
Abstract
The use of adverse effect data from animals as the gold standard in regulatory toxicology has a long tradition dating back to the 1960s. It has also been increasingly criticised, based on both scientific and animal welfare concerns, and yet, animal studies remain the gold standard in most areas of toxicology to this very day. In the 1980s, when the first generation of non-animal methods were evaluated as alternatives to animal testing, it was logical to compare the ‘new’ data obtained with historical animal data. This worked reasonably well for simple endpoints, such as skin and eye irritation, but became problematic for the more complex systemic endpoints, since in these cases, the in vivo effects are not directly comparable to those observed in in vitro systems. While the need to redefine the gold standard is not new, there is still no consensus on how to do so. We propose a consistent principle that avoids the need for animal reference data, while also ensuring an equivalent or better level of protection. We argue that the gold standard can be redefined, or rather bypassed, by focusing on risk management outcomes rather than the outputs of animal methods. This allows us to more efficiently protect human health and the environment, ensuring the safe use of chemicals while also identifying less hazardous chemicals for use as substitutes. We describe how this might work out for two main contexts of use: classification and labelling, and risk assessment. This has implications for the implementation of the EU Commission Roadmap toward the phasing out of animal testing in chemical safety assessments.
Introduction
The use of adverse effect data from animals as the gold standard in chemical toxicology has a long tradition. It dates back to the 1960s when legislation was first introduced in Europe and North America to mandate and standardise the safety testing of chemicals. 1 The reliance on animal data also has a long tradition of criticism, based on scientific and animal welfare concerns, 2 and yet, animal studies remain the gold standard in most areas of toxicology to this very day. In the 1980s and 1990s, the first generation of non-animal methods were being developed and evaluated as replacement alternatives to animal testing. It was therefore logical to compare the ‘new’ data obtained with data obtained through traditional animal studies. Examples included in vitro tests for mutagenicity, acute systemic toxicity, and skin and eye irritation. This worked reasonably well for simple endpoints, such as skin and eye irritation, but quickly became problematic for the more complex systemic endpoints. In the latter cases, the in vivo effects are not directly comparable to those observed in in vitro systems, the outputs of which are typically upstream mechanistic events, which might lead to one or more adverse effects traditionally reported in in vivo studies. At the beginning of the 2000s, the concept of the prediction model (PM) was introduced as a means of standardising not only the generation of in vitro data but also their interpretation for regulatory purposes. 3 The PM was originally proposed as a means of interpreting the data of both standalone tests and tests in combination (test batteries), but the concept was later re-expressed in terms of a Data Interpretation Procedure (DIP) associated with a defined approach (DA). 4
The concept of the PM had a direct impact on the way in which non-animal methods were evaluated, especially in the context of formal validation studies. There was a subtle but significant shift in research design from a straightforward comparison (measures of correlation) to an assessment of predictive performance. While the former could be used as a line of evidence for the relevance of a non-animal method, the latter morphed into a pass–fail criterion. An example is the use of in vitro cytotoxicity methods to assess acute oral systemic toxicity. Comparisons of cell death with acute lethality (oral LD50 value) started with the Multicentre Evaluation of In Vitro Cytotoxicity (MEIC) study, organised by the Scandinavian Society of Cell Toxicology. 5 The correlations were modest at best, but work continued to standardise some of the methods. Around 20 years later, when a standardised (and reproducible) method — the 3T3 Neutral Red Uptake cytotoxicity assay — was evaluated for its predictive ability, it was found to be useful in a weight-of-evidence approach to identify non-toxic chemicals (LD50 > 2000 mg/kg). However, it was considered likely to underpredict chemicals acting via specific mechanisms of action not captured by the 3T3 test system, or those that first require biotransformation in vivo. 6
Another important historical development was the introduction and elaboration of the Adverse Outcome Pathway (AOP) concept. 7 An AOP is a theoretical construct that acts as a bridge between the mechanistic relevance of one or more non-animal methods and their regulatory relevance. More specifically, mechanistic data generated by non-animal methods can be extrapolated to an Adverse Outcome that is the actionable information in (current) decision making. AOPs and networks of AOPs are increasingly being elucidated, in the hope that they will inform quantitative prediction models — sometimes referred to as quantitative AOPs. 8
With hindsight, while the PM concept provides a valid means of exploring the translatability of in vitro outputs, it has been misused in validation studies through its application to methods that were never going to ‘succeed’ as standalone approaches. The question, though, is: What does success look like? Experience has shown that when the relevance of a non-animal method is judged narrowly in terms of its predictivity, there are simply too many uncertainties in the extrapolation (prediction) process, and thus this approach is simply setting the scene for failure. Increasing the complexity of the modelling approach also has its limitations. The risk is that we continue to develop more and more sophisticated ways of translating in vitro data (e.g. in vitro to in vivo extrapolation using PBK models) without breaking out of the current assessment paradigm. This is arguably a key factor hindering the acceptance of non-animal methods, and consequently their uptake into regulation.
As toxicological databases evolved and became more amenable to data mining and statistical analysis, several studies started to analyse the inherent variability in the animal data, i.e. the reproducibility of the animal studies themselves. Examples include skin sensitisation, acute toxicity and repeat-dose toxicity.9–12 This is valuable information in its own right and can be used to challenge the confidence we have in animal studies. However, the insights have mostly been used to set expectations for the predictivity of non-animal methods — in other words, a non-animal method should not be expected to be more accurate in predicting the output of an animal study (the observed effect) than is one animal study when predicting another. The problem with this reasoning, however, is that it reinforces the use of animal data as the gold standard and ties us even closer to the current assessment paradigm.
The workaround is to step back from predictivity assessments and focus instead on risk management outcomes rather than the outputs of methods. In other words, we can judge new methods on the basis of their mechanistic relevance, reproducibility and importantly their ability to inform the right decisions, without trying to predict the outputs of dated and highly variable animal studies. This is the principle of ‘equivalent protection’. As non-animal methods can be standardised to the point of maximising reproducibility, the debate around these methods should therefore shift to their utility (protective capacity) in decision making.
In this Comment article, we reflect on how the principle of equivalent protection might work for two main contexts of use — classification and labelling, and risk assessment — which underpin the entire EU regulatory framework.
Implications for classification and labelling
In the context of Chemicals 2.0, we have previously presented classification and labelling as the basis for hazard-based risk (so-called ‘generic risk’) management, with a view to eliminating or minimising exposure to the chemicals of highest concern. 13 This is in line with the current EU approach, the only difference being that we proposed hazard classes based on upstream events rather than adverse outcomes, since this allows a wide range of adverse effects to be captured more efficiently and effectively, and simplifies the regulatory system.
It is proposed that a new classification scheme (see Figure 1),
13
based on bioactivity and systemic availability, would lead to the same levels of protection but without the need to predict adverse effects in animal studies. The rationale is that chemicals classified as high concern (in the red zone) would be banned or severely restricted — i.e. to use these substances, authorisation for specific uses would be required. Meanwhile, the medium concern chemicals (orange zone) could be used with restrictions, following chemical-specific risk assessments for their intended uses (for example, maximum allowable concentration limits in finished products). The low concern chemicals (green zone) could be used without restriction, enabling the innovation of safe and sustainable materials. It is important to note that these risk management consequences are specific to a given region (the EU) and may diverge across sectors due to socioeconomic and political considerations. However, the way in which the underlying levels of concern are expressed could be harmonised. A new classification scheme for chemicals based on three levels of concern (High, Medium and Low). ADME = absorption, distribution, metabolism and excretion; NAM = new approach methodology (in the narrow sense of non-animal method). (Adapted from Berggren and Worth
13
).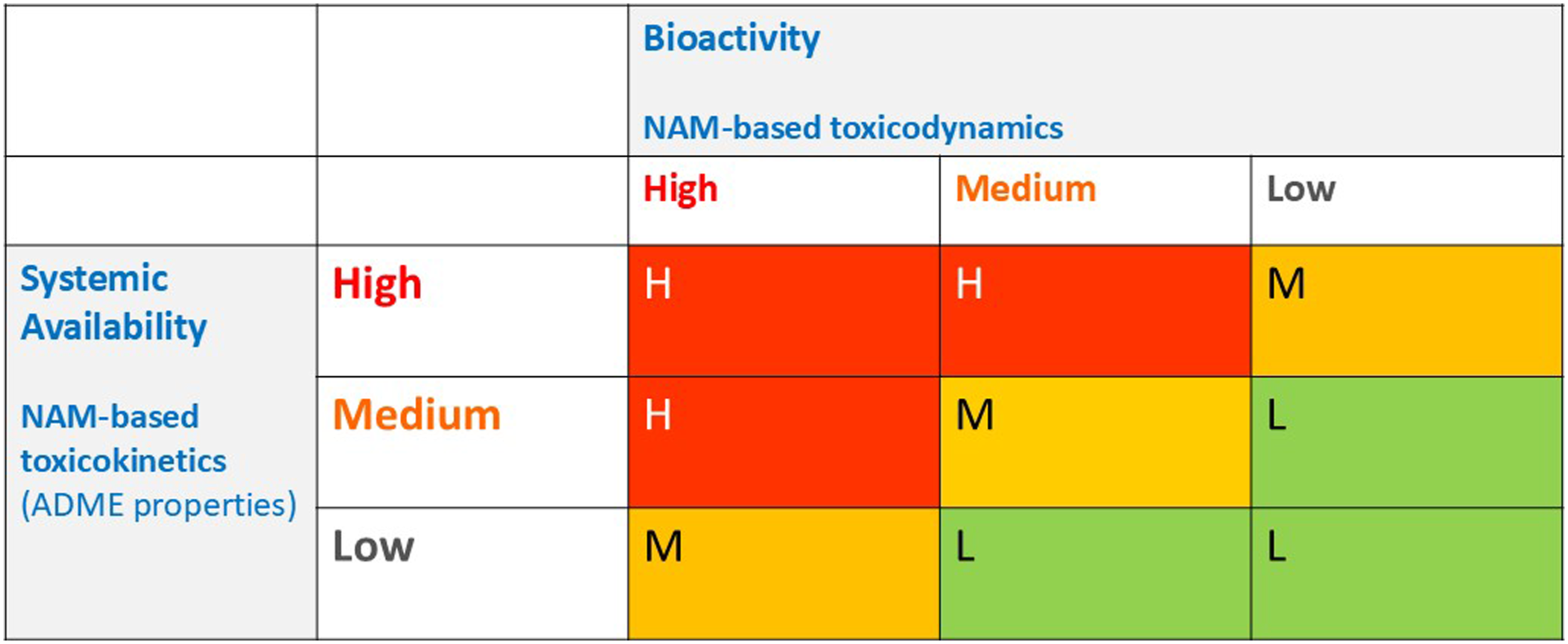
The feasibility of this approach is now being explored by the European Partnership for Alternative Approaches to Animal Testing (EPAA) through its Designathon for systemic toxicity. During the pilot phase of this challenge, 23 teams submitted prototype solutions to the challenge of defining potential New Approach Methodologies (NAMs) for bioactivity and systemic availability, for use in the classification of chemicals as low, medium or high concern for systemic toxicity. Further information on the prototype solutions and the list of 150 reference chemicals used can be found at the EPAA website. 14 Representatives from these teams met and discussed the outcome of the pilot phase during a workshop held in March 2024. Breakout sessions explored technical aspects of the proposed NAMs, as well as the similarities and differences in the suggested potential approaches to classification put forward by the participants. The next phase of the challenge will build on the work done so far and further explore the areas of chemical space, biological space, and classification strategies. It is hoped that the EPAA Designathon will be an informative activity in the frame of the EU Commission Roadmap 15 toward the phasing out of animal testing in chemical safety assessments. The roadmap is a response to the European Citizens Initiative, Save cruelty-free cosmetics — Commit to a Europe without animal testing. 16
Implications for risk assessment
In the context of Chemicals 2.0, we previously proposed that chemicals with intrinsic properties considered to be of medium concern (orange) should undergo a risk assessment with a view to determining safe exposure levels. 13 The challenge then for chemicals in this category is to find one or more analogues that have already been risk assessed and appropriately managed, and to use any restrictions in place for the analogues to inform the risk management of the new chemical. We emphasise the use of the word ‘inform’ here, reflecting the fact that read-across, as traditionally practised, is an element within a weight-of-evidence (WoE) approach, which includes consideration of information already available on the substance.
We are therefore proposing to use read-across to inform risk management, but with a twist — that is, to read across regulatory conclusions and restrictions rather than adverse outcomes. Exactly how this would work in practice is an open question. One possibility could be to read across specific concentration limits and associated conditions of use. Another would be to group substances into permissible concentration ranges — a kind of ‘exposure banding’ approach. Again, these risk management consequences are region-specific and possibly sector-specific, but this does not exclude the possibility of defining underlying levels of concern in a consistent manner.
A simple and illustrative example of the first possibility would be allyl nonanoate, a cosmetic ingredient used as a perfuming agent. This chemical, and structurally similar esters of allyl alcohol, are listed in Annex III of the Cosmetic Products Regulation with the following restriction: “Use only when the level of free allyl alcohol in the ester is less than 0.1%.” 17
To support read-across, an increasing array of user-friendly computational tools are becoming publicly available. 18 For example, the ToxEraser tool has been designed as a read-across tool to support substitution, making reference to existing safety measures. 19
If this way of applying the principle of equivalent protection is broadly supported, then the next step would be to develop practical guidance on how to carry out the read-across for different chemistries, properties of concern and exposure scenarios. Additional considerations might include the treatment of aggregate and combined exposures — and, of course, how the read-across fits within a broader WoE approach. Key questions to be addressed in any read-across approach relate to: a) the choice of relevant physicochemical, toxicokinetic, toxicodynamic and possibly use-related properties; b) the choice of similarity metric (quantitative measure of similarity); and c) the grouping algorithm — i.e. how the properties and similarity metric are deployed to form the groups (risk categories).
Although sophisticated algorithms exist for grouping chemicals, we believe that this last step should be as simple and interpretable as possible. The starting point is an exercise in reverse engineering, grouping chemicals with similar risk management measures, and identifying the properties leading to the final risk management decision. Ideally, it should be possible to relate discrete intervals of the selected properties with the relevant risk categories (for example, by means of a decision tree). Overall, the choices should be informed by calibrating the read-across approach based on chemicals that have already been assessed (to ensure that the same risk management outcomes are achieved), before applying it to chemicals that are not yet risk-managed. Finally, the approach should also be documented in a transparent manner, following existing guidance (e.g. the OECD Guidance on grouping of chemicals
20
).
Concluding remarks
While the principles of validation established in the 1990s remain valid today, there is a need to update their practical application to accelerate the uptake of non-animal methods. The need to expedite both the validation and acceptance of non-animal methods is recognised in the context of the EU Commission Roadmap toward the phasing out of animal testing in chemical safety assessments. 15 In this Comment article, we argue that the continued reliance on animal data as the gold standard is an impediment to progress in validation and consequently acceptance.
The solution we propose is conceptually simple — to adopt the principle of ‘protection’ rather than ‘prediction’, where protection is understood in terms of the risk management decisions that have already been taken for a large number of chemicals on the market. Of course, this relies on the assumption that the decisions taken to date are the ‘correct’ ones, even in the face of unreliable animal data. We believe this assumption is reasonable, as in many cases, the risk management measures were triggered by undesirable effects on health or the environment resulting from the use of the chemical, which were then considered sufficiently reduced by the risk management taken. Whether the risk management measures were sufficiently protective is difficult to establish, but assuming that this was the case, then it should be possible to develop a new paradigm that ensures protection even more efficiently, whilst also avoiding over protection (and consequent loss of useful chemicals).
In the context of Next Generation Risk Assessment (NGRA), an increasing number of case studies in the scientific literature (e.g. Wood et al. 21 ) advocate for the use of points of departure or the bioactivity–exposure ratio (BER) as a means of operationalising protection, based on the seminal work of Paul Friedman et al. 22 Typically, an in vitro point of departure is translated to an external safe exposure level by PBK modelling. The approach has been shown to be protective in the sense that the points of departure extrapolated from non-animal methods are, in the vast majority of cases, numerically less (i.e. are more conservative) than the corresponding points of departure based on animal studies. This is a valid scientific approach, but if we are pedantic about terminology, then it represents another approach to prediction, which is only indirectly related to protection. Importantly, it does not allow us to escape from the current assessment paradigm.
The aim of this Comment article is not to trigger discussion on whether there is a need to replace the so-called ‘gold standard’ in toxicology, which we take as self-evident, but rather on how to apply the principle of equivalent protection. Our focus has been on two broad key contexts of use that are most relevant to the EU regulatory framework — hazard classification and risk assessment. Just as the EPAA Designathon is exploring novel strategies for classification and hazard-based risk management, we propose that a feasibility study is warranted to extend this thinking to the problem of risk assessment. This could be another activity for the EPAA, which could be framed in the context of the Commission’s roadmap toward phasing out animal testing. The alternative way forward, i.e. continuing to focus on the prediction of outputs rather than outcomes, could be a strategic mistake. It could be a serious impediment not only to the replacement of animal studies in chemical safety assessments but also to the transition to a more efficient and effective regulatory system.13,23
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the European Commission Joint Research Centre.
Disclaimer
This article reflects the views of the authors and does not necessarily reflect those of the European Commission.