
Abstract
Objective:
To evaluate the effectiveness of lurasidone as maintenance treatment for schizophrenia.
Method:
Adults experiencing an acute exacerbation of schizophrenia initially received 12–24 weeks of open-label treatment with lurasidone (40–80 mg/d, flexibly dosed). Patients who maintained clinical stability for ⩾12 weeks were randomized in double-blind fashion to placebo or lurasidone (40–80 mg/d, flexibly dosed) for an additional 28-week treatment period. The primary efficacy endpoint was time to relapse (based on Kaplan–Meier survival analysis).
Results:
A total of 676 patients enrolled in the open-label phase; 285 met protocol-specified stabilization criteria and were randomized to lurasidone (N=144) or placebo (N=141). During the open-label phase, mean Positive and Negative Syndrome Scale total score decreased from 90.1 to 54.4 in patients who met clinical stability criteria and were randomized. In the double-blind phase, lurasidone significantly delayed time to relapse compared with placebo (log-rank test, p=0.039), reflecting a 33.7% reduction in risk of relapse (Cox hazard ratio (95% confidence interval), 0.663 (0.447–0.983); p=0.041). Probability of relapse at the double-blind week 28 endpoint (based on Kaplan–Meier analysis) was 42.2% in the lurasidone group and 51.2% in the placebo group. Minimal changes in weight, lipid, glucose, and prolactin were observed throughout the study.
Conclusions:
This multicenter, placebo-controlled, randomized withdrawal study demonstrated the efficacy of lurasidone for the maintenance treatment of patients with schizophrenia.
Keywords
Introduction
Schizophrenia is a debilitating mental illness involving psychotic relapses that can worsen the course of the disease, increase the potential for treatment nonresponse, and reduce the likelihood that patients will achieve long-term remission (Emsley et al., 2013; Kane, 2007; Tandon et al., 2009). Long-term treatment with antipsychotic agents is generally necessary to control the symptoms of schizophrenia, minimize risk for relapse, and enable sustained remission (Kane, 2007; Tandon et al., 2010).
Lurasidone is an atypical antipsychotic agent approved for the treatment of adult patients with schizophrenia in the United States and the European Union, as well as other countries. Lurasidone acts as an antagonist with potent affinity for dopamine D2 and serotonin 5-HT2A and 5-HT7 receptors (Ishibashi et al., 2010). In addition, lurasidone has moderate affinity at α2A and α2C adrenergic receptors (as an antagonist) and 5-HT1a receptors (as a partial agonist), and no appreciable affinity for histamine H1 receptors or muscarinic M1 receptors (Ishibashi et al., 2010).
The efficacy of lurasidone in the treatment of patients with schizophrenia was demonstrated in a series of short-term, placebo-controlled studies (Loebel et al., 2013a; Meltzer et al., 2011; Nakamura et al., 2009; Nasrallah et al., 2013; Ogasa et al., 2013). A 12-month, double-blind, non-inferiority study of lurasidone compared with quetiapine XR provided evidence for the long-term effectiveness of treatment with lurasidone in patients with schizophrenia (Loebel et al., 2013b), which was further supported by data from two six-month, open-label extension studies (Citrome et al., 2014; Stahl et al., 2013) and a 12-month, randomized, double-blind safety study with an active comparator arm (Citrome et al., 2012).
The objective of this study was to evaluate the efficacy of lurasidone as a maintenance treatment for patients with schizophrenia using a well-established, placebo-controlled study design.
Methods
This double-blind, placebo-controlled, randomized, withdrawal study (ClinicalTrials.gov identifier: NCT01435928) was conducted between October 2011 and August 2013 at 71 sites in the United States (n=45), Slovakia (n=7), Russia (n=6), Serbia (n=6), France (n=3), South Africa (n=3), and Italy (n=1). All patients provided informed consent prior to enrollment. Study procedures were approved by institutional review boards associated with each site, and study conduct was consistent with the International Conference on Harmonization Good Clinical Practice guidelines and with the ethical principles of the Declaration of Helsinki.
Study entry criteria
Patients aged 18–75 years, inclusive, diagnosed with schizophrenia (based on DSM-IV-TR criteria), and experiencing an acute exacerbation were eligible for enrollment. Entry criteria included a Positive and Negative Syndrome Scale (PANSS) (Kay et al., 1987) total score of ⩾80 with a score ⩾4 (moderate severity) on one or more positive subscale items, and a Clinical Global Impressions-Severity (CGI-S) (Guy, 1976) scale score of ⩾4 (moderate severity). Patients were excluded from study participation if they were diagnosed with another Axis I or II disorder that had been a primary focus of treatment during the previous three months, if they had a history of treatment resistance to antipsychotic agents, or if they showed evidence of current or recent substance abuse or suicidal ideation.
Concomitant medication
Antipsychotic medications other than lurasidone were not allowed. Treatment with antidepressant medications and/or mood stabilizers was permitted during the study in patients who had been taking a stable dose for at least 30 days prior to the open-label stabilization phase baseline; initiation or increases in dosage of these medications during the study was prohibited. Benztropine was permitted up to 6 mg/d as needed for movement disorders; alternate medications allowed for acute extrapyramidal symptoms (EPS) were biperiden (up to 16 mg/d), trihexyphenidyl (up to 15 mg/d), or diphenhydramine (up to 100 mg/d). Propranolol was permitted up to 120 mg/d, as needed, for akathisia. Investigators were instructed to use benzodiazepines sparingly; sedative-hypnotics (e.g. eszopiclone, zolpidem) were permitted, as needed, for insomnia. Anxiolytics, sedatives, or hypnotics were not to be administered within eight hours of a psychiatric assessment.
Study medication adherence
Assessment of adherence to study medication was based on tablet counts from returned blister packs at study visits. Adherence was calculated by dividing the actual number of doses taken by the number that should have been taken within a specified time period and multiplying by 100. Patients who missed more than 25% of scheduled doses or took more than 125% of scheduled doses were considered non-adherent.
Study design
Open-label stabilization phase
Acutely ill patients were enrolled in a 12- to 24-week open-label treatment phase (Figure 1). Patients were treated with lurasidone at a starting dose of 40 mg/d; flexible dosing from 40 mg/d to 80 mg/d was allowed after three days up until the last four weeks of the stabilization period, during which no dose adjustments were permitted. Study medication was taken once daily in the evening, with a meal or within 30 minutes after eating. Patients were assessed weekly during the open-label phase in order to evaluate clinical stability. Safety assessments included treatment-emergent adverse events, movement disorder signs or symptoms (using the Simpson–Angus Scale (SAS) (Simpson and Angus, 1970), the Barnes Akathisia Rating Scale (BARS) (Barnes, 1989), and the Abnormal Involuntary Movement Scale (AIMS) (Guy, 1976)), suicidal ideation (using the Columbia–Suicide Severity Rating Scale (C-SSRS) (Posner et al., 2011)), laboratory measures, vital signs, and electrocardiogram (ECG).
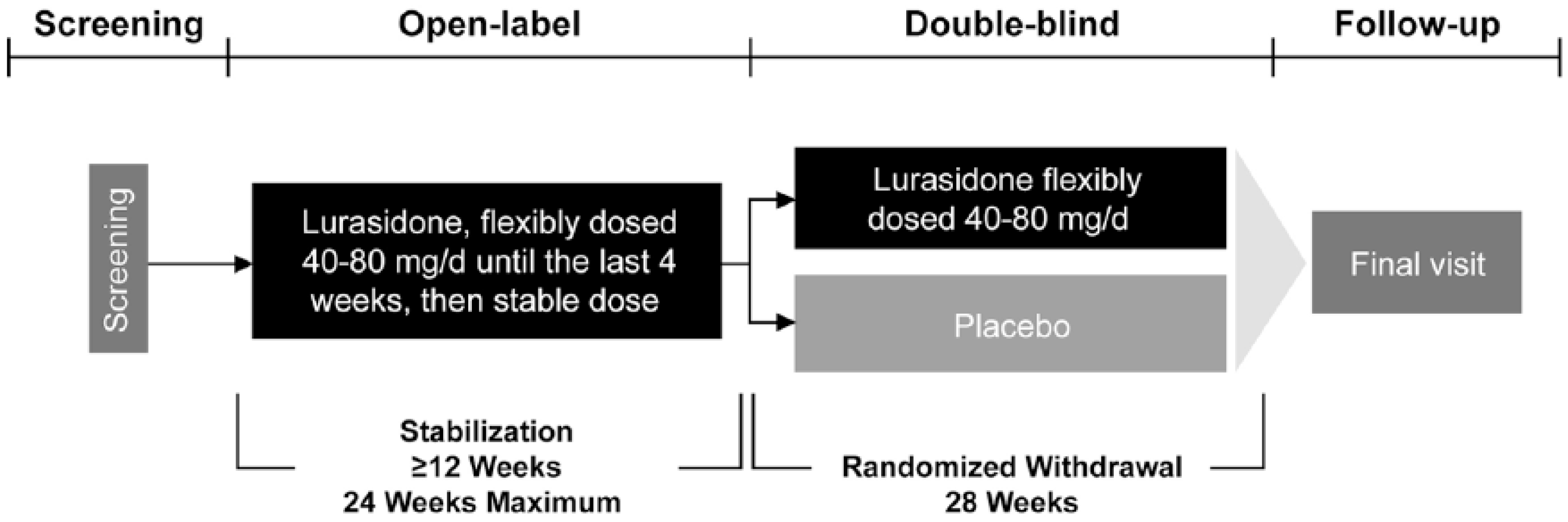
Study design.
In order to enter the 28-week, double-blind, randomized withdrawal phase of the study, patients were required to achieve and maintain clinical stability for at least 12 weeks during the open-label stabilization phase and to have remained on a stable dose of lurasidone for four weeks prior to randomization. Clinical stability was defined as a PANSS total score ⩽70, with PANSS item scores ⩽4 on all positive subscale items and the item for “uncooperativeness”, (item G8), and a CGI-S score <4. Two excursions (defined as a PANSS total score up to a maximum of 80 and/or a CGI-S score of 4 and/or PANSS positive subscale item score of 5) after initial attainment of these stability criteria were permitted per protocol, except during the last four weeks of the open-label stabilization phase. Patients who achieved 12 weeks of clinical stability were immediately eligible to enter the double-blind phase of the study and were not required to complete the full 24 weeks of the open-label phase. Patients who failed to maintain 12 consecutive weeks of clinical stability during 24 weeks of open-label treatment (or had not yet achieved clinical stability after the first 12 weeks) were discontinued from the study and referred for appropriate follow-up care.
Double-blind, randomized withdrawal phase
The objective of the double-blind phase was to assess the efficacy of continued treatment with lurasidone versus switching to placebo for preventing relapse in patients who had maintained clinical stability with lurasidone for at least 12 weeks. Patients who met the criteria for clinical stability during the open-label stabilization phase were randomized at baseline of the double-blind phase via an interactive voice/web response system to receive lurasidone or identically-matched placebo (in a 1:1 ratio) for up to 28 weeks. None of the investigators, study staff, or patients had access to the randomization assignment. The initial dose of lurasidone in the double-blind phase of the study was the same as the final open-label dose; adjustments within the range of lurasidone 40 mg/d–80 mg/d were then permitted (dose increase at weekly intervals and dose reduction as needed for tolerability).
The primary efficacy endpoint was time to relapse (based on Kaplan–Meier survival analysis), with relapse defined as ⩾1 of the following during the double-blind phase:
An increase of ⩾25% from double-blind baseline in PANSS total score and CGI-S worsening of ⩾1 point for two consecutive visits no more than ten days apart.
At any single visit, a PANSS item score of ⩾5 (moderately severe) on hostility or uncooperativeness, or a PANSS item score of ⩾5 on two or more items of unusual thought content, delusions, conceptual disorganization, or hallucinatory behavior.
Initiation of supplemental treatment with an antipsychotic agent other than lurasidone, an increased dose of an antidepressant or mood stabilizer, an increase in lorazepam (or benzodiazepine equivalent) dose by ⩾2 mg/d for at least 3 days, or electroconvulsive therapy.
Insufficient clinical response or exacerbation of underlying disease reported as an adverse event, as determined by the study investigator.
Deliberate self-injury or repeated aggressive behavior, active suicidal or homicidal ideation or attempt.
Psychiatric hospitalization due to worsening schizophrenia.
Secondary efficacy measures during the double-blind phase included change from double-blind baseline in PANSS and CGI-S scores. Safety assessments in the double-blind phase were similar to those in the open-label phase.
Study visits were conducted weekly for the first two weeks of the double-blind phase and then every two weeks thereafter. Patients were contacted by telephone between scheduled visits to monitor clinical symptoms and adverse events, and an unscheduled visit was made as early as possible for patients who appeared to be symptomatic.
Statistical methods
The safety populations in the open-label and double-blind phases comprised those patients who received at least one dose of study medication in each phase of the study, respectively. The intent-to-treat (ITT) population used for the efficacy analysis in the double-blind phase was the same as the double-blind safety population in this study.
A Kaplan–Meier survival analysis for time to relapse by treatment group in the double-blind phase was performed. The primary efficacy analysis utilized an unstratified log-rank test (with treatment as a fixed effect) for time to relapse in the ITT population. As a confirmatory analysis, the hazard ratio of time to relapse and corresponding 95% confidence interval (CI) values were calculated for lurasidone versus placebo groups using a Cox proportional hazards model.
Based on assumed relapse event rates (at the week 28 endpoint) of 30% with lurasidone and 50% with placebo, it was determined that 98 relapse events would be required to detect this drug-placebo difference with 90% power using a log-rank test, assuming two interim analyses and the final analysis (using the Haybittle–Peto method) and an overall two-sided alpha level of 0.05. Thus, the study was to be terminated after a total of 98 relapse events had occurred. Two pre-specified unblinded interim analyses were performed during the double-blind phase by an external, independent data safety monitoring board after approximately 50% and 75% of the planned total number of relapse events had taken place (49 and 74 events, respectively). Due to the interim analyses, the nominal p value for statistical significance was adjusted for the primary efficacy analysis to ensure an overall type I error rate of 0.05. Based on information fractions of 50%, 75%, and 100% for the interim and final analyses, the p value for statistical significance was established at 0.0119 for the two interim analyses, 0.042 for the final analysis of the primary endpoint (log-rank test of time to relapse), and 0.05 for all secondary endpoints.
Time to all-cause discontinuation during the double-blind phase was analyzed in the same manner as time to relapse. Other secondary efficacy measures assessed during the double-blind phase were evaluated utilizing mixed-model repeated-measures (MMRM) analysis that included treatment, visit (as a categorical variable), pooled center, double-blind baseline score, and a treatment-by visit interaction term. An unstructured covariance matrix was used for within-patient correlation. Analysis of covariance (ANCOVA) with the last observation carried forward (LOCF) was also utilized to evaluate secondary efficacy outcomes. The number needed to treat (NNT) to prevent one additional relapse was derived for the lurasidone groups as follows: NNT=1/(lurasidone relapse rate – placebo relapse rate).
Results
A total of 676 patients enrolled in the open-label phase; 285 patients (42.2%) met protocol-specified criteria for stabilization and were randomized to lurasidone (N=144) or placebo (N=141); two patients completed the open-label phase but were not randomized (Figure 2). For patients who met clinical stability criteria and were randomized, demographic and clinical characteristics at baseline of the double-blind phase were similar among the lurasidone and placebo groups (Table 1).
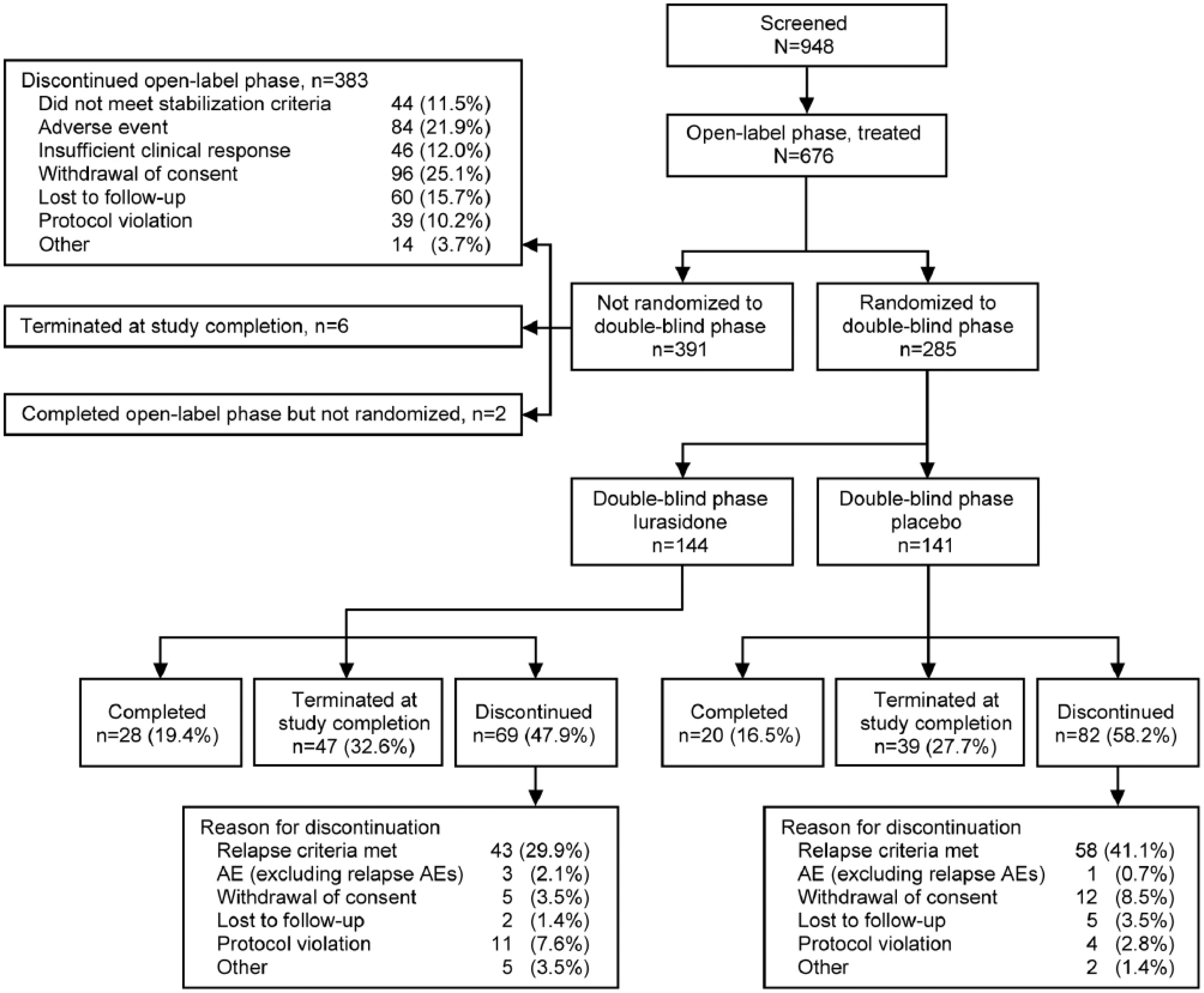
Patient disposition. AE: adverse event.
Patient demographics and baseline clinical characteristics. a
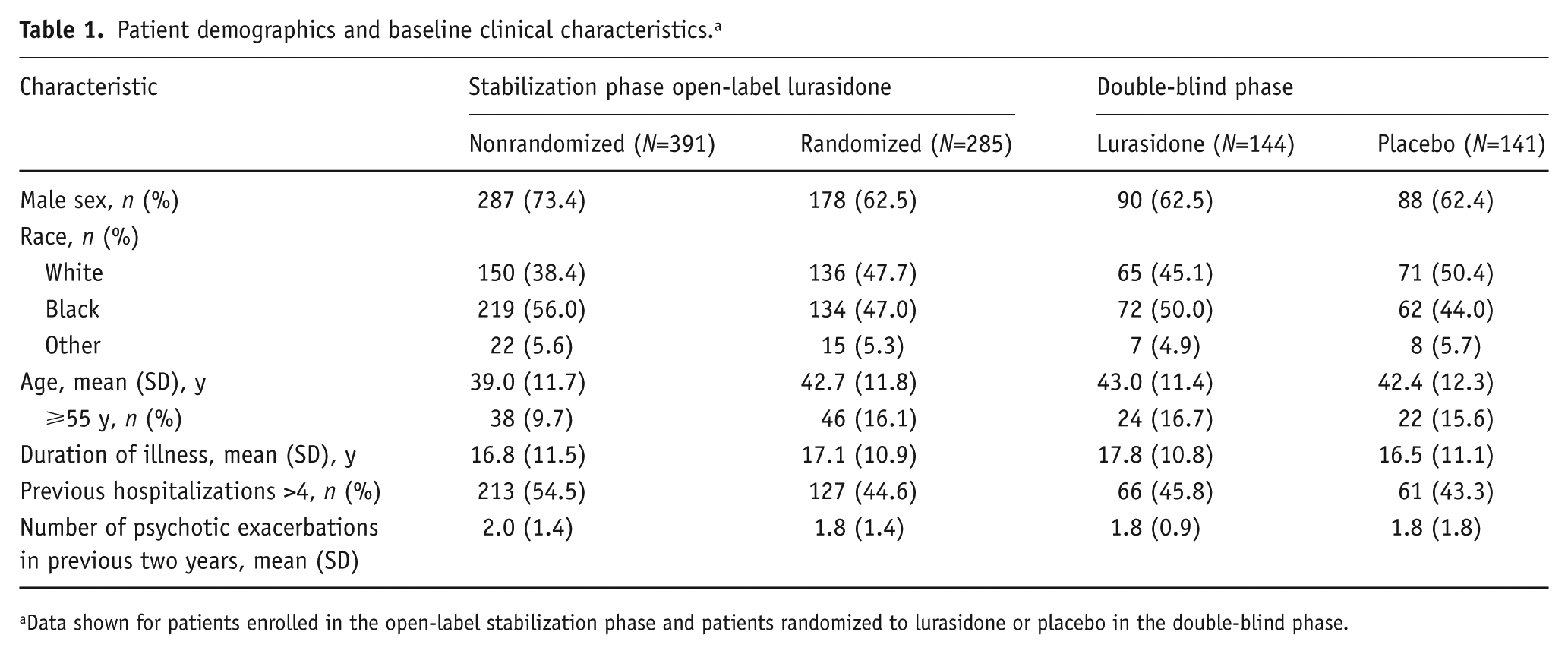
Data shown for patients enrolled in the open-label stabilization phase and patients randomized to lurasidone or placebo in the double-blind phase.
Open-label stabilization phase
Among patients who discontinued from the open-label phase, the most common reasons were withdrawal of consent (24.7%), adverse events (21.6%, including 10.0% with worsening of schizophrenia), lost to follow-up (15.4%), insufficient clinical response (11.8%), and failure to meet stabilization criteria (11.3%; Figure 2).
For patients who met clinical stability criteria and were randomized, the mean lurasidone dose during the open-label phase was 67.7 mg/d (the modal daily dose was 80 mg in 73.2% of patients), and the mean open-label treatment exposure was 17.8 weeks. During the open-label phase, mean PANSS total score decreased from 90.1 to 54.4 and mean CGI-S score from 4.5 to 2.7 in patients who met clinical stability criteria and were randomized. Symptomatic improvement was observed within the first one to two weeks of open-label treatment with lurasidone, with a mean time to attainment of initial stabilization of 5.2 weeks.
Double-blind, randomized withdrawal phase
During the double-blind phase, the mean daily dose of lurasidone was 78.9 mg/d; the modal daily dose was 80 mg in 78.5% of patients. The most commonly used concomitant medications were anxiolytics (26.4% of patients taking lurasidone and 25.5% taking placebo), antidepressants (23.6% and 27.7%, respectively), and sedatives/hypnotics (13.2% and 23.4%, respectively). Anticholinergic agents were given to 10.4% of patients treated with lurasidone and 13.5% receiving placebo.
Neither of the two protocol-specified unblinded interim analyses for time to relapse demonstrated a significant difference between lurasidone and placebo at the adjusted value of p<0.0119 (log-rank test); therefore, the study continued as planned and was terminated after 101 relapses occurred. Lurasidone significantly delayed time to relapse compared with placebo (log-rank test, p=0.039) (Figure 3), reflecting a 33.7% reduction in risk of relapse (Cox model hazard ratio (95% CI), 0.663 (0.447–0.983); p=0.041). At the week 28 endpoint of the double-blind phase, the Kaplan–Meier estimate for probability of relapse was 42.2% for patients receiving lurasidone compared with 51.2% for the placebo group (NNT=12).
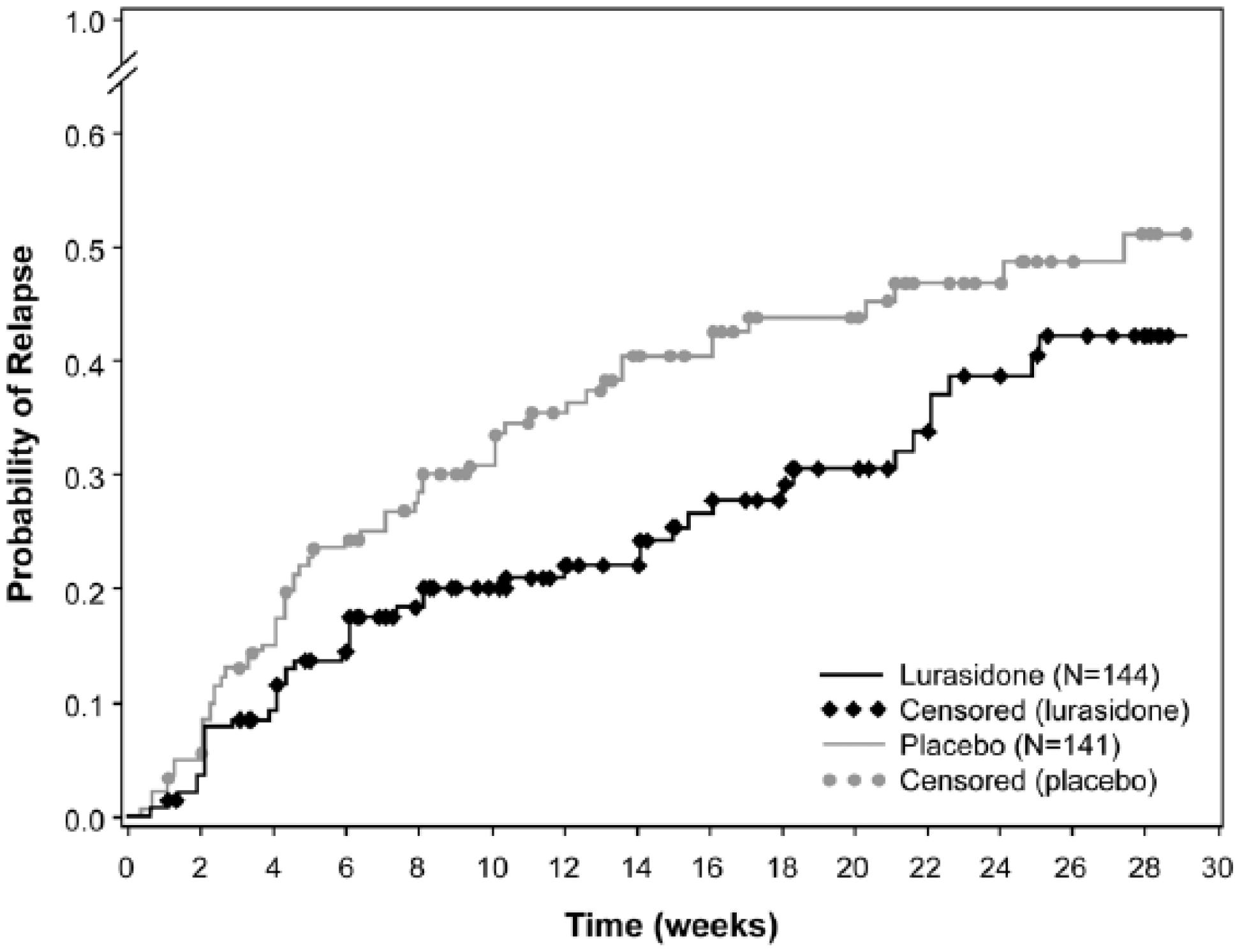
Kaplan–Meier survival curve of time to relapse. Time to relapse was censored at the time of study completion or early termination for patients who discontinued from or completed the double-blind phase without experiencing a relapse event.
Fewer patients discontinued, for any reason, from the lurasidone group (47.9%) than from the placebo group (58.2%) during the double-blind phase. The probability of all-cause discontinuation at the week 28 endpoint based on the Kaplan–Meier survival analysis was lower for lurasidone (58.2%) than for placebo (69.9%), but the between-group comparison for time to all-cause discontinuation was not statistically significant (log rank test, p=0.070). During the double-blind phase, 29.9% of patients in the lurasidone group and 41.1% in the placebo group discontinued from the study due to an observed relapse event (Table 2). The most common reason for relapse for patients in the lurasidone group were insufficient clinical response (criterion #4) and PANSS item scores (criterion #2); for patients in the placebo group, the most common reason for relapse were PANSS item scores (criterion #2), PANSS total/CGI-S score (criterion #1), and insufficient clinical response (criterion #4). Among patients who experienced a relapse, psychiatric hospitalization due to worsening schizophrenia was reported in 9.3% of lurasidone-treated patients and 12.1% of patients receiving placebo.
Summary of relapse by criterion.
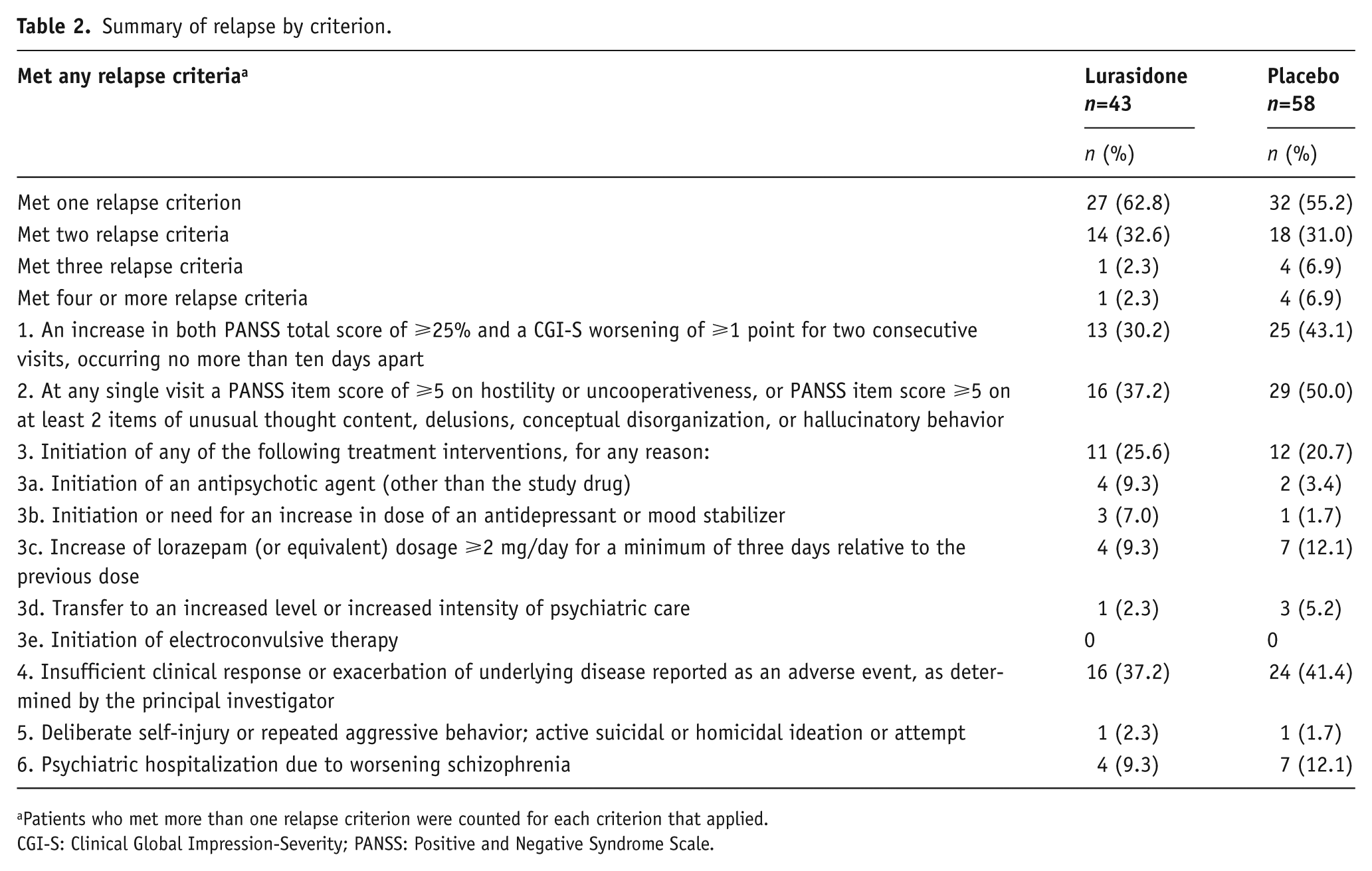
Patients who met more than one relapse criterion were counted for each criterion that applied.
CGI-S: Clinical Global Impression-Severity; PANSS: Positive and Negative Syndrome Scale.
Patients receiving placebo demonstrated significantly greater worsening in PANSS total and CGI-S scores over the double-blind phase compared to patients receiving lurasidone. Least squares mean change (ANCOVA at week 28 LOCF) was +12.4 in the placebo group versus +8.3 in the lurasidone group on the PANSS (p=0.029) and +0.7 versus +0.4 on the CGI-S (p=0.015) (Figure 4). MMRM analysis found that between-group differences for lurasidone versus placebo were significant for the overall PANSS total (p=0.019) and CGI-S (p=0.002) models; however, they did not reach statistical significance at the week 28 endpoint.
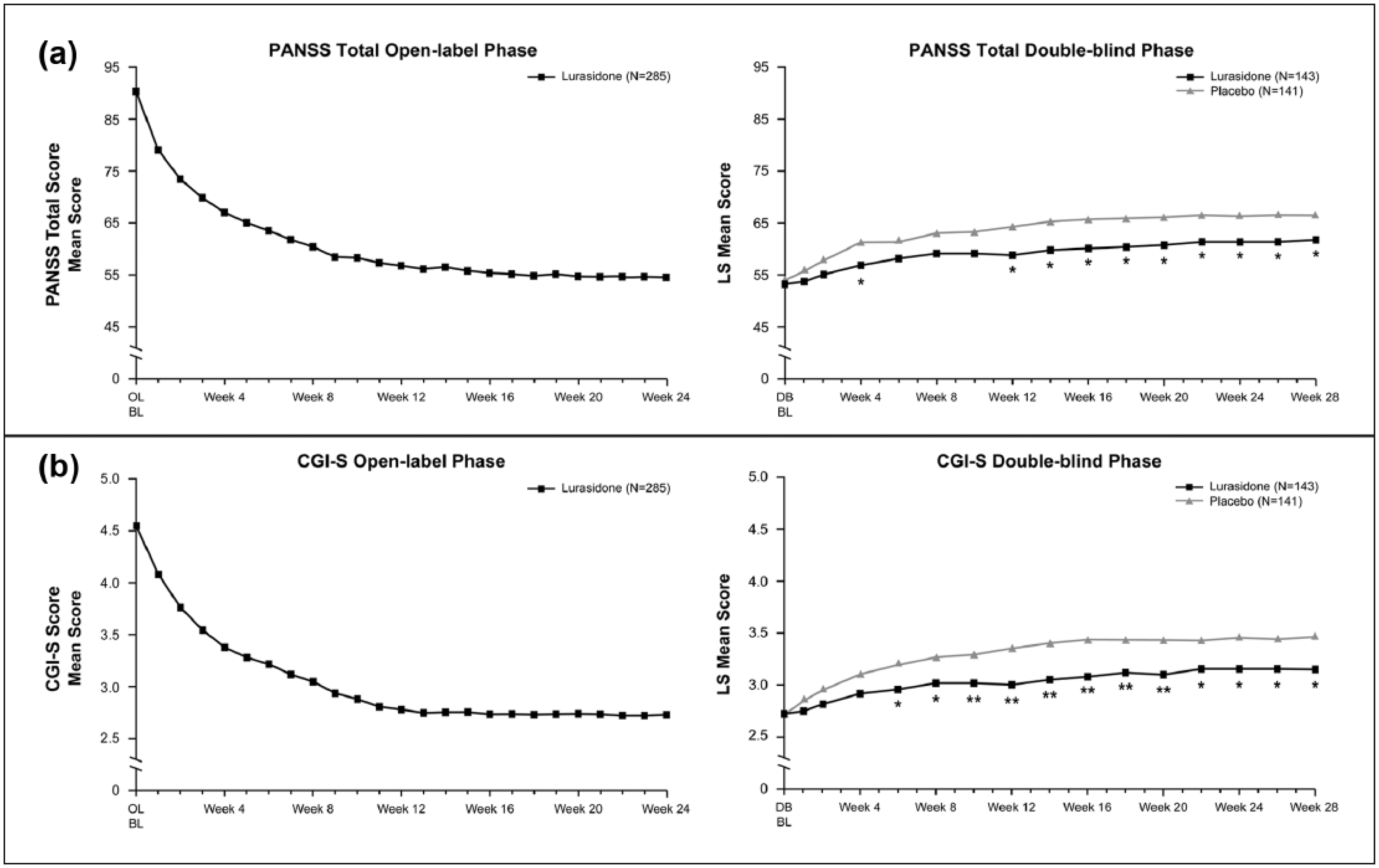
Change in PANSS total score (a) and CGI-S score (b) in the open-label phase and double-blind phase (ANCOVA, LOCF) among patients who met criteria for clinical stability and were randomized. *p<0.05. **p<0.01. ***One patient in the lurasidone group did not have a post-baseline assessment. ANCOVA: analysis of covariance; BL: baseline; CGI-S: Clinical Global Impression-Severity; DB: double-blind; LOCF: last observation carried forward; LS: least-squares; OL: open-label; PANSS: Positive and Negative Syndrome Scale.
Efficacy differences were observed between US and non-US sites. In the double-blind phase, 200 patients (70.2%) were randomly assigned at US sites and 85 patients (29.8%) at non-US sites. Compared with placebo, lurasidone significantly delayed time to relapse in the non-US subgroup (log-rank test, p=0.010) but not the US subgroup (log-rank test, p=0.414).
Study medication adherence
Open-label medication adherence data were available for 91.2% of patients who completed the open-label stabilization phase and were randomized. Among these patients, mean adherence during the open-label period was calculated as 99.9%. In the double-blind phase, study medication adherence data were available for 96.5% of patients treated with lurasidone and 90.8% of patients receiving placebo. Among these patients, mean adherence in the double-blind phase was calculated as 99.9% for lurasidone and 100.0% for placebo. Two patients receiving lurasidone and one patient receiving placebo were deemed non-adherent to study medication in the double-blind phase (all with >125% of scheduled doses).
Safety
During the open-label phase, adverse events were experienced by a similar proportion of patients who achieved clinical stability and were randomized into the double-blind phase (73.3%) compared to those who were not randomized (73.1%). The most common adverse events (⩾ 10%) among all patients in the open-label phase were akathisia (13.9%), headache (11.4%), and nausea (10.2%) (Table 3). In the double-blind phase, the percentage of patients reporting any adverse event was similar for the lurasidone (53.5%) and placebo (54.6%) groups; the incidence of EPS-related adverse events in the double-blind phase was low (4%) and comparable between the groups. The discontinuation rate due to adverse events (including the adverse event-related relapse criterion of worsening of schizophrenia) during the double-blind phase was 13.9% for lurasidone and 15.6% for placebo. Among patients who were treated with lurasidone in both the open-label and double-blind phases, the most common adverse events (⩾10%) were akathisia (16.7%), insomnia (12.5%), headache (11.8%), nausea (11.1%), and anxiety (11.1%) (Table 3).
Incidence of treatment-emergent adverse events (TEAEs) reported in ⩾5% of lurasidone-treated patients. a
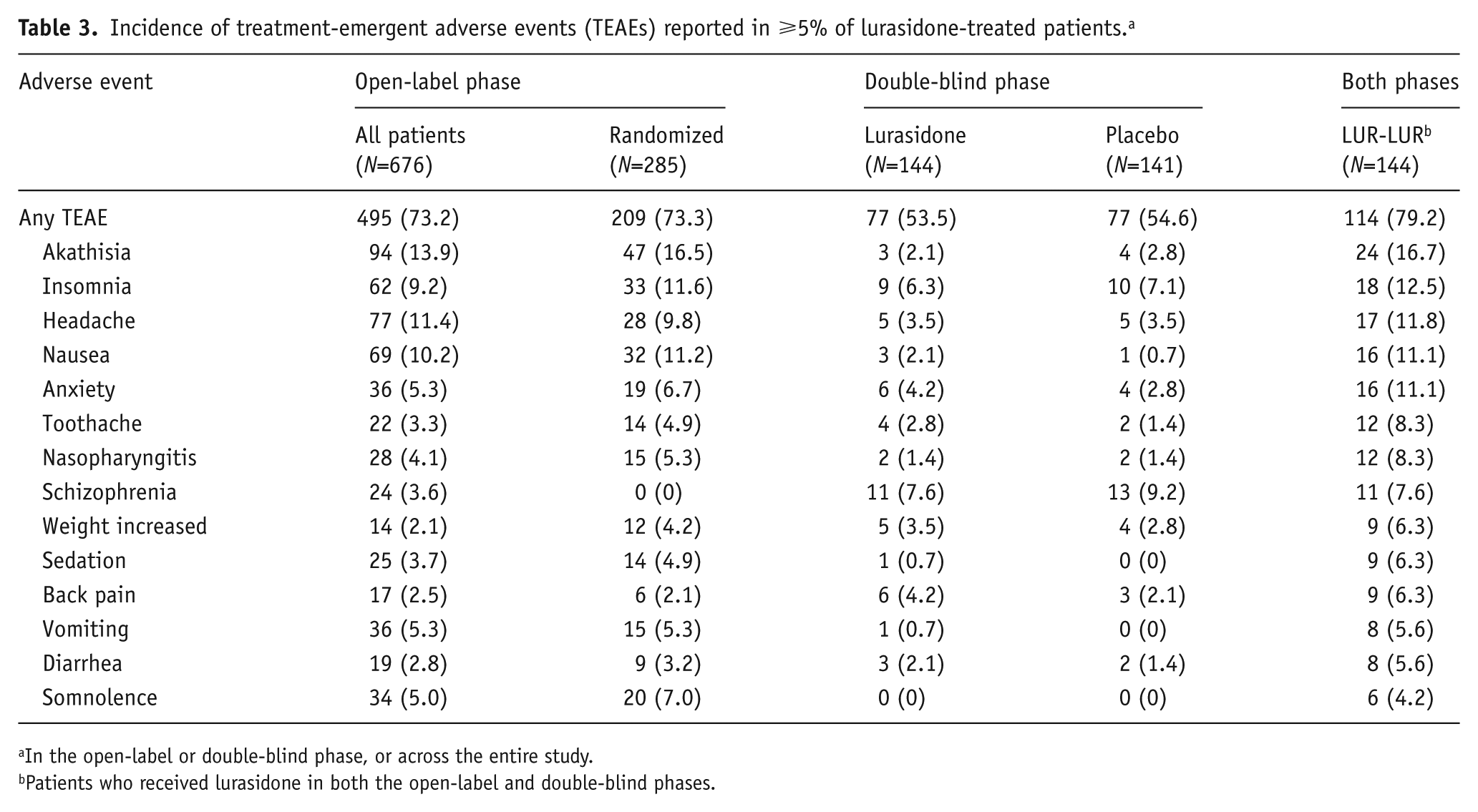
In the open-label or double-blind phase, or across the entire study.
Patients who received lurasidone in both the open-label and double-blind phases.
Serious adverse events were reported in 8.7% of patients during the open-label phase, and consisted primarily of schizophrenia (3.0%) or psychotic disorder (1.6%). In the double-blind phase, serious adverse events were more common in the placebo group (7.8%) compared with the lurasidone group (4.2%), with schizophrenia (2.8% for placebo and 0.7% for lurasidone) and psychotic disorder (1.4% in each group) as the most common. Almost half of enrolled patients (46.2%) had a history of previous suicidal ideation or behavior (based on the C-SSRS). During the study, suicidal ideation emerged in 6.3% of patients during the open-label phase and in 1.4% of patients randomized to lurasidone, compared with 3.5% of patients randomized to placebo in the double-blind phase; there were no suicide attempts. One death (sudden cardiac death) occurred during the open-label phase of the study; this death was judged by the investigator to be unrelated to study medication.
Minimal changes in weight, lipids, glucose, and prolactin were observed. In patients treated with lurasidone in both the open-label and double-blind phases, mean weight change was -0.6 kg (LOCF); weight gain ⩾7% and weight loss ⩾7% were experienced by a similar proportion of patients (17.4% and 16.7%, respectively); and median change in metabolic parameters was -1.0 mg/dL for total cholesterol, -1.0 mg/dL for triglycerides, and 0.0 mg/dL for glucose (LOCF). Change in movement disorder signs or symptoms (as measured by SAS, BAS, and AIMS scores) were generally absent to mild in patients treated with lurasidone.
Discussion
The efficacy of lurasidone for the maintenance treatment of patients with schizophrenia was demonstrated in this multicenter, placebo-controlled, randomized withdrawal study. Forty-two percent of patients who entered the study completed the open-label stabilization phase, demonstrating that maintenance of stability with lurasidone was feasible over a three-month period. In the double-blind phase, treatment with lurasidone (40–80 mg/d) significantly delayed time to relapse and reduced the risk of relapse by 33.7% compared with placebo. The results of this study are consistent with findings of a previous 12-month, double-blind study, which showed that long-term treatment with lurasidone (40–160 mg/d) was associated with a relatively low probability of relapse, and demonstrated the non-inferiority of lurasidone relative to quetiapine XR (200–800 mg/d) for relapse prevention in patients with schizophrenia (Loebel et al., 2013b). The results of the current study, taken together with the findings of the above-noted study (Loebel et al., 2013b), support the use of lurasidone as maintenance treatment for patients with schizophrenia across the dose range of 40–160 mg/d.
A key strength of the current study is that clinical stability, defined using rigorous criteria, was demonstrated for at least 12 weeks during the open-label stabilization phase. This extended stabilization period increased the likelihood that subsequent emergence of symptoms represented recurrence of illness and not merely symptom fluctuation in patients who were not clinically stabilized. The requirement of a minimum of 12 weeks of clinical stability prior to randomization was consistent with regulatory agency guidance regarding the design of relapse-prevention studies in schizophrenia (Committee for Medicinal Products for Human Use (CHMP), 2012; Department Health and Human Services et al., 2005) and was similar to other maintenance treatment studies that included a stabilization period (Beasley Jr et al., 2003; Kane et al., 2011; Kramer et al., 2007; Peuskens et al., 2007).
The magnitude of effect observed with lurasidone in the present study (relapse rate at the double-blind week 28 endpoint of 42.2% for lurasidone and 51.2% for placebo based on Kaplan–Meier analysis (NNT=12), and 29.9% for lurasidone and 41.1% for placebo based on observed cases (NNT=9)) was somewhat smaller than findings reported in a recent meta-analysis of placebo-controlled studies of antipsychotic agents for relapse prevention in patients with schizophrenia, which found observed relapse rates at 7–12 months of 27% for active medication and 64% for placebo (NNT=3) (Leucht et al., 2012).
There is substantial variation in relapse criteria among published studies of maintenance treatment in schizophrenia (Arato et al., 2002; Beasley Jr et al., 2003; Csernansky et al., 2002; Kane et al., 2011; Kramer et al., 2007; Peuskens et al., 2007; Pigott et al., 2003), which makes cross-study comparison among antipsychotic agents difficult to interpret. This study utilized a number of relapse criteria; meeting any one criterion constituted a protocol-defined relapse. The rate of psychiatric hospitalization in this study, which may be considered an indicator of more severe relapse, was relatively low and favored lurasidone over placebo. The inclusion of non-hospitalization relapse criteria in this study could have introduced variability into the primary outcome assessment, potentially obscuring differences between lurasidone and placebo.
In contrast to most previous studies of maintenance treatment for schizophrenia (Arato et al., 2002; Beasley Jr et al., 2003; Csernansky et al., 2002; Kane et al., 2011; Peuskens et al., 2007; Pigott et al., 2003), this study enrolled acutely ill patients (rather than clinically stable patients) and required them to achieve and maintain clinical stability (as defined by stringent criteria) before entering the randomized withdrawal (i.e. relapse prevention) phase. We note that the 28-week double-blind phase was shorter than in some studies of other antipsychotic agents (Arato et al., 2002; Csernansky et al., 2002). This may have limited the number of relapses that occurred, particularly in the placebo group, compared with other studies.
Detection of differences between investigational agents and placebo in clinical trials of patients with schizophrenia has become increasingly problematic as treatment effect sizes have declined over time (Agid et al., 2013; Kemp et al., 2010; Khin et al., 2012; Rutherford et al., 2014). Consistent with these long-term trends, relapse rates in this study were lower in the placebo group and higher in the lurasidone group compared with results of the Leucht et al. (2012) meta-analysis. Consistent with prior analyses of regional differences in schizophrenia trials (Chen et al., 2010; Khin et al., 2012), time to relapse in this study strongly favored lurasidone compared to placebo for participants outside the United States; however, a significant treatment effect was not observed for participants in the United States. The reasons for this disparity in relapse rates by region are not known but warrant further investigation. However, we note that subgroup analyses (including those based on geographic region) are subject to a number of methodological concerns, including diminished sample size, reduced power, and increased variability, which increase the risk of spurious findings (Chen et al., 2010).
A meta-analysis of randomized, controlled trials of 15 anti-psychotic agents in the treatment of patients with schizophrenia found relatively small differences between medications in terms of efficacy, but substantial differences in side effects (Leucht et al., 2013). The results of the present study are consistent with this meta-analysis and with previous studies (Citrome, 2011, 2012) in which lurasidone was effective in the treatment of schizophrenia and was associated with a favorable safety profile. One limitation of this study was the restricted lurasidone dose range. Only lower doses (40–80 mg/d) of lurasidone were used, which may have reduced the ability of lurasidone to prevent relapse for some patients in this study. The lurasidone dose range selected for this study was based on the initial FDA-approved dose range of 40–80 mg/d (Sunovion Pharmaceuticals Inc., 2010). More than 70% of patients in both the open-label and double-blind phases of the present study received lurasidone dosed at 80 mg/d, indicating that a higher daily dose may have been needed to achieve and maintain clinical stability for some patients. The currently approved dose range for lurasidone in the treatment of patients with schizophrenia is 40–160 mg/d.
Additional study limitations, beyond the restricted dose range for lurasidone, include the lack of serum drug concentration measurement and limited information regarding medication adherence. Although medication adherence rates appear to be high based on pill counts, the extent of adherence to study medication based on serum concentration, which may be a more sensitive measure of adherence, is unknown.
In conclusion, this multicenter, placebo-controlled, ran-domized withdrawal study demonstrated the efficacy and safety of lurasidone for the maintenance treatment of patients with schizophrenia. Maintenance of efficacy was established by 12 weeks of clinical stability (rigorously defined) during open-label treatment with lurasidone. Patients who were randomized to continued treatment with lurasidone in the double-blind phase demonstrated significantly delayed time to relapse and reduced frequency of relapse compared with patients randomized to placebo. The safety profile of lurasidone in the present long-term study was consistent with prior studies, with minimal changes in weight, lipid parameters, and measures of glycemic control.
Footnotes
Acknowledgements
Nancy Holland, PhD, Synchrony Medical Communications, LLC, provided medical writing and editorial assistance for this manuscript under the direction of the authors. Financial support for this writing and editing assistance from Synchrony Medical Communications, LLC, was provided by Sunovion Pharmaceuticals, Inc., Marlborough, MA, USA.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Rajiv Tandon has served as an honorary consultant for Sunovion Pharmaceuticals, Inc.
Josephine Cucchiaro, Debra Phillips, David Hernandez, Yongcai Mao, Andrei Pikalov, and Antony Loebel are employees of Sunovion Pharmaceuticals, Inc.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Clinical research was sponsored by Sunovion Pharmaceuticals, Inc. and Takeda Pharmaceuticals International, Inc. The sponsors were involved in the study design, collection, and analysis of data. The interpretation of results and the decision to submit this manuscript for publication in the Journal of Psychopharmacology were made by the authors independently.