
Abstract
Objectives
A randomized, placebo-controlled, multicenter study was conducted to evaluate the safety and efficacy of polidocanol endovenous microfoam (1%, Varithena® [polidocanol injectable foam], BTG International Ltd.).
Methods
Patients (n = 77) with symptomatic, visible varicose veins were randomized to treatment with either Varithena 1% or placebo.
Results
Varithena provided greater mean changes from Baseline in patient-reported assessments of symptoms (e.g., heaviness, achiness, swelling, throbbing, itching [HASTI®] score 30.7 points vs 16.7 points, p = 0.0009, primary endpoint; and modified Venous Insufficiency Epidemiological and Economic Study-Quality-of-Life/Symptoms [m-VEINES-QOL/Sym; p < 0.001]), physician-assessed VCSS, and physician- and patient-assessed appearance compared with placebo.
The HASTI score correlated highly with the modified-VEINES-QOL/Sym and Chronic Venous Insufficiency Questionnaire-2 scores (r = 0.7 to > 0.9, p ≤ 0.001). Adverse events included contusion, incision-site hematoma, and limb discomfort. Venous thrombus adverse events were reported as mild and generally resolved without sequelae.
Conclusions
Varithena provided significantly greater symptom relief and improvement in leg appearance compared with placebo. Adverse events were generally mild and transient. (www.clinicaltrials.gov [NCT00758420]).
Keywords
Introduction
Varicose veins are a manifestation of chronic venous insufficiency. 1 Women have a higher prevalence of varicose veins and more commonly have symptoms.2,3 Heaviness in the legs, achiness, swelling, throbbing, and itching (HASTI™ symptoms) have been identified as symptoms most important to patients.4–6
Traditional management of chronic superficial venous disease includes compression stockings, surgery, endovenous heat ablation techniques (radiofrequency and laser ablation), and liquid sclerotherapy or physician-compounded foam (PCF) treatment. 7
For over 15 years, physicians have compounded liquid sclerosants into foam using the techniques described by Cabrera.8–12 Chemical foams, made by mixing gas and sclerosant, have different properties depending on the mode of preparation and the gas/air ratio. 13 PCFs are typically made using nitrogen-rich room air. There have been case reports of significant neurological adverse events (AEs) attributed to gas embolism due to large, persistent nitrogen bubbles arising from PCFs.14–18 While cerebral gas embolism is not the only cause of neurological event following sclerotherapy, it is a preventable cause. Other described causes include thrombotic embolization crossing a right-to-left shunt with the neurological event occurring several days after intervention, and vasoactive substances such as endothelin and histamine.19,20
Unlike PCFs, polidocanol endovenous microfoam (PEM 1%, approved in the USA as Varithena® [polidocanol injectable foam], BTG International Ltd.) is made with a very low concentration of nitrogen (<0.8%) plus carbon dioxide and oxygen. Varithena was designed following the Cabrera patent in 1996, with subsequent improvements, which anticipated the problems with PCFs and provided solutions by using physiological absorbable gases. 21 Additionally, Varithena is dispensed from a proprietary canister device, giving it superior in vitro foam characteristics compared with PCF, including more consistent bubble size, better cohesiveness, and longer contact with the endothelium. 13 Here, we report the results of a multicenter trial of the efficacy and safety of Varithena 1%.
Methods
The trial was a randomized, single-blind, placebo-controlled, parallel-group study conducted at 5 USA investigator sites from October 2008 through August 2009. Patients were assessed at Baseline and Weeks 1, 4, 8, and 12 post-treatment.
The study was conducted according to the principles of the Declaration of Helsinki. The protocol and informed consent form were approved by Schulman Associates Institutional Review Board, Cincinnati, Ohio, and WFUHS IRB, Winston Salem, North Carolina. Patients provided written informed consent.
Patients
Adults (18 to 65 years) with visible and symptomatic varicose veins and Clinical-Etiology-Anatomy-Pathophysiology (CEAP) clinical classification C2-5, with saphenofemoral junction (SFJ) incompetence and reflux (>5 sec) in either the GSV or major accessory veins, were enrolled. Patent foramen ovale (PFO) was initially an exclusion criterion; however, the US FDA agreed with the removal of this exclusion criterion after another study showed that patients with known PFO had no subclinical injury from Varithena-derived intracerebral bubbles. 22
Treatment
Patients were randomized on a 1:1 basis to receive a single treatment with study drug, i.e., either Varithena 1% or placebo (agitated saline, ≤15 mL). Randomization was via a computer-generated random allocation sequence prepared by United BioSource Corporation (Blue Bell, PA), implemented using sealed envelopes. The maximum volume of Varithena was initially 30 mL and was later reduced to 15 mL (described further in Safety assessments). Patients who received placebo could receive open-label Varithena 1% in the same leg after the Week 12 assessments.
Todd et al. 23 previously described the methods by which treatment was delivered and patient blinding was achieved. Under ultrasound guidance, the incompetent GSV was marked and cannulated mid-thigh, and study drug was administered in stages with the leg elevated—from cannulation point to the SFJ, from cannulation to the distal GSV, and finally into visible varicosities. Injection of the GSV distal to the point of cannulation and of visible varicose veins could be made through the original GSV access or by separate access with one or more butterfly needles placed under ultrasound guidance. The volume of foam injected (not to exceed the maximum) was that required to fill the veins to be treated and was monitored on ultrasound. Patients had post-procedure compression therapy with short stretch bandages and eccentric compression pads and 30–40 mm Hg compression stockings for 48 hours followed by compression stockings only for an additional 12 days. Patients and independent observers remained blinded through Week 12.
Patient-reported assessments
Schedule of assessments used in efficacy evaluations.
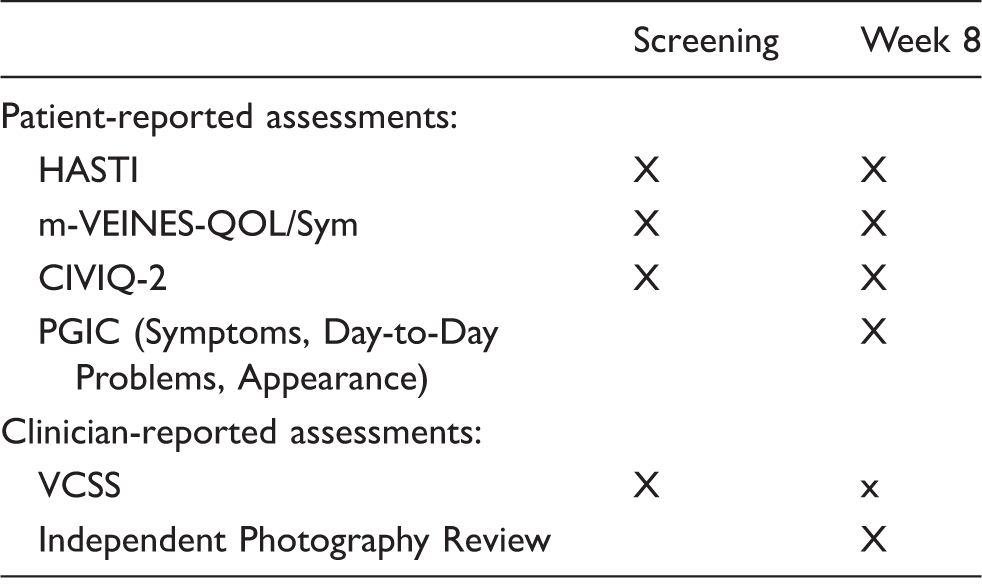
HASTI: heaviness, achiness, swelling, throbbing, itching; m-VEINES-QOL/Sym: modified Venous Epidemiological and Economic Study-Quality-of-Life/Symptoms; CIVIQ-2: Chronic Venous Insufficiency Questionnaire 2; PGIC: Patient Global Impression of Change.
For the current study, the original VEINES-QOL/Sym questionnaire was modified (m-VEINES-QOL/Sym) by changing the recall period from 4 weeks to 1 week and modifying the response options accordingly, both for the number of available response options and the language of the response options. 24
Concurrent with this study, qualitative research in patients with varicose veins revealed that heavy legs, achy legs, swelling, throbbing, and itching (HASTI) are the symptoms most relevant and important to them.4–6 The current study contributed to the psychometric validation of the VVSymQ® instrument and the VVSymQuick® instrument, both of which query the patient regarding HASTI symptoms.4–6,27 The HASTI score for varicose veins was derived from the answer to the question “How often have you had any of the following leg problems?” for each of the five HASTI symptoms (heavy legs, achy legs, swelling, throbbing, itching) using a paper questionnaire and 1-week recall period.4–6 The possible responses were “All of the time” (assigned a numeric value of 1), “Most of the time” (2), “A good bit of the time” (3), “Some of the time” (4), “A little of the time” (5), and “None of the time” (6). Higher scores indicated least vein symptoms.
The Patient Global Impression of Change (PGIC) was a single-item questionnaire on which patients rated their impression of post-treatment change. 28 It was administered for (1) appearance of veins, (2) leg symptoms, and (3) day-to-day problems. Response options were “Much improved” (score +3), “Moderately improved”(+2), “Improved in a small but important way” (+1), “About the same, hardly any change” (0), “A little worse” (−1), “Moderately worse” (−2), and “Much worse” (−3).
Clinician-reported assessments
A blinded assessor rated patients on the VCSS. 29 For the independent photography review (IPR) of appearance, three blinded dermatologists compared standardized photos from Screening and Week 8, randomly presented for each patient. The IPR score was the improvement in appearance at Week 8 compared with appearance at Screening, scored on a 7-point scale. Response options were, Week 8 is “Slightly improved” compared with Screening (+1), Week 8 is “Moderately improved” compared with Screening (+2), and Week 8 is “Much improved” compared with Screening (+3), Screening is “Much improved” compared with Week 8 (score –3), Screening is “Moderately improved” compared with Week 8 (−2), Screening is “Slightly improved” compared with Week 8 (−1), Screening and Week 8 are “About the same” (0). The median IPR score was used.
Physiologic response to treatment was determined by duplex ultrasound at Weeks 4 and 12. A “duplex ultrasound responder” was a patient who had either: (1) elimination of reflux through the SFJ or (2) complete occlusion of the GSV or treated major accessory vein.
Safety assessments
AEs were recorded at all visits. The severity of the AE was assessed by the investigator using standard regulatory definitions applicable to clinical trials, these being mild (transient symptoms, no interference with the patient’s daily activities), moderate (marked symptoms, moderate interference with the patient’s daily activities), and severe (considerable interference with the patient’s daily activities, which is unacceptable). A serious AE was defined as any untoward medical occurrence that results in death, life-threatening illness, in-patient hospitalization or prolongation of existing hospitalization, persistent or significant disability/incapacity, congenital abnormality/birth defect, or an important medical event, including DVT.
Detailed per-protocol duplex ultrasound assessments of the deep veins (at Screening and Weeks 1, 4, and 12) were completed using repeated compression and color flow from the malleoli upward; these were more comprehensive than that described in the 2008 Intersocietal Commission for the Accreditation of Vascular Laboratories (ICAVL). All thrombi detected in the deep and muscular calf veins were reported, as were all extensions of thrombus from the SFJ into the common femoral vein.
Prior clinical experience with Varithena 1% included injection volumes of ≤60 mL. The current study originally allowed ≤30 mL of Varithena. Based on the occurrence of five venous thrombi among the first 19 patients treated with Varithena 1%, and a trend suggesting higher occurrence of venous thrombi with higher volumes of Varithena 1%, the protocol was amended to specify ≤15 mL of Varithena. Studies show that most patients can be effectively treated with 15 mL.21,23,30 An independent Venous Thromboembolic Event Review Board (VTERB) of independent experts was convened to provide overview of venous thrombi in all clinical studies in the Varithena program and to advise on the clinical significance, diagnosis, treatment, monitoring, and prevention of venous thrombi. All venous thrombi were adjudicated by the VTERB. In the ongoing review of the venous thrombus AEs during the US phase 3 clinical trials, three distinct location patterns were recognized for the thrombi: (1) non-occlusive thrombi extending from the GSV into the common femoral vein, also known as “common femoral vein thrombus extension,” equivalent to EHIT; 31 (2) thrombi limited to the gastrocnemius or soleal veins, ie, isolated gastrocnemius and soleal vein thromboses (IGSVT);32,33 and (3) DVT, which was further classified into proximal DVTs (femoral and/or popliteal veins) or distal DVTs (thrombi in the anterior tibial, posterior tibial and/or popliteal veins). This classification scheme was applied to the venous thrombi in this study (see Safety section).
Venous thrombi were to be managed according to standard study site protocol. Sites were instructed to refer to the American College of Chest Physicians (ACCP) Recommendations for Treatment of Venous Thromboembolic Disease (VTE). 34 Additionally, per protocol, in patients with a venous thrombus, a duplex ultrasound was performed 1 and 2 weeks following detection, and then monthly until the thrombus was resolved or stable.
Placebo group patients who elected open-label Varithena 1% after the Week 12 assessments were monitored for 4 more weeks, with duplex ultrasonography performed at Weeks 1 and 4 post-treatment.
Analyses
The primary efficacy endpoint was updated from m-VEINES-Sym score change at Week 8 to HASTI symptoms score change at Week 8. Both HASTI and m-VEINES-Sym score results are presented.
Secondary endpoints included change in m-VEINES-Sym, m-VEINES-QOL, CIVIQ-2, PGIC-Symptoms, VCSS, PGIC-Appearance, and IPR scores. For PGIC and IPR scores, treatment group differences were analyzed using the Cochran-Mantel-Haenszel Chi square statistic with rank scores stratified by site. The absolute changes from Screening (considered to be Baseline) in HASTI, m-VEINES-Sym, m-VEINES-QOL, and CIVIQ-2 scores were analyzed by time point using analysis of covariance (ANCOVA) with treatment group and site as class variables and Baseline score as a continuous covariate.
Duplex ultrasound response rates at Weeks 4 and 12 were compared between groups using the Cochran-Mantel-Haenszel Chi square statistic and were stratified by site.
To assess construct validity of the HASTI score, Baseline scores and score changes at Week 8 were correlated between the HASTI score and the m-VEINES-Sym, m-VEINES-QOL, CIVIQ-2, and VCSS scores using Pearson correlations.
A sample size of 70 was chosen, providing 80% chance of demonstrating statistical significance at 5% level using a two-sided t-test, assuming a difference of 6.8 points (standard deviation of 10) between Varithena 1% and placebo in the change from baseline in m-VEINES-Sym at Week 8. Target recruitment was 37 patients per arm to allow for lost patients.
Results
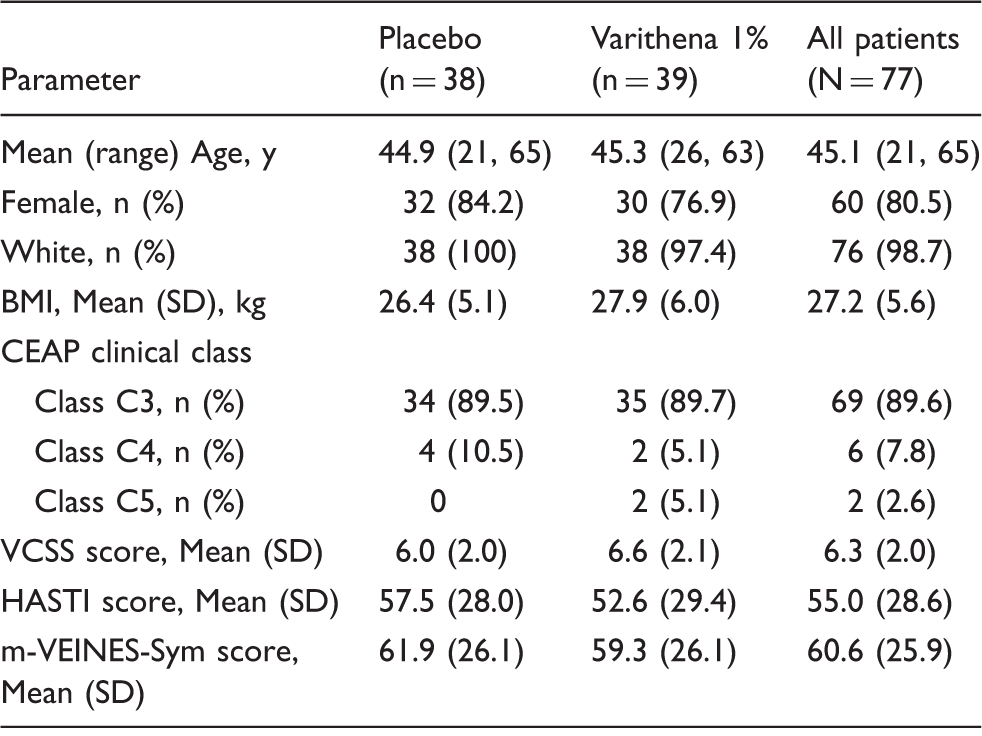
Patient disposition is shown in Figure 1. Seventy-seven patients were randomized (38 placebo, 39 Varithena 1%); all completed the blinded portion of the study and were included in the efficacy and safety evaluations. Thirty-four placebo group patients opted to receive open-label Varithena 1%, resulting in 73 patients who received Varithena 1%. As the maximum volume of Varithena permitted was originally 30 mL, the mean total volume of blinded Varithena 1% injected was 16.51 mL (range 7 to 30 mL). For patients enrolled after the protocol amendment that reduced the maximum permitted volume of Varithena 1% from 30 mL to 15 mL, the mean total volume of blinded Varithena 1% injected was 12.60 mL (range 7 to 15 mL). The mean total volume of open-label Varithena was 13.50 mL (range 4 to 15 mL). Baseline characteristics were similar between groups, as shown in Table 2.
Patient disposition. Patient demographics at Baseline. HASTI: heaviness, achiness, swelling, throbbing, itching; VEINES-Sym: modified Venous Epidemiological and Economic Study-Symptoms.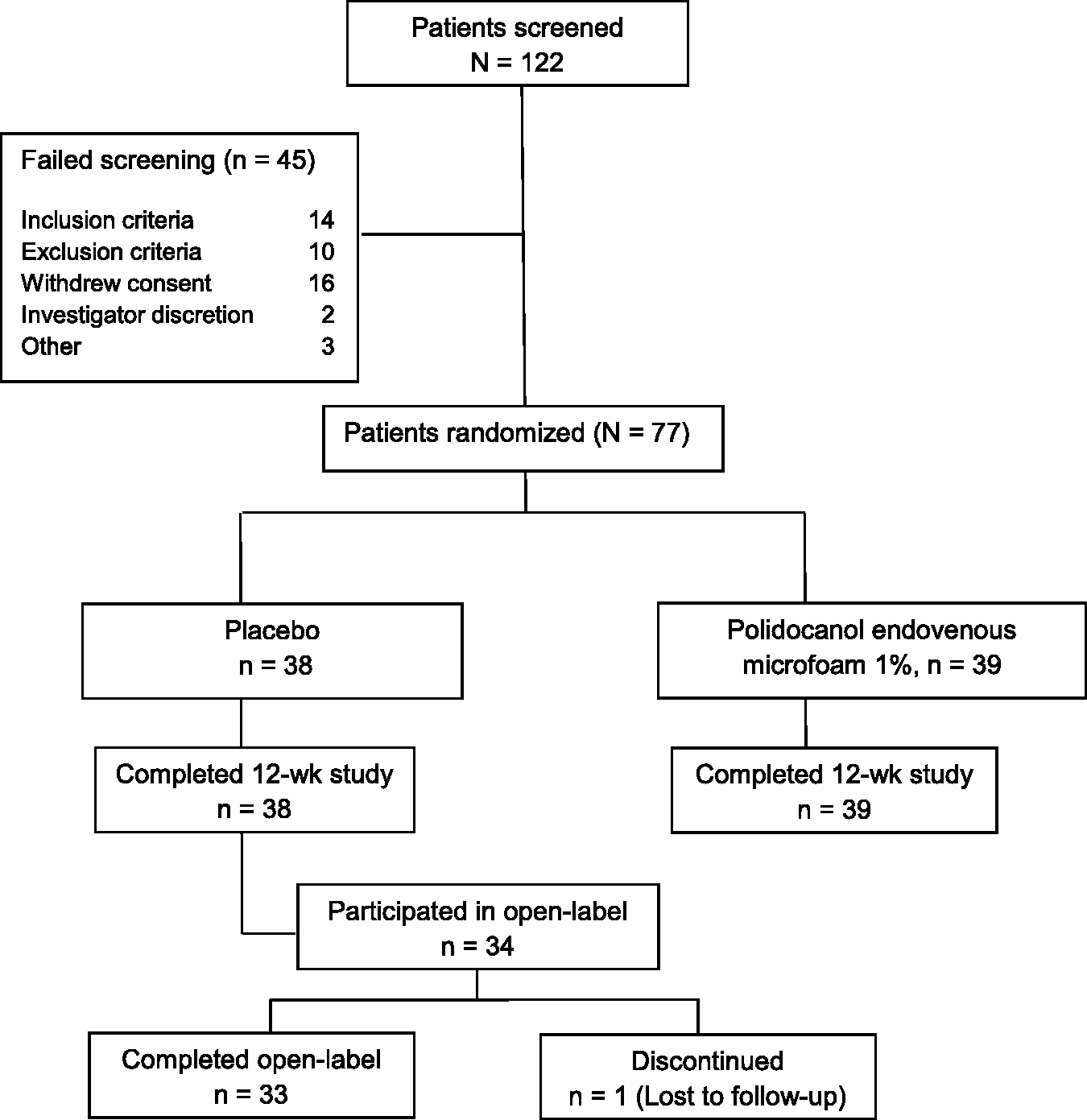
Efficacy
Varithena provided significantly greater improvement than did placebo at Week 8 in HASTI symptoms score (change from baseline 30.7 points versus 16.7 points; p = 0.0009) (Table 3, Figure 2), as well as m-VEINES-Sym/QOL, CIVIQ-2, PGIC-Symptoms, and PGIC-Day-to-Day Problems scores (p < 0.02) (Table 3). VCSS scores improved a mean 3.4 points (SD 2.5) with Varithena vs a mean 0.7 points (SD 2.4) for placebo.
Absolute value of mean change in score from Baseline at Week 8 for endpoints with a scale of 0–100. Change from Baseline in efficacy scores at Week 8 and between-group comparison. HASTI: heaviness, achiness, swelling, throbbing, itching; m-VEINES-QOL/Sym: modified Venous Epidemiological and Economic Study-Quality-of-Life/Symptoms; CIVIQ-2: Chronic Venous Insufficiency Questionnaire 2; PGIC: Patient Global Impression of Change. Higher scores indicate better outcome. Raw scores were transformed to final summary scores (Lamping et al.
24
). Least square mean and standard error (SE) from ANCOVA model with treatment group and site as class variables and corresponding Baseline score from the questionnaire as a continuous variable. Lower scores indicate better outcome. Raw scores transformed to final summary scores (CIVIQ User's GUIDE26). Mean (SD). p Values from the Cochran-Mantel-Haenszel Chi square statistic with rank scores stratified by site. Clinician-assessed instrument. n = 38.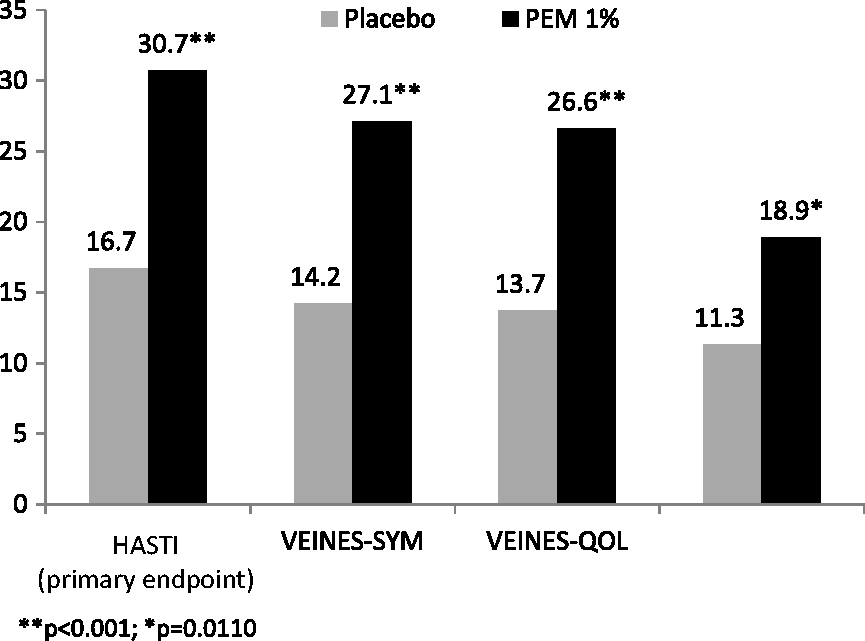
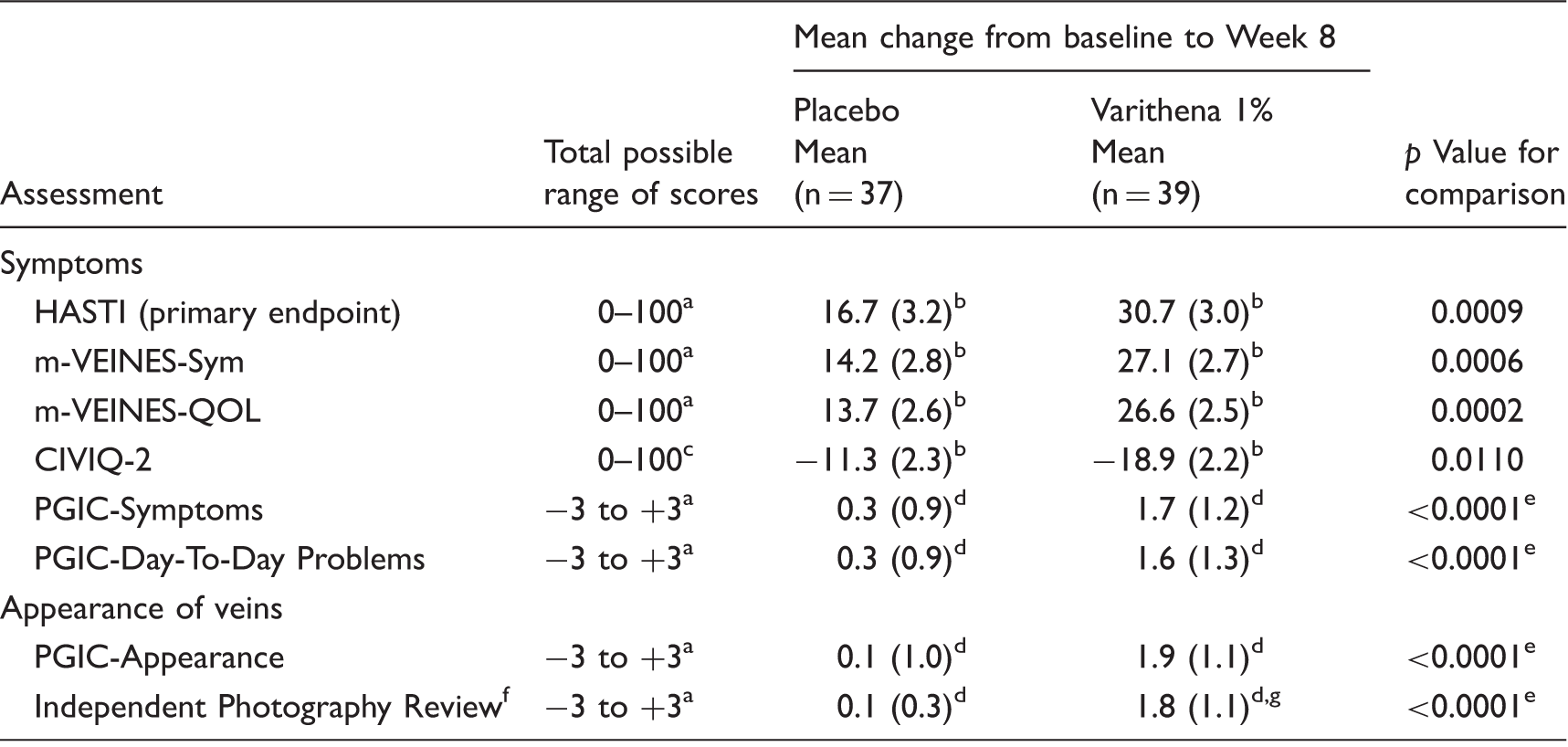
Varicose vein appearance improved more with Varithena than with placebo at Week 8 (PGIC-Appearance score change 1.9 points versus 0.1 points [p < 0.0001]; IPR score change 1.8 points versus 0.1 points [p < 0.0001]) (Table 3).
Duplex ultrasound response was nearly universal in the Varithena group and absent in the placebo group at Week 4 (90% vs 0% of patients, p < 0.0001).
Correlation of the HASTI scores with other instrument scores
Correlations between HASTI scores and other symptoms-based instrument scores at Baseline and correlations for the score changes at Week 8.

HASTI: heaviness, achiness, swelling, throbbing, itching; m-VEINES-QOL/Sym: modified Venous Epidemiological and Economic Study-Quality-of-Life/Symptoms; CIVIQ-2: Chronic Venous Insufficiency Questionnaire-2; PGIC: Patient Global Impression of Change.
Negative correlations arise from the fact that lower scores for VCSS and CIVIQ-2 indicate better status, whereas higher scores on the HASTI, m-VEINES-QOL/Sym, and CIVIQ-2 instruments indicate better status.
p ≤ 0.001.
Safety
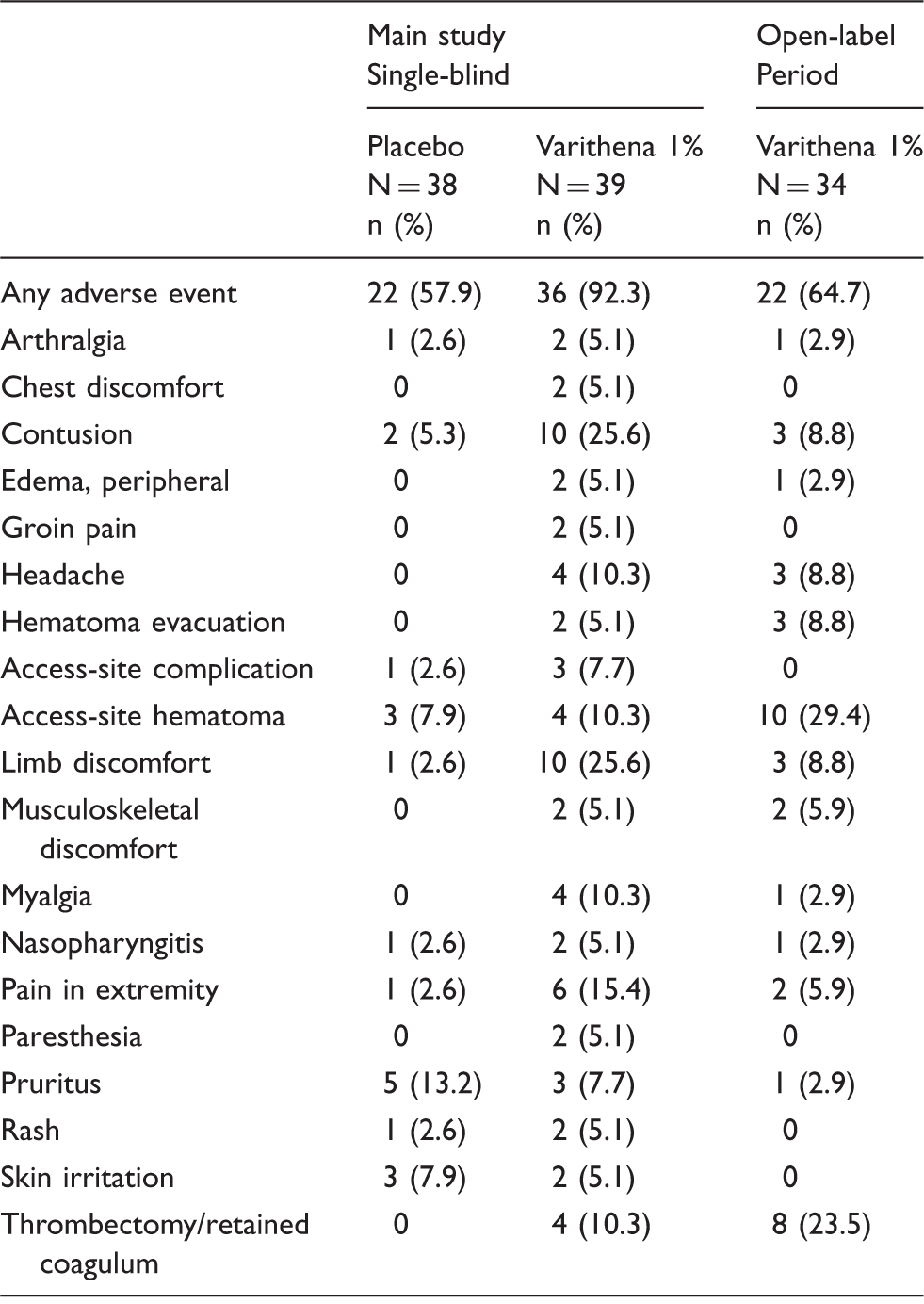
Adverse events that occurred in ≥5% of patients (placebo or Varithena 1%).
All venous thrombus AEs are reported in Table 6.
Venous thrombus adverse events in patients treated with Varithena 1%.

No patients had venous thrombus event during the placebo treatment period.
Common Femoral Vein Thrombus Extension (CFVTE): non-occlusive thrombi extending from the GSV into the common femoral vein, also known as “common femoral vein thrombus extension,” equivalent to EHIT.
Proximal DVT: thrombi in the femoral and/or popliteal veins.
Distal DVT: thrombi in the anterior tibial, posterior tibial and/or peroneal veins.
Isolated gastrocnemius and soleal vein thromboses (IGSVT): thrombi only in the gastrocnemius or soleal veins.
Serious AEs occurred in two patients. One patient in the placebo group had a transient ischemic attack on Day 13 that lasted two days and resolved without sequelae. One patient in the Varithena group had sick sinus syndrome that resulted in the placement of a pacemaker. Both events were considered by the investigator to be unrelated to study drug.
The majority of AEs (78%) resolved without sequelae by the final study visit. There were no pulmonary emboli diagnosed, and no neurological events in Varithena-treated patients.
Discussion
Varithena 1% demonstrated statistically significant differences from placebo in improving varicose veins symptoms on all patient-reported outcome instruments (HASTI, m-VEINES-QOL/Sym, CIVIQ-2, PGIC-Symptoms, and PGIC-Day-to-Day Problems). The physician-reported VCSS also improved more with Varithena than with placebo. Similarly, patients and physicians rated a greater improvement in appearance at Week 8 after treatment with Varithena 1% than after placebo (p < 0.0001).
One limitation of the study was that no adjustments were made for multiplicity of analyses. Another limitation was the modest sample size; however, patient blinding proved successful and, despite the sample size, the results proved statistically significant.
These results aligned with findings from previously published randomized, placebo-controlled trials of Varithena conducted in patients with SFJ incompetence and symptomatic and visible varicosities.23,30 In the VANISH-1 and VANISH-2 studies, Varithena was superior to placebo in decreasing the patient-reported VVSymQ symptoms scores, improving patient and clinician assessments of appearance, and improving the revised VCSS and VEINES-QOL/Sym scores at Week 8 (p ≤ 0.0001).
The HASTI instrument showed excellent correlation with the other patient-reported outcome instruments for Baseline scores and for the change in score at Week 8 (|r| = 0.78 to 0.97, p ≤ 0.001), 35 demonstrating that this succinct five-item instrument adequately captured the symptom information contained in the longer instruments. The high correlations demonstrate the construct validity of the HASTI instrument because the scores changed as would be expected after treatment. As might be expected, the HASTI symptoms score correlated modestly with VCSS, which reflects disease severity from both physician-interpreted and patient-reported assessments {Vasquez 2007}.29 The HASTI instrument is being further developed into an instrument that may be appropriate for use in everyday clinical practice (VVSymQuick).
Treatment with Varithena 1% was generally well tolerated. The AE profile was consistent with previously reported clinical research performed with Varithena 1%.23,30 The most common AEs were as anticipated with this type of intervention, including access-site hematoma, limb discomfort, and contusion. There were no serious drug-related AEs. All venous thrombus AEs were asymptomatic and mild, and were only detected on detailed ultrasound examination performed as part of the clinical trial. Importantly, there were no reported neurological AEs or pulmonary emboli. Compared with foam made with room air, Varithena may provide the benefit of absorbability because of the absence of nitrogen. The neurologic disturbances reported with foam sclerotherapy, though recently linked hypothetically to the release of endothelin-1 19 and histamine, 20 might also be related to gas embolisms that originate from the foam.16,36,37 Unlike the DSS and Tessari methods of foam production, Varithena produces no large bubbles. 13
Conclusions
Proprietary polidocanol endovenous microfoam (Varithena 1%) is an important addition to the currently available treatment modalities for varicose veins. Patients with varicose veins who received treatment with Varithena 1% demonstrated significantly greater symptom relief and improvement in visible vein appearance compared with those who received placebo. Varithena 1% was generally well tolerated; AEs were generally mild. Venous thrombi were detected only on protocol-defined ultrasound scans; all were mild and asymptomatic. All resolved or stabilized, regardless of whether or not they were treated with anticoagulants. The HASTI patient-reported outcome instrument is simple, with only five elements, is specific to varicose vein symptoms, and is being developed into a new clinical tool. It demonstrated sensitivity to treatment and moderate correlation with the VCSS and strong correlation with other established QOL instruments.
Footnotes
Acknowledgements
The authors acknowledge David Wright, MB, FRCS, for his participation in the design and execution of the Varithena clinical program; Elizabeth Orfe and Ellen Evans for the scientific and operational management of the study; John Ilgenfritz (United BioSource Corporation, Blue Bell, PA) and Claire Daugherty for statistical support; Tuli Ahmed (JetStream Clinical, LLC, work funded by Biocompatibles, Inc., a BTG International group company), Thomas King, and Irene Durham for medical writing assistance; and David J Goldberg, MD, Hackensack, NJ; David Bank, MD, Mount Kisco, NY; and Robert Weiss, MD, Hunt Valley, MD, for independent review of standardized digital photography. DW, EO, EE, CD, TK, and ID are current or former employees of BTG International group companies.
Authors’ notes
Varithena 013 Investigator Group: Kathleen Gibson (principal investigator), Lake Washington Vascular Surgeons, Overlake Medical Tower Office, 1135 116th Ave. NE, Suite 305, Bellevue, WA; Lowell Kabnick, New York University Langone Medical Center, New York, NY; Nick Morrison, Morrison Vein Institute Ltd., 8575 East Princess Drive, Suite 223, Scottsdale, AZ; Gilly Munavalli, Dermatology, Laser and Vein Specialists of the Carolinas, Midtown Medical Plaza, 1918 Randolph Road, Suite 550, Charlotte, NC; and John D. Regan, WFUBMC Interventional Radiology, Medical Center Boulevard, Winston Salem, NC. Varithena®, HASTI™, VVSymQ®, and VVSymQuick™ are trademarks of Provensis Ltd, a BTG International group company.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KG was the principal investigator and is a paid consultant for the sponsor. LK has received funding from the sponsor for his work as an investigator and consultant.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was sponsored by Provensis Ltd (a BTG International group company). Varithena is manufactured by BTG Biocompatibles Ltd on behalf of Provensis Ltd.
Ethical approval
The protocol and informed consent form were approved by Schulman Associates Institutional Review Board, Cincinnati, Ohio, and WFUHS IRB, Winston Salem, North Carolina.
Guarantor
KG.
Contributorship
KG drafted versions of the manuscript, enrolled patients, and interpreted data. LK interpreted data. KG and LK reviewed and edited the manuscript and approved the final version.