
Abstract
This work describes the relevance of toxicology studies of environmental chemicals, with a focus on phthalates, for a hypothesis that certain human male reproductive disorders and diseases have a common etiology of disturbance of normal development in utero. The “Testicular Dysgenesis Syndrome” hypothesis in humans has parallels in male reproductive tract abnormalities and microscopic lesions reported for phthalate toxicity in rats. Additionally, this work describes the histological findings of abnormal testicular development (testicular dysgenesis) in rats as compared to those in humans, as well as potential findings in rats at different ages, from the embryo to the adult.
The Testicular Dysgenesis Syndrome Hypothesis in Humans
The Testicular Dysgenesis Syndrome (TDS) hypothesis states that a suite of male reproductive disorders seen in infant boys and adult men all have a common origin of impaired development of the fetal testis (Figure 1). 1 This hypothesis, as originally put forth, involved four male reproductive disorders, many of which have been shown to be increasing in incidence in Western countries: testicular cancer, poor fertility/sperm quality, cryptorchidism, and hypospadias. 1 A more recent addition to this hypothesis is decreased anogenital distance (AGD), for a total of five disorders. 2 These disorders can occur separately, or as a suite, and having one disorder may be a risk factor for the others. It may be most common to have just a single disorder, such as low sperm count. Less common and more severe would be the whole suite of disorders within an individual. Regardless of where an individual may be on the TDS spectrum, there may be histological lesions of dysgenesis in the testis, which will be described herein, concomitant with one or more of the other disorders. 1 Cryptorchidism in the infant is associated with poor semen quality and a 2- to 5-fold increased risk of testicular cancer in the adult. Early surgical treatment of cryptorchidism does not appear to significantly reduce the risk of later onset testicular cancer, suggesting a common origin in fetal life (see review by Rodprasert et al). 3
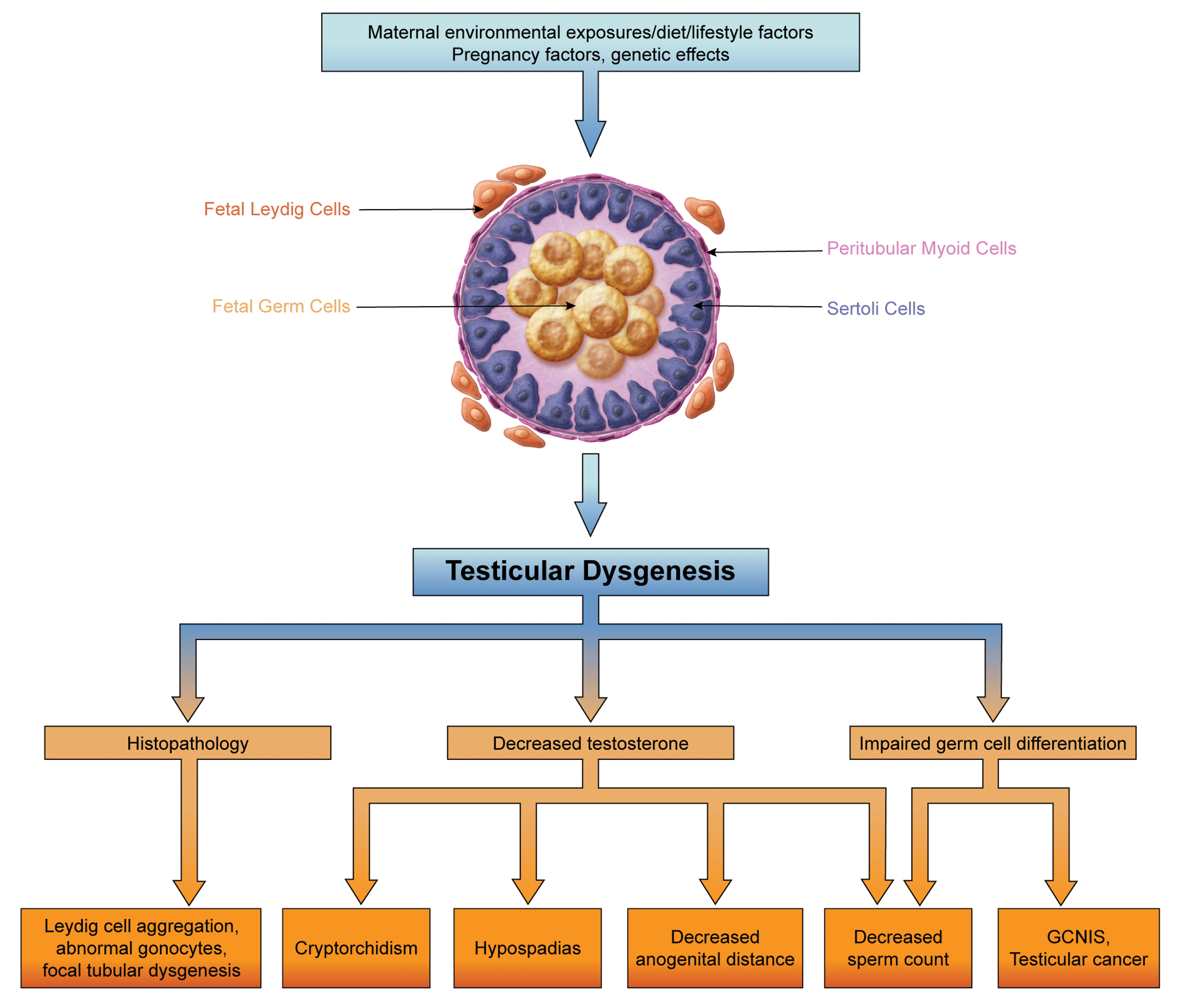
The Testicular Dysgenesis Syndrome (TDS) hypothesis states that events occurring during fetal development of the testis are related to increasing trends in certain human reproductive disorders/diseases. These include cryptorchidism and hypospadias in infants, decreased anogenital distance in both infants and adults, and decreased sperm counts and testicular cancer in adults. In addition to these clinical manifestations, there are microscopic lesions in the testis, including abnormal Leydig cell aggregation, persistent and/or multinucleated gonocytes, and focal seminiferous tubule dysgenesis. Based on Skakkebaek et al, 1 Skakkebaek et al, 2 and van den Driesche et al. 4
The most well-documented increase in a TDS disorder is testicular cancer, over 95% of which are testicular germ cell tumors (TGCT) in young adult males, usually between 20 and 34 years of age. In the United States, incidence rates have been increasing since at least the 1950s, with new cases increasing on average by 0.7% each year over 2009 to 2018. 5 The incidence rates of testicular cancer have increased in many Western countries, suggesting that lifestyle or environmental factors may be involved. The young age of onset of this cancer is further support that an exposure to an environmental factor during early or fetal life may be involved. Second, an increasing body of evidence indicates that human male fertility or sperm counts have declined over the past several decades, with no sign of stabilizing. 6- 8 Overall, these studies report similar declines of ∼1% per year in North America and Europe, but not in non-Western countries, with a ∼50% decline in sperm concentration over the past 70 years. 6- 8 Also, the proportion of men with low sperm count (less than 40 million sperm/mL of ejaculate) appears to be increasing, from ∼15% in the 1930s to ∼40% in the 1990s to 2000. 9 Increases in testicular cancer in young adult men and declines in sperm count over the relatively short time scale observed further suggest that lifestyle and/or environmental factors, rather than genetic factors, may be at play. Next, some studies indicate that the two most common developmental abnormalities of male genitals in human infants, cryptorchidism and hypospadias, may be also increasing in prevalence over time, again mainly in Western countries. 10,11
The most recent human male reproductive tract disorder included in the TDS hypothesis is decreased AGD. 2 Anogential distance is a sexually dimorphic measure of perineal length in many mammals. Shorter AGD has been reported in boys with hypospadias and cryptorchidism, linking normal genital development and AGD in humans. 12 In men, shorter AGD has been linked to lower total sperm count and poor semen quality. 13,14 Also, childless men who have been evaluated for infertility tended to have a significantly shorter AGD (31.8 vs 44.6 mm) than men who had fathered a child. 13 Decreased male AGD at birth is a permanent change, and decreased AGD has been shown in rats to be a barometer of androgen action during a critical developmental window. 15
The Masculinization Programming Window Is a Vulnerable Developmental Period
This critical time frame for male reproductive system development, the masculinization programming window (MPW), is considered to be embryonic day (E) 15.5 to E18.5 in rats and believed to likely be between 8 and 14 weeks of gestation in humans. 15 This is a sensitive window for normal development of all male reproductive organs. In rats, insufficient androgen action and/or production by the fetal Leydig cells during the MPW, due to exposure to the antiandrogenic agents dibutyl phthalate (DBP) or flutamide, for example, causes decreased AGD, cryptorchidism, hypospadias, decreased penile length, and decreased seminal vesicle weight. 15,16 In men, impaired germ cell differentiation in utero is proposed to lead to impaired spermatogenesis in adults and development of the precursor lesion to TGCT, germ cell neoplasia in situ (GCNIS), as well as TGCT. 2 In rats, maldevelopment of the seminiferous cords due to exposure to antiandrogens during the MPW leads to the histologic lesion of focal tubular dysgenesis, as described later. 17
Environmental Chemicals as a Potential Etiology of TDS
Genetic defects (eg, aneuploid states such as Klinefelter syndrome), pregnancy factors (eg, intrauterine growth restriction), and maternal lifestyle factors or environmental exposures, or a combination of these factors, have been implicated in TDS disorders (Figure 1). However, pregnancy factors and genetic abnormalities account for only a fraction of TDS cases. 1 Given the increasing trends in industrialized countries of TDS disorders, environmental exposures are a suggested cause of TDS. Initially, TDS in men was hypothesized to be due to in utero exposure to xenoestrogens. 18 This “estrogen hypothesis” was partially based on findings in men whose mothers took the synthetic estrogen diethylstilbestrol during pregnancy in the 1940s to 1970s. The increase in TDS cases is now instead hypothesized to be induced by environmental exposures to endocrine disrupting compounds that interfere with androgen production and signaling.
Apart from observations in rats, additional supporting evidence that TDS in human males may be caused by in utero exposure to environmental antiandrogens also stems in part from over 40 years of observational studies of male reproductive system maldevelopment in wildlife exposed to environmental chemicals (reviewed in Edwards et al 19 ). In wildlife, TDS may manifest as demasculinization (maldevelopment or underdevelopment) of the male reproductive system. A well-known example is demasculinization in alligators in Lake Apopka, Florida, USA, after a spill of dichlorodiphenyltrichloroethane, which is metabolized to the potent antiandrogen dichlorodiphenyldichloroethylene. 20 The alligators had reduced penile length (24%) and plasma testosterone concentrations (70%) as compared to similarly sized alligators at an uncontaminated lake. 20 In a domestic species, Lea et al 21 reported a decline in semen quality with a concurrent increase in cryptorchidism over 26 years in a population of stud dogs from a UK guide dog breeding program. These associations occurred even after removal of stud dogs with the poorest semen quality, no change in mean age, and with low coefficients of inbreeding in the five breeds. Within the same geographic area, the researchers detected polychlorinated biphenyls and diethylhexyl phthalate (DEHP) in adult dog testes and commercial dog foods at concentrations that have been shown to perturb reproductive function in other species, suggesting a potential link to environmental chemical exposure. 21
Phthalate Toxicity
“Phthalate Syndrome” in Rats
TDS-like findings have been well-characterized in experimental studies of rats exposed in utero to phthalates. Phthalates, a class of chemicals found in adhesives, flexible plastics, industrial solvents, personal care products, food, and food packaging and other sources are known to cause reproductive and developmental toxicity in rodents, especially in males. 22,23 Male rats exposed during the MPW to phthalates, such as DBP or DEHP, exhibit a spectrum of reproductive abnormalities comprising a “Phthalate Syndrome.” 23 These include absence or malformation of reproductive organs, hypospadias, cryptorchidism, reduced AGD, decreased spermatogenesis, retained nipples, and histological lesions of testicular degeneration/atrophy, multinucleated gonocytes, and focal seminiferous tubule dysgenesis. 4,16,17,23 -27 In utero phthalate exposure in rats is a model for human TDS, as it causes the TDS disorders of cryptorchidism, hypospadias, decreased sperm counts, and decreased AGD, but it does not induce testicular germ cell cancer. 16
In rats, prenatal phthalate exposure during the MPW adversely decreases androgen production in the fetal testis, disturbing development, and leading to cryptorchidism, hypospadias, and reduced AGD in the newborn, but also has lasting reprogramming effects in adulthood. Exposure of pregnant rat dams to 500 mg/kg/d DBP between E13.5 and E21.5 caused a 50% to 70% reduction in intratesticular testosterone in male fetal rats between E17.5 and E21.5. 28 This was associated at E21.5 with an approximate 40% reduction in the number of adult Leydig stem cells, which was maintained into adulthood (postnatal day [PND] 75), even though eventual adult Leydig cell numbers were not reduced either before (PND25) or after (PND75) puberty. Despite having similar numbers of adult Leydig cells as controls, adult male rats that had been exposed in utero to DBP had significantly lower blood testosterone levels and significantly elevated blood luteinizing hormone (LH) levels, consistent with “compensated adult Leydig cell failure.” 28 Similarly, infertile men with idiopathic oligospermia may have normal or low-normal serum testosterone levels with slightly elevated LH levels, indicating a similar compensation of dysfunctional Leydig cells. 29 The adult effect in rats after in utero phthalate exposure may occur through epigenetic-induced changes. The potential mechanism in male rats may be due to phthalate-induced reduced expression of steroidogenic acute regulatory protein via increased histone methylation of its proximal promoter. 28 The decrease in androgen levels during in utero exposure thus may not only impair male reproductive development in the fetus and newborn, but also reprogram Leydig cell function and testosterone production in the adult via epigenetic changes.
Phthalates in Humans
Epidemiological studies have shown pervasive human exposure to phthalates. 22 Epidemiological data of adult exposure to phthalates and adverse effects of reproductive health in men have been reviewed by Radke et al, 30 who reported robust evidence supporting a link between phthalate exposure (urinary metabolites) observed in adult men and decreased AGD, semen quality, testosterone, and fecundity (time to pregnancy), especially for DEHP and DBP. It is obviously challenging to link chemical exposures of mothers to decreased spermatogenesis or TGCT in their sons two to three decades later. However, there is some evidence for a link between exposures of mothers to environmental chemicals and TDS disorders in infant boys. Swan et al 31 reported a significant association of four phthalate metabolites in maternal urine during the first trimester of pregnancy with short AGD: body weight in sons, and that short AGD: body weight was associated with increased likelihood of cryptorchidism and smaller penis size, a sort of “phthalate syndrome” in boys.
Germ Cell Abnormalities in Rats and Humans
Rats rarely get testicular cancer as observed in men (seminoma or nonseminoma TGCT). However, similar fetal germ cell abnormalities occur in both rats in phthalate toxicity and humans with TDS disorders (Table 1). Gonocytes, the fetal germ cells that are the precursors of spermatogonia, are large, round cells with abundant cytoplasm and large nuclei at the center of seminiferous cords. They normally migrate between peripherally located Sertoli cells, contact the basement membrane, and then transform into spermatogonia. In rats, this transition from gonocytes to spermatogonia occurs between birth and PND7 and gonocytes normally disappear by ∼PND10. 32 In humans, gonocytes are abundant in the center of seminiferous cords by 10 weeks of gestation, migrate between the Sertoli cells and contact the basement membrane and transition to “intermediate cells” at 10 to 22 weeks of gestation, and then to fetal spermatogonia by 22 weeks of gestation. 33 Then postnatally, the fetal spermatogonia transition to type A (adult dark) spermatogonia between 3 and 9 months of age. 33
The Spectrum of Microscopic Features in the Testis in Testicular Dysgenesis Syndrome in Man and Phthalate Toxicity in the Rat.
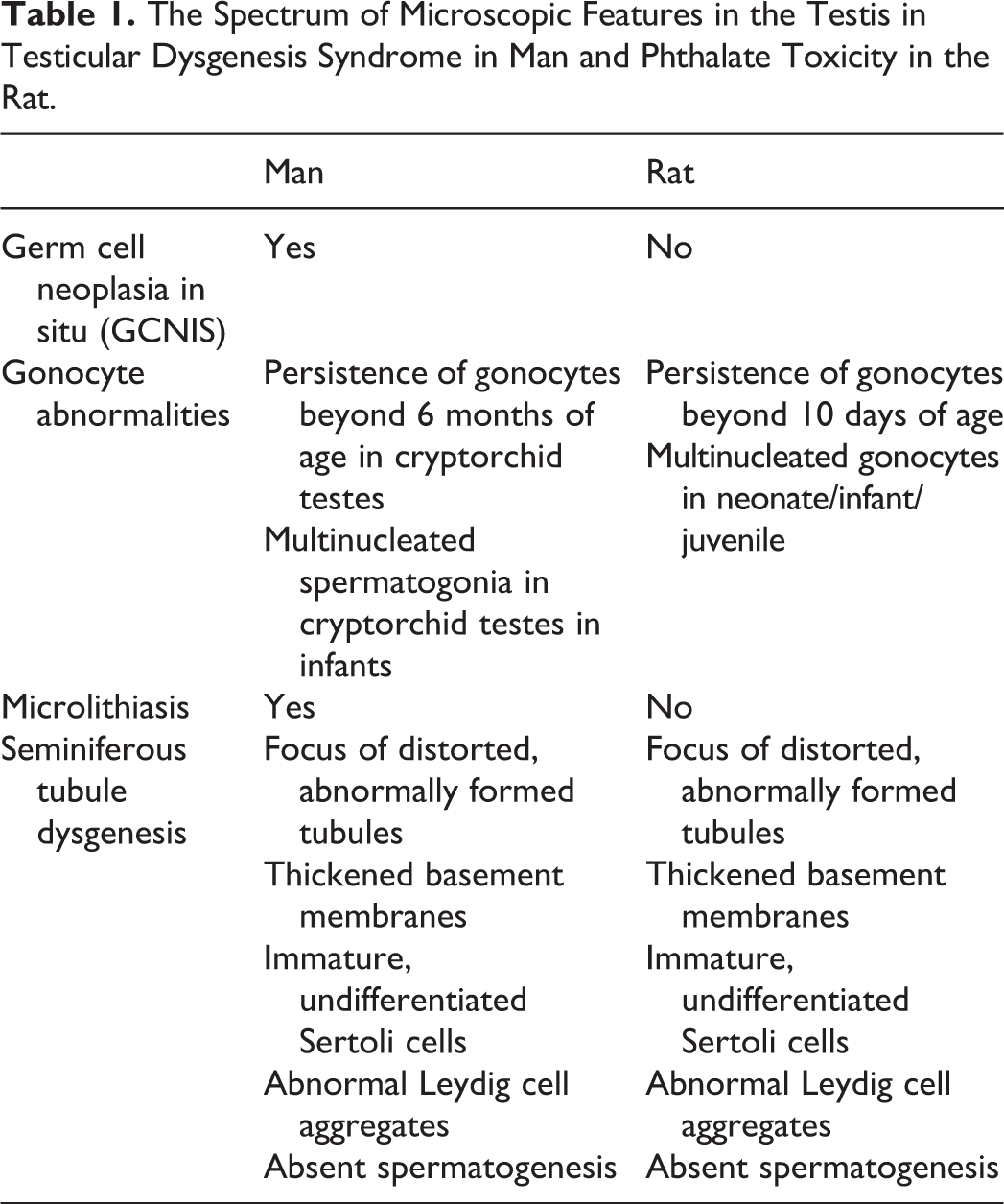
Gonocyte abnormalities are seen in humans as abnormal persistence beyond 6 months of age or as multinucleated fetal-type spermatogonia in infant boys with cryptorchid testes (Table 1). 33,34 Hutson et al 33 reported delayed or interrupted migration and transformation of gonocytes into type-A spermatogonia, with persistence of large numbers of gonocytes in the center of cords, well beyond 6 months of age, as well as decreased numbers of type-A spermatogonia in biopsies from cryptorchid testes from boys undergoing corrective surgery (orchidopexy). In another study of boys undergoing orchidopexy, biopsies revealed that 8% of the boys had multinucleated spermatogonia, with bizarre nuclei and a fetal appearance. 34 Notably, 2 of the 163 boys had the precursor lesion to TGCT, called GCNIS. The GCNIS lesions, first identified as carcinoma in situ in 1972, are large, undifferentiated fetal gonocyte-like cells, with abundant clear cytoplasm and large nuclei present along thickened, hyalinized basement membranes of one or more seminiferous tubules, with patchy involvement in the testis. 35 Germ cell neoplasia in situ is proposed to develop in the fetus or shortly after birth, reflecting abnormal germ cell differentiation and a fetal or early life origin of adult disease.
In utero phthalate exposure causes a disorder of germ cells that manifests as abnormal persistence of fetal germ cells (gonocytes) and the presence of multinucleated gonocytes in the juvenile rat (Table 1 and Figure 2). In a soon to be published US National Toxicology Program (NTP) report of a 2-year DEHP study in rats that included in utero exposure (beginning on gestation day 6), abnormal gonocyte persistence and multinucleated gonocytes were observed in rats that died (cause of death unknown) at 23 and 24 days old (Figure 2). 36 Since gonocytes normally complete the migration between Sertoli cells and transition to spermatogonia by PND10, this represents abnormal persistence. In addition to delayed or interrupted migration and abnormal persistence of gonocytes, the testes from these juvenile rats exhibited multinucleated gonocytes in rare tubules (Figure 2). Multinucleated gonocytes (typically 2-4 nuclei, but up to 13) have been previously reported after in utero phthalate exposure in other studies, as early as gestation day 17. 37
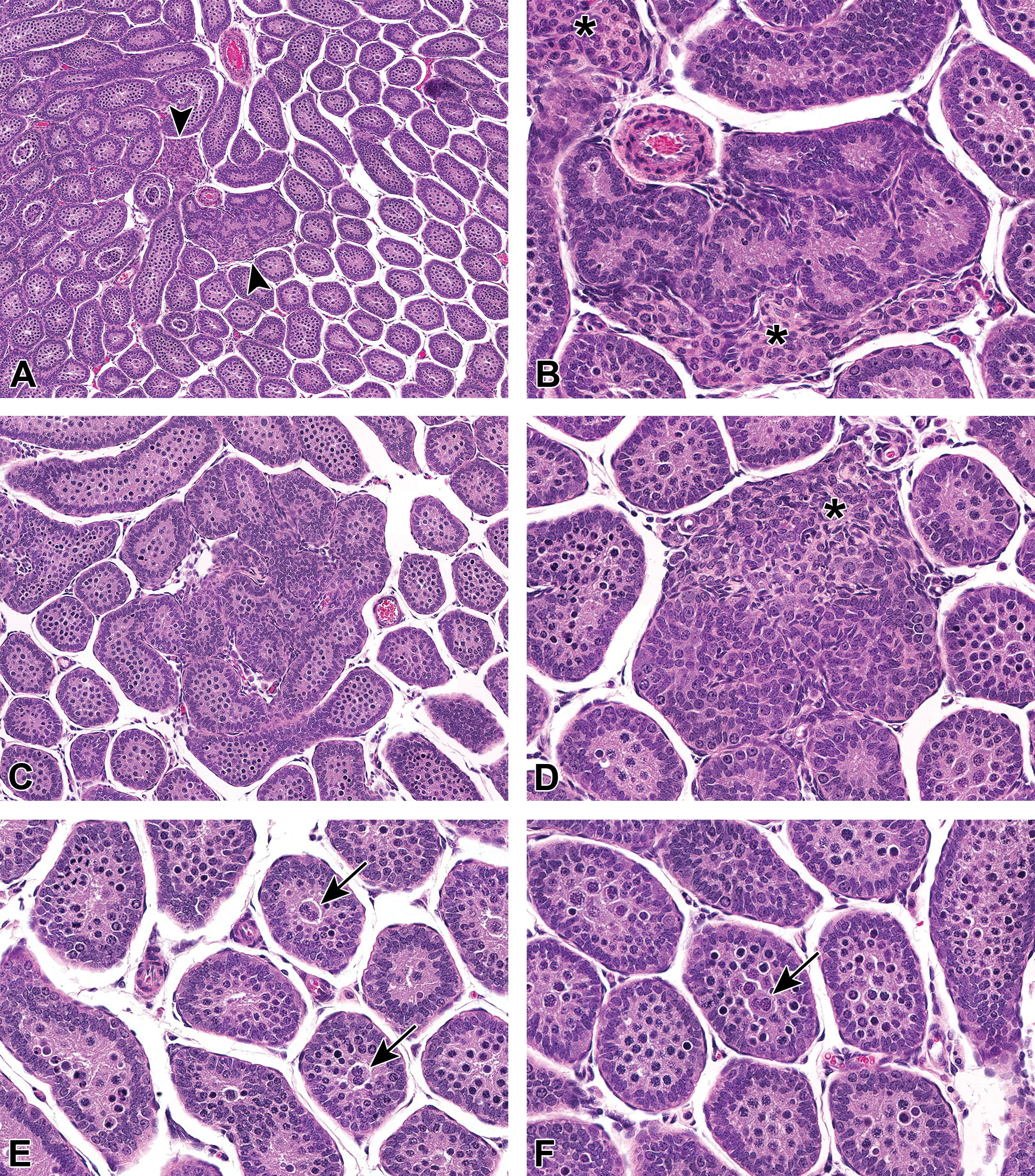
Examples of seminiferous tubule dysgenesis in juvenile rats (PND23 or 24) from a US National Toxicology Program 2-year study of diethylhexyl phthalate with perinatal exposure (beginning on gestation day 6). A, Focal seminiferous tubule dysgenesis near the center of a section of the testis, associated with abnormal Leydig cell aggregation (arrowheads). B, At higher magnification, the tubular dysgenesis lesion is comprised almost solely of Sertoli cells. Abnormal Leydig cell aggregation (*) is adjacent to the malformed tubules. C, Seminiferous tubule dysgenesis that does not feature prominent, abnormal Leydig cell aggregation and contains some germ cells. D, It is difficult to ascertain the tubular borders in this focus of dysgenesis, and it is possible that the adjacent abnormal Leydig cell aggregate (*) contains ectopic cells. E and F, Multinucleated germ cells (arrows) in the center of tubule lumens, concomitant in testes that also contain tubular dysgenesis lesions. The persistence of gonocytes in the tubular lumen beyond PND10 is also abnormal. H&E. H&E indicates hematoxylin and eosin; PND, postnatal day.
Seminiferous Tubule Dysgenesis in Rats and Humans
In addition to the presence of gonocyte abnormalities, the microscopic lesion of focal seminiferous tubule dysgenesis also has parallels in humans with TDS and in rats exposed in utero to phthalates (Table 1). In utero phthalate exposure during the MPW in rats causes the histologic lesion of focal seminiferous tubule dysgenesis, seen as one or more nodular foci (up to the entire section) of malformed, distorted seminiferous tubules with thickened and hyalinized basement membranes, and typically (but not always) accompanied by abnormal Leydig cell clustering (Figures 2 and 3). 16,37- 39 Dysgenesis lesions are inactive in spermatogenesis and contain immature-appearing Sertoli cells with disorderly distribution within the tubule and small, elongated, and occasionally cleaved nuclei lacking the prominent nuclei and tripartite nucleoli seen in the more orderly arranged, mature Sertoli cells (Figure 3). Areas of focal tubular dysgenesis are often a minor component of the testis, so it is possible to miss them if sampling is inadequate. Sertoli-cell only tubules containing immature-appearing Sertoli cells, often lacking a lumen, are also common in DBP-exposed testes and may represent an additional manifestation of dysgenesis in rats, as well as in men. 16,40 In the soon to be published US NTP studies of DBP and DEHP that included exposure beginning on gestation day 6, 18% or 20% of male rats exposed to 10,000 ppm of DBP or DEHP, respectively, had seminiferous tubule dysgenesis. 36,41 The dysgenesis lesions were typically seen in one of three sections of the testis, usually the section also containing the rete testis. 24,41 Testes containing seminiferous tubule dysgenesis lesions from the 2-year-old rats exposed in utero and throughout life to DBP also commonly had germinal epithelium atrophy, interstitial edema, and an unusual fibrosis and/or sperm granuloma in the rete testis region, which has been previously reported. 24,42
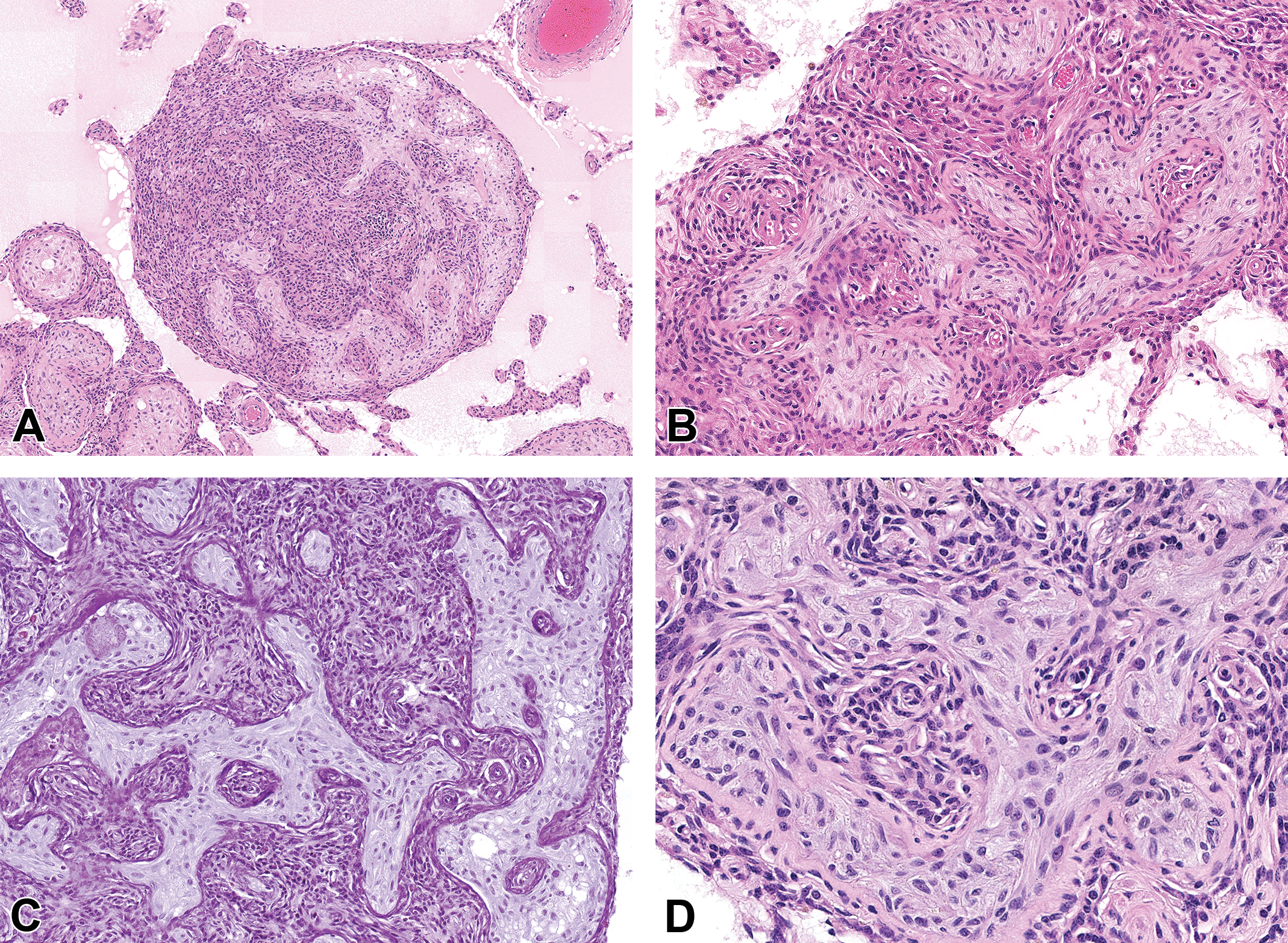
Examples of seminiferous tubule dysgenesis in 2-year-old rats from US National Toxicology Program 2-year studies of diethylhexyl phthalate (A and B) or dibutyl phthalate (C and D) with perinatal exposure (beginning on gestation day 6). A, A nodular lesion of malformed seminiferous tubules closely associated with a large aggregation of spindloid Leydig cells. The surrounding interstitium is edematous, with nearby Sertoli-cell-only tubules with thickened basement membranes. H&E. B, Seminiferous tubule dysgenesis with malformed tubules that are surrounded by aggregated, spindloid Leydig cells. H&E. C, When tubular dysgenesis lesions are stained with Periodic Acid-Schiff/hematoxylin (PAS/H), the irregular, thickened tubular borders are especially prominent, with invaginations that appear as “islands” of Leydig cells inside tubules. PAS/H. D, At high magnification, the Sertoli cells within the malformed tubules appear immature, with disorderly distribution within the tubule and small, elongated, and occasionally cleaved nuclei. They lack the prominent tripartite nucleoli and large nuclei of mature Sertoli cells. The tubular borders are irregularly thickened and hyalinized. H&E. H&E indicates hematoxylin and eosin.
Interestingly, similar focal histological testicular dysgenesis is also reported in testicular biopsies from patients with TDS disorders, including infertile men, men with cryptorchidism, and in the contralateral or ipsilateral testis in men with TGCT (in either scrotal or cryptorchid testes; Table 1). 43,44 The Leydig cell aggregation associated with the abnormal, undifferentiated tubules may represent what is referred to as Leydig cell micronodules in humans. 40 Additional features in testes from men that, to the author’s knowledge, have not been reported in rats are the presence of lamellar microliths in either the abnormally formed or Sertoli-cell-only tubules or the presence of the precursor lesion to testicular cancer, GCNIS lesions (Table 1). 40
Potential Pathogenesis and Timeline of the Spectrum of Lesions Seen With Dysgenesis in Rats
The Leydig cell clumps in focal dysgenesis lesions appear to represent aggregation rather than proliferation. The malformed tubules in dysgenesis lesions are often surrounded by aggregates of Leydig cells that are small, spindle-shaped, and contain scant cytoplasm, resembling fetal Leydig cells rather than adult Leydig cells or the cells with abundant eosinophilic cytoplasm typical of focal Leydig cell hyperplasia or adenomas (Figure 3). 24,37 Based on stereology, the actual number of Leydig cells is not increased in testes with dysgenesis lesions, further evidence that the associated Leydig cells represent an abnormal distribution rather than hyperplasia. 38 Indeed, Lara et al 17 demonstrated that the fetal Leydig cells abnormally aggregate toward the center of the testis at E17.5, two days after initiation of DBP exposure at the start of the MPW on E15.5, then progress in severity to E21.5, and the aggregations persist postnatally and throughout life. 38 This Leydig cell aggregation is considered the first sign of dysgenesis in the rat testis (Figure 4).
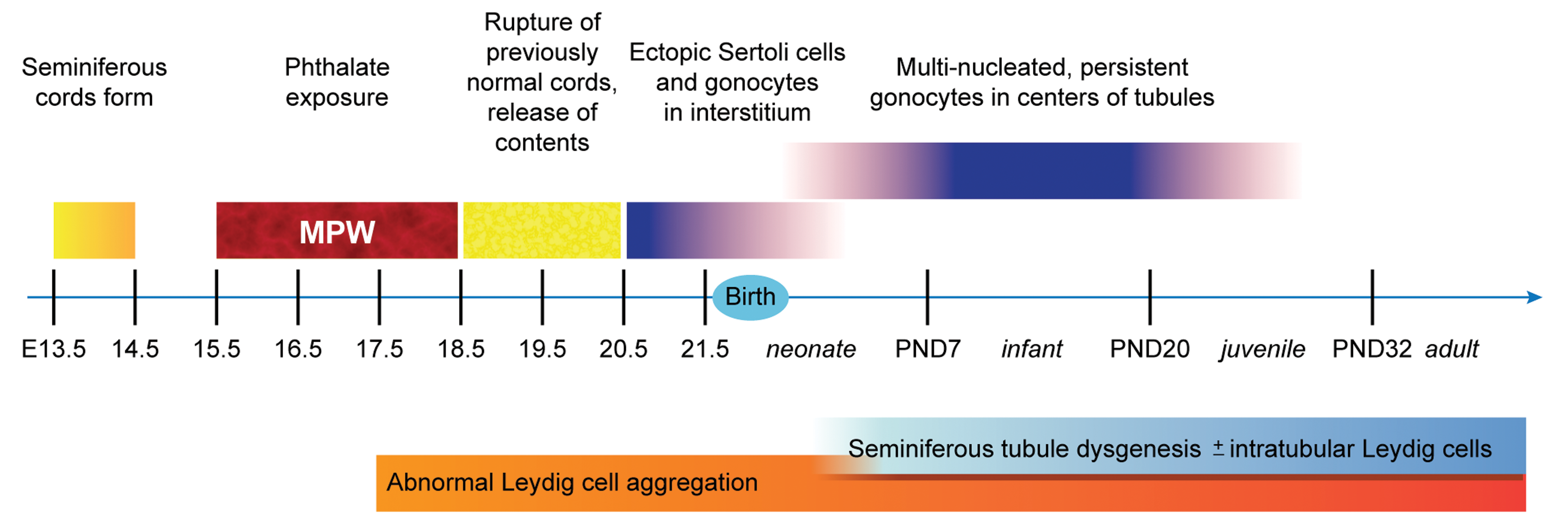
Recent work by Lara et al 17 sheds light on how histologic signs of dysgenesis may arise. During development in the rat, the seminiferous cords form between E13.5 and E14.5 (Figure 4). The peritubular myoid cells normally surround each seminiferous cord, separating it from the interstitium. Together with the Sertoli cells, the peritubular myoid cells contribute to the production of the basal lamina. Even when exposure to DBP commenced via the dam on E13.5, seminiferous cords were still seen as normally formed two days later at E15.5. 17 Then at E18.5, whether DBP exposure began at E15.5 or E17.5, Lara et al 17 demonstrated, with the use of triple staining for Sertoli cells, gonocytes, and peritubular myoid cells, that the seminiferous cords in exposed rats appeared to rupture, releasing their contents (ie, Sertoli cells and gonocytes), along with scattered peritubular myoid cells, into the interstitium. Subsequently, another feature of testicular dysgenesis in rats exposed during the MPW to phthalates is the presence of ectopic cells: Sertoli cells and gonocytes in the interstitium and Leydig cells within tubules (Figure 4). 16,17,38 The ectopic Sertoli cells and gonocytes appear in late gestation and disappear sometime early in postnatal life, but the ectopic intratubular Leydig cells may persist throughout postnatal life (Figure 4). 16,17,4,39 The actual histologic lesion of seminiferous tubule dysgenesis, malformed/misshapen tubules, is not evident in embryonic testes and only becomes evident in the neonatal period, during the first seven days of life (Figure 4). 16
To summarize, based on current knowledge, it appears that phthalate exposure during the MPW leads to the first sign of dysgenesis, abnormal clustering of Leydig cells, beginning at E17.5 (Figure 4). This abnormal aggregation progresses in severity for the rest of gestation and then persists throughout postnatal life. The second sign of dysgenesis is rupture of previously normal seminiferous cords beginning at E18.5, resulting in ectopic cells in the interstitium (Sertoli cells and gonocytes, which disappear by early postnatal life) and in the seminiferous tubules (Leydig cells, which may persist throughout postnatal life; Figure 4). Sometime between late gestation and up to at least the juvenile stage, persistent, multinucleated gonocytes that fail to migrate normally to become spermatogonia may be seen, and these then disappear at some point (Figure 4). Then in the neonatal rat, the abnormal tubules of seminiferous tubule dysgenesis become evident and then persist throughout postnatal life (Figure 4).
Conclusions
The animal model of phthalate exposure in rats provides a window into the potential genesis of the Testicular Dysgenesis Syndrome hypothesis in humans. Both phthalate toxicity in rats and TDS in humans feature abnormalities of external genitalia (cryptorchidism, hypospadias, decreased AGD), decreased spermatogenesis, and microscopic findings of abnormal gonocyte persistence and seminiferous tubule dysgenesis. Despite some differences (ie, the absence of testicular germ cell cancer and its precursor lesion in rats), phthalate toxicity in rats is a useful tool for the potential understanding of a suite of male reproductive disorders in humans.
Footnotes
Acknowledgments
The author thanks Beth Mahler (EPL, Inc.) and David Sabio (EPL, Inc.) for figure preparation and Drs Susan Elmore (NIEHS) and Mark Cesta (NIEHS) for their critical manuscript reviews.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the NIH, National Institute of Environmental Health Sciences.