
Abstract

Genetic variants identified with next-generation sequencing (NGS) could help in accurate diagnosis, provide insights into aspects of disease biology, 1 and, thus, lead to improved care. Psychiatric symptoms and syndromes overlap with rare genetic disorders associated with intellectual disability (ID) 2 and autism spectrum disorders (ASDs) 3 at one end and with neurodegenerative disease 4,5 at the other end. These rare variants may be shared within family members. 6 Hence, the study of rare variants of moderate-to-large effects becomes important, as they may highlight the dysfunction of disease-associated pathways.
Rare variants associated with disease syndromes may arise de novo and may be related to founder effects, population bottlenecks, genetic drift, or natural selection pressures. Therefore, they also offer insights into the inherent mutability of genes, genetic repair mechanisms, and population diversity. 7
We reviewed the records of 11 individuals presenting to the psychiatric services in our hospital, who also underwent targeted sequencing through a genetic analysis service, after informed consent. In general, genetic analysis was requested because of clinical suspicion based on rapid disease progression, atypical disease course, younger age of onset, or significant family history. Specific indications for each case are mentioned in the respective sections.
Case Presentations
Both the gene and the variant identified were known genetic causes; identification helped zero down contribution to the patient’s phenotype. Gene identified was known to be related to the patient’s phenotype, but the variant identified was novel, or a new molecular interacting partner(s) of a causative gene was found Neither the gene nor the variant identified had previously been specifically implicated in the patient’s phenotype but may have biological and heuristic significance.
List of Variants Detected by Targeted Sequencing in the Patients Manifesting Psychiatric Syndromes
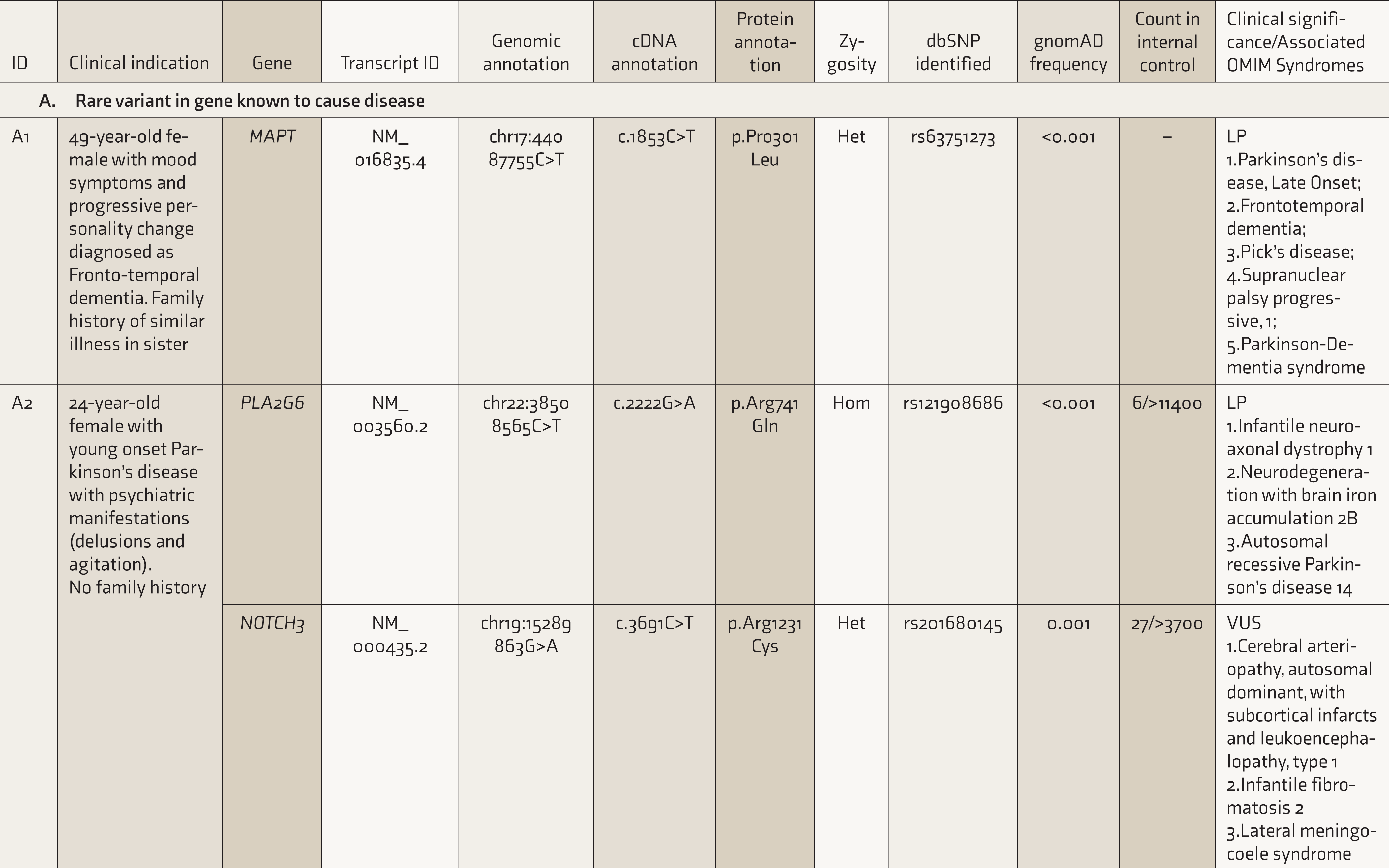


Het: Heterozygous; Hom: Homozygous; LP: Likely Pathogenic; VUS: Variant of Uncertain Significance; NA: Not available.
* This patient was tested for mutations in all 79 exons of the dystrophin gene using multiplex ligation-dependent probe amplification (MLPA). The patient shows deletion of the DMD gene involving exons 45-48. As per the Leiden muscular dystrophy pages the deletion is an in-frame deletion and likely to be a case of Becker muscular dystrophy.
Rare Variant in a Gene Known to Cause Disease: Case A1
Clinical Presentation
A 49-year-old female on treatment for hypothyroidism, working successfully as an executive, presented with behavioral changes in the form of jocularity and disinhibition, and then developed hyperorality, sleep disturbances, restlessness, and repetitive behaviors, over the past 3 years. The initial diagnosis had been a mood disorder, but the symptoms progressed over a few months, with a deterioration of speech and memory. Echolalia, bruxism, and mouthing behavior were observed on examination. A parent and an uncle had been diagnosed with late-onset dementia, and a sibling had similar symptoms. Magnetic resonance imaging showed frontal and anterior temporal lobe atrophy. Frontotemporal dementia (FTD) was diagnosed, and treatment was started with a combination of donepezil (20 mg/day) and trazodone (50 mg/day).
Indication for NGS
Presence of a young-onset, rapid progression of a dementia syndrome, with a significant family history.
Findings From NGS
A likely pathogenic, heterozygous missense variant; c.1853C>T in the gene MAPT (MIM *157140) was detected. The variant leads to the proline residue at position 301 to be substituted with leucine (p.Pro301Leu).
Biological Relevance of Findings
Mutations in the MAPT gene are a known cause of FTD (MIM #600274), and it is one of the three genes responsible for most cases of familial FTD, in addition to C9orf72 and GRN. The specific variant seen in this case has been reported in very few families throughout the world. There is only one other report from Asia documenting this variant, in a Chinese pedigree 8 ; but within FTD cases, this mutation is relatively common. 9
The tau protein molecule can be broadly subdivided into an amino-terminal projection domain, a microtubule-binding domain, and a carboxy-terminal domain. The p.Pro301Leu (P301L) mutation seen in this subject disturbs the conserved PGGG motif within the microtubule-binding domain encoded by exon 10 of the gene. The proline residue at position 301 is critical for the structure of the protein. In vitro functional assessment had shown that this mutation (P301L), in exon 10, has the highest propensity for aggregation than other MAPT mutations. 10 More recent evidence shows that this particular mutation gives rise to multiple diverse pathological conformations of tau protein. 11 Dementia could be a consequence of this process.
New Variant in a Gene Known to Be Associated with the Disease, and Its Plausible Biological Consequences: Case B1
Clinical Presentation
A 19-year-old male presented with insidious onset of symptoms from the age of 11 years, characterized by skin picking, obsessions of contamination, pathological doubt, checking and contrast behavior, need for symmetry, need to repeat, and magical thinking. There were also sudden anger outbursts and aggression toward family members, with significant assaultive behavior. The individual, the younger of two siblings, of nonconsanguineous parents had features of inattention, hyperactivity and impulsivity, oppositional behavior, and conduct symptoms, from early childhood. A family history of major depression and anankastic personality traits in a parent and schizophrenia-like illness in a grandparent were noted. On examination, a body mass index of 38 kg/m 2 and multiple cafe-au-lait spots (five measuring more than 2 cm in diameter), trichilemmoma in the chest, and axillary/inguinal freckling were seen. Blood investigations revealed high total cholesterol and LDL cholesterol but were otherwise unremarkable. The diagnoses of obsessive-compulsive disorder (OCD), attention deficit hyperactivity disorder (ADHD), oppositional defiant disorder, conduct disorder in the home context, neurofibromatosis type 1 (NF1), and metabolic syndrome were made. Treatment with lithium (1050 mg/day), escitalopram (40 mg/day), and chlorpromazine (50 mg/day) was initiated.
Indication for NGS
Presence of a diagnosable neurocutaneous syndrome (Neurofibromatosis; MIM #162200), a neurodevelopmental disorder (ADHD; MIM #143465), and early-onset OCD (MIM #164230) along with a family history of psychiatric illness.
Findings From NGS
Two possible pathological variants were seen. A novel variant was detected in the Neurofibromin 1 gene (NF1; MIM *613113), categorized as “likely pathogenic” (c.701_ 702delTG; p.Tyr235ProfsTer6). Further sequencing revealed the de novo origin of the mutation, as it was absent in both parents of the patient. The second variant in the TRIO gene (MIM *601893) was categorized as a “variant of unknown significance” (c.5267A>G, p.His1756Arg).
Biological Relevance of Findings
Mutations in the NF1 gene lead to increased susceptibility to cancers (leukemia and neurofibromatosis). NF1 is also associated with several neurobehavioral phenotypes, such as ID, ASD, and ADHD. 12 This gene has the highest rate of de novo mutations in the human genome, with ~3000 disease-causing mutations reported. In this case, the specific mutation is a novel, two-base deletion in the gene’s coding sequence, causing the substitution of tyrosine at position 235 to proline and a termination codon downstream (six codons from this mutation), producing a truncated NF1 protein.
Transgenic fly models of NF1 display excessive grooming, 13 a well-established phenotype of OCD in model systems. 14 The downstream effect of NF1 relies upon signaling proteins; GTP-bound forms of these proteins act as molecular switches, turning on downstream effectors. One such GTP-based “switching system” commonly used in cells is the Rho signaling pathway. This patient also has a variant detected in the TRIO gene that encodes a GDP-GTP exchange factor in this particular signaling pathway. The TRIO gene is also associated with neurodevelopmental phenotypes, such as ID and ASD. 15
The case highlights the complex phenotypes associated with NF1 gene mutations and raises the possibility of multigenic contributions to the final disease manifestation.
Variant(s) in the Gene(s) Not Directly Implicated in Disease But Suspected to Be of Biological Significance: Case C1
Clinical Presentation
A 23-year-old female presented with gradual behavior change, from the age of 15 years, characterized by decreased social interaction, occasional smiling to self, ambivalence, lack of interest toward any activities, feelings of depersonalization, and, eventually, poor self-care and severe neglect. There was no history suggestive of delusions, hallucinations, or pervasive mood changes, and no significant medical history. There was a family history of psychosis and completed suicide in both a sibling and an aunt. There was also a history of multiple first trimester abortions in the mother. Developmental milestones were attained at the appropriate age, but the academic performance was poor from middle school, and significant help was required from the parents to complete assignments. Premorbidly, the parents reported someone who was slow to warm up, aloof and distant, and with few friends. There was a history of treatment with several antidepressants and antipsychotics with minimal improvement. The physical examination was unremarkable, except for a stilted gait. A current diagnosis of simple schizophrenia on a back-ground of schizoid traits, with a possibility of pervasive developmental disorder, was made.
Indication for NGS
Atypical course and psychiatric symptomatology, significant autism-like features, and a history of psychosis in the sibling.
Findings from NGS
Two variants were detected in genes previously thought to be important for susceptibility to autism. A variant in the CIC gene (MIM *612082) was detected, classified as a “variant of unknown significance” (c.4551G>T; p. Lys1517Asn). A partial deletion at chr15q11.2 involving the genes TUBGCP5 (MIM *608147) and NIPA1 (MIM *608145) was also reported.
Biological Relevance of Findings
CIC protein functions as a transcription repressor, along with another protein, ATXN1 (MIM *601556). Behavioral changes in mice heterozygous for CIC gene knock-out suggest that it has important functions in the medial amygdala and hypothalamus in modulating social interactions. Truncating variations in CIC gene in clinical cases were found in patients with a spectrum of phenotypes, including developmental delay/ID, ADHD, ASD, and seizures. 16
A partial deletion of the genomic region on chr15q11.2 was also detected, with one copy of TUBGCP5 and NIPA1 genes deleted in the patient’s genome. Reports of genetic disturbances in the 15q11.2 BP1-BP2 region, which encompasses these genes, have been known to increase the susceptibility to neuropsychiatric or neurodevelopmental problems, including ASDs. 17 The loss of one copy of these genes or haploinsufficiency, 18 on chromosome 15, along with the variant in CIC, may be responsible for the clinical presentation in this case.
Discussion
The cases described highlight the diverse contexts where genetic testing may be ordered in clinical psychiatry. Targeted sequencing is intended to detect rare, possible pathogenic variants in genes already linked to disease. The classification of variants is based on American College of Medical Genetics and Genomics guidelines, 19 which define variants as pathogenic, benign, or of uncertain significance. These guidelines do caution that the “variant category does not imply 100% certainty” and that these may be reclassified as more data accumulates. 20 Given that our understanding of rare variant contributions to psychiatric disorders is evolving, it may be useful to report plausible links to disease syndromes.
We detected rare, possible damaging variants in several instances, which raise several interesting issues on gene defects and their downstream biology. In Case A1, the rare variant was in the MAPT gene, a well-established genetic link to FTD. The presence of NF1 gene mutation detected in Case B1 adds to the complex array of symptoms already reported with these mutations. It even suggests using statins (lower RAS activity) to target repetitive behaviors in individuals with neurofibromatosis and autism. 21
Case A3 exemplifies the coexistence of schizophrenia and Becker’s muscular dystrophy (BMD; MIM #300376), which has been occasionally observed. 22 In this person, the deletion is in the distal part of the DMD gene involving exons 45-48, which are part of the brain-expressed isoform. Consequently, the dystrophin protein expressed in different parts of the brain would be impacted, contributing to the neuropsychiatric syndrome. Interestingly, Dp 140 is expressed in the fetal stage and may also have a neurodevelopmental role. 23
In another person (Case B3), a mutation was detected in the OPA1 gene (MIM *605290), which has been associated with parkinsonism and dementia along with chronic progressive external ophthalmoplegia. 24 However, for this case, the monozygotic twin also harbored the same variant but was not symptomatic; hence the role of this variant needs to be interpreted with caution. A person who presented with features of deficit schizophrenia (Case C2) showed a variant in ABCD1 (MIM *300371), which has been associated with X-linked adrenoleukodystrophy (MIM #300100). 25 The mother had a similar deficit state, but the brother had a history of paranoid symptoms. Attenuated behavioral symptoms in females and more severe symptoms, including psychotic symptoms, in males are known to occur in X-linked adrenoleukodystrophy. Another individual (Case B4), who developed progressive supranuclear palsy (PSP) variant of PD on the background of long-standing schizophrenia, had a mutation in the TENM4 gene (MIM *610084). Missense mutations in this gene have been recently implicated as possible candidates for schizophrenia in a Han Chinese population in both familial and sporadic cases. 26 Missense mutations are also associated with essential tremor, 27 but the relation to PD/PSP variant is hitherto unknown.
It must be noted that in two persons (Cases D1 and D2), no variants were found that could contribute to the phenotype, despite a high index of suspicion based on family history and rapid disease progression, highlighting that widespread diagnostic testing for psychiatric conditions is not recommended.
Conclusion
There are several ways that genetic factors can shape the phenotype in those diagnosed with a psychiatric disorder. We suggest that NGS techniques can be a helpful addition when there are expediting factors to consider, such as an early onset, strong family history of mental illness, complex/atypical presentations, and minor physical anomalies or neurocutaneous markers. Most importantly, they provide novel insights into disease biology that can further elucidate the mechanisms underlying psychiatric syndromes.
Supplemental Material
Supplemental material for this article is available online.
Footnotes
Acknowledgements
We would like to acknowledge CSIR Institute of Genomics and Integrative Biology, New Delhi where clinical exome sequencing for one of the cases has been performed.
Ethical Statement
Written informed consent was obtained from the patient or their caregiver for publication of this case report at the time of sample collection for targeted sequencing.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Salary support for RS, RKN, AGS, and PP was provided by the Accelerator program for Discovery in Brain disorders using Stem cells.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.