
Abstract
Ocular medical devices (MDs) represent a very wide and promising field of human ophthalmology. In preclinical studies evaluating the safety and/or performance of these ocular MDs, the choice of histologic technique and the focus of the histopathologic evaluation method should take into consideration the following aspects: the specific guidelines possibly associated with the MD or combination product, the ocular compartment in contact with the MD and its specificities, and last the nature of the biomaterial used in the MD. Following a brief overview of animal models, this short review will present the different types of ocular MDs and will present the specificities of the histologic technique and the histopathologic evaluation related to ocular MDs.
This short review will present the different types of ocular medical devices (MDs) and will focus on the specificities of the histologic technique and the histopathologic evaluation related to ocular MD. Comparative anatomy of the eye will not be reviewed here, since exhaustive bibliography is available (Vezina 2013). Of particular interest is a review (Lloyd, Faragher, and Denyer 2001) which underlines the interactions between the ocular anatomy and the different types of ocular implants. This can be complemented with more recent updates on ocular biomaterials (Williams 2014; Li and Mai 2017).
The choice of the histologic technique and the method of histopathologic evaluation will first depend on the nature and indication of the ocular MD, and therefore the ocular compartment that will be in contact with the device following application, injection, or implantation. These criteria will determine the corresponding guidelines. International Organization for Standardization (ISO) 10993-1 is the basic horizontal International Standard for biological evaluation of MDs and serves as a framework for planning biological evaluation tests. ISO 10993-10 assesses possible contact hazards from device-released chemicals that may produce skin and mucosal irritation, eye irritation, and delayed contact sensitization. ISO 10993-6 assesses local tissue effects following device implantation. In addition, usage tests for specific devices are defined in vertical standards, such as ISO 9394: 2012 for contact lenses, ISO 11979: 2006 for intraocular lenses (IOL), ISO 16672: 2015 for ocular endotamponades, and ISO 15798: 2013 for ophthalmic viscosurgical devices (OVDs). Therefore, both horizontal and vertical standards should be taken into consideration when designing preclinical studies for assessment of ocular MD.
The biomaterial constitutive of the ocular MD will also influence fixation and the histologic technique. It should also be added that some implants consist of drug-delivery systems that will therefore be considered as a drug and evaluated as such. On the contrary, implants that do not deliver any pharmacologically active drugs should be considered as MDs and accordingly evaluated. This is the case for IOL (physical mode of action), glaucoma drainage devices (mechanical mode of action), and retinal implants (electrical mode of action) that will be detailed hereafter.
Ocular MDs will be presented following a short section about animal models. Although an exhaustive list will not be provided here, the main types of ocular MD will be presented and classified according to their ocular compartment of application, injection, or implantation.
Animal Models for the Evaluation of Ocular MDs
The rabbit is the most commonly used species for preclinical studies evaluating MD (Werner, Chew, and Mamalis 2006), mainly because of the request by several guidelines. However, the porcine species (either domestic pigs or minipigs) are increasingly used in view of the closer resemblance with the human eye. Useful reviews list the different animal models for ocular studies and help in choosing the most appropriate species (Shrader and Greentree 2018; Tsonis 2008).
Of note, some guidelines referring to the preclinical evaluation of ocular MD lack clarity regarding the appropriate animal model. Recently, a new version of the ISO 16672 standard for ocular endotamponades was released (ISO 16672: 2015). In particular, this updated version had the following sentence added in the intraocular implantation test (paragraph 6.2.3): “Due to differences between the vascularised human retina and the avascular rabbit retina especially for non-aqueous substances a suitable animal model has to be validated.” This request of an animal model other than the rabbit appeared to be based on an inexact statement since the rabbit retina is not avascular. This erroneous statement likely comes from the differences described between the rabbit and human vascularization of the retina. The human retinal vasculature is known as holangiotic, meaning that retinal vessels are present in the whole retinal surface, similar to what is observed in the fundus of nonhuman primates. The rabbit retinal vasculature is known as merangiotic, meaning that retinal vessels are present in a limited area of the retina, known as the visual streak, and extending from the nasal to temporal side in a horizontal plane. Besides this difference, it therefore appears that the rabbit remained a valuable model since the vascular area of the retina remains in direct contact with endotamponades agents injected in the posterior chamber.
Topically Applied Ocular MD
Eye drops are classified as ocular MD when they do not contain pharmacologically active substances. When alternative in vitro methods cannot be used, ocular irritation tests are conducted according to the ISO 10993 part 10 (2010). Histopathologic analysis is not requested as per this standard, although it is occasionally performed when there are unexpected results. In this case, standard ocular histopathology is performed, based on examination of the cornea through vertical sagittal sections of the eyeball, as well as the other ocular compartments and ocular adnexa. Classical fixation by immersion in Davidson’s solution is preferred for rodent eyes. Immersion fixation in Davidson’s solution should not exceed 24 hr for rodent eyes and 48 hr for larger eyes, in order to prevent artifacts caused by glacial acetic acid. This short fixation in Davidson’s solution should be followed either by transfer to 70% ethanol before histological processing or by long-term storage in 10% neutral buffered formalin (NBF). Optimal fixation of rabbit eyes is achieved when internal ocular structures are directly exposed to the fixative solution (Schafer and Render 2013). This is why the window technique is recommended for species larger than rodents. In this case, a 1:1 mixture of 10% NBF and 4% glutaraldehyde appears as the best preservative option (Bolon et al. 2013). The incision or window should not be done on a freshly enucleated eye but should follow a 1-hr fixation to allow for globe hardening.
Contact lenses and contact lens care products are evaluated according to the ISO 9394: 2012 standard, which recommends a 22-day study in the rabbit with daily application. Although this standard requests histopathologic evaluation of the cornea, conjunctivae, iris, and lens, it does not provide any mandatory evaluation method. The evaluation method should therefore be based on the horizontal standard for irritation tests (ISO 10993, part 10), similar to what was described above for eye drops. Neither microscopic observation nor ultrastructural surface analysis of the contact lens is requested.
Injected Ocular MD
Ocular MD can be injectable substances that are nonpharmacologically active, such as endotamponade agents. Ocular pathological conditions affecting the retina (retinal detachment, diabetic retinopathy) or the vitreous itself (vitreous hemorrhage) may require surgical removal of the vitreous (vitrectomy). Immediately following vitreous removal, there is temporary or permanent replacement by an intraocular endotamponade agent which is intended to hold the retina in place. Conventional intraocular endotamponade agents include gases (perfluoropropane or sulfur hexafluoride) that disappear progressively and spontaneously as well as silicone oil that must be removed a few weeks after the vitrectomy procedure. The guideline specific for ocular endotamponade agents (ISO 16672: 2015) refers to the standard ISO 10993 (parts 1 and 6) for the biological evaluation. For this particular type of intravitreal injection, histopathologic evaluation should rely on the examination of several vertical step sections of the eyeball since the article-related effects can be restricted to specific areas, especially the lower part of the eye. It is very important that histologic trimming minimizes the blading sections through the eyeball in order to prevent histologic artefacts, especially retinal detachment. For that purpose, embedding in deep cassettes is preferred since it allows embedding of an entire half of a rabbit globe after only one sagittal or parasagittal section.
OVDs represent a class of nonpharmacologically active surgical implants with viscous and/or viscoelastic properties that are used during surgery of the anterior segment. OVDs are designed to create and maintain space, to protect intraocular tissues, and to manipulate tissues during surgery. OVDs are tested according to the ISO 15798: 2013, which recommends a 7-day intraocular implantation test in the rabbit with no mandatory histopathologic evaluation. The general requirements for implantation tests outlined in ISO 10993-6 should also apply.
Implanted Ocular MD
Orbital Implants
Orbital implants are used since early 1980s for cosmetic purposes during reconstructive surgery following enucleation or evisceration. Implantation of a solid implant is required after eyeball removal in order to maintain the orbital volume. It is then associated with an ocular prosthesis for aesthetic purposes. The first orbital implants were nonporous spheres made of silicone or polymethyl methacrylate (PMMA). They are now less frequently used than porous spherical implants made of hydroxyapatite (HA, synthetic or coralline), polyethylene (PE), or aluminum oxide (ceramic). Porous implants allow for development of a foreign-body giant-cell reaction and consecutive fibrovascular ingrowth through the implant pores. A histopathologic study in explanted humans demonstrated that implant colonization was complete by 11 months following implantation of either HA or PE implants (Tambe et al. 2009). However, fibrovascular ingrowth was shown to start as early as 4 weeks following implantation (Shields et al. 1991).
Standard paraffin embedding can generally be used for orbital implants, following removal of the orbital content. However, HA orbital implants require either preliminary decalcification or plastic embedding. Plastic embedding is used for ceramic orbital implants. A specific stain such as McNeal tetrachrome or modified Paragon can be useful for plastic-embedded HA implants since they permit differentiation between the implant material and the newly formed bone spicules that are frequently observed with this type of implant (Sekundo and Seifert 1998).
Histopathologic evaluation should focus on the extent and quality of the tissue ingrowth within the implant, since fibrovascular ingrowth is the prerequisite for implant integration. In particular, the cellularity and neovascularization of the colonizing tissue are key factors to obtain implant integration and minimize the risk of implant extrusion, migration, or infection.
Intracorneal Implants and Keratoprostheses
Intracorneal implants are positioned within the corneal stroma and are used to enhance the corneal function, while keratoprostheses are real corneal substitutes that focally replace the cornea in its full thickness (Lloyd, Faragher, and Denyer 2001).
Intracorneal implants comprise intracorneal inlays (mainly for presbyopia) and intrastromal rings or ring segments (mainly for myopia). Intracorneal inlays were initially made of soft polymers, typically PMMA or polysulfone. Multiple small fenestrations were then added to these implants in order to prevent disturbance of the nutrient flow throughout the corneal stroma. More recently, hydrogel polymers were also developed to better preserve the metabolic gradient in the cornea (Arlt et al. 2015). Intrastromal rings are implanted in the deep corneal stroma to modify the corneal curvature and are classically made of PMMA. Paraffin-based histology is generally chosen for these implants (Andreghetti et al. 2013) and a double stain with hematoxylin and eosin and periodic acid of Schiff allows for adequate corneal staining. Histopathologic evaluation should focus on the thickness of the corneal epithelium that can be adversely thinned due to the presence of the implant. When the device is removed from the corneal implantation site, the histologic analysis of the cornea can be supplemented with surface analysis of the explanted device. Surface analysis can be provided by standard scanning electron microscopy (SEM) for a qualitative evaluation of surface alteration or by SEM coupled with identification by energy dispersive X-ray spectroscopy (SEM-EDX) for a quantitative evaluation of possible surface deposits.
Keratoprostheses are used in patients presenting with a severely damaged or diseased cornea and following corneal graft failure. They are implanted intrastromally in continuity with the remaining part of the cornea. Keratoprostheses generally are made of a central clear optical part (PMMA) and a peripheral skirt (titanium, PMMA, polytetrafluoroethylene, etc.). The most widely used is the Boston Type I keratoprosthesis made of a PMMA optical part and a titanium skirt that is designed to sandwich a fresh donor graft. Other keratoprostheses may have both parts made of soft polymers, poly (2-hydroxyethyl methacrylate). The outer skirt is designed to integrate with the corneal tissue by the invasion of keratocytes, while the central zone remains optically clear.
An alternative technique known as osteo-odonto-keratoprosthesis (OOKP) is known as the “tooth in eye surgery” and consists of an autograft of tooth root–alveolar bone complex following a complex multistage procedure (Avadhanam, Smith, and Liu 2015). During the first step, the prosthesis is prepared by extraction of a tooth root-alveolar bone complex from the patient and central insertion of a PMMA cylindrical optical part. This OOKP is then subcutaneously implanted for 3 months in the patient’s cheek in order to gain adequate tissue colonization and vascularization. After 3 months, the OOKP is explanted and reimplanted in the eye by anchoring it to the sclera and covering it with a buccal mucosal graft. Two recent reviews give a thorough overview of the different types of keratoprostheses (Avadhanam, Smith, and Liu 2015; Salvador-Culla and Kolovou 2016).
Whatever the type of keratoprosthesis, the histologic technique should rely on plastic embedding of the implanted cornea, especially when the peripheral part of the keratoprosthesis is made of titanium. Of note, the PMMA optical part does not allow for plastic embedding using PMMA; therefore, an alternative plastic embedding medium should be used, such as epoxy resins.
Scleral Buckles
Scleral buckles are implants designed to correct large retinal detachments by attaching the detached retina to the choroid. First developed buckles were resorbable (e.g., tendon graft or porcine gelatin). Since they did not provide a long-lasting effect, they were recently replaced by nonabsorbable buckles made of soft polymers (PE, silicone) or acrylate-based hydrogels. Of particular interest, the silicone buckles elicit a sustained encapsulation reaction which facilitates its surgical removal. Silicone buckles consist of a thin band of polymer that encircles the eyeball and permanently fix the choroid and detached retina together. The histologic technique is facilitated by the fact that the implant is peripherally located around the eyeball and therefore easily detected and trimmed. However, multiple sections should be examined to ascertain the retinal attachment.
Glaucoma Drainage Devices
Glaucoma drainage devices, or shunts, are designed to reduce intraocular pressure in open angle glaucoma by draining the excessive aqueous humor from the anterior segment to posterior ocular compartments. They can be implanted either in the subconjunctival space or between the choroid and sclera (suprachoroidal shunts). While the former are generally visible, the latter might not be detected easily at external macroscopic examination. Histologic trimming can therefore be a challenge when the section has to be along the longitudinal axis of the implant.
Most devices consist of a flexible tube that shunts aqueous humor to a posterior explant plate. This explant plate is designed to maintain an artificial subconjunctival space as a reservoir for drained aqueous humor. However, the biomaterials used can be as varied as silicone, polypropylene, PMMA, titanium, or stainless steel. The histologic technique should therefore be first chosen based on the biomaterial nature: shunts bearing metal will need to be plastic embedded (Nyska et al. 2003; De Feo et al. 2009), while shunts made of soft polymers can be paraffin embedded.
Histopathologic evaluation should focus on the following adverse effects that can occur with glaucoma shunts: device migration, scleral protrusion, secondary retinal degeneration, and so on. More recent devices are made of porous silicone and they are intended to be fully colonized by host tissue (Figure 1). However, this tissue reaction should not be associated with fibrous encapsulation of the implant that would prevent proper diffusion of the aqueous humor through the biomaterial.
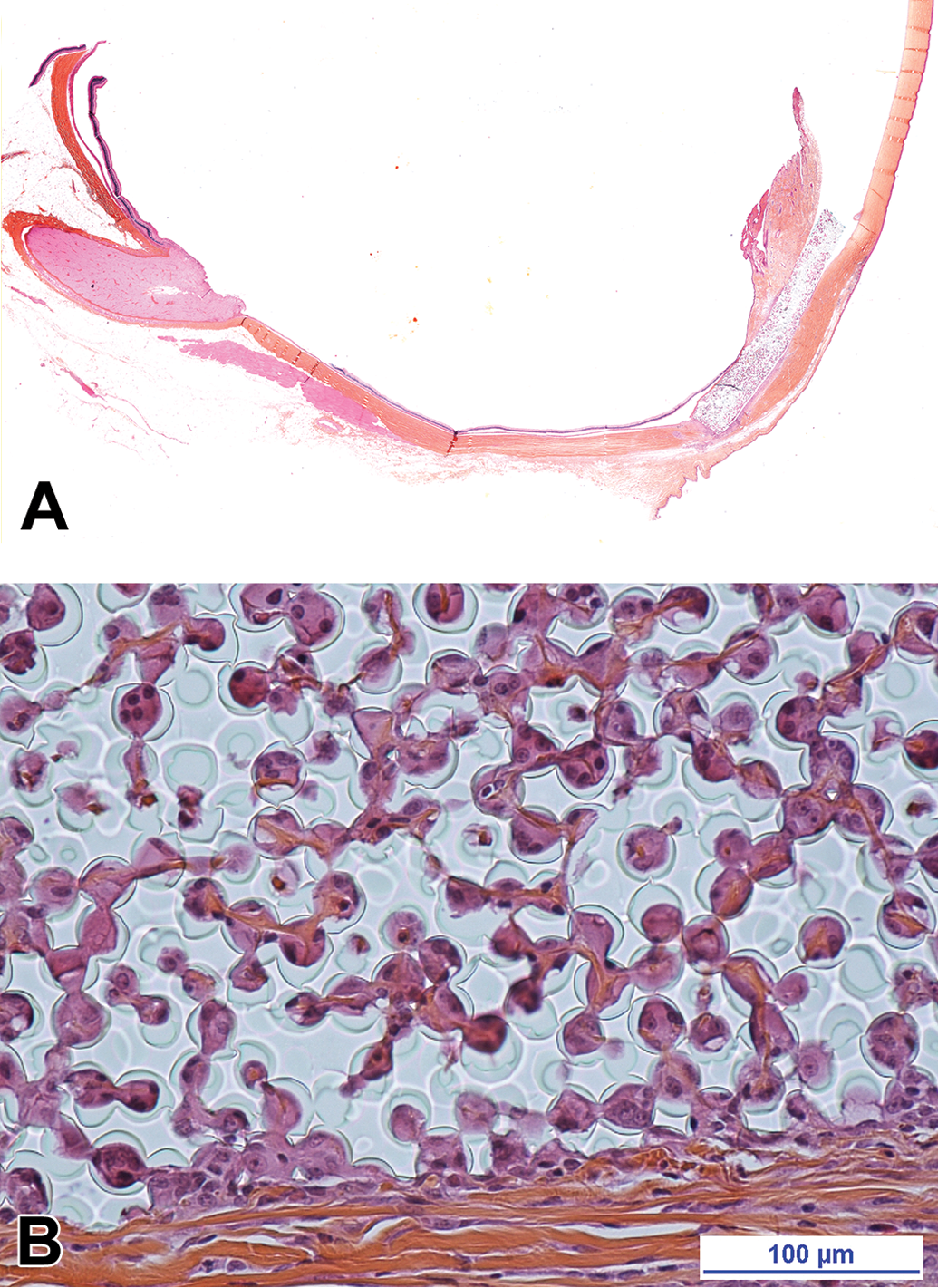
(A) Porous silicone drainage device implanted in a rabbit eye for 26 weeks (safranin, hematoxylin, and eosin stain). (B) The porous silicone implant is fully colonized by host cells and tissues (safranin, hematoxylin, and eosin stain).
IOL
In cataract patients, IOL are implanted in the capsular bag following removal of the opacified crystalline lens. They usually consist of a small central optic with side structures called haptics to hold the lens in place (Figure 2A). Of note, the current ISO 11979 standard requests the IOL to be removed from freshly enucleated eyes. Half of the explanted IOLs are then subjected to SEM analysis of surface deposits and changes, coupled with EDX analysis for detection of calcium and phosphate elements to detect surface calcification; the other half of the explanted IOLs are used for analysis of optical properties. The remaining ocular tissues after explant of the IOL are examined microscopically. This request in the standard was likely due to the technical difficulties in histologic processing of acrylate IOL. However, IOL removal from freshly enucleated eyes clearly provides suboptimal histology. It is our opinion that half of IOL-implanted eyes should have the IOL removed and analyzed as per the ISO request. The other half should be left untouched and histologically evaluated with the IOL in place in order to get robust biocompatibility results. Histological processing of this type of implant, if left in place for processing, requires equatorial dissection with the IOL maintained in the anterior pole. This trimming should be done following complete fixation via the window technique. Following short fixation in a 1:1 solution of 10% NBF and 4% glutaraldehyde, a 2 × 2 mm aperture or a 5 mm slit is created in the sclera just caudal to the equator, to allow for internal fixation. Hardening of the eyeball is accomplished by transfer of the globe to 10% NBF for at least 24 hr before trimming. After equatorial dissection, vertical parasagittal trimming is performed through the anterior half cup of the eyeball to obtain two unequal paraffin blocks: the “two-thirds” block will allow evaluation of the central part of the IOL, while the “one-third” block will allow evaluation of the IOL haptics (Figure 2B). A central block is also taken from the posterior half cup to allow evaluation of the posterior segment and the retina.
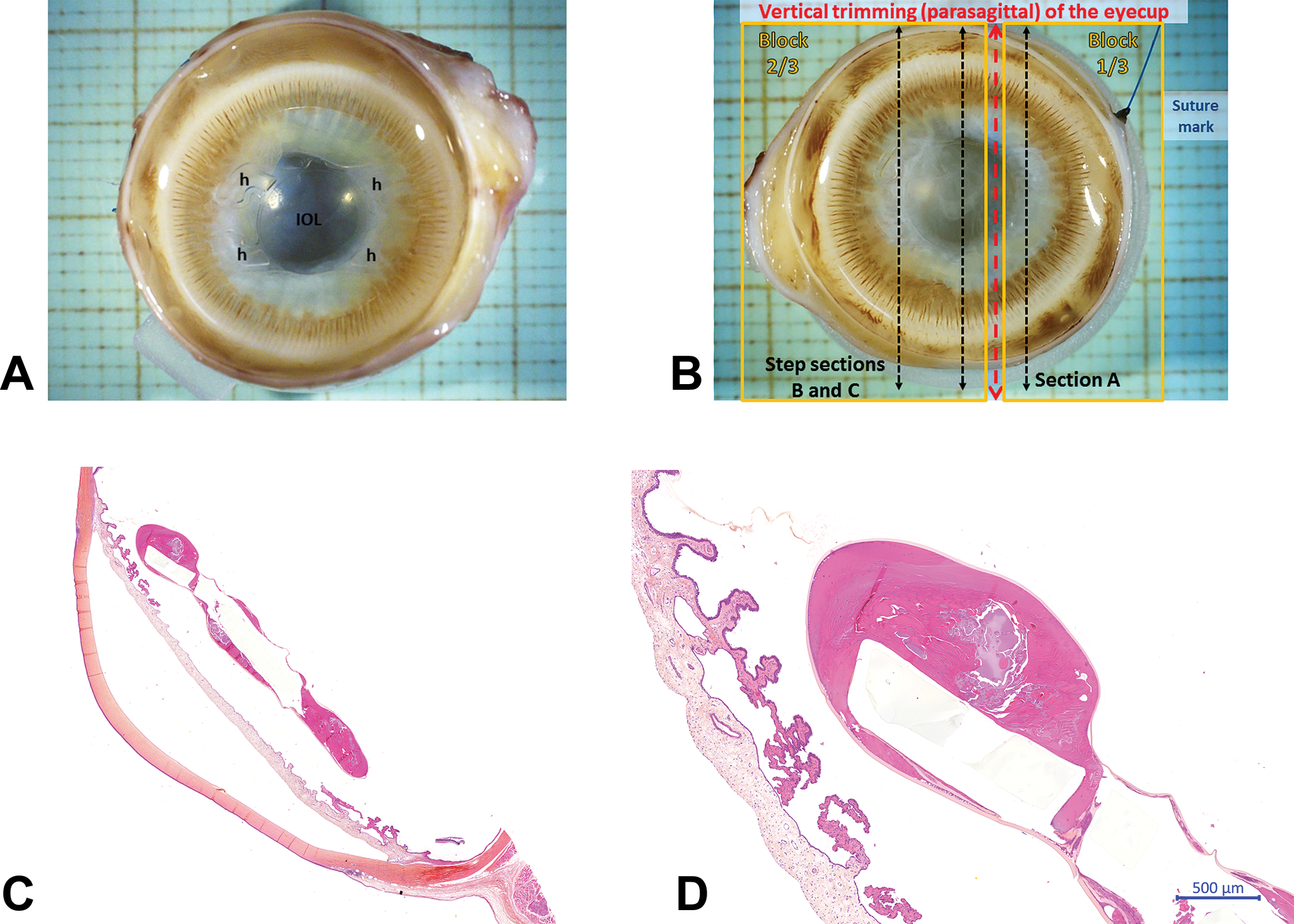
Intraocular lens (IOL) implanted in a rabbit eye for 12 weeks. (A) Macroscopic observation of the implanted IOL with the four haptics (H) visible. (B) Trimming scheme. (C) Microscopic observation at low magnification of the implanted IOL, following paraffin embedding and safranin–hematoxylin–eosin staining. (D) High magnification of the implanted IOL showing secondary cataract around the implanted IOL.
Histopathologic evaluation of IOLs is determined by the study purpose. Generally, an IOL will first be evaluated through a short, 3- to 6-weeklong performance study, which is sufficiently long to evaluate the performance in terms of lens epithelial cell regrowth (secondary cataract). A longer study, generally six-month duration, will assess the biocompatibility of the used biomaterial. The short performance study and the longer biocompatibility study will not evaluate the same histopathologic parameters: the performance study will focus on the influence of the IOL on the prevalence and/or severity of secondary cataract (Figure 2C and D). The biocompatibility study will focus on more classical histopathologic parameters such as absence of adverse inflammatory, degenerative changes, or necrotic changes. It should be noted, however, that performance and biocompatibility are frequently tightly related: an IOL that is not biocompatible can induce adverse changes that will in turn enhance the occurrence and/or severity of secondary cataract.
Drug Delivery Devices
Drug delivery devices are combination products that associate a biomaterial with a drug designed to be progressively released in a specific ocular compartment. Because of the difficulties for pharmacologically active agents to gain access to ocular compartments, delivery devices represent a wide and growing category of devices and the most recent advances are regularly reviewed (Morrison and Khutoryanskiy 2014; Kuno and Fujii 2011). Many ocular delivery devices are applied topically, such as eye drops, ophthalmic gels, and ointments, or also via small implants that are inserted in the inferior or superior cul-de-sac. Contact lenses can also be loaded with pharmacologically active substances. All these topical devices are evaluated through classic ocular irritation studies.
Other ocular delivery devices are injected or implanted in the posterior segment (vitreous). Drug-eluting IOL generally contain antibiotics, while intravitreal implants often contain glucocorticoids. A classic example is the Retisert® nonresorbable implant made of silicone that is used to treat posterior uveitis. Following surgical implantation, this device releases fluocinolone acetonide for 30 months and then has to be removed and replaced.
As for all combination products, the testing strategy will depend on the classification of the delivery system. Most of these systems rely on a device whose only role is to release the pharmacologically active substance. Since the device does not have a role per se (like a glaucoma shunt that has a mechanical filtering effect), it is classified as a drug and tested as such. On the contrary, a drug-eluting IOL will be considered an MD since the primary mode of action remains the functional restoration of the cataractous lens while the antibiotic coating of the IOL is only an ancillary aimed at preventing secondary bacterial infection.
Retinal Prostheses
Retinal prostheses are designed for sight restoration of patients with retinal degeneration. The prostheses work by electrically stimulating the remaining functional neurons of the damaged retina. These implants are epiretinal (lying on the retina, Figure 3A), subretinal (in the subretinal space), or suprachoroidal (between the choroid and the sclera). Epiretinal prostheses are generally associated with a pair of glasses that either captures images or provides energy to the retinal implant. Images are converted into stimulation signals either in the implant itself or by a pocket computer in the glasses and then transmitted via the electrode arrays of the implant to the retinal cells.

Epiretinal prosthesis implanted in a domestic pig eye for 4 weeks. (A) Macroscopic observation of the implanted prosthesis following dissection of the posterior eyecup. (B) Microscopic observation of the implanted prosthesis following plastic embedding and modified Paragon staining (inset: high magnification of the retina, modified Paragon).
All currently evaluated retinal prostheses have metallic electrode arrays which require plastic embedding of the implanted retina (Figure 3B). The trimming scheme should include both the implanted retinal area and unimplanted retina. Histopathologic evaluation should focus on retinal integrity since these prostheses rely on functional retinal neurons. Evidence of retinal detachment should also be carefully evaluated. Good histologic technique is necessary to allow the pathologist to distinguish between genuine retinal detachment that can be a consequence of the implantation procedure and artifactual detachment related to trimming. Finally, the optic nerve should also be carefully evaluated since degenerative changes can be observed following implant-related retinal degeneration.
Conclusion
Ocular MD represents a very wide and promising field of human ophthalmology. In preclinical studies evaluating the safety and/or performance of these MD, the choice of histologic technique and the focus of the histopathologic evaluation should take into consideration the following aspects: the specific guidelines possibly associated with the MD, the ocular compartment in contact with the MD and its specificities, and last the nature of the biomaterials used in the MD. Figure 4 summarizes the different types of ocular MD and their associated guidelines and should help in determining the most adequate histologic technique.
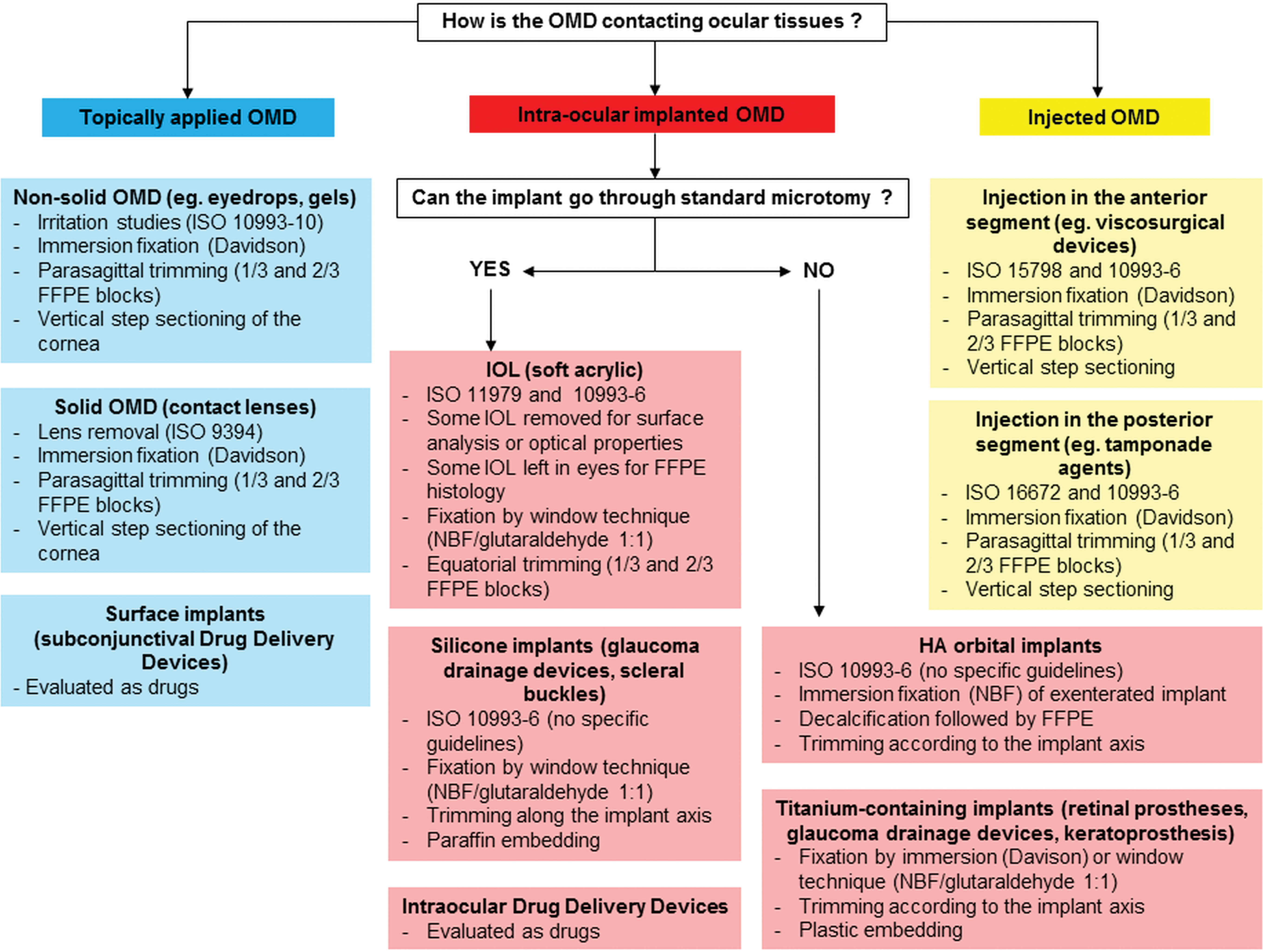
Summary of the different types of ocular medical device associated guidelines for evaluation and specificities of the histologic technique (FFPE: Formalin-Fixed Paraffin-Embedded, HA: Hyaluronic Acid, IOL: Intraocular Lens, NBF: Neutral Buffered Formalin, OMD: Ocular Medical Device).
Footnotes
Acknowledgments
The author would like to thank Karen S. Regan, DVM, DACVP, DABT, for the valuable proof reading and James A. Render, DVM, PhD, DACVP, who gave very helpful advices for the design of preclinical ocular studies conducted at North American Science Associates.
Author Contributions
The author contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential, real, or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.