
Abstract

Respected Sir,
Methylmalonic acidemia (MMA) is an inborn error of metabolism due to a deficit in methylmalonyl-CoA mutase. Patients typically present within the first year of life with developmental delay, seizures, encephalopathy, and stroke. A sporadic late-onset form, cblC (Cobalamin C) disease, 1 presents with behavioral abnormalities and is often misdiagnosed. 2 Rett Syndrome (RTT) is a disorder found almost exclusively in females and characterized by a period of apparently normal neuropsychic development followed by progressive deterioration of acquired skills. “Typical” RTT presents with neuroregression, autism, stereotypies, and gait anomalies. However, multiple atypical variants with peculiarities in presentation and course have been recognized, making the diagnosis difficult. Guidelines use the same main criteria applied to typical RTT but with a lower threshold, complemented by supportive criteria. 3 The current view of RTT is that it is a neurodevelopmental disorder, not a degenerative one, despite many cases presenting in late childhood.
We report a case of MMA masquerading clinically as atypical RTT, with novelty in presentation and progression.
Case Presentation
A girl aged five years and five months presented with loss of acquired speech, difficulty walking, and repetitive behaviors. She was born at term and had attained milestones at age-appropriate intervals until age 5. At play school, she could recite poems and write alphabets, and had adequate social skills. Consequent to a urinary tract infection (UTI), she began to display loss of speech and motor milestones over seven months. Her mother initially noticed her inability to interact and maintain eye contact, followed by difficulty walking and loss of hand function. She lost bowel and bladder control and was unable to feed without assistance. Consequently, she was forced to discontinue play school. On presentation, her vocabulary was disyllabic, and she could stand only with support. She displayed repetitive self-injurious behavior, lacked reciprocal communication, and had minimal eye contact. On physical examination, head circumference was 45 cm (<0.1 percentile value for age), indicative of microcephaly. Both lower limbs showed spasticity and exaggerated deep tendon reflexes. There was no evidence of scoliosis or breathing difficulties. qEEG revealed multifocal spike and wave complexes interspersed with pseudo-periodic quiescence, suggestive of impending encephalopathy. MRI brain showed bilateral symmetrical T2 Flair signal changes in the external capsule and perisylvian region.
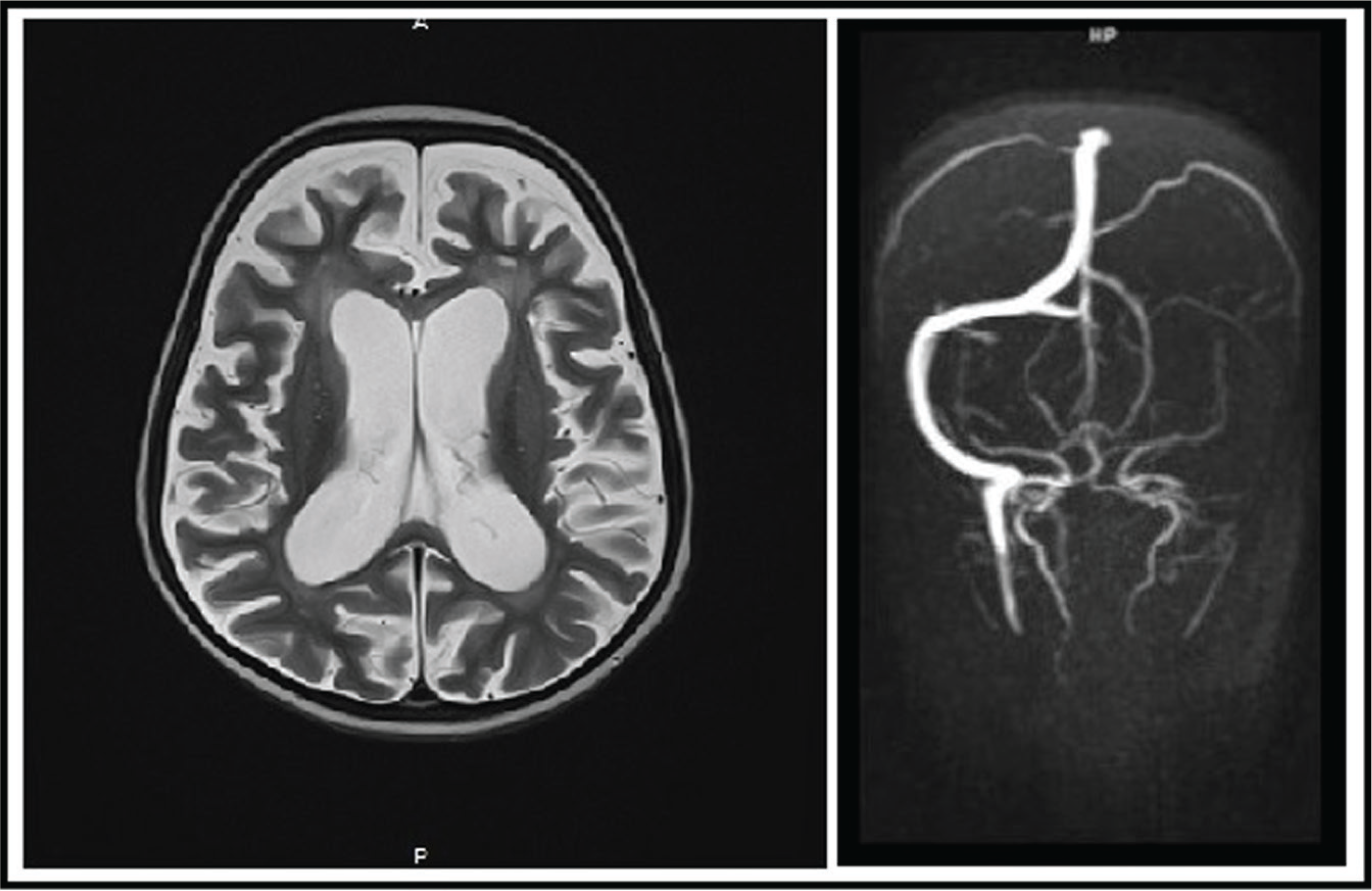
(a) MRI Brain showing global cerebral atrophy. (b) MRA Brain showing thrombosis of the left transverse and sigmoid sinus, and left IJV.
Applied behavioral analysis, IQ assessment, and evaluation of communication revealed profound impairment in socio-adaptive functioning and moderate autism.
Alternative and augmentative communication skills, skills training, and occupational therapy were imparted. The patient showed rapid improvement in her activities of daily living and communication. She regained the ability to feed independently and, following physiotherapy, could walk with aid.
She maintained well for two months. However, after repeated bouts of UTI, she displayed similar symptoms of developmental regression and had multiple focal seizures. Serology revealed leucopenia (TLC = 3200/cu.mm), methylmalonic acidemia (64.4mol/L), hyperhmocysteinemia (171µmol/L), and elevated lactate levels (42 mg/dL). Megaloblastic anemia was also found, with Hb = 5.2gm/dL and macrocytic erythrocytes with hypersegmented neutrophils on peripheral smear.
Repeat MRI brain revealed diffuse cerebral atrophy and bilateral diffusion restriction in the caudate and putamen, with Magnetic Resonance Angiography (MRA) Brain revealing thrombosis of the left transverse and sigmoid sinus, and left internal jugular vein.
She was managed symptomatically for MMA and epilepsy, after which she had reduced frequency of seizures, attained neck support, and could sit for a while.
Discussion
With a history of neuroregression and autism on presentation, our case was clinically suggestive of RTT. Additionally, the course broadly resembled that of RTT, where an initial rapid destructive phase was followed by a symptom plateau. 4 On closer inspection, however, the case did not conform to the existing subtypes of atypical RTT, thereby establishing our clinical dilemma.
Investigating further, neuroimaging showed cerebral atrophy and lesions in the globus pallidus, suggesting a possible underlying neurometabolic defect leading to neurodegeneration. 5 Serological tests revealing MMA and hyper-homocysteinemia confirmed these suspicions. Thus, aberrant neurometabolism was at the root of a case presenting with neuroregressive symptoms.
Both RTT and MMA are incompletely understood entities with a wide array of overlapping and confounding clinical symptoms. A high index of clinical suspicion ought to be exercised, as early diagnosis and multimodal management can halt the otherwise relentless progression of these multi-systemic diseases.
Our case, showing neurodegeneration underlying the Rett phenotype, opens the door for discovering additional variants with similar pathomechanism. In light of our findings, a relook at the current diagnostic criteria for atypical RTT would not be amiss.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient/guardians has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patient/guardians understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.