
Abstract
Peripheral neuropathy associated with chronic occupational and deliberate overexposure to neurotoxic organic solvents results from axonal degeneration in the central and peripheral nervous system. Human and experimental studies show that axonopathy is triggered by the action of neuroprotein-reactive γ-diketone metabolites formed from exposure to certain aliphatic solvents (n-hexane, 2-hexanone) and aromatic compounds (1,2-diethylbenzene, 1,2-4-triethylbenzene, 6-acetyl-1,1,4,4-tetramethyl-7-ethyl-1,2,3,4-tetralin). Neuroprotein susceptibility is related primarily to their differential content of lysine, the ∊-amino group of which is targeted by γ-diketones. Specific neuroprotein targets have been identified, and the sequence of molecular mechanisms leading to axonal pathology has been illuminated. While occupational n-hexane neuropathy continues to be reported, lessons learned from its experimental study may have relevance to other causes of peripheral neuropathy, including those associated with aging and diabetes mellitus.
Introduction
Repeated workplace overexposure to n-hexane, an inexpensive organic solvent that has been widely used in industry, is an established cause of a self-limiting distal symmetrical peripheral neuropathy underpinned neuropathologically by central–peripheral distal axonal degeneration. 1,2 Other neurological phenomena associated with workplace exposure to n-hexane and solvent mixtures containing n-hexane include reduced balance control, 3 impaired temperature sensation (cold allodynia), 4 changes in color vision, 5,6 maculopathy and optic neuropathy, 7,8 and abnormal or reduced sense of smell. 9 High-dose n-hexane overexposure, which alone or in a solvent mixture that has been used to induce euphoria, can precipitate a subacute onset of neuropathy resulting in severe muscle weakness and wasting. 10 -15
Peripheral neuropathy arising from repeated exposure to n-hexane was demonstrated 50 years ago in printing plants, sandal and shoe-making shops, and furniture factories in Asia, Europe, and the United States. 10,16 -22 The problem has continued into the 21st century, with reports of n-hexane neuropathy among American automotive technicians, 23 shoe factory workers in Turkey, 24 screen printers in India, 25,26 electronic industry workers in Japan and South Korea, 27,28 and in a Chinese medicine plant in Taiwan. 29 -32 Additionally, between 1996 and 2004, there were 16 published reports of occupational n-hexane neuropathy in mainland China (Fujian, Guangdong, Heilongjiang, Henan, Jiangsu, and Liaoning) and, through 2009, 137 employees in a Suzhou electronic factory received hospital treatment for n-hexane neuropathy. 33 Sixty patients were treated for n-hexane intoxication in Guangdong Province between January 2017 and January 2018. 34 The main causes of chronic n-hexane poisoning (females > males) in an electronic enterprise of Guangdong’s Shenzhen City in 2017 were: long working hours, poor ventilation of workshops, unsatisfactory personal protective equipment, inadequate occupational health management, and an imperfect occupational health examination mechanism. 35
n-Hexane is metabolized by cytochrome P4502E1 (CYP2E1) to compounds with progressively greater neurotoxic potency, including 2-hexanol, 2-hexanone, and 2,5-hexanedione, 36 -38 a 1,4-diketone (γ-diketone) that binds directly with the ∊-amino group of lysine to form a 2,5-dimethyl pyrrole adduct (Figure 1). 40 -42 Pyrrole formation is an absolute requirement for neurotoxicity resulting in peripheral neuropathy. 43 -45 Similarly, 1,2-diethylbenzene, a solvent used in ion exchange resins and in powderless etching, appears to be metabolized to 1,2-diacetylbenzene (1,2-DAB), 46,47 which reacts with lysine to form a benzo-fused pyrrole or isoindole. The isoindole and pyrrole adducts form oxidized dimers and polymers that are seen as blue-violet and orange-colored pigments, respectively (Figure 1). Thus, rodents treated systemically with 1,2-DAB develop a bluish discoloration of skin, eyes, and internal organs, including nerves, spinal cord and brain, which reflects the reaction between the γ-diketone and proteins throughout the body. Calculations based on the density functional theory indicate the chromophore formed from the reaction of 1,2-DAB with proteins is likely to be composed of dimers of oxidized isoindoles. 48 A yellow discoloration results from systemic treatment of rodents with the aliphatic γ-diketone, 2,5-hexanedione (2,5-HD), which produces the lowest number of adducts in (rat) serum and the highest in the liver, where it has potential toxicity. 42
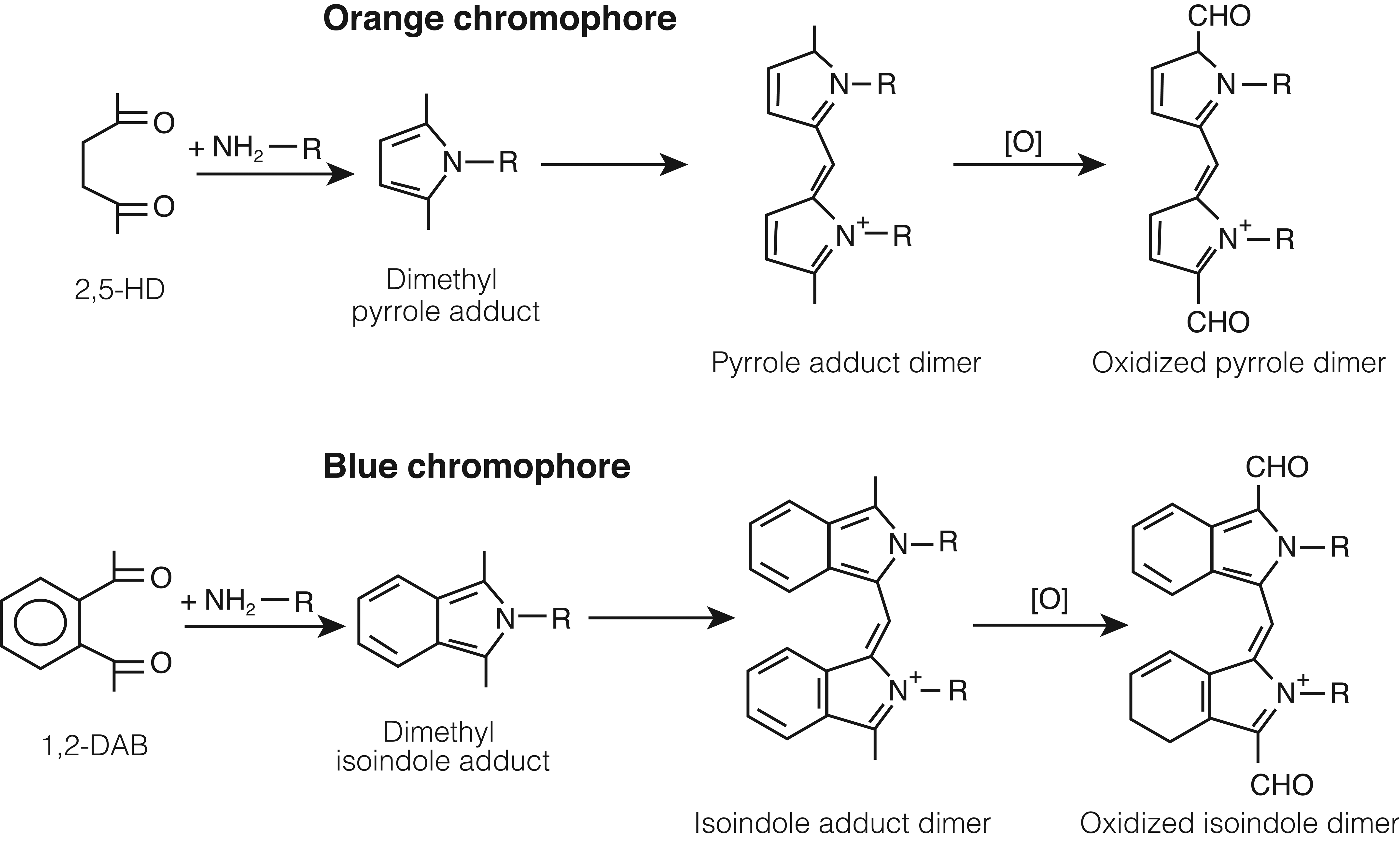
γ-Diketone reactivity with lysine. Reaction of 2,5-hexanedione (2,5-HD, upper) and 1,2-diacetylbenzene (1,2-DAB, lower) with lysine to form dimethyl-pyrrole and -isoindole adducts, respectively, which form dimers that oxidize and polymerize to cross-link targeted proteins. The colored pigments that result appear to be associated with the dimer formation. Antioxidants block 1,2-DAB-induced cytotoxicity of SH-SY5Y cells. 39
Whereas 2,5-HD and 1,2-DAB are protein-reactive, chromogenic, and induce central–peripheral axonopathy, closely related isomers of the aromatic γ-diketone (eg, 1,3-DAB) and the aliphatic γ-diketone (eg, 2,4-HD) do not react with proteins to form chromogens and do not affect nerve fibers in the central nervous system (CNS) and peripheral nervous system (PNS). 49 -53 It can therefore be inferred that protein (lysine) reactivity is directly related to γ-diketone neurotoxic potential, and chromogenicity is a biomarker of neurotoxic action. Unknown is whether the same molecular mechanism underlies the ability of 2,5-HD to inhibit proliferation of neuroprogenitor cells in primary neuronal cultures, disrupt murine hippocampal neurogenesis, 54 and impair neurodevelopment of chick embryos. 55
The neurotoxic potency of γ-diketones increases stepwise from 2,5-HD, 3-methyl-2,5-HD, 3,4-dimethyl-2,5-HD (DMHD) and 1,2-DAB (Figure 1), presumably because the molecular configuration stepwise increases access of the reactive 1,4-diketo group to the target ∊-amino groups of protein lysines. 50,56 Similarly, 1,2-diethylbenzene (but not 1,3-DEB) and 1,2,4-triethylbenzene (but not 1,3,5-TEB), the parent compounds of 1,2-DAB and 1,2,4-DAB, respectively, induce peripheral neuropathy in rodents. 53 Other aromatic compounds, such as the polycyclic musk 6-acetyl-1,1,4,4-tetramethyl-7-ethyl-1,2,3,4-tetralin (AETT), formerly used in fragrances and as a food additive, yield a metabolite that is protein-reactive, forms a bluish chromogenic pigment, and induces nerve fiber damage in systemically treated rodents. 57,58 By contrast, neither tetralin nor its α-tetralol metabolite form a chromogen in vitro or induce axonal degeneration in mice treated by repeated intraperitoneal injection with either compound. 59 With the exception of tetralin, other aromatic hydrocarbons reported in early studies to cause blue-green-colored urine have not been studied for chronic neurotoxic potential (Table 1).
Chromogenic Organic Solvents as Reported by Gerarde 60 From His Experimental Animal Studies Using a Single Subcutaneous Injection of the Listed Substances of Unknown Purity in a Dose of Approximately 5 mL/kg.a
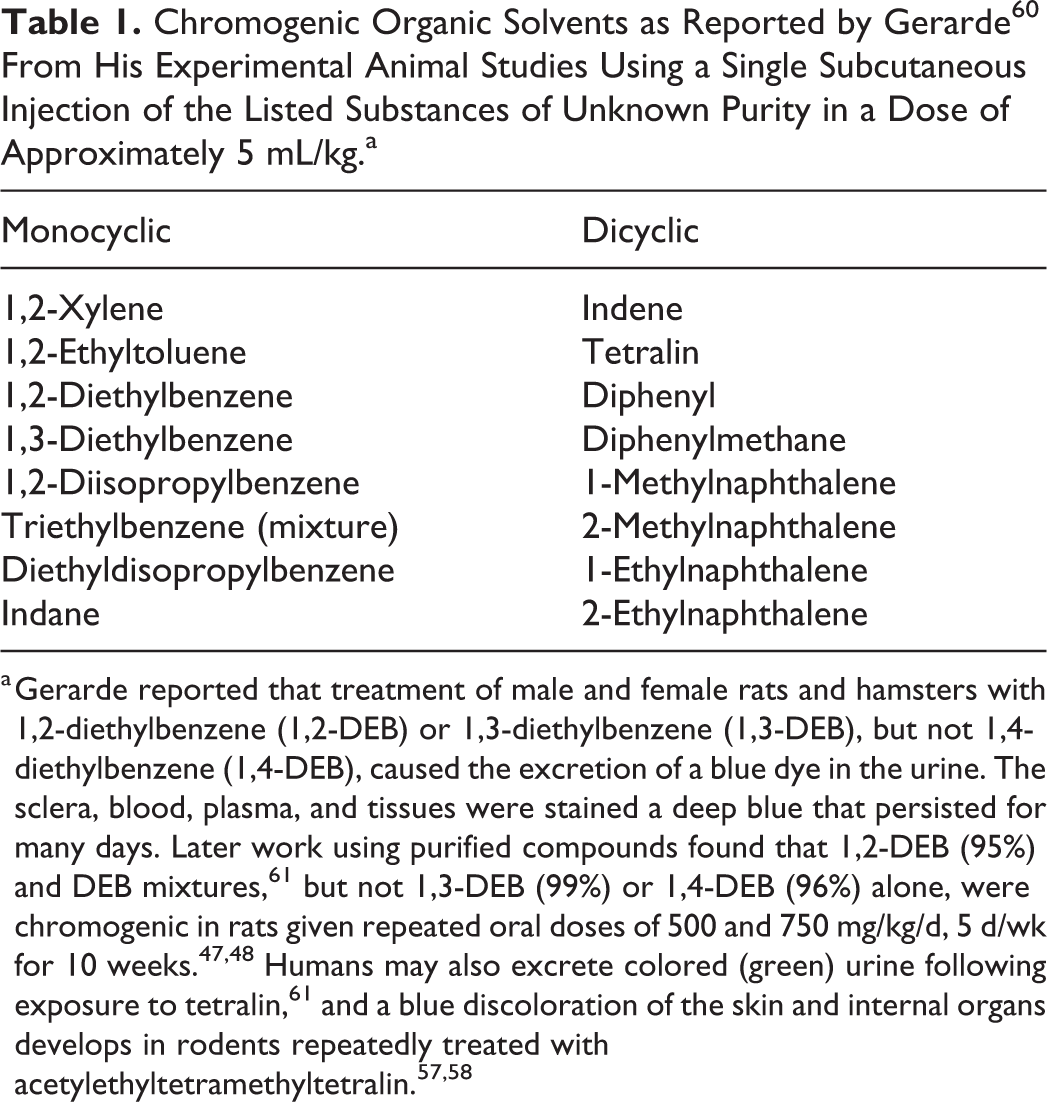
a Gerarde reported that treatment of male and female rats and hamsters with 1,2-diethylbenzene (1,2-DEB) or 1,3-diethylbenzene (1,3-DEB), but not 1,4-diethylbenzene (1,4-DEB), caused the excretion of a blue dye in the urine. The sclera, blood, plasma, and tissues were stained a deep blue that persisted for many days. Later work using purified compounds found that 1,2-DEB (95%) and DEB mixtures, 61 but not 1,3-DEB (99%) or 1,4-DEB (96%) alone, were chromogenic in rats given repeated oral doses of 500 and 750 mg/kg/d, 5 d/wk for 10 weeks. 47,48 Humans may also excrete colored (green) urine following exposure to tetralin, 61 and a blue discoloration of the skin and internal organs develops in rodents repeatedly treated with acetylethyltetramethyltetralin. 57,58
The neuropathologic lesion induced by repeated systemic treatment with γ-diketones is one in which large-diameter and elongate nerve fibers in the CNS (mostly ascending and descending spinal cord tracts), as well as the PNS, initially develop axonal changes. 62 Small-diameter and unmyelinated fibers are less susceptible but also exhibit degenerative changes. Experimental studies with rodents show that n-hexane, 2-hexanone, and 2,5-HD each induces degenerative changes in the distal regions of limb nerves and ascending and descending spinal cord tracts, 21,62 with comparable changes in the optic nuclei and hypothalamus. 63,64 Characteristic nerve fiber changes consist of focal axonal swellings filled with 10 nm neurofilaments (NFs) that commonly develop on the proximal sides of nodes of Ranvier, thereby resulting in retraction of paranodal myelin followed by localized demyelination and remyelination, with distal, retrograde (“dying-back”) axonal degeneration. 21
Neurofilaments are synthesized in nerve cell bodies and transported distally (anterograde) along the axon at a rate of approximately 1 mm/d during which they are continuously exposed to a blood-borne neurotoxic agent. The spatiotemporal evolution of these pathological changes has been observed directly in organotypic cultures composed of structurally and functionally coupled explants of spinal cord, spinal ganglia, and striated muscle during continuous treatment (2.8 mM) with n-hexane or metabolites (2-hexanol, 2,5-hexanediol, 2-hexanone, 5-hydroxy-2-hexanone, and 2,5-HD) and 2,4-HD as a negative control. 65 Comparable treatment with the most potent lysine-reacting aliphatic γ-diketone, 3,4-dimethyl-2,5-hexanedione (DMHD), 66 induced giant axonal swellings filled with NFs in more proximal regions of peripheral nerve fibers in culture, followed later by distal, retrograde axonal degeneration. 65
The neuropathological picture is modified by the rate of systemic intoxication (dose, frequency, and duration) and the timing of examination. For example, axonal atrophy is prominent in peripheral nerves and ascending (gracile fasciculus and spinocerebellar tract) and descending (corticospinal and rubrospinal tracts) spinal tracts of rats treated by gavage with very high (up to 400 mg/kg/d) doses of 2,5-HD, 67,68 probably because the chemical assault rapidly arrests the anterograde transport of NFs, the density and subunit composition of which regulate axon diameter. 69 This is particularly evident when animals are treated with 1,2-DAB, a compound with markedly greater neurotoxic potency than 2,5-HD. 50,51 Animals treated with 1,2-DAB display massive proximal NF swellings, some of which develop intraspinally just distal to the initial axonal segment of the motor neuron. 50 When proximal axonal accumulation occurs in spinal roots, marked localized demyelination and remyelination results as a secondary response to axonal swelling and then attenuation, respectively. Indeed, the extent of demyelination and remyelination was so extensive in the spinal roots of AETT-treated animals that it was erroneously interpreted as a primary demyelinating disease. 52
The molecular basis of these compound-dependent patterns of γ-diketone-induced nerve fiber pathology appears to lie in their differential neurotoxic potency and, by extension, degree of protein adduction and polymerization. Highly potent neurotoxic γ-diketones, such as 1,2-DAB and DMHD, rapidly degrade 1 or more of the 3 proteins (NF heavy [NF-H], NF medium [NF-M], and NF-light [NF-L]) that make up the formed NF and/or the transport system on which anterograde NF transport depends. By contrast, lower potency γ-diketones, such as 2,5-HD, appear to complex with and degrade axonal proteins more slowly, such that the NFs are able to move anterograde along the axon until further transport is impeded in distal regions of the nerve fiber where they accumulate proximal to nodes of Ranvier. The rapid segregation of axonal microtubules from NFs induced by intrafascicular injection of 2,5-HD into the sciatic nerve in vivo may perturb the transport of NFs, 70 which then become disorganized and later accumulate in focal axonal swellings. The proposed relationship between the chemical structure of γ-diketones, relative potency, and the resulting location of focal axonal swellings with amassed 10 nm NFs is shown in Figure 2.
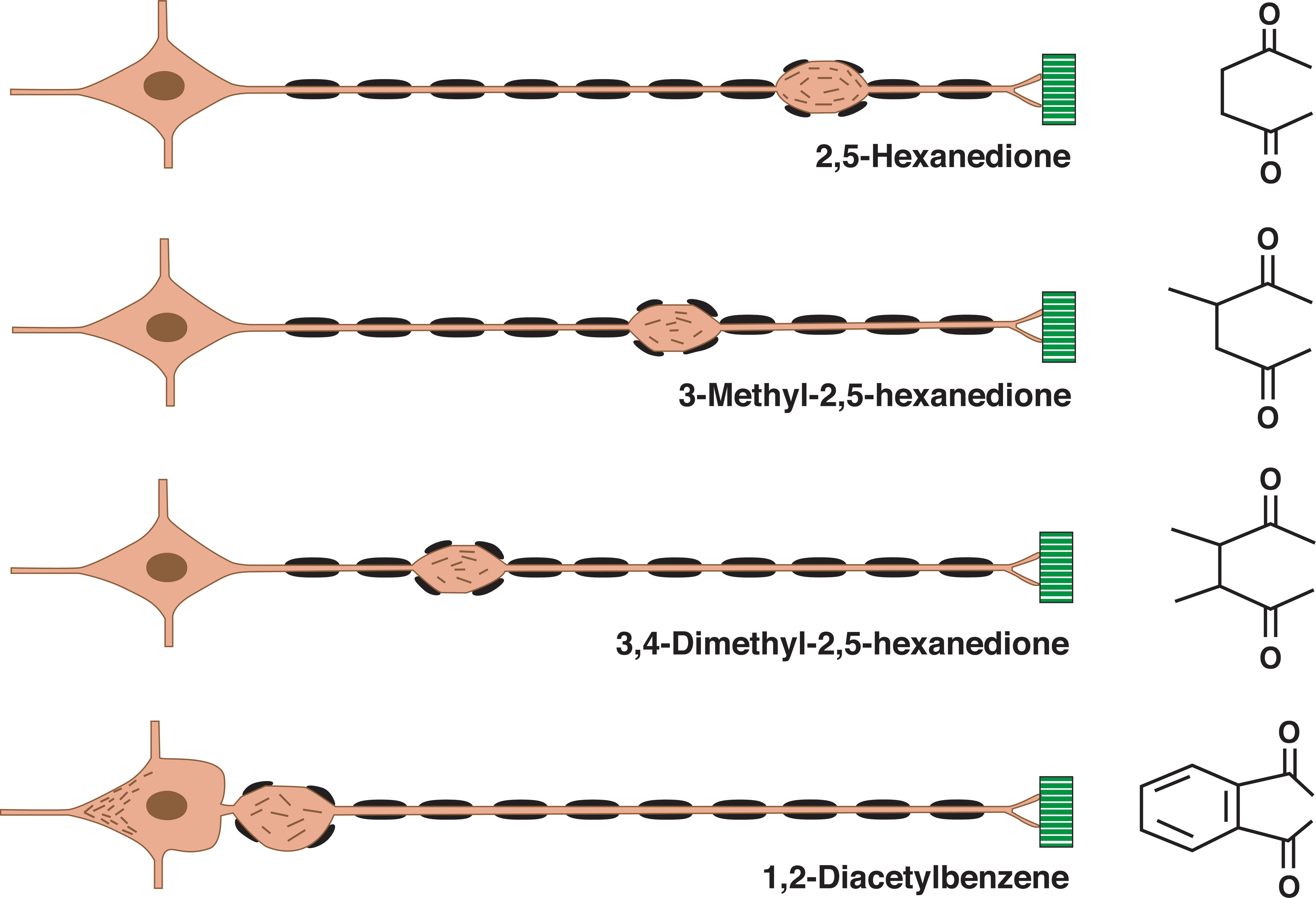
Nerve fiber response to γ-diketones. Diagram of the location of initial focal axon (orange) swellings and associated secondary myelin (black) retraction in an elongate, large-diameter peripheral nerve fiber originating from the neuron soma (left) and terminating at sites of muscle (green) innervation. γ-Diketone potency and associated neuroprotein reactivity increase from 2,5-hexanedione (2,5-HD, upper) to 1,2-diacetylbenzene(1,2-DAB, lower), where neurofilaments accumulate both in the neuronal soma, distal to the initial segment, and in proximal spinal roots (as shown). The differential spatial “rigidity” (ability to rotate in space) of the 1,4-diketo groups may dictate the protein reactivity and, hence, neurotoxic potency of the illustrated γ-diketones. This diagram illustrates the principle that the toxic potential of the γ-diketone dictates how far neurofilaments are transported from the neuron cell body along the nerve fiber before they accumulate in focal axonal swellings; the greatest transport distance representing the least potent (2,5-HD) and, hence, the slowest developing effect. The most potent γ-diketone, 1,2-DAB, rapidly reacts with axonal proteins, such that anterograde transport of neurofilaments is arrested before or shortly after their exit from the neuron cell body. This figure is constructed from the author’s examination of single teased nerve fibers, and light and electron micrographs of cross-sectioned nerves, from peripheral nerve fibers undergoing γ-diketone axonopathy. 2,52,53,62,63,65,71 -73
Assessment in vitro of the chromogenic reaction of amino acids with DAB isomers demonstrates that 1,3-DAB fails to react with lysine, ornithine, glycine, γ-aminobutyric acid, or
Chromogenic Reaction of Selected Amino Acids With Neurotoxic (1,2-DAB) and Non-Neurotoxic (1,3-DAB) Isomers of Diacetylbenzene.a
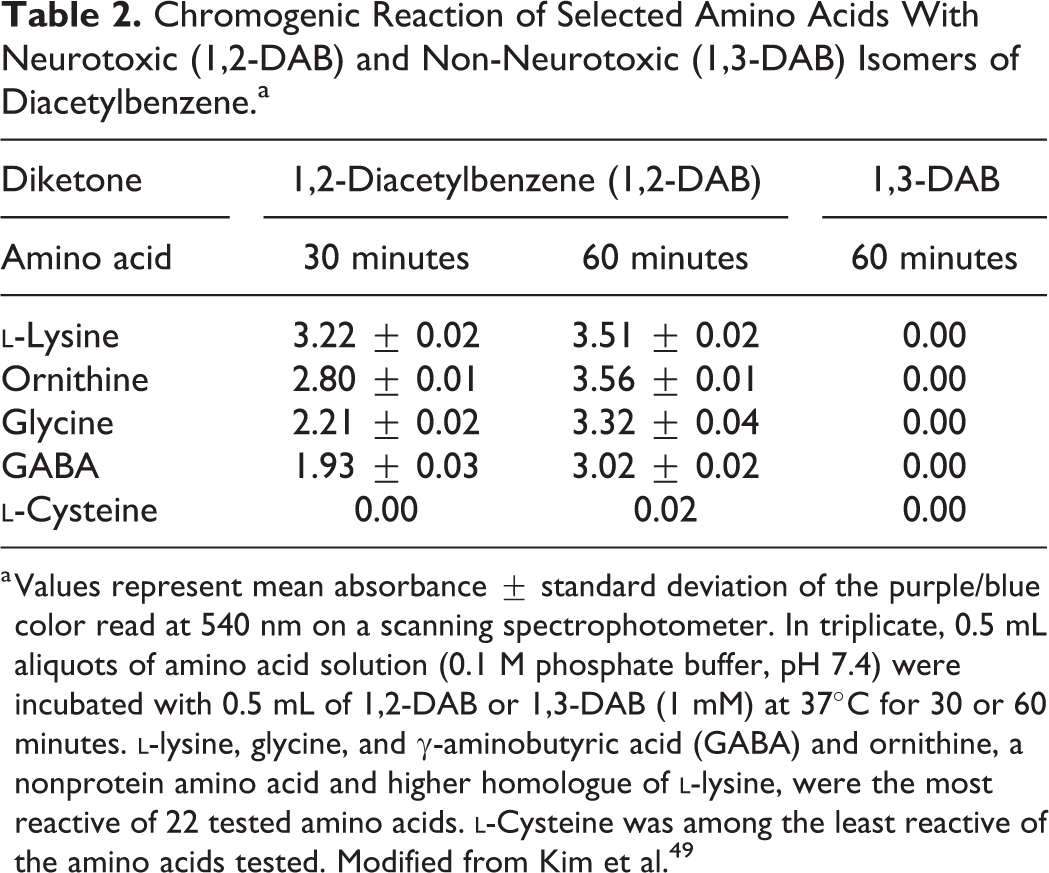
a Values represent mean absorbance ± standard deviation of the purple/blue color read at 540 nm on a scanning spectrophotometer. In triplicate, 0.5 mL aliquots of amino acid solution (0.1 M phosphate buffer, pH 7.4) were incubated with 0.5 mL of 1,2-DAB or 1,3-DAB (1 mM) at 37°C for 30 or 60 minutes.
Total Lysine Content of Axonal Proteins.a
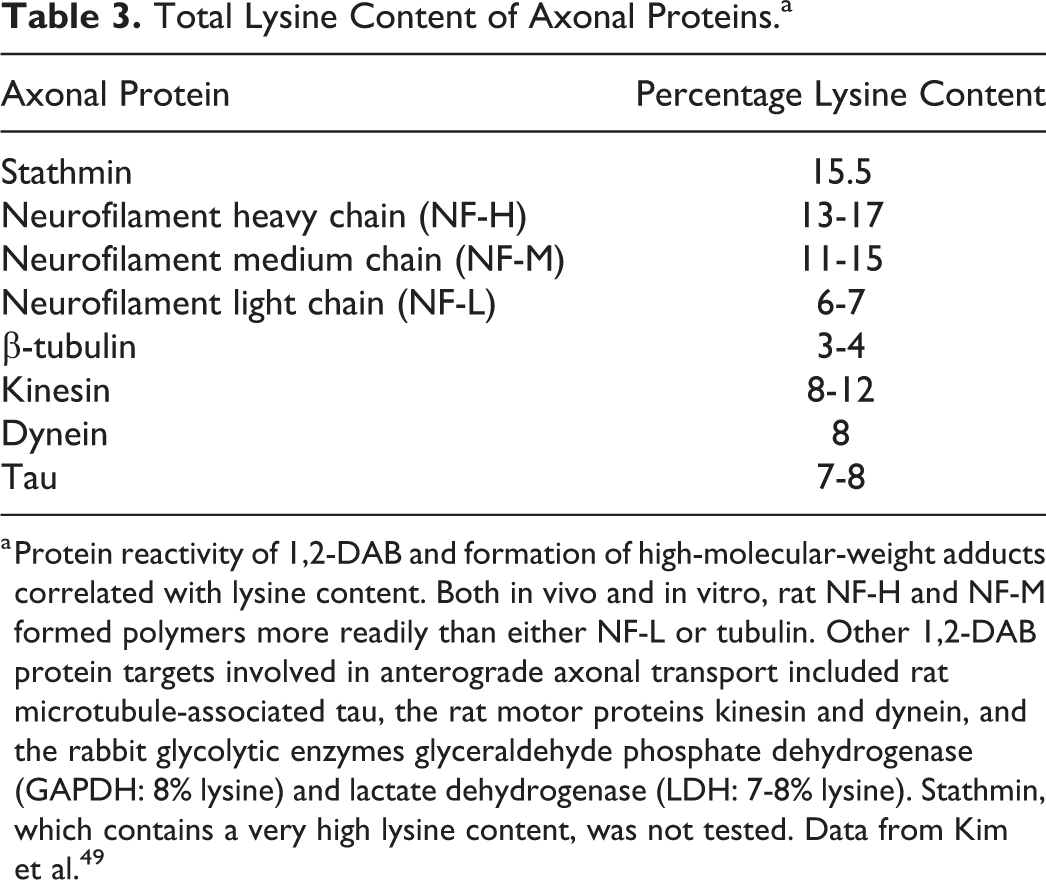
a Protein reactivity of 1,2-DAB and formation of high-molecular-weight adducts correlated with lysine content. Both in vivo and in vitro, rat NF-H and NF-M formed polymers more readily than either NF-L or tubulin. Other 1,2-DAB protein targets involved in anterograde axonal transport included rat microtubule-associated tau, the rat motor proteins kinesin and dynein, and the rabbit glycolytic enzymes glyceraldehyde phosphate dehydrogenase (GAPDH: 8% lysine) and lactate dehydrogenase (LDH: 7-8% lysine). Stathmin, which contains a very high lysine content, was not tested. Data from Kim et al. 49
γ-Diketone-Induced Changes in Brain Gene Expression
Toxicogenomic studies have determined changes in brain gene expression following single intraperitoneal treatment of male C57BL/6 mice with 1,2-DAB (50 mg/kg) versus 1,3-DAB (50 mg/kg) or vehicle (2% acetone in saline). One-hour posttreatment, significant (ie, <1.5-fold, q < 0.1) 1,2-DAB-upregulated genes included transcription factors/regulator (Zic1, Fus, Junb, Klf4, Mapk8ip); those involved in cell cycle progression/proliferation (Rif1 homologue, Junb, Btg2, Mapk8ip); transmitter-related function (Gabt4, BQ746452 [similar to Gabbr1], Cplx2); protein degradation (Ube2 s, AK034214 [similar to Dirc2]); lipid metabolism (BB32327894 [similar to Gpam]), and microtubule-binding (Eml2, which negatively regulates microtubule polymerization, and AK038316, similar to Dock9, a member of a family of genes coding for proteins that regulate the actin cytoskeleton). At 6 hours posttreatment, 1,2-DAB upregulated genes included transcription factors/regulator (Dsip1, Mafk), those involved in cell cycle progression/proliferation (Pmaip1, Nfkbia, Igfbp1, Dsipl, Mafk, Cdkn1a [P21]), apoptosis regulating (Pmaip1, Angpt14, Dsip1, Cdkn1a (P21)), lipid metabolism (Tts-2.2 homologue), inhibitor of protein degradation (Ct1a2b), and neuronal antioxidant (Mt2). These findings demonstrate a complex and temporally evolving pattern of brain transcriptional perturbation in response to systemic diketone treatment.
There is sparse information on changes in gene expression associated with long-term treatment with n-hexane and 2,5-HD, its protein-binding neurotoxic metabolite. Neurofilament subunit gene expression was modestly reduced (approximately 20%) in the dorsal root ganglia of rats with 2,5-HD neuropathy. 79 Chronic treatment of rats with 2,5-HD reportedly increased cellular apoptosis in the spinal cord of rats by downregulating nerve growth factor expression and subsequently repressing the phosphoinositide 3-kinase/Aktsignaling pathway. 80 However, long-term systemic treatment of rats with 2,5-HD reportedly increased the percentage of proliferating neural stem and progenitor cells in the granule cell layer of the hippocampus and the subgranular zone. 81 Neuropathological changes were found in the mammillary body and visual nuclei of cats treated with low levels of 2,5-HD for prolonged periods. 63
n-Hexane inhalation altered the methylation status of promoters of genes associated with ovarian cell apoptosis and steroid hormone biosynthesis, 82 and treatment with 2,5-HD increased apoptosis in human ovarian granulosa cells. 83,84
γ-Diketone-Induced Changes in Neuroprotein Abundance
Early CNS Protein Changes
The brain proteome was examined in mice recovering from a single intraperitoneal injection of 1,2-DAB, 1,3-DAB, or DAB vehicle (Table 4). Marked 1,2-DAB-associated changes were noted in the relative abundance of brain proteins involved in microtubule interaction, other structural proteins, and cell energy metabolism. The greatest change was seen for actin α-2. Association of actin filaments with microtubules is important for various cellular processes, such as cell division, migration, vesicle and organelle transport, and axonal growth. 85 Actin α-2 has at least 10 lysine-acetylation sites, and lysine-acetylated actin can interact with cyclase-associated protein to form an inhibitor of inverted formin 2 (INF2). Significantly, dominant missense mutations in INF2 are linked to Charcot-Marie-Tooth disease, an inherited human axonal neuropathy. 86,87
DAB Neuroproteome Relative Abundance of Brain Proteins of Young Adult C57/BL6 Mice 168 Hours After a Single Intraperitoneal Injection of 1,2-DAB (50 mg/kg Body Weight), 1,3-DAB (50 mg/kg), or DAB Vehicle (2% Acetone in Saline).a
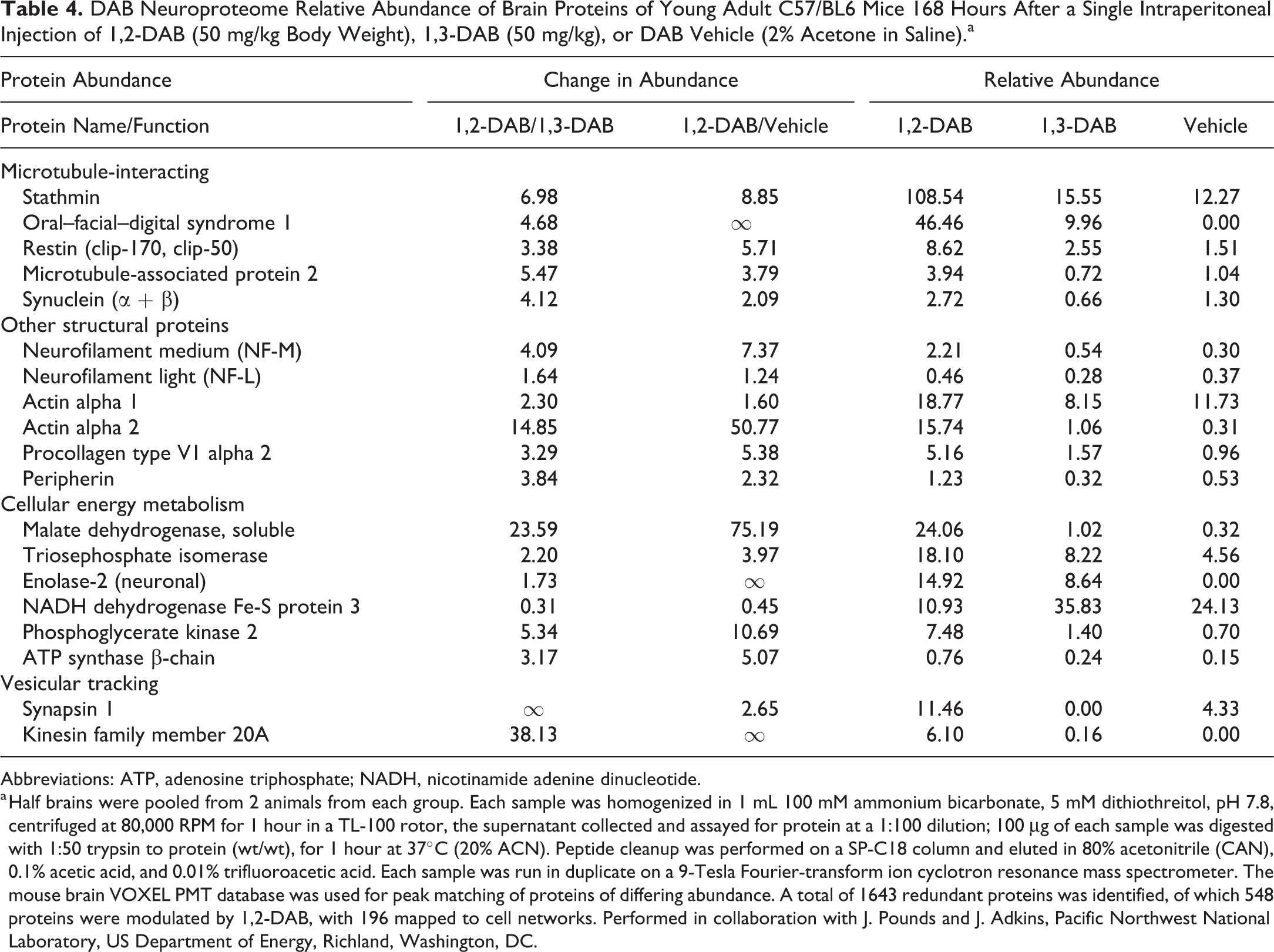
Abbreviations: ATP, adenosine triphosphate; NADH, nicotinamide adenine dinucleotide.
a Half brains were pooled from 2 animals from each group. Each sample was homogenized in 1 mL 100 mM ammonium bicarbonate, 5 mM dithiothreitol, pH 7.8, centrifuged at 80,000 RPM for 1 hour in a TL-100 rotor, the supernatant collected and assayed for protein at a 1:100 dilution; 100 μg of each sample was digested with 1:50 trypsin to protein (wt/wt), for 1 hour at 37°C (20% ACN). Peptide cleanup was performed on a SP-C18 column and eluted in 80% acetonitrile (CAN), 0.1% acetic acid, and 0.01% trifluoroacetic acid. Each sample was run in duplicate on a 9-Tesla Fourier-transform ion cyclotron resonance mass spectrometer. The mouse brain VOXEL PMT database was used for peak matching of proteins of differing abundance. A total of 1643 redundant proteins was identified, of which 548 proteins were modulated by 1,2-DAB, with 196 mapped to cell networks. Performed in collaboration with J. Pounds and J. Adkins, Pacific Northwest National Laboratory, US Department of Energy, Richland, Washington, DC.
It seems reasonable to posit that 1,2-DAB attacks actin’s lysine sites, thereby blocking the formation of INF2 and contributing to the genesis of γ-diketone axonopathy. There was also a markedly increased abundance in 1,2-DAB versus 1,3-DAB or vehicle of CLIP-1 (CAP-GLY domain-containing linker protein), a microtubule plus-end-associated protein that interacts with dynein and binds tightly to formins to accelerate actin filament elongation. 88,89 1,2-Diacetylbenzene versus 1,3-DAB also showed some increased abundance of microtubule-associated protein 2 and α-synuclein, which facilitates the formation of short, mobile transportable microtubules that play an important role in axonal transport. 90 These changes in protein expression should be confirmed with additional proteomic studies.
Brains of mice 1 week after a single 1,2-DAB treatment showed increased abundance of stathmin (Table 4) and proteins involved in stathmin regulation: cyclin-dependent kinase 5 (cdk5), RAS-related C3 botulinum substrate 1 (rac1), heat shock protein conjugate 70 (hsv70), and protein phosphatase 2a (pp2a). These brains also showed changes in stathmin-regulated genes, including cyclin-dependent kinase inhibitor 1a (Cdkn1a, P21), epidermal growth factor (Egf), and Krüppel-like factor 4 (Klf4). Stathmin is a ubiquitous, brain-enriched phosphoprotein with a very high lysine content that binds to tubulin and inhibits microtubule assembly, an activity regulated by phosphorylation. 91,92 Stathmin is of special interest in relation to γ-diketone axonopathy because aged stathmin knockout mice also develop axonal degeneration and secondary demyelination. 93 Additionally, incubation of mouse brain and testes homogenates with 1,2-DAB (1-10 mM) reduced the intensity of the native stathmin band in a concentration-dependent manner, with corresponding increases in high-molecular-weight protein adducts. By contrast, under similar conditions, 1,3-DAB had no effect on stathmin, and adducts were not observed. Stathmin is therefore of potential mechanistic relevance to γ-diketone axonopathy. 94
Later CNS Protein Changes
Neuroprotein studies have been performed on the lumbosacral spinal cord of rats with severe γ-diketone neuropathy. 1,2-DAB significantly altered the expression of protein disulfide isomerase, an enzyme involved in protein folding, and gelsolin, an actin-capping and -severing protein. Thirty-four proteins were markedly modified by 2,5-HD, of which NF-L, gelsolin, protein disulfide isomerase, glutathione S-transferase, nicotinamide adenine dinucleotide (reduced) dehydrogenase 1α, pyruvate kinase, and fatty acid synthase were also modified by 1,2-DAB. 2,5-Hexanedione induced a 1,2-DAB-like proteomic signature by altering the expression levels of proteins involved in maintaining the physical integrity of axons (reduced), in controlling redox and protein-folding mechanisms (reduced), and in supporting energy metabolism (increased). 51 While the spinal cord proteome also suggested a reduction in α-II spectrin (Spna2), a key protein in the maintenance of axonal integrity, degradation of Spna2 by calpain- and/or caspase is reportedly not central to the pathogenesis of 1,2-DAB axonopathy. 95
Perturbation of energy metabolism resulting in reduced adenosine triphosphate (ATP) has been postulated to be linked to the etiology of central–peripheral axonopathy. 96 2,5-Hexanedione reduced the rate of ATP synthesis in isolated brain mitochondria 97 and pyruvate-restored ATP deficits in cat nerves treated with 2,5-HD. 98 Consistent with this hypothesis, recovery from systemic treatment with 1,2-DAB was associated with a very marked increase in brain abundance of soluble malate dehydrogenase and, to a lesser extent, other proteins involved in energy metabolism, including glycolysis (triosephosphate isomerase, phosphoglycerate kinase 2, enolase 2) and the electron transport chain (nicotinamide adenine dinucleotide dehydrogenase Fe-S protein 3; ATP synthase β-chain; Table 4). Oxidative stress and resulting tissue damage have been attributed to γ-diketone toxicity of liver, kidney, brain, and SY5Y neuroblastoma cells. 39,99,100
Normal Aging and Diabetes Mellitus
Are neurotoxic γ-diketones physiological metabolites and might they participate in other types of peripheral neuropathy, including those associated with the aging process and diabetes mellitus? One study of 31 normal subjects with no known n-hexane exposure found low levels (<0.006 mg/L) of free 2,5-HD in urine. 101 A second study reported that healthy subjects without occupational exposure to n-hexane had detectable levels of 2,5-HD in the blood (6-30 μg/L) and urine (0.17 and 0.98 mg/L), only a minimal part of which was considered to have derived from exposure to hydrocarbon-polluted air. 102 However, a third study found that 1.3% of 1200 normal subjects with no known occupational exposure to n-hexane had blood levels of the neurotoxic alkane above the method detection limit. 103 A fourth investigation of urine samples from 123 healthy Italian subjects recorded a 2,5-HD reference value of 0.795 mg/L for men and 0.627 mg/L for women. 104 A fifth very large study of healthy Chinese subjects (n = 8235) with no occupational exposure to n-hexane or 2-hexanone showed a median urine 2,5-HD concentration of 0.171 mg/L for males and 0.147 mg/L for females, with increasing 2,5-HD excretion with the advance of age. 105 In a sixth study, investigation of 208 male and female subjects revealed a median level of urinary pyrrole adducts of unstated origin of 0.91 nmol/mL. 106 Finally, with respect to diabetes mellitus, a small study of serum samples from normal individuals, and from subjects with type 2 diabetic neuropathy, revealed similar qualitative profiles of volatile metabolites (low nanogram concentration), including relatively high and low concentrations, respectively, of 2-hexanone and 3-heptanone, both of which can undergo ω-oxidation to form γ-diketones. 107 Also present in similar concentrations in normal and diabetic sera was 2-butanone (methyl ethyl ketone), a compound that potentiates the neurotoxic potency of n-hexane and 2-hexanone. 108 -111 Since degenerative nerve fiber changes with the advance of aging resemble those seen in the early stages of distal axonopathies, 112 and central–peripheral axonopathy underpins type 2 diabetic neuropathy, 71 it will be important to determine the origin of γ-diketones and precursors with neurotoxic potential in normal subjects and whether they contribute to nerve fiber changes in aged subjects and those with diabetic neuropathy.
Conclusion
Structure-activity studies 52,113 show that γ-diketone metabolites of certain aliphatic and aromatic solvents selectively react with proteins throughout the body to form chromogenic polymers. Despite this global assault and widespread formation of protein adducts (in serum, liver, kidney, and brain), 114 other than the nervous system, few tissues (testes, liver, and kidney) undergo degenerative/atrophic changes in γ-diketone-treated animals. 99,115 The special susceptibility of CNS and PNS neurons to γ-diketones appears to be related to their requirement for transport of proteins over very long distances (ie, from cell soma via the axon to the nerve terminal). Although protected by blood–brain/nerve barriers, γ-diketones not only cross these regulatory interfaces but can also increase their permeability. 116 Elongated, large-diameter nerve fibers are affected first, probably because of the large volume of NF and other proteins undergoing anterograde transport. However, increasing the γ-diketone potency and dosage results in more rapid protein adduction, such that NFs are arrested shortly after commencing their transport from sites of synthesis in neuronal somata. This results in the formation of focal giant axonal swellings containing masses of disoriented NFs, while more distal regions of axons undergo atrophy because they fail to receive a normal complement of NFs. Distal, retrograde axonal degeneration then ensues by a proposed mechanism in which trapped mitochondria release stored calcium and trigger calcium-activated proteases that effectively sever the axon distal to the site of NF accumulation. 72
The exact targets of γ-diketones are not clear. Microtubules are unequivocally impacted by γ-diketones, either directly or indirectly, or both. In vitro, 2,5-HD-treated tubulin had a decreased lag time and an increased maximal velocity of microtubule assembly. 115 Systemic 2,5-HD treatment causes longitudinal aggregation of axonal microtubules, sometimes in isolated clusters and sometimes decorating the perimeter of mitochondria, an event that occurs within minutes of intraneural γ-diketone injection 70 and followed by the development of NF-laden axonal swellings days later. 73 This seems to be associated with loss of the 3- to 5-nm thick cross-bridges formed by MAP 2 or tau on axonal microtubules 117 or from the carboxy-terminal tail domains of phosphorylated NF-H and NF-M that laterally interconnect intermediate NFs. 118 -120 Neurofilament medium (and NF-H?) contains a variable lysine–serine–proline (KSP) repeat subdomain flanked by 2 highly conserved subdomains, the number of tri-, tetra-, and pentapeptide KSP repeats dispersed along the subdomain increasing in proportion to axon length and caliber. 121 Given the high affinity of γ-diketones for lysine residues, their reactivity with high lysine content NF-H and NF-M, the exceptional vulnerability of long and large-diameter axons in systemically treated rodents, and the earlier and greater involvement of lower relative to upper extremities in γ-diketone neuropathy, the NF sub-units NF-H and NF-M are also intimately involved in the pathogenesis of γ-diketone axonopathy. Additionally, NF-H knockout mice are refractory to the γ-diketone-like giant NF swellings induced by systemic treatment with β, β′-iminodipropionitrile, which suggests that NF-H is a key mediator of this type of axonopathy. 122
Normal anterograde axonal transport of NF subunits appears to be dependent on myosin-mediated interactions within the actin cortex that lines the inner axolemmal surface. 123 Actin is also involved in microtubule-dependent vimentin transport. 124 Actin contains 19 lysines of variable reactivity, of which K325 and K327 are most reactive with acetic anhydride. 125 Given the very large increased production of brain actin following systemic treatment with 1,2-DAB, actin lysines are also likely to be important γ-diketone targets. Stathmin, which binds to and sequesters monomeric tubulin, thereby preventing microtubule polymerization, has a very high lysine content and is thus a prominent target for γ-diketones, a position supported by the increase in brain abundance of stathmin after systemic treatment with 1,2-DAB. Serine 63, a key phosphorylation site for stathmin-induced destabilization of microtubules, is next to lysine 62, 92,126 which might be an important site for γ-diketone attack. Stathmin binds directly along the microtubule wall, such that a pool of this protein is immediately available to participate in protofilament dissociation when the moving plus end of a depolymerizing microtubule reaches stathmin molecules. 127 Other axonal proteins with significant lysine components are the motor proteins dynein and kinesin, although it is noteworthy that increased brain abundance of these neuroproteins was not apparent after systemic treatment with 1,2-DAB.
The foregoing description of γ-diketone neuropathy represents one of the most detailed analyses of the molecular mechanisms underlying a central–peripheral distal axonopathy, a pattern of nervous system damage seen in a number of genetic, metabolic, nutritional, and toxic neuropathies. The molecular mechanisms involved in the neurotoxic actions of γ-diketones may therefore have relevance in comparable neuropathologic states, such as those associated with aging and diabetes mellitus.
Footnotes
Acknowledgments
The experimental studies reviewed here were variously performed by students, technicians, postdoctoral fellows, and faculty colleagues at Albert Einstein College of Medicine, Oregon Health & Science University, Oregon State University, Pacific Northwest National Laboratory, and University of Houston; in surname alphabetical order: Joshua Adkins (proteomics), Monica Bischoff (animal studies), Alan Cranson (toxicogenomics), Max Deinzer (mass spectrometry), David Dixon (computation), Dickran Houropian (neuropathology), Seyed Hashemi (protein studies), Robert Kayton (electron microscopy), Min-Sun Kim (neuropathology), Michael Lasarev (biostatistics), Valerie Palmer (animal studies and toxicogenomics), the late Edith Peterson (tissue culture), Michael Politis (animal studies), Colin Poole (chromatography), Joel Pounds (proteomics), Mohammad Sabri (protein studies), Herbert Schaumburg (neurology and neuropathology), Mary Seelig (tissue culture), Dean Sproles (toxicogenomics), Arnold Sterman (neuropathology), Sarah Trimpkin (mass spectrometry), Desire Tshala-Katumbay (neuropathology and protein studies), Bellina Veronesi (tissue culture), Joy Zagoren (animal studies), Chang-Guo Zhan (computation studies), and the late Albert Zlatkis (chromatography). The author thank Chen Xiao for discussion. Ann Hubbs kindly reviewed the article.
Declaration of Conflicting Interests
The author declares no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author discloses receipt of the following financial support for the research, authorship, and/or publication of this article: The toxicogenomic and proteomic studies reported here for the first time were supported by grant U19 ES011384 from the National Institute of Environmental Health Sciences.