging should be conducted in order to corroborate or not the presence of activated microglia in MDD in a non-invasive manner. Thus, an increased density of activated microglia was observed post mortem in the anterior midcingulate cortex, dorsolateral prefrontal cortex and mediodorsal thalamus of suicidal patients with affective disorders (Steiner et al., 2008).
More recently, an increased gut permeability or ‘leaky gut’ theory was described as a possible contributor to the peripheral and central production of pro-inflammatory cytokines by microglia in a subgroup of depressed patients. The investigated subjects were diagnosed with MDD and presented specific symptoms which have been correlated to increased levels of IgM and IgA to lipopolysaccharide (LPS) of enterobacteria in chronic fatigue syndrome (Maes et al., 2007). The observed symptoms were pain, muscular tension, fatigue, concentration difficulties, failing memory, irritability, stress and irritable bowel, among others. In summary, depressed patients demonstrated elevated serum IgM and IgA levels against LPS of gram-negative enterobacteria, as compared with healthy controls. Increased IgM and IgA levels indicate an increased gut permeability, allowing invasive enterobacteria to cause a systemic and central inflammation (Maes et al., 2008; Slyepchenko et al., 2017).
Elevated pro-inflammatory cytokines and hypothalamic–pituitary–adrenal (HPA) dysfunction in major depressive disorder
Numerous studies have indicated that MDD is accompanied by elevated levels of inflammatory biomarkers, such as the pro-inflammatory cytokines interleukin (IL)-1β, IL-6, IL-18, tumor necrosis factor alpha (TNF-α), interferon-gamma (INF-γ) (Alboni et al., 2010; Dowlati et al., 2010; Hasler, 2010; Krogh et al., 2014; Loftis et al., 2010; Maes, 2011; Messay et al., 2012; Najjar et al., 2013; Pace and Miller, 2009; Raison and Miller, 2011; Rawdin et al., 2013; Steiner et al., 2011; Sukoff Rizzo et al., 2012; Zunszain et al., 2011) and the acute phase proteins such as C-reactive protein (CRP) (Jokela et al., 2015; Valkanova et al., 2013) Munzer et al. (2013) even suggested that, besides, for example, stress hormones and psychopathological measures, cytokines may serve as biomarkers for individualized treatment of depression (Munzer et al., 2013). Thus, animal studies have shown that systemic exposure to inflammatory challenges, such as LPS, not only causes a systemic inflammation but also induces a central inflammatory response in the brain, which is reflected by activation of microglia (Qin et al., 2007).
The pro-inflammatory cytokines produced during activation of microglia might have an effect on central serotonin levels and affect the HPA axis (Figure 1). The immune and neuroendocrine systems act together in order to restore and maintain physiological homeostasis during inflammation and other harmful stimuli that might induce systemic cytokine production (Beishuizen and Thijs, 2003). Therefore, it has been suggested that abnormalities in the HPA axis might play a key role in the development and recurrence of depression. Increased cytokine production may contribute to the development of depression directly via activation of the HPA axis or indirectly through cytokine-induced glucocorticoid receptor resistance (Anders et al., 2013). The release of TNF-α and IL-6 increases the production of corticotrophin-releasing hormone, adrenocorticotropic hormone and cortisol by acting directly on hypothalamic and pituitary cells (Dowlati et al., 2010). Cytokines might also increase glucocorticoid receptor resistance through several signaling pathways, including activation of the p38 mitogen-activated protein kinase (MAPK) and by stimulating changes in the expression of glucocorticoid receptors (Anders et al., 2013; Pace et al., 2007). The high levels of circulating stress hormones in the CNS might affect the neurotransmitter homeostasis, the neuronal growth factor synthesis and ultimately, disturb the functioning of neuronal circuits of the limbic system (Guloksuz et al., 2014). HPA hyperactivity has been associated with the pathophysiology of suicidal behavior, excessive activity of the noradrenergic system and dysfunction of the serotonergic system (Kim et al., 2008; Steiner et al., 2008).
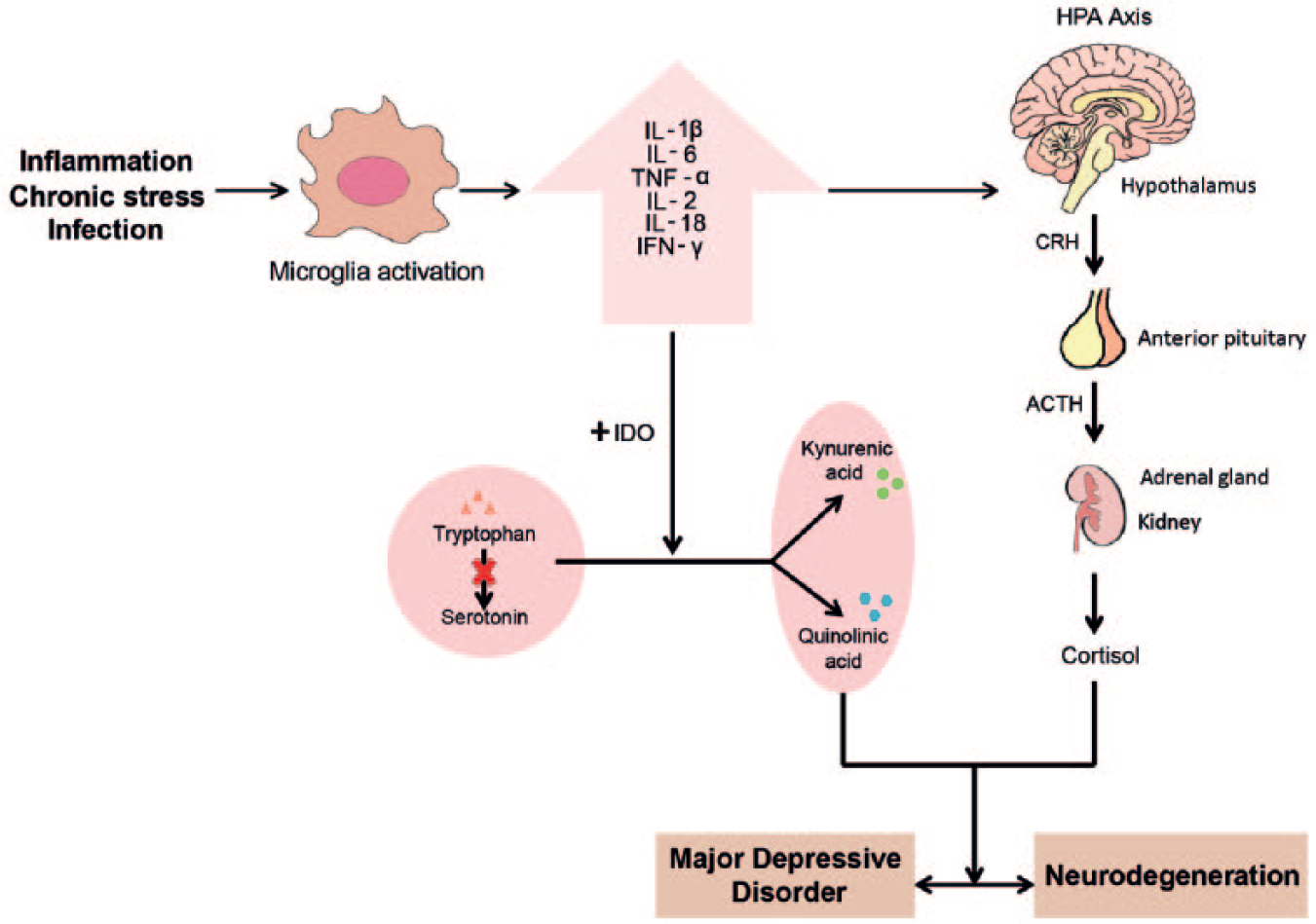
Hypothesis of immune involvement in the pathophysiology of major depressive disorder. Inflammatory, infectious and stressful challenges might trigger the activation of the resident microglia. Activated microglia produce pro-inflammatory cytokines that can contribute to neurodegeneration and depressive disorders through the hyper-activation of the HPA axis and the increase in indoleamine-2,3-dioxygenase (IDO) enzyme activity. Hyper-activation of the HPA axis leads to the increase of corticotrophin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH) and cortisol that disturb neurotransmitter homeostasis (mainly noradrenergic and serotonergic systems) and the neuronal growth factor synthesis. IDO decreases the synthesis of serotonin by switching the balance between the production of serotonin from tryptophan and the production of kynurenic acid (KYN) and quinolinic acid (QUIN). Depletion of serotonin leads to depressive symptoms. QUIN acts as a neurotoxin, gliotoxin, pro-inflammatory mediator and can also alter the integrity of the blood–brain barrier (BBB).
Pro-inflammatory cytokine effects on neurotransmitter metabolism
The link between pro-inflammatory cytokines and decreased serotonergic synthesis has already been extensively explored. It was hypothesized that during inflammation, pro-inflammatory cytokines such as IL-1β, IL-2, IL-6, INF-γ and TNF-α (Anderson et al., 2013; Kim et al., 2012; Myint and Kim, 2003) increase the activity of IDO and reduce the production of serotonin. IDO catalyzes tryptophan (TRP) catabolism through the kynurenine pathway (Dobos et al., 2012; Myint, 2012; Myint et al., 2013), producing kynurenic acid (KYN), quinolinic acid (QUIN) and nicotinamide adenine dinucleotide (NAD+) (Guillemin, 2012; Zunszain et al., 2011). Substantial evidence demonstrates that a pro-inflammatory scenario leads to increased and unbalanced production of tryptophan catabolites (TRYCATs) that play a major role in the development and maintenance of MDD. A recent meta-analysis by Ogawa and colleagues (2014) demonstrated convincing evidence for lowered plasma TRP levels in patients with MDD. The study included MDD patients (n = 744) and healthy controls (n = 793), and found a highly significant decreased levels of TRP in depressed patients vs controls (p < 0.001). A secondary analysis using only data of unmedicated MDD patients (n = 156) and controls (n = 203) demonstrated an even more pronounced difference in TRP levels in unmedicated patients, when compared with controls (p < 0.001). These data suggest that psychotropic therapy (antidepressants, antipsychotics and benzodiazepines) reduced the difference in TRP levels between groups (Ogawa et al., 2014). Decreased levels of TRP and consequent depletion of serotonin results in the development of depressive symptoms, as proposed by the classic monoamine depletion hypothesis. IDO induction may have evolved as a mechanism for the maintenance of NAD+, which is the final product of the IDO and TRP catabolism pathway. NAD+ is important for the induction of sirtuins, which contribute to many of the processes that are deregulated in depression including neurogenesis, circadian rhythms and mitochondrial regulation (Anderson et al., 2013). Despite the evidence that suggest a role of TRYCATs in depression, one should keep in mind that TRYCATs have also been associated with the psycho-somatic symptoms that accompany depression. Since depression and somatization shared common pathways, it may be difficult to discriminate between these effects (Maes and Rief, 2012).
QUIN, a product formed in the TRYCAT pathway, is an endogenous N-methyl-D-aspartate (NMDA) receptor agonist, while KYN is an NMDA antagonist. QUIN is a neurotoxin and responsible for the generation of reactive oxygen and nitrogen species (ROS and RNS, respectively). A disrupted balance between KYN and QUIN production is observed in the neurotoxicity associated with several inflammatory brain diseases such as Alzheimer’s disease, Parkinson’s disease and major psychiatric disorders. Activated microglia and infiltrating macrophages are the major source of QUIN in the brain and it is involved in the deleterious pathophysiological cascade within the CNS (Guillemin, 2012). An aberrant NMDA receptor stimulation associated with pro-inflammatory cytokines may suppress brain-derived neurotrophic factor translation, neurogenesis, provoke changes in brain volume, along with dendritic atrophy and synaptic loss (Savitz et al., 2015; Swardfager et al., 2016). The atrophy of the hippocampus in patients with MDD has been demonstrated, not only by imaging techniques such as magnetic resonance imaging (MRI) but also in post mortem studies (Stockmeier et al., 2004). QUIN also increases glutamate release to neurotoxic levels, inducing oxidative and nitrosative stress (O&NS) in MDD (Slyepchenko et al., 2016). O&NS damages lipids, proteins and the DNA, as demonstrated through lipid peroxidation, DNA strand breaks, increased protein carbonyl formation and disruption of mitochondrial function (Bryleva and Brundin, 2017; Morris et al., 2017). Inflammatory responses are often accompanied by O&NS, as reviewed in detail by Maes et al. (Maes et al., 2011). Under normal conditions, the level of ROS are balanced by an antioxidant defense system. However, when there is an unbalanced condition between oxidants and antioxidants, a state of oxidative stress is achieved. Recently, a meta-analysis confirmed the association between depression and oxidative stress, measured mainly in plasma or serum of depressed patients and healthy controls (Palta et al., 2014). It is also known that lower levels of antioxidants, such as co-enzyme Q10, glutathione, ascorbic acid, vitamin E, zinc and polyunsaturated fatty acids are regularly detected in the blood of depressed patients (Maes et al., 2011; Slyepchenko et al., 2016), supporting the notion of an oxidative-stress state in this population.
Other potential harmful effects of inflammatory cytokines on neurotransmitter function are due to the disruption of tetrahydrobiopterin (BH4). BH4 is an essential enzyme co-factor for phenylalanine hydroxylase, tryptophan hydroxylase and tyrosine hydroxylase which are rate-limiting enzymes for the synthesis of serotonin, dopamine and norepinephrine, respectively (Miller et al., 2013). Moreover, BH4 is also an enzyme co-factor for the conversion of arginine to nitric oxide (NO) through nitric oxide synthase (NOS) (Haroon et al., 2012). Inflammatory cytokines stimulate the production of NO, increasing the utilization of BH4 and thus decreasing neurotransmitter synthesis (Miller et al., 2013).
TNF-α is a specific pro-inflammatory cytokine that has received attention as a potential modulator of the serotonin transporter (SERT or 5-HTT) and consequently 5-HT uptake and brain availability. The first study to demonstrate in vitro the capacity of TNF-α to increase the expression of SERT in mouse brain cell lines was conducted in 2006 (Zhu et al., 2006). Afterwards, another study showed that prolonged in vitro treatment with TNF-α enhances SERT expression and activity in both glial and neuronal cells, suggesting that the p38 MAPK pathway could be involved (Malynn et al., 2013). Therefore, it was hypothesized that under conditions of chronic inflammation, increased levels of pro-inflammatory cytokines such as TNF-α would enhance SERT-mediated 5-HT uptake and significantly impact the available extracellular 5-HT. Since astrocytes rapidly degrade 5-HT following uptake, enhanced astrocyte uptake might affect the turnover rate of this neurotransmitter, resulting in decreased total brain 5-HT (Malynn et al., 2013). In a proof-of-concept study conducted by Cavanagh et al. (2010), six patients with rheumatoid arthritis were treated with adalimumab (a TNF-α inhibitor) and tested the hypothesis that TNF-α blockade would alter SERT activity in the brain of the patients, through single photon emission tomography (SPECT). In addition, depressive severity was evaluated through the HDRS. SPECT scans were conducted 14 days before the start of the treatment and repeated 4 days after the last treatment. There was a significant decrease in SERT density (p = 0.03), with five of the patients exhibiting a 20% decrease. Depressive scores improved in all subjects. This represents one of the first in vivo studies suggesting the link between TNF-α blockade and SERT modulation (Cavanagh et al., 2010)
All the aforementioned pathways have detrimental effects in clinical depression and additionally play a role in chronic depression. Pro-inflammatory cytokines, TRYCATs and O&NS together may contribute to a state called neuroprogression, related to neurodegeneration, reduced neurogenesis, neural plasticity and apoptosis (Maes et al., 2012).
Major depressive disorder as a comorbidity to pro-inflammatory medical conditions: circumstantial evidence
Several inflammatory diseases have also been associated with higher risks of development of depression and this might provide further clues for our understanding of the underlying mechanism of MDD. Patients with a myocardial infarction (MI), for example, have a prevalence of depressive disorder that is about three times higher than in the general population (Liu et al., 2013). MI triggers an inflammatory cascade that leads to increased pro-inflammatory cytokines in plasma. These cytokines can be transported across the blood–brain barrier and promote the activation of microglia (Rana et al., 2010). Conventional antidepressants generally have limited effect in MI patients (Liu et al., 2013), probably due to the presence of neuroinflammation as a result of the chronic elevated immune profile. Autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis are also associated with a higher prevalence of depression. The association might be explained by two hypotheses: (1) chronic stress derived from long-term use of corticosteroids impairs corticosteroid-receptor signaling, therefore, the severe clinical condition and the inadequate adaptation to stress cause persistent hyper-secretion of stress hormones; (2) persistent elevation of pro-inflammatory cytokines due to the chronic inflammation leading to neuroinflammation through the aforementioned pathways (Postal and Appenzeller, 2015). Obesity has also been linked to the development of depression via the elevated inflammatory profile associated with the disorder (Pan et al., 2012). This relation might be partially explained by the fact that adipocytes in the white adipose tissue secrete cytokines, mainly IL-6 and TNF-α, that are referred to as adipocytokines (Shelton and Miller, 2010). The secretion of the pro-inflammatory markers might lead to an immune activation and be a risk factor for the development of MDD. In fact, this theory has been supported by a meta-analysis conducted by Luppino et al. (2010) showing a clear bidirectional association between depression and obesity: obese people have a 55% increased risk of developing depression over time, while depressed people had a 58% increased risk of becoming obese. Depression’s causal role in obesity might be due to neuroendocrine disturbances, through a long-term activation of the HPA axis and release of cortisol, along with an unhealthy lifestyle (Luppino et al., 2010).
In summary, there are circumstantial evidences that links (neuro)inflammation to MDD, in particular: (1) microglia activation that occurs in a number of neuropsychiatric conditions (Frick et al., 2013); (2) pro-inflammatory conditions like obesity, MI and autoimmune diseases that are often accompanied by depression (Raison, 2014); (3) presence of neuroinflammation during a major depressive episode in MDD patients visualized through PET imaging (Setiawan et al., 2015); (4) significant microgliosis in depressed patients that committed suicide (Steiner et al., 2008); (5) elevated profile of pro-inflammatory cytokines in the blood of depressed patients as compared with controls (Howren et al., 2009); (6) development of ‘depressive-like behavior’ in rodents systemically exposed to inflammatory conditions, exhibiting elevated levels of activated microglia (Wohleb et al., 2012, 2014).
Anti-inflammatory treatment for major depressive disorder with non-steroidal anti-inflammatory drugs
Cyclooxygenases in neuroinflammation: pre-clinical studies
As previously discussed, MDD appears to be associated with elevation of pro-inflammatory cytokines both in peripheral blood and the brain, at least in a subpopulation of the patients. These pro-inflammatory cytokines can trigger an inflammatory cascade in the brain, which includes the induction of cyclooxygenases (COXs) that are key enzymes in the production of prostaglandins (Harden et al., 2015). Based on this observation, one could hypothesize that treatments targeting the enzymes cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) could have a beneficial effect in the subgroup of depressed patients with elevated levels of pro-inflammatory cytokines. Indeed, elevated COX-2 messenger ribonucleic acid (mRNA) expression was found for the first time in peripheral blood of patients with recurrent depressive disorder by Gałecki et al. (2012) (Gałecki et al., 2012). Both COX isoforms catalyze the same reactions: oxidation of arachidonic acid (AA) to yield prostaglandin G2 (PGG2), followed by a peroxidase reaction which converts PGG2 to prostaglandin H2 (PGH2). In these reactions, reactive oxygen species are also produced that can cause severe cell damage. PGH2 is transformed into PGE2, PGF2α, PGD2, PGI2 and TXB2 by specific terminal synthases (Aïd and Bosetti, 2011). PGE2 is the main prostaglandin implicated in the inflammatory response, pain, fever and autonomic functions (Blais et al., 2005). Furthermore, COX-1 and COX-2 are both expressed in the brain. COX-2 is detected in synaptic dendrites and excitatory terminals, mainly in cortex, hippocampus and amygdala, whereas COX-1 is expressed by microglia and perivascular cells (Maes, 2012).
COX-1 has been shown to support the inflammatory process and facilitate pro-inflammatory upregulation of prostaglandins in animal models of neuroinflammation (Aïd and Bosetti, 2011). Indeed, Choi et al. (2008) demonstrated that mice deficient of COX-1 showed less neuron degeneration, less microglia activation and lower expression of pro-inflammatory cytokines and PGE2 after exposure to LPS via lateral ventricle injection than wild-type mice. Likewise, inhibition of COX-1 with SC-560 (COX-1 selective inhibitor) showed similar effects as the genetic deletion of COX-1 (Choi et al., 2008).
In contrast to COX-1, COX-2 can have either a neurotoxic or anti-inflammatory role depending on inflammatory stimuli. Results of pre-clinical studies, mainly with celecoxib (a COX-2 selective inhibitor) treatment are contradictory. In a model of chronic unpredictable stress in rats, celecoxib treatment was administered for 21 days. The depressive behavior in the stressed rats was reversed by the NSAID and PGE2 concentrations decreased relative to untreated controls (Guo et al., 2009). Another well-known rat model of depression, olfactory bulbectomy (OBX), was used to evaluate the antidepressant effect of celecoxib treatment for 14 days. Behavioral alterations of OBX rats were reversed by the drug, whereas pro-inflammatory cytokines IL-1 and TNF-α levels in the pre-frontal cortex and hypothalamus decreased, probably by reduction of systemic PGE2 synthesis (Myint et al., 2007). Also, the hypothesis that aging contributes to behavioral impairment and increases in the pro-inflammatory markers in the hippocampus was tested by Casolini et al (2002), using rats aged 12- and 24-months old. Chronic treatment with celecoxib for 4 months reduced the levels of IL-1β, TNF-α and PGE2 in the hippocampus, and lower corticosterone levels in the 12-month-old rats (beginning of the aging process). This experiment also demonstrated a possibility for improvement of cognitive impairment and the inflammatory state at the beginning of the aging process (Casolini et al., 2002). However, COX-2 might have also a neuroprotective function in response to an inflammatory challenge. Genetic deletion of COX-2 enhanced the vulnerability towards an LPS challenge, resulting in increased neuronal damage in the hippocampus, increased activation of scavenger receptor A mRNA (specific marker for phagocytic microglia) and increased the expression of TNF-α, IL-6 and IL-1β, as compared with wild-type mice. Furthermore, inhibition of COX-2 by chronic administration of celecoxib for 6 weeks caused an increase in IL-1β levels in the brain of wild-type mice exposed to LPS, as compared with non-treated LPS-exposed mice (Aid et al., 2008).
Taken together, these data suggest that the enzyme COX-1 mainly has a pro-inflammatory role in the brain, whereas COX-2 could be involved in both pro- and anti-inflammatory responses. Interestingly, curative treatment with COX-2 selective inhibitors in (neuro)inflammatory animal models have shown mostly beneficial outcomes by decreasing inflammatory markers in the brain and reversing behavioral alterations, suggesting that there might be a possible application for patients with depression and elevated pro-inflammatory profile (data summarized in Table 1). Also, attenuating the pro-inflammatory role of COX-1 seems to be a good strategy to avoid activation of microglia and the support for the neuroinflammatory process. Further animal studies with selective COX-1 inhibitors still need to be conducted in order to obtain a better understanding of their role in neuroinflammation and putative therapeutic implications.
Summary of results obtained in pre-clinical studies applying NSAID treatment in (neuro)inflammation models. The table presents the animal model used, number of subjects (n), duration of treatment, NSAID (selectivity), type of treatment (preventive or curative) and final outcome (beneficial/not beneficial).
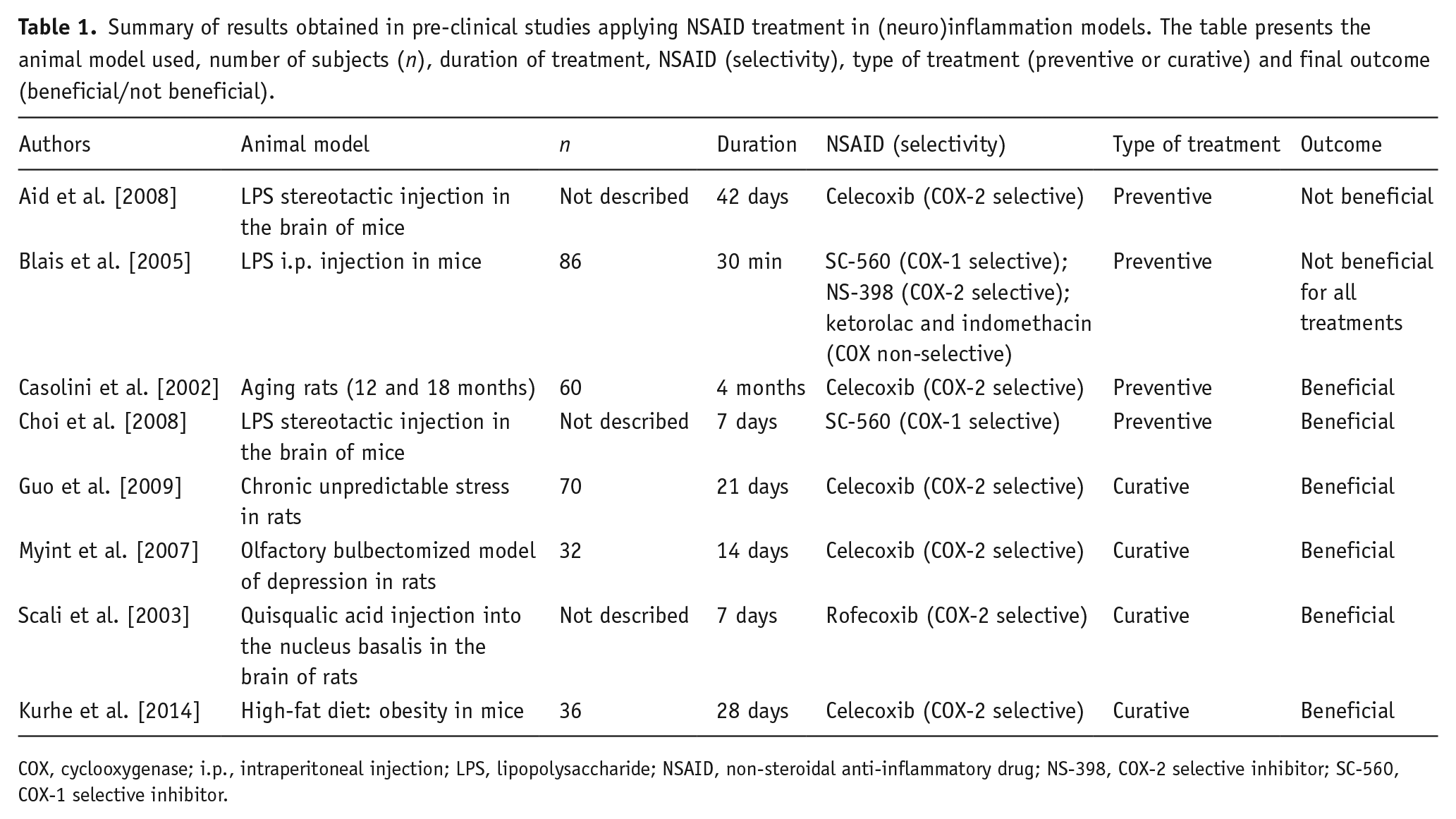
COX, cyclooxygenase; i.p., intraperitoneal injection; LPS, lipopolysaccharide; NSAID, non-steroidal anti-inflammatory drug; NS-398, COX-2 selective inhibitor; SC-560, COX-1 selective inhibitor.
NSAID monotherapy for major depressive disorder: clinical studies
NSAIDs demonstrated promising results in clinical trials for depression, mainly involving patients with inflammatory disease comorbidities. In patients with osteoarthritis, depression is 2–3 times more prevalent than in age-matched controls (Iyengar et al., 2013). In a study including pooled data from five randomized, multicenter, double-blind, placebo-controlled trials on 1497 patients with osteoarthritis, subjects were screened for MDD with the standard patient health questionnaire-9 (PHQ-9) and were divided into three treatment groups: ibuprofen/naproxen (non-selective COX inhibitors), placebo or celecoxib, administered for a duration of 6 weeks. Both groups using NSAIDs (ibuprofen/naproxen or celecoxib) showed a trend towards a reduction in depressive symptoms in patients with osteoarthritis, based on the PHQ-9 scores (Iyengar et al., 2013). A possible limitation of this study that might have affected the results was the celecoxib dosage. The recommended therapeutic dose is 400 mg/day, while the patients included in this study received only 200 mg/day.
A study that evaluated the efficacy of anti-inflammatory treatment for depressive symptoms alleviation not linked to inflammatory comorbidities demonstrated that it might not have any beneficial effect. An investigation of the therapeutic benefits of COX inhibitors in late-life depression was performed in 2528 participants over 70-years old with or without significant depressive symptoms, which were screened and randomized to receive celecoxib, naproxen or placebo for 12 months. Only 449 patients were considered depressed at baseline according to their score on the Geriatric Depression Scale (GDS). After the treatment with either drug, the GDS score was not reduced (Fields et al., 2012). Even though the sample size of this study was big and the treatment period was long, a critical measure of the inflammatory markers was not performed. Thus, it is conceivable that some of the patients included in this study might not have an elevated immune profile and therefore, would not have any benefit from the therapy with NSAIDs.
An epidemiological study called ‘The Health in Men Study’ published two papers regarding the usage of aspirin in older men (aged 69–87 years old) as prevention for the development of depression. The first study evaluated 5556 patients, of which 4461 (89.9%) had a cardiovascular disease. A 5-year follow up revealed that aspirin did not reduce the odds of developing depression in late life. One possible explanation is that aspirin might lead to greater medical complications due to bleeding, increasing the risk of small cerebrovascular lesions that contribute to a higher incidence of depression (Almeida et al., 2010). The second study evaluated a sample of 3687 patients to access the relationship of high plasma homocysteine (tHcy), which is associated with higher risk of cardiovascular events, and the onset of depression. The study confirmed that high tHcy is associated with an increased risk of depression, with an odds ratio (OR) of 1.80 (95% confidence interval (CI) = 1.39–2.35) and that the usage of aspirin is associated with a decrease in the risk of depression among these patients, with an OR of 0.60 (95% CI = 0.20–1.79) (Almeida et al., 2012). Another study in 345 female subjects evaluated the risk of developing depression in relation to the usage of aspirin during a 10-year follow-up. Estimated rates of MDD were 1.7 (95% CI = 0.4–6.9) per 1000 person-years for subjects using aspirin and 12.2 (95% CI = 7.9–19.0) per 1.000 person-years for the non-users. This study suggests that exposure to aspirin seems to be associated with a reduced risk of developing MDD (Pasco et al., 2010).
As summarized in Table 2, available data from a limited number of studies suggest that curative treatment of MDD with NSAIDs can have a beneficial effect on the relief of depressive symptoms, whereas data on the preventive treatment with COX inhibitors are still inconclusive. A meta-analysis conducted by Köhler et al. (2014) demonstrated that monotherapy treatment with celecoxib had borderline significance in the relief of depressive symptoms (Köhler et al., 2014). Future clinical randomized clinical trials using NSAID treatment monotherapy should be better structured, including only patients with elevated levels of inflammatory markers in the blood or cerebral spinal fluid (CSF) in combination with questionnaires for scoring depressive symptoms. NSAID monotherapy should not be encouraged in the absence of inflammation and neither used as replacement of conventional antidepressants. Moreover, a better description of adverse effects in the studies is paramount. An advantage of anti-inflammatory treatment strategies is that definitive readouts of target engagement are available. Since the majority of anti-inflammatory therapies have an effect on inflammatory markers, it can be easily determined through blood/plasma samples if the treatment is indeed acting on the desired target (Miller and Raison, 2015) and terminated as soon as the inflammation is resolved. Complementary diagnostic tools such as brain imaging of inflammatory biomarkers (e.g. TSPO) through PET would be desirable, since the relationship between central and peripheral inflammation in MDD is still understudied. Only when the effects on both depressive scores and inflammatory markers are known, a clear conclusion can be reached about the link between inflammation reduction and depression alleviation. Treatment with NSAIDs in a subgroup of patients with low depressive symptoms and immune deregulation/inflammatory comorbidities (Köhler et al., 2014) seems to be the best approach for future research. Nevertheless, indiscriminate use of anti-inflammatory treatments for MDD patients without inflammation might be harmful, since inflammatory cytokines play a pivotal role in learning and memory, as well as in neural integrity, neurogenesis and synaptic pruning (Yirmiya and Goshen, 2011). Thus, a maximum safe treatment length is yet to be established.
Résumé of the outcome of clinical studies relating the usage of non-steroidal anti-inflammatory drug (NSAID) and depression. The table shows the number of subjects (n), type of subjects, duration of treatment/prevention, NSAID (selectivity), type of treatment (preventive/curative) and final outcome (beneficial/not beneficial).
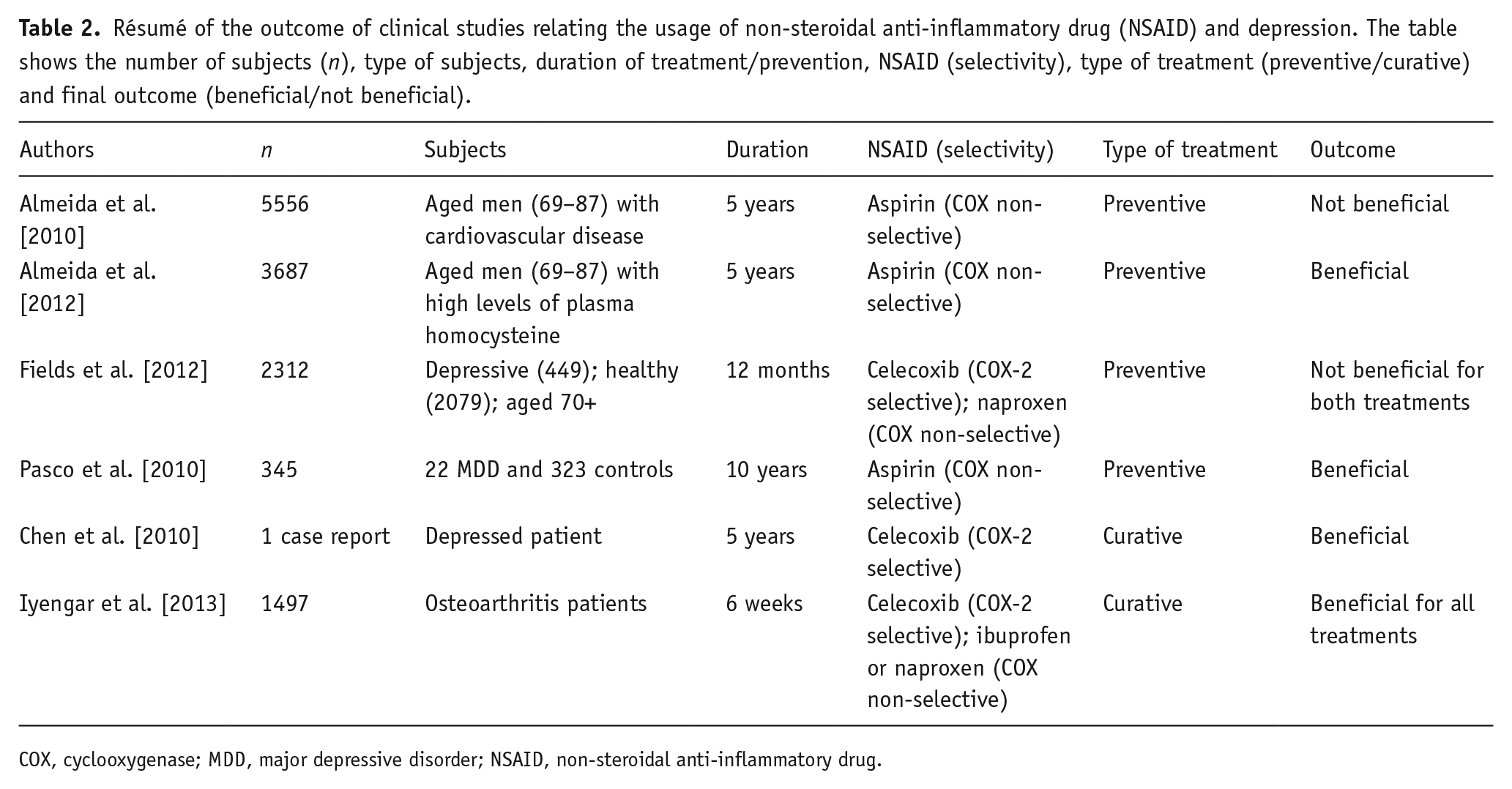
COX, cyclooxygenase; MDD, major depressive disorder; NSAID, non-steroidal anti-inflammatory drug.
Side effects of non-steroidal anti-inflammatory drugs
Considering the possible beneficial aspects of NSAID treatment for depression, one should be aware of the possible side effects. In a review by Funk and FitzGerald (2007), COX-2 inhibition was associated with an increased susceptibility to thrombosis, hypertension and atherosclerosis due to a thrombotic effect by inhibition of prostacyclins derived from endothelial COX-2 (Funk and FitzGerald, 2007; Schjerning Olsen et al., 2013). A meta-analysis conducted by Kearney et al. (2006) extracted data from randomized controlled trials regarding the risk of vascular events associated with the usage of selective COX-2 inhibitors vs placebo, and vs traditional NSAIDs (n = 145,373). In the comparison between selective COX-2 inhibitors vs placebo, a 42% higher incidence of vascular events occurred in COX-2 users as compared with placebo. A twofold increase in MI was also observed. When evaluating stroke incidence, no difference between groups was found. Importantly, 121 trials were long-term trials (mean of 139 weeks) while 112 were short-term trials (mean of 11 weeks). Two thirds of vascular events occurred in nine long-term trials; therefore, the hazards of cardiovascular events emerged after a year to 18 months of selective COX-2 inhibitors’ chronic usage. With regard to selective COX-2 inhibitors vs traditional NSAIDs, no significant difference was found regarding the risk of vascular events (Kearney et al., 2006). The increased risk of vascular events might be due to an increased Th1 immune response (pro-inflammatory) when selective COX-2 inhibitors are chronically used. This hypothesis was based on the atherosclerotic plaques scenario, where selective COX-2 inhibitors might provoke macrophage accumulation at the inflamed arterial endothelial site. This response leads to increased production of pro-atherogenic cytokines, attracting lymphocytes and macrophages that will exacerbate the inflammation, increase plaque instability and vulnerability to rupture, embolization and consequent MI (Padol and Hunt, 2010). Still, this immunological modulation is a slow process, occurring after 12 months of chronic therapy (Kearney et al., 2006; Padol and Hunt, 2010). This possible mixed anti-inflammatory and pro-inflammatory effect of COX-2 deserves consideration before chronic usage recommendation in clinical practice (Scher and Pillinger, 2009).
Additionally, NSAIDs are well recognized for causing peptic ulceration and ulcer complications. Cohort studies have estimated that the total risk of hospitalization for gastrointestinal complications associated with NSAID use are between 1.3 and 2.2 events per 1000 patients. Protective strategies as co-prescription of a protective drug such as misoprostol or a proton pump inhibitor can be applied to reduce those events (Hawkey, 2003). Selective COX-2 inhibitors present an advantage in this matter as the incidence of clinically significant ulcers were reduced by 54% and ulcer complications by 57%, as compared with non-selective NSAIDs (Bombardier et al., 2000).
In summary, if the inflammatory hypothesis for MDD is confirmed, the implication would be that anti-inflammatory strategies might hold promise for the treatment of depressed patients with chronically elevated inflammatory biomarkers or inflammatory comorbidities. The effectiveness of the treatment must be followed by measurements of peripheral inflammatory biomarkers associated with depressive scores and the obtained data should be used to determine the required treatment length. Nonetheless, NSAIDs are far from being a panacea for depression and might benefit only a subgroup of depressed patients.
The anti-inflammatory effect of antidepressants
Effect of antidepressants on cytokine levels
Although antidepressants have been used in therapy for depression for more than 50 years, the mechanism of action of most of these drugs still remains a mystery. Antidepressant drugs usually act on the monoaminergic systems, although they can have different mechanisms of action. Some antidepressant drugs were also found to elicit anti-inflammatory and neuroprotective effects, which might be partly due to their influence on cytokine production (Obuchowicz et al., 2014). The influence on the cytokine production might be related to antidepressant action on cyclic adenosyl monophosphate (cAMP), serotonin metabolism, the HPA axis or through a direct action on neurogenesis (Taraz et al., 2013).
The silencing of over-activated glia by antidepressants could stop neuroinflammation and may therefore be beneficial not only for MDD, but also for other CNS diseases. The effects of antidepressant drugs on cytokine levels have been investigated in vitro in cell cultures. A recently published study by Obuchowicz et al. (2014) evaluated the anti-inflammatory effects of imipramine, a tricyclic antidepressant (TCA), and fluoxetine, a selective serotonin reuptake inhibitor (SSRI), on IL-6, IL-1β and TNF-α secretion by primary mixed glial cultures stimulated with LPS. Even though both drugs were able to decrease the levels of the pro-inflammatory cytokines, only imipramine prevented morphological changes and activation of microglia (Obuchowicz et al. 2014). In another study, Xia et al. (1996) tested the inhibitory effect of the antidepressants imipramine, clomipramine (TCAs) and citalopram (SSRI) on the release of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α by stimulated human lymphocytes and monocytes. All antidepressants exerted inhibitory effects on cytokine release and also increased the levels of cAMP (Xia et al. 1996). In another in vitro study, clomipramine, sertraline (SSRI) and trazodone (heterocyclic antidepressant) were able to decrease IFN-γ levels and increase the production of the anti-inflammatory cytokine IL-10 (Maes et al., 1999). Using the same methodology, Maes et al. (2005) found that fluoxetine (SSRI) also has immunomodulatory effects on the secretion of cytokines, such as TNF-α and IFN-γ (Maes et al., 2005). Nonetheless, conflicting results have been published by Munzer et al. (2013), who tested the effect of escitalopram (an SSRI), citalopram and mirtrazapine (serotonin and noradrenaline reuptake inhibitors, SNRIs) on the secretion levels of cytokines IL-1β, IL-2, IL-4, IL-6, IL-17, IL-22 and TNF-α in stimulated whole blood of 15 depressed patients ex vivo. A T cell (OKT3) and a B cell (5C3) stimulant was used in the whole blood to induce cytokine production in vitro. Curiously, citalopram increased the levels of IL-1β, IL-6, TNF-α, and IL-22, whereas mirtrazapine increased IL-1β, TNF-α and IL-22 and escitalopram decreased IL-17. The differences in cytokine production levels might be due to distinctive therapeutic effects between the drugs (Munzer et al., 2013).
Pre-clinical studies have yielded interesting results for antidepressants that decrease cytokine levels, mainly TCAs and SSRIs. Alboni et al. (2013) demonstrated promising effects with imipramine and fluoxetine. Both imipramine and fluoxetine were able to reduce the levels of IFN-γ, IL-6 and increase the expression of the anti-inflammatory cytokine IL-4 in the hypothalamus of healthy male Sprague-Dawley rats after a 28-day treatment (Alboni et al., 2013). Moreover, imipramine was also able to decrease mRNA levels for IL-6 in brain microglia in a rat model of social defeat, which was accompanied by a reversal in social avoidance behavior (Ramirez et al., 2015). In a model of MI, rats were either treated with saline or escitalopram for 2 weeks. Plasma levels of IL-1β, TNF-α and PGE2 were significantly decreased after treatment (Bah et al., 2011).
Several studies have explored the anti-inflammatory effect of antidepressants in clinical studies, as summarized in Table 3. The class of antidepressants that was mostly tested was the SSRI, followed by TCA and SNRI. Most studies showed anti-inflammatory effects of antidepressants. For instance, Brunoni et al. (2014) recently published the results of a trial with 103 depressed patients treated with sertraline for 6 weeks. The levels of IL-2, IL-4, IL-6, IL-10, IL-17, IFN-γ were found to be significantly decreased after the treatment (Brunoni et al., 2014). ‘The Netherlands Study of Depression and Anxiety’ evaluated IL-6 and TNF-α levels in depressed patients and found that SSRIs were able to decrease IL-6 levels, but not TNF-α (Vogelzangs et al., 2012). The effect on IL-6 is in agreement with other studies published (Basterzi et al., 2005; Hannestad et al., 2011; Sluzewska et al., 1995; Taraz et al., 2013; Yoshimura et al., 2009). Summarizing, SSRIs have an effect on cytokine levels, mainly on IL-6, TNF-α and IL-1β. On the other hand, when evaluating the effects of venlafaxine (SNRI) on TNF-α and IL-1β in 12 MDD patients after 8 weeks of treatment, the HDRS score decreased at least 50% compared with baseline, whereas no decrease was observed in pro-inflammatory cytokine levels. (Piletz et al., 2009). This might be due to two factors: the length of the study was too short to observe any effects on cytokine levels, or because of the known pro-inflammatory effect of norepinephrine on innate immune cells (Thayer and Sternberg, 2010). However, the small number of patients completing the trial (n = 12) might be a major limitation of the study. The authors mentioned that the power analysis performed revealed that an n = 12 would be sufficient to detect an antidepressant-induced effect on pro-inflammatory cytokines assuming the agent would act as an SSRI as reported by Leo et al. (2006). SSRIs and SNRIs have different mechanisms of action, and therefore the same antidepressant-induced effect on pro-inflammatory cytokines should not be expected. Also, the sample size of the study of Leo et al. (2006) was of 46 MDD patients and 46 age-matched healthy controls. Therefore, the obtained data by Piletz et al. (2009) should be interpreted with care. The limited duration of the treatment with antidepressant drugs applied and the small number of subjects evaluated seem to be general limitations of the studies published until now.
Summary of clinical studies that assessed the anti-inflammatory effect of antidepressants on cytokines in MDD. The table describes the type and number of subjects investigated, antidepressant used, duration of treatment, cytokines assessed and final outcome of the study.
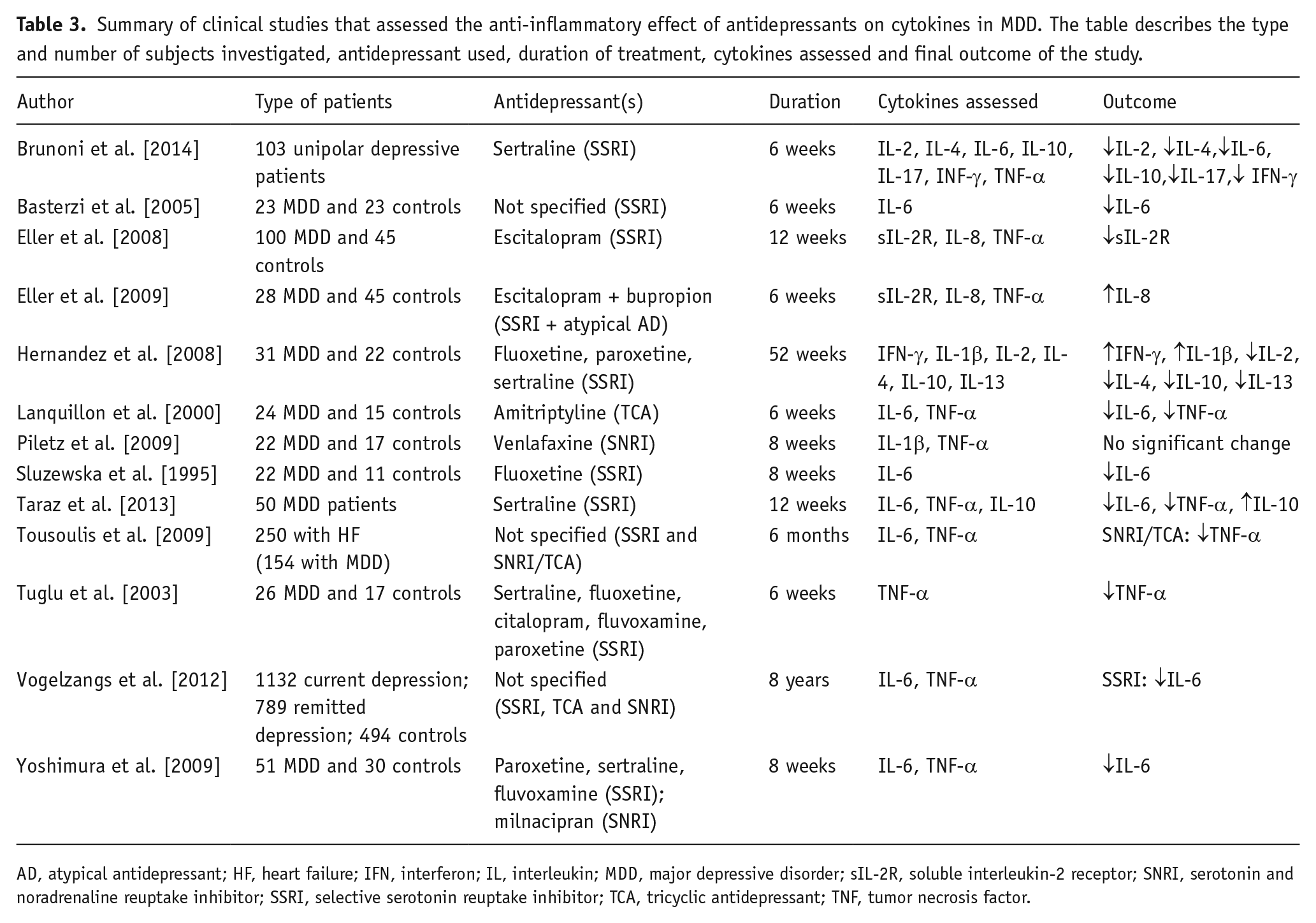
AD, atypical antidepressant; HF, heart failure; IFN, interferon; IL, interleukin; MDD, major depressive disorder; sIL-2R, soluble interleukin-2 receptor; SNRI, serotonin and noradrenaline reuptake inhibitor; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant; TNF, tumor necrosis factor.
Even though promising results were obtained in vitro and in pre-clinical studies with TCAs, limited clinical trials have been performed with this class of antidepressant. Tousoulis et al. (2009) conducted a clinical study in 154 patients that suffered from heart failure and developed MDD. A total of 120 patients received an SSRI and 34 patients were treated with a TCA/SNRI. The ones that were treated with TCA/SNRI had lower levels of TNF-α and CRP compared with the SSRI group (Tousoulis et al., 2009). The underlying mechanism of action that can explain how TCAs affect pro-inflammatory cytokines is largely unknown. It is hypothesized that antidepressants (mainly SSRIs and TCA) decrease pro-inflammatory cytokines levels through the cAMP pathway (Figure 2). 5-HT (5-hydroxytryptamine or serotonin) increases intracellular cAMP levels via G protein-coupled serotonin receptors that can stimulate adenylyl cyclase, which results in inhibition of the protein kinase A pathway and a reduction in the expression of cytokines (Xia et al. 1996). For imipramine in particular, the effect might be explained by the down-regulation of microglial activation (Ramirez et al., 2015).
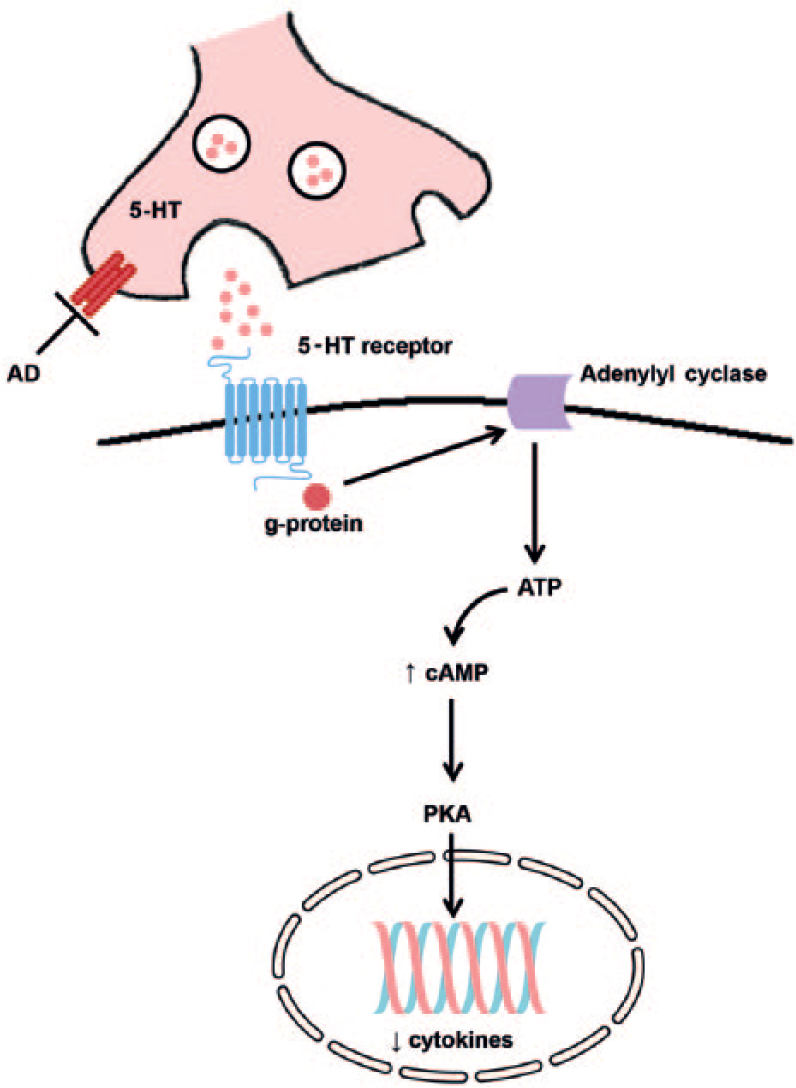
Possible anti-inflammatory mechanism of action of antidepressants. Antidepressants that increase the levels of serotonin (i.e. SSRIs) might exert their anti-inflammatory effects by cAMP-mediated pathways. 5-HT increases intracellular cAMP levels via 5-HT receptors linked to G protein-mediated stimulation of adenylyl cyclase, leading to a reduction in the expression of cytokines via inhibition of the protein kinase A (PKA) pathway.
Possible biomarkers for major depressive disorder: can we predict response to therapy?
As already suggested, pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α, could be useful biomarkers for investigating the presence or level of inflammation during MDD screening. Moreover, studies have demonstrated that the levels of pre-treatment biomarkers can be useful predictors of treatment response.
Lanquillon et al. (2000), performed a study in 24 MDD patients and 15 controls, using a 6-week treatment protocol with amitriptyline (TCA) and evaluating immunological parameters such as IL-6 and TNF- α in whole blood. After the treatment period, patients were classified as responders and non-responders according to HAM-D scale. Patients who responded to treatment had significantly lower levels of IL-6 than controls (p < 0.05) or non-responders (p < 0.05) on baseline measurements. Moreover, regardless of treatment outcome, IL-6 levels in both responders and non-responders returned to control levels after 6 weeks of treatment. TNF-α levels were elevated in responders and non-responders at baseline, as compared with controls (p < 0.01). After treatment, the decrease in TNF-α levels was only significant in the responder group (p < 0.05). The data suggest that IL-6 levels can predict response to therapy, whereas TNF-α paralleled the clinical response (Lanquillon et al., 2000). Another study conducted by O’Brien et al. (2007) investigated the differences in the levels of IL-6, IL-8, IL-10, soluble IL-6 receptor (which may act as an agonist of IL-6) and TNF-α between depressive patients who were SSRI-treatment resistant, former SSRI-treatment-resistant patients that are now euthymic due to therapy change and healthy controls. The depressed patients who were SSRI non-responders had marked activation of pro-inflammatory cytokines with high levels of IL-6 and TNF-α. Currently euthymic patients with a prior history of SSRI resistance had pro-inflammatory cytokine levels similar to healthy subjects (O’Brien et al., 2007). Eller et al. (2008) also found that higher levels of TNF-α are predictive for non-response in depressed patients (Eller et al., 2008). Yoshimura et al. (2009) found a correlation between high baseline levels of IL-6 with refractory depression (Yoshimura et al., 2009). Therefore, it seems that suppression of pro-inflammatory cytokines may be necessary for clinical recovery from depression and that reduction in inflammation might not happen in depressed patients who fail to respond to treatment.
Increased pro-inflammatory cytokines are known to induce acute phase (inflammatory) response in the liver, increasing the levels of CRP (Miller et al., 2009) and lower albumin and zinc in depression (Maes, 1995; Roomruangwong et al., 2016), suggesting that these could be useful biomarkers of depression. An interesting study conducted by Raison et al. (2013) tested if infliximab, a monoclonal antibody directed against TNF-α, would improve the mood of treatment-resistant depressive patients, since this pro-inflammatory cytokine has been associated with depression and poor treatment response. The study randomized 60 medically healthy adults with treatment-resistant major depression, to either receive three infusions of infliximab (5 mg/kg) or three saline infusions at baseline, week 2 and 6. Clinical assessments of depressive scores were made through HAM-D, the Clinical Global Impression – Severity scale, and inflammatory status (hs-CRP, and TNF and its soluble receptors I and II), which were conducted at baseline and weeks 1, 2, 3, 4, 6, 8, 10 and 12. At the end of the trial (week 12), neither differences in HAM-D score between groups, nor significant interactions between treatment and time were found. A significant effect of time on decreased the HAM-D score was observed in both groups (p < 0.05). However, when analyzing baseline hs-CRP values to predict response to infliximab, a plasma hs-CRP concentration greater than 5mg/L was found to be the point at which infliximab-treated patients exhibited a greater decrease in HAM-D score than placebo patients (Raison et al., 2013). The effect size of participants with hs-CRP levels higher than 5mg/L was 0.41, which is in line with the efficacy of antidepressants against placebo in most studies (Raison, 2016).
When selecting immunological biomarkers, one should take into consideration classical markers that already have standardized assays for clinical application, such as CRP (Dantzer et al., 2011), albumin and zinc. Pro- and anti-inflammatory cytokines should be determined when possible, even though the assays are not standardized yet and are more restricted to research trials. Ideally, the biomarkers should be measured through peripheral and central (CSF) samples.
Side effects of antidepressants
Despite all the aforementioned benefits of antidepressants, the treatment is not free from risks and side effects. In a study to assess self-discontinuation of antidepressants, 313 patients were interviewed to investigate the reason for stopping the treatment. Side effects were reported by 20% of the patients as the main cause of discontinuation (Samples and Mojtabai, 2015). Premature self-discontinuation is associated with relapse and incomplete response to the treatment. The key side effects associated with antidepressant use vary by class of the antidepressant and might vary also by medication within each class (Kemp, 2014). Even though TCAs are very effective in treating depression, they also act on other receptor systems, including histaminic, cholinergic, adrenergic and postsynaptic serotonin receptors, leading to significant and sometimes intolerable side effects. Moreover, TCAs have a narrow therapeutic index and at higher doses, they might cause seizures and death due to slowing of intraventricular conduction, leading to complete heart block or ventricular re-entry arrhythmias (Ferguson, 2001). SSRIs, on the other hand, are better tolerated and significantly less toxic in an overdose than TCAs. The most common side effects related to SSRIs are gastrointestinal like nausea, activation syndrome, sexual dysfunction, body weight gain, insomnia and serotonin syndrome (Miskovic, 2015). Antidepressants that also act on the noradrenergic system (i.e. SNRIs) are associated with significantly greater increase in blood pressure and heart rate than SSRIs (Kemp, 2014) and also with gastrointestinal effects, activation syndrome, insomnia and sexual dysfunction (Miskovic, 2015). In order to decrease self-discontinuation, it is important to choose an efficient antidepressant with the profile of side effects that will not hamper the quality of life of the patient.
Augmentative anti-inflammatory strategies
Non-steroidal anti-inflammatory drugs
For those patients who fail to respond to the initial antidepressant therapy, alternative treatment approaches are switching medication, augmentation or combination therapies (Mendlewicz, 2008). Augmentation strategies involve the usage of agents that are non-standard antidepressants to enhance the therapeutic effect of a known antidepressant (Akhondzadeh et al., 2009). Augmentation with COX-2 inhibitors such as celecoxib has been studied in clinical trials with promising results. For instance, a double-blind, randomized, placebo-controlled study was conducted in 40 patients, using celecoxib + reboxetine (a norepinephrine reuptake inhibitor) or placebo + reboxetine, as treatment for 6 weeks. A decrease of 55% in the HDRS score was observed after treatment with celecoxib + reboxetine, compared with a 33% decrease after treatment with reboxetine + placebo. Interestingly, 45% of the patients in the celecoxib group showed complete remission after 6 weeks, compared with 20% in the placebo group (Müller et al., 2006). Abbasi et al. (2012) conducted a clinical trial assessing the cytokine profile and depressive symptoms in a group of patients treated with sertraline + celecoxib and in a group treated with sertraline + placebo; 40 patients were randomly assigned to each group and the HDRS score as well as the levels of IL-6 were measured at baseline and after 6 weeks of treatment. At the end of week 6, the HDRS score was reduced by 96% in the celecoxib group compared with a 50% reduction in the placebo group. Also, the IL-6 levels in serum were significantly more reduced in the celecoxib group compared with the placebo group (Abbasi et al., 2012). Celecoxib was also successfully used in a 6-week combination treatment with another SSRI, fluoxetine, in 40 depressive patients. The combination of celecoxib with fluoxetine was able to reduce the HDRS score with 90%, while placebo + fluoxetine reduced only 50% (Akhondzadeh et al., 2009).
Since the add-on strategy with NSAIDs, mainly celecoxib, could be a potentially effective augmentation, a meta-analysis of the data from randomized clinical trials was performed by three authors. Faridhosseini et al. (2014) analyzed data from four randomized clinical trials that used celecoxib as augmentation therapy for unipolar depression (n = 160 patients). Celecoxib or placebo was used in combination with sertraline, reboxetine or fluoxetine and the effect on HDRS score was evaluated. The pooled OR of treatment response of celecoxib vs placebo was 6.6, 95% CI = 2.5–17, p < 0.0001; pooled OR of remission response was 6.6, 95% CI = 2.7–15.9, p < 0.0001; pooled OR in means of HDRS score decrease at week 6 was 3.43, 95% CI = 1.9–4.9, p < 0.0001. In summary, celecoxib in a daily dose of 400 mg/day as an add-on therapy to antidepressants is effective in MDD. No adverse effects attributable to celecoxib were observed (Faridhosseini et al., 2014). Köhler et al. (2014) included 10 randomized clinical trials, from which only 4 were add-on therapy with NSAID (i.e. celecoxib) or placebo with an SSRI or SNRI. A significant improvement in depressive symptoms was observed when compared with placebo in four trials (n = 132); standardized mean difference (SMD) was −0.82, 95% CI = −1.17 to −0.46, p < 0.001). In addition, add-on treatment improved remission with OR 7.89, 95% CI = 2.94–21.17, p < 0.001 and response (three trials, n = 92 patients) with OR 6.59, 95% CI = 2.24–19.42, p < 0.001). No evidence of increased gastrointestinal or cardiovascular adverse effects was reported; the length of trials ranged from 6–8 weeks (Köhler et al., 2014). Rosenblat et al. (2016) preformed a meta-analysis on data from randomized clinical trials focused on bipolar depression. Only two studies used NSAIDs (aspirin or celecoxib) and compared the efficacy with placebo (n = 53). Pooled effects size revealed an SMD of 0.02, 95% CI = −0.52–0.56, p > 0.05. Add-on treatment strategy had no significant difference in depressive symptoms (HDRS) or young mania rating scale (YMRS). This result might be attributable to the low level of add-on NSAID studies in bipolar disorder and therefore, it might be underpowered and less robust than other meta-analyses (Rosenblat et al., 2016).
Based on these data, it seems that adjuvant treatment with COX-2 inhibitors might be a good strategy to improve responsiveness of unipolar depressive patients. Even though the clinical studies performed until now focused mainly on the reduction of depressive symptoms as the outcome parameter, effects of treatment on immunological biomarkers (e.g. cytokines, CRP) should be an outcome parameter as well, especially when SSRIs and NSAID combination treatment is applied. More clinical studies with larger samples size and improved study design should be conducted in order to have more substantial evidence for the additional efficacy of add-on NSAID treatment.
Moreover, when assessing the adverse effects of add-on NSAID treatment in the reported trials, no relevant adverse effects were found. This might be attributable to the short treatment period that ranged from 6–8 weeks. This period might be too short to identify any adverse effects. Recently, a study was performed on a Korean population to investigate the risk of intracranial hemorrhage among patients treated either with the combination of antidepressants and NSAIDs (n = 2,072,613) or antidepressants alone (n = 2,072,613), during a 30-day follow up after the first antidepressant prescription. An increased risk of intracranial hemorrhage was found when combined therapy was used (hazard ratio 1.6, 95% CI = 1.32–1.85). The risk of intracranial hemorrhage was greater in men than women (2.6, 95% CI =1.93–3.42 vs 1.2, CI 95% = 0.89–1.57). No significant difference was found between antidepressant classes (Shin et al., 2015). Even though the study provides alarming data, further research is required to replicate the data in different ethnic groups. No explanation was given concerning the higher risks in men than women, or if the risk increases with aging. Another study by Bak et al. (2002) was conducted to investigate association of SSRI usage with ischemic stroke or intracerebral hemorrhage, since previous published data demonstrated that SSRI attenuated platelet activation and decreased the risk of thromboembolism formation (Chu et al., 2016). The results suggest that SSRIs are not a risk factor for intracerebral hemorrhage and are probably not associated with decreased risk of ischemic stroke. In a secondary analysis, an investigation of current associations of SSRI and NSAIDs, only five events of intracerebral hemorrhage were described among 659 total intracerebral hemorrhage events (Bak et al., 2002). Nevertheless, in primary psychiatric care, caution must be taken when prescribing antidepressants for current NSAID users with other associated risk factors (e.g. anti-platelet aggregation therapy).
Non-pharmacological anti-inflammatory strategies
Non-pharmacologic augmentation strategies such as exercise and mind–body therapies (MBTs) have demonstrated anti-inflammatory effects in several diseases, such as heart failure (Pullen et al., 2008) and depression (Lavretsky et al., 2011). MBTs include meditation, yoga, progressive relaxation and Tai Chi. (Muehsam et al., 2017). These practices have been described as able to regulate emotional and affective response to stress and therefore, influence the immune system. A meta-analysis conducted by Morgan et al. (2014) demonstrated a significant effect of MBTs in decreasing CRP as compared with controls (Morgan et al., 2014). Evidence also suggests that regular endurance exercise decreases inflammatory markers, mainly CRP (Haaland et al., 2008). Although MBTs and exercise are non-pharmacological augmentative strategies, both are able to decrease stress levels and modulate immunity, increasing the quality of life of MDD patients.
Besides MBTs other lifestyle changes, such as dietary interventions, could have a beneficial effect on depression as well. For example, nutraceuticals like zinc and omega-3 fatty acids were shown to have beneficial effects on depression (Sarris et al., 2016). These nutrients may exert their effect by interaction with inflammatory pathways (Johnson, 2015).
Concluding remarks
Several theories are available to explain the pathophysiology of MDD, mainly focusing on the involvement of environmental factors associated with genetic and biochemical components (Maciel et al., 2013). Inflammatory deregulation as an etiological factor appears to be a plausible hypothesis to explain why 30–50% of the depressive patients do not respond to conventional therapy. The presence of continuous stimuli that increase the levels of pro-inflammatory cytokines in the brain can cause neurotransmitter imbalance, neuroinflammation and neurodegeneration. Intervention in this process could be an important aim to prevent further damage in the CNS. The levels of inflammatory biomarkers, mainly IL-1β, IL-6, TNF-α and hs-CRP in the serum of depressed patients could be useful biomarkers, along with evaluation of depressive scores. This assessment would be important to guide physicians in patients’ classification (e.g. depression with elevated immune profile), selection of the best treatment to be applied and as an indicator of possible treatment resistance to conventional antidepressant drugs.
Concerning treatment, SSRIs were shown to have an anti-inflammatory effect in clinical trials, as they proved potent agents able to decrease the levels of pro-inflammatory cytokines and to diminish depressive symptoms in a subset of patients. Another benefit regarding the usage of SSRI is their higher tolerability in patients. More studies need to be conducted to further elucidate the anti-inflammatory mechanism action of SSRIs and other classes of antidepressants, such as TCAs. Imipramine in particular, shows an interesting effect in vitro and in pre-clinical studies; besides decreasing the pro-inflammatory cytokines, it prevented morphological changes and activation of microglia, thus limiting neuroinflammation. This effect might be useful to prevent further neurodegeneration associated with chronic activation of microglia not only in MDD, but in other CNS diseases associated with neuroinflammation. In general, the best results seem to be obtained with the combination of antidepressants with NSAIDs when initial antidepressant therapy fails (i.e. augmentative therapy). NSAIDs alone have shown best antidepressant results when administered as curative treatment to patients with an inflammatory comorbidity.
Even though considerable lines of research support the immunological hypothesis of depression, the main treatment targets have remained limited to the monoaminergic system. This is mainly due to the fact that the evidence for successful treatment of MDD with anti-inflammatories is still insufficient to adapt treatment guidelines for the subgroup of depressed patients with increased cytokine levels. In the present review, we have summarized the evidence for the benefits of anti-inflammatory treatment for MDD, either as single treatment or as augmentative therapy, and highlighted the anti-inflammatory effect that some antidepressants exert by themselves. However, before anti-inflammatory therapies for MDD can be applied in regular patient care, prospective studies on the efficacy of the anti-inflammatory treatment that combine the evaluation of depressive symptoms with quantification of inflammatory biomarkers are much needed. With substantial proof of efficacy, these alternative approaches can be applied in patient-tailored therapy-selection strategies, thus striving for an improved quality of life, especially for depressive patients with an immunological deregulation profile and treatment resistance to conventional therapy.
Footnotes
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.