
Abstract
The U.S. Food and Drug Administration Center for Devices and Radiological Health (FDA/CDRH) has recently published several in vivo test guidance documents that mention refinements, reductions, or replacement animal testing strategies to facilitate the leveraging of data from large animal safety tests for conventional rodent testing. In response to the recently enacted Food and Drug Administration Safety and Innovation Act Section 907, which facilitates expedited access to novel therapies commonly described as Breakthrough Therapy Designation, FDA/CDRH has discussed efficient regulatory strategies for first-in-human investigation, including early feasibility study guidance. Large gains in humane care and translational research could also be attained by examples in FDA’s Guidance for the Use of International Organization for Standardization 10993-1, which states that large animal safety studies may be considered as replacement rodent tests if the scientific principles, methods, and end points (SPME) are considered and applied. This article discusses SPME for the replacement of conventional rodent testing by the inclusion and integration of clinical, diagnostic, and pathologic data obtained from well-designed large animal studies. The recommendations include consideration for study designs that utilize methods for an overall more comprehensive interrogation of animal systems.
Keywords
The purpose of the International Organization for Standardization (ISO) 10993 series of testing is to render a reasonable evidence of safety about regional or systemic responses to the test device. It should be remembered that ISO 10993-1 describes general principles governing the biological evaluation of medical devices, including testing within the framework of a risk management process. In particular, section 6.2.1 dictates the test procedures taken into account that while the protection of humans is the primary goal of the risk management program, a secondary goal is to ensure animal welfare and to minimize the number and exposure of the test animals (International Standards Organization 2009).
A research animal replacement, refinement, and reduction of test strategy demands that key personnel from industry and U.S. Food and Drug Administration (FDA) who are involved in the submission and review of safety data to regulatory authorities consider integration of clinical, toxicological, and veterinary methodology. The goal of reducing animal use in medical device testing is not new and indeed there has been a significant push to develop and validate new and improved test methods, both in vivo and in vitro. A recent article by Myers (2017) comprehensively outlines the current regulatory landscape with respect to ongoing efforts to replace animal models with validated in vitro tests. This article discusses the apparent contrast between the professed desire by regulatory bodies such as FDA and the European Union for replacement of animal biocompatibility tests with the reality of reluctance and lack of demonstrable progress in accepting new methods. Myers (2017) presents validated in vitro tests that in principle satisfy ISO 10993 standards for irritation, sensitization, pyrogenicity, and acute toxicity including some whose sensitivity and accuracy are superior to currently advocated in vivo tests. This article does acknowledge that there are some biocompatibility tests such as thrombogenicity and clinically relevant implantation that still rely on animal data. Where animal data are still required by the regulatory process, there should remain a significant and sustained effort by the medical device and regulatory communities to reduce the animal burden.
In vivo test data prepared for the Center for Devices and Radiological Health (CDRH) of the U.S. FDA are often but not always submitted and reviewed by separate teams of corporate and regulatory personnel. Information intended to support the biological reactivity of medical devices is obtained through testing in accordance with the scientific principles and methodology of the ISO 10993 series of tests based on characterization of the device by type of tissue and contact duration. The biocompatibility standards set forth in ISO 10993 are specific and provide limited latitude for interpretation. In contrast, data obtained from large animal studies to support the safety of a medical device are typically reviewed by scientific experts with reference to applicable device-specific guidance and risk assessments. The availability and scope of such reference documents for a particular device or device class is more variable, and their application in the review of safety data is therefore more open to variability in reviewer-specific interpretation. Moreover, sponsors and regulatory reviewers might differ in their view of specific recommended acute or chronic safety tests utilizing animal species such as rabbits, dogs, small ruminants, and swine. One example is with respect to levels of departure from identical animal cohorts. Specifically, a particular safety study reviewer might strongly advocate for the use of traditional sex-matched cohorts with identical study numbers regardless of the device being tested or the type of data being sought. However, a well-designed large animal study with more clinically relevant routes of exposure or administration could offer safety data of equivalent or superior quality without needing identical cohorts in number or sex. In the authors’ view, such considerations for appropriate, clinically relevant large animal safety study design may reduce the need for overall numbers and controls while preserving the utility of testing.
This pattern of segregated preparation and review and allegiance to identical scientific methods used in ISO 10993 in vivo tests can miss opportunities of animal refinement, reduction, and replacement (3R) as advocated in ISO 10993-1:2009 as well as FDA/CDRH’s more recent final biocompatibility guidance and draft cross-center animal studies guidance (U.S. FDA 2016).
More importantly, the methods of examination and system interrogation commonly utilized in large animal research care and use, with emphasis on principles of modern veterinary observation and documentation, generally provide higher level of scientific principles, methods, and end points (SPME) intensity, duration, and a more robust biological data down to an organ system and tissue level of detail. These more clinical veterinary-specific methodological and study design attributes could theoretically make for a better opportunity to interrogate the safety of the medical device because they stem from efforts at enhanced frequency and intensity of monitoring to detect adverse events in animal research subjects. Enhancement in the comprehensiveness and reliability of medical device safety testing is a shared objective in the overall mission to protect human health.
Complimentarily, FDA/CDRH leadership is seeking ways to study and make available breakthrough technologies and to provide human clinical research trial approvals for iterative technology where imperfect biologic systems exist (U.S. Congress 2012; U.S. FDA 2013). CDRH’s recent Strategic Priorities also align with more efficient use of postmarket data to streamline regulatory decision-making (U.S. FDA/CDRH 2017).
An effort at aligning 3R and facilitating Breakthrough Technology or first-in-human (FIH) technologies seems attainable by consolidating all in vivo test requirements: those deemed necessary from the ISO 10993 series and those targeted for large animal safety testing into a systematic and comprehensive animal test plan whenever possible.
Importantly, the referenced biocompatibility guidance emphasizes that large animal replacement testing may be utilized if the SPME from the ISO 10993 test were considered and applied.
The purpose of this article is to provide logistical approaches to the clinical relevance and potential applicability of large animal tests where opportunity exists to replace classic rodent tests from the ISO 10993 series and enrich the correlative value to the data for the determination of safety for human subjects by applying sound considerations for SPME.
Initial Considerations
Overall Rationale for Leveraging Large Animal Studies
Before any nonconventional test strategy is attempted, the rationale for why this may be acceptable should be discussed with FDA through FDA’s presubmission and feedback program (U.S. Administration 2014).
Within the context and spirit of presubmission planning between FDA and industry, Annex B of ISO 10993-1:2009 Section B.2 provides general guidance for the biological evaluation of medical devices using a risk management process, including the provision of a rationale for the tests selected. Most medical devices involving any significant risk necessarily evoke one or more large animal safety tests to address those key risks. At this juncture of the risk management process, the ability to consider end points and methodology to replace rodent biocompatibility test requirements can be considered.
Assimilation of Experts
An appropriate risk management process using Annex B of ISO 10993-1:2009 and recommendations from the 2016 FDA biocompatibility guidance would ideally involve participation of a multidisciplinary team, including FDA, industry, and outside experts. This group would assess the risks for which the animal testing is intended to address and determine whether the proposed replacement strategy results in any areas of insufficiency. If any areas are deficient, the group would determine whether they are acceptable, given the other available information the company is providing to FDA. Currently, this multidisciplinary process is variable at FDA and depends heavily on the categories of information sent to FDA, the experience of the lead reviewer in convening predecisional meetings, and FDA’s internal efforts to adequately discuss and determine risk of the sponsor’s biocompatibility and large animal test plans to provide feedback to the sponsor. It also often requires the FDA human physician or clinical team member to render expertise. Specifically, it is essential to review proposed testing plans within the context of the device’s intended use and intended treatment population so that any interpretive insufficiencies or voids in information are identified and their potential implications for the human clinical population clearly articulated. Typically, FDA parcels sections of a submission according to the information submitted. Thus, a team clinician may not be assigned if there is no clinical data to review. There may also be insufficient internal FDA discussion of similar or identical deficiencies developed independently by multiple expert reviewers that are simultaneously conveyed to the sponsor despite their redundancy. Thus, if a particular expert is deemed necessary by the sponsor for the review of the proposed replacement testing, it is suggested that this opinion be conveyed to FDA in the cover material of the presubmission.
Numbers of Animals
One perceived deficiency to the replacement of rodent biocompatibility testing is an objection to the general omission of sex-match controls in large animal studies, for instances, in which those considering the value of a replacement test prefer identical cohorts to conventional rodent tests. As an example, the review experts may feel that there should be identical numbers of nulliparous ewes and rams in the large animal study; however, the test site might determine that such a construct would be particularly burdensome as large animal housing of intact males can raise per diem costs due to the need for separation to minimize male–male conflict. Indeed, the majority of large animal studies in the authors’ experience use one sex and rarely have safety issues that are disparate between chronic rodent safety tests (implant, systemic toxicity) and large animal tests. Accordingly, those in favor of reduced animal use should be prepared to provide a scientific rationale for why the risk analysis for the particular device is reasonably addressed in the replacement testing if both sexes are not studied equally.
There is also a paucity of information about the translational importance of including identical sex cohorts in preclinical trials. While it is generally accepted that such inclusion represents good science, proponents of increased numbers of animals in large animal studies have a difficult task to demonstrate that this additional animal burden has translated to greater predictive value for research or marketing submissions or that gender difference in safety outcomes would reasonably be detected even in cohorts of the size recommended for rodents (Sandberg 2014). In addition, the numbers of animals in either large animal or rodent tests are not generally statistically powered to detect meaningful differences in gender outcomes; and therefore, this perceived deficiency should be weighed against the overall cost of redundancy, particularly if a large human trial will be suitably powered. This is consistent with respect to CDRH’s recently stated strategic priorities (U.S. FDA/CDRH 2017).
The use of positive and negative controls
Large animal study design may compare a marketed predicate to a test device or if the systems evaluation is possible through well-understood acceptance criteria for tissue reactivity or physiologic response, instead may review outcomes based on well-understood acceptance criteria for each of the proposed end points. Another area in which reviewers and the public may have difficulty applying SPME is in whether the large animal testing methodology should include positive control groups for the evaluation of systemic responses.
It is important to note that one major reason for which positive and negative controls are needed in rodent biocompatibility testing is that rodent tests for systemic evaluation use an extract of the test article. Exposure to the test article via an extract is indirect and rather nonclinically relevant and so any effects seen must be compared to effects from controls. Further, the route of exposure can itself be irritating (e.g., peritoneal injection). Overcoming these limitations therefore requires the design of adequate negative and positive control cohorts.
Such requirements can be impractical and unnecessary in a large animal test where the exposure is clinically relevant and direct.
Regardless of the route of exposure selected in the large animal study, a sound leveraging argument should include an explanation to FDA about why identical test cohort construct may be acceptable in the large animal replacement test.
Duration of Study
Other objections regarding the ability to leverage large animal studies for conventional rodent tests are often related to the duration of large animal studies and the implications for interpretation of chronic effects such as carcinogenicity. The relative age of the large animals versus rodents, principally in chronic systemic toxicity testing and chronic implant testing, is often a topic of concern for FDA. The longest animal testing for medical devices is usually two years and typically only for devices with novel materials. For most quadrupeds that comprise the available pool of large animal models, this represents at best 1/5 of their life span. For rats and mice, the same testing duration comprises significantly more of their life span such that it has theoretical applicability to biological responses over mid and geriatric life. These concerns generally arise with respect to a need to know about carcinogenic potential. A rationale may be suitable for a shorter percentage of the overall animal life span if other explanations are offered. For example, FDA has provided an option to provide chemical characterization and risk assessment of leachables and extractables in the referenced 2016 Biocompatibility Guidance (U.S. FDA 2016) that may suffice to explain why carcinogenicity concerns may not be an issue. Other rationales are again centered in FDA’s strategy to streamline regulatory decisions through collecting more safety information from the target species (human) in the postmarket period (U.S. FDA/CDRH 2017).
Individual Tests for Consideration
Systemic Toxicity (Acute, Subchronic, Chronic, and Pyrogenicity): ISO 10993-11
Perhaps the most important safety characteristic of a device is its absence of toxic systemic effects on all timescales. A device and its constituent materials can potentially impact the macro- and microstructure and function of organs as well as generalized physiologic homeostasis beyond the site of device–body contact. Toxic effects can be seen at any point along a continuum from acute (e.g., anaphylaxis) to chronic (e.g., renal insufficiency secondary to particulate accumulation). Device systemic toxicity testing is performed in accordance with ISO 10993-11, with rodent and lagomorph models historically preferred on the basis of study numbers and sex-matching. However, ISO 10993-11 states there is no absolute criterion for selecting a particular animal species for systemic toxicity testing (International Standards Organization 2017b). Therefore, the opportunity exists to leverage appropriate large animal testing for systemic toxicity. Indeed, in many cases, a large animal model with the device in a clinically relevant site and used as clinically intended can provide more accurate data regarding expected human systemic toxicity. For example, a drug-eluting stent implanted in a sheep vessel the same size and location as its intended human clinical implantation site would yield substantially more clinically relevant and translatable data than would an extract of the device injected intraperitoneally to a rodent.
Methods of Testing
ISO 10993-11 summarizes the various routes of administration for toxicity testing, including implantation, intramuscular, subcutaneous, intravenous, and intraperitoneal. The standard states that the most relevant route of administration shall be used (International Standards Organization 2017). It follows, logically, that toxicity testing for a particular device should be performed using device components and exposure routes that reflect the intended clinical use of the device and that failure to do so can lead to findings that are inconclusive or confounding. However, in practice, it is the authors’ experience that rodent toxicity testing routinely utilizes intraperitoneal administration of test articles regardless of the nature of the device being tested. In addition to not being clinically relevant for most medical devices currently in development, intraperitoneal injection can potentially result in inappropriate injection location (i.e., into an abdominal organ or hollow viscus), variation in substance absorption and metabolism, induction of peritonitis or sclerosis, and animal pain that are not necessarily attributable to the test device or components (Claasen 1994; Turner 2011). Variability in operator technique can have a significant effect on outcome, for example, in the development of peritoneal fibrous adhesions. Fibrous adhesions can occur in both test and control groups, confounding results for both sets of animals. The use of intraperitoneal injection also necessitates comparatively large numbers of rodent subjects as device extraction requires polar and nonpolar solvents as well as positive and negative controls. In contrast, a large animal model with the device placed in a clinically identical or relevant location and dedicated, objective clinical monitoring provides accurate, trustworthy, and relevant toxicity data while reducing the overall animal burden.
Evaluation of Testing
Evidence of systemic toxicity is obtained through three major routes in any animal model: clinical evaluation, clinical pathology, and anatomic pathology. Clinical evaluation ideally yields in-life information regarding an animal’s overall clinical status including behavior, appetite and water intake, presence of overt signs of pain or distress, and signs of systemic compromise (e.g., dyspnea, seizure activity, hemorrhage, and profound lameness). Clinical pathology provides time point and trend data for hematologic and biochemical parameters that can be compared to individual baseline as well as accepted species- and breed-specific standards. Anatomic pathology permits the gross and histologic assessment of major and minor organs, including those with direct device contact as well as downstream and end-organ targets. Taken together, these data sets create an overall clinical picture of animal health and systemic stability following treatment with a device and can reveal evidence of toxicity throughout the observation period.
Clinical Evaluation
A major limitation of traditional rodent and lagomorph toxicity testing models is the perceived lack of need or the real lack of training or staffing to adequately and meaningfully evaluate in-life health and to correlate this with clinical pathologic monitoring. Often, animals are merely observed, and this task is delegated to husbandry or facility staff who have limited or no veterinary training. While these individuals can report on the vitality, food and water intake, and weight change of study animals, subtler clinical signs and direct physical examination findings are not obtained and are at risk of being missed altogether. In the context of the 3R effort, this can mean the loss of a substantial amount of useful animal data that might ultimately require the use of more animals or repeating a study.
The use of a large animal model with a clinically relevant device and implant location allows the sponsor to draw accurate and meaningful conclusions about systemic toxicity findings and increases confidence in the absence of toxicity. Daily evaluation of animals by trained veterinary staff (veterinarian and veterinary technician) following the problem-oriented veterinary medical record format provides consistent, reliable, and thorough data regarding in-life health and potential clinical manifestations of underlying toxicity provided adequate case report forms are kept. Physiologic parameters (heart rate, respiratory rate, and temperature), physical and neurologic functions (posture, gait, mentation, and cranial and peripheral nerves), thoracic auscultation, abdominal palpation, rectal and urogenital evaluation and palpation, and external surface assessment and palpation are all routine components of the veterinary subjective, objective, assessment, and plan (SOAP; Ettinger 2010; Hampshire 2015; U.S. FDA 2010). This is in stark contrast to a rodent or lagomorph model with a single pathologic evaluation at termination and minimal commentary on the preceding in-life period. Importantly, the SOAP provides a standardized documentation for the identification of abnormalities, their assessment and diagnosis, and a plan for further evaluation, treatment, and any change to the animal’s participation in the study.
Table 1 compares and contrasts differences between the standard clinical signs and evaluative criteria and the large animal veterinary SOAP. A sample SOAP is provided in Table 1.
Comparative Clinical Reporting and Evaluative Criteria Large Animal (Veterinary Subjective, Objective, Assessment, and Plan [SOAP]) versus Rodent (International Organization for Standardization [ISO] 10993-11).
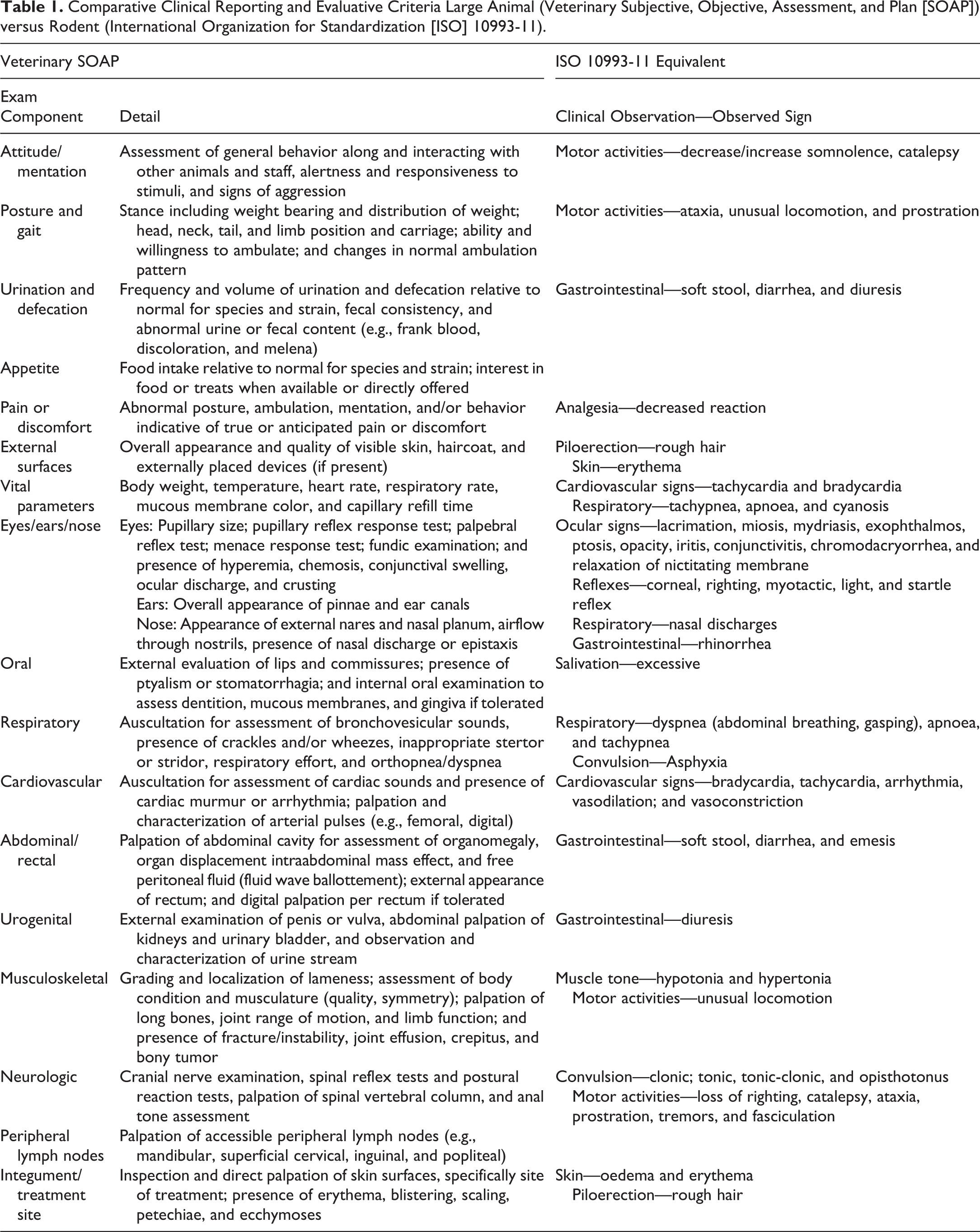
In the large animal evaluation, depending on the device being tested, complete physical examination can also be augmented by other noninvasive diagnostic techniques such as ultrasound, echocardiography, advanced imaging (Computerized Tomography, MRI), and fluoroscopy/angiography for interim time-point assessment of device functionality, vascular and hemodynamic status, and tissue changes.
Physiologic Monitoring
Dedicated physiologic monitoring is particularly useful for assessment of device pyrogenicity. During the index procedure and periprocedural anesthetic recovery period, standard of care includes frequent monitoring (i.e., every 15 min) of animal body temperature. This initial period up to several hours of initial device exposure encompasses the time frame in which a device-related systemic pyrogenic reaction and febrile response would be expected to be seen and is theoretically superior to a rabbit pyrogen test which relies on abstract and somewhat arbitrary criteria for test repetition in the case of questionable test results (Ogoina 2011; Cartmell 2002). Briefly, the rabbit pyrogen test involves the intravenous (ear vein) injection of an extract solution of the test article into a cohort of three rabbits and monitoring for individual body temperature increase over 3 hr. Diligent temperature monitoring in a clinically relevant animal model, alone or in combination with periprocedural clinicopathologic evaluation, should therefore detect pyrogenic effects.
Annex G of 1SO 10993-11 provides information about materials-mediated pyrogenicity and expressly states that it is not necessary to test all new medical devices for in vivo pyrogenicity but rather suggests that materials containing substances that have previously elicited a pyrogenic response and/or new chemical entities should be tested. In the authors’ experience, although the FDA guidance Attachment A recommends the test “for consideration,” it is unusual for FDA to exempt this requirement.
However, no other test in the ISO 10993 series seems more arbitrary or wasteful of animal life and research burden when applied to medical devices. The traditional scientific rationale is arguably inapplicable since this particular test was developed in accordance with U.S. Pharmacopeia (2014) under Section 51 in response to recommendations to test biologics in Section 1041.
The test as applied to rabbits requires test article extraction and is not a clinically relevant route of exposure or without added distress during restraint and injection (Parasuraman 2017).
Specifically, the rabbit pyrogen test initially utilizes 3 rabbits. The methods are to extract the test article in normal saline at 50°C for 72 hr, cool the extract, warm the extract to 37°C, and inject 10 ml/kg of the extract via an ear vein. A baseline temperature is obtained followed by additional temperature measurements taken every 30 min for 3 hr. The evaluation criteria are as follows: If no rabbits show a temperature increase of at least 0.5°C above the baseline temperature value, the test article passes the test and is considered free of pyrogens; If any animal shows a temperature increase of at least 0.5°C, the test is continued using an additional 5 rabbits; and If no more than three of the now 8 test article recipients were to show temperature increases of more than 0.5°C and if the sum total of the eight individual animals’ largest temperature increases does not exceed 3.3°C, then the test article passes the test and is considered to have passed the pyrogen test.
The SPME in a large animal medical device study are arguably superior because the route of administration is identical to the intended human setting under usual and ordinary contexts of use, the methods of detection are more frequent and the duration of observation for fevers is longer in modern large animal settings with acceptable monitoring practices and testing usually occurs under sedation, anesthesia, or in the early perioperative period during additional physical examinations under gentle restraint following acclimation to restraint. The end point (fever) is the same.
One potential confounder in studies intended to leverage large animal testing to replace pyrogen testing is the use of nonsteroidal anti-inflammatory analgesics which may suppress febrile responses and should be utilized in a timing strategy that would not confound the interpretation of body temperature.
A second potential confounder is lack of attention to the provision of heating apparatus during surgery to avoid artificially lowering body temperature due to hypothermic responses under anesthesia.
The FDA guidance for pyrogen testing also makes the distinction between materials-mediated and endotoxin-mediated pyrogenic responses with the recommendation that endotoxin-mediated responses be addressed in the sterility section of regulatory submission. This supportive test information is usually ex vivo and is exclusive of the arguments made for materials-mediated animal test replacement strategies.
Clinical Pathology
Modern veterinary standard of care in large animal safety protocols includes clinical pathologic monitoring for hospitalized patients. Blood work and urinalysis are indispensable for detecting organ-specific and general systemic markers of inflammation, infection, and organ dysfunction. These tests also provide the basis for identifying trends in response to treatment. Additionally, many interventional and peripheral vascular device applications require hematologic monitoring of antiplatelet and/or anticoagulant therapy during and chronically following the index procedure. Although traditionally required at baseline and termination, clinical pathology data obtained at interim time points can validate and bolster clinical observation and eventual anatomic pathological findings as well as provide insight into the pathogenesis and progression of systemic toxic responses.
Obtaining blood samples from a smaller set of large animals is arguably also more technically and economically feasible than from a larger cohort of small animals. Additionally, in many large animal models, reference ranges and normal values have been established to facilitate more direct comparison with human hematologic and biochemical parameters.
Consistent with the initial considerations in large animal replacement testing, it is important to bear in mind that in studies that place both a test and control device in the same animal, clinical pathologic abnormalities consistent with systemic toxicity cannot alone be cleanly attributed to either test or control. In such cases, local tissue response can still be differentiated histopathologically, but laboratory values must be interpreted in light of the inherent confounding effect. If all are within expected reference range, it can be assumed that neither device had an adverse physiologic effect, but if there are abnormalities, a rodent study design with clear separations test or control recipients is recommended.
Table 2 characterizes hematology, serum chemistry, and urine analysis values commonly obtained in large animal studies at baseline, interim, and term. The values are similar for rodents; ordinarily at baseline and term, and in some test protocols, samples may be pooled in each cohort in order to compensate for minimum blood volumes needed.
Routinely Measured Hematology, Serum Chemistry, and Urinary Values.
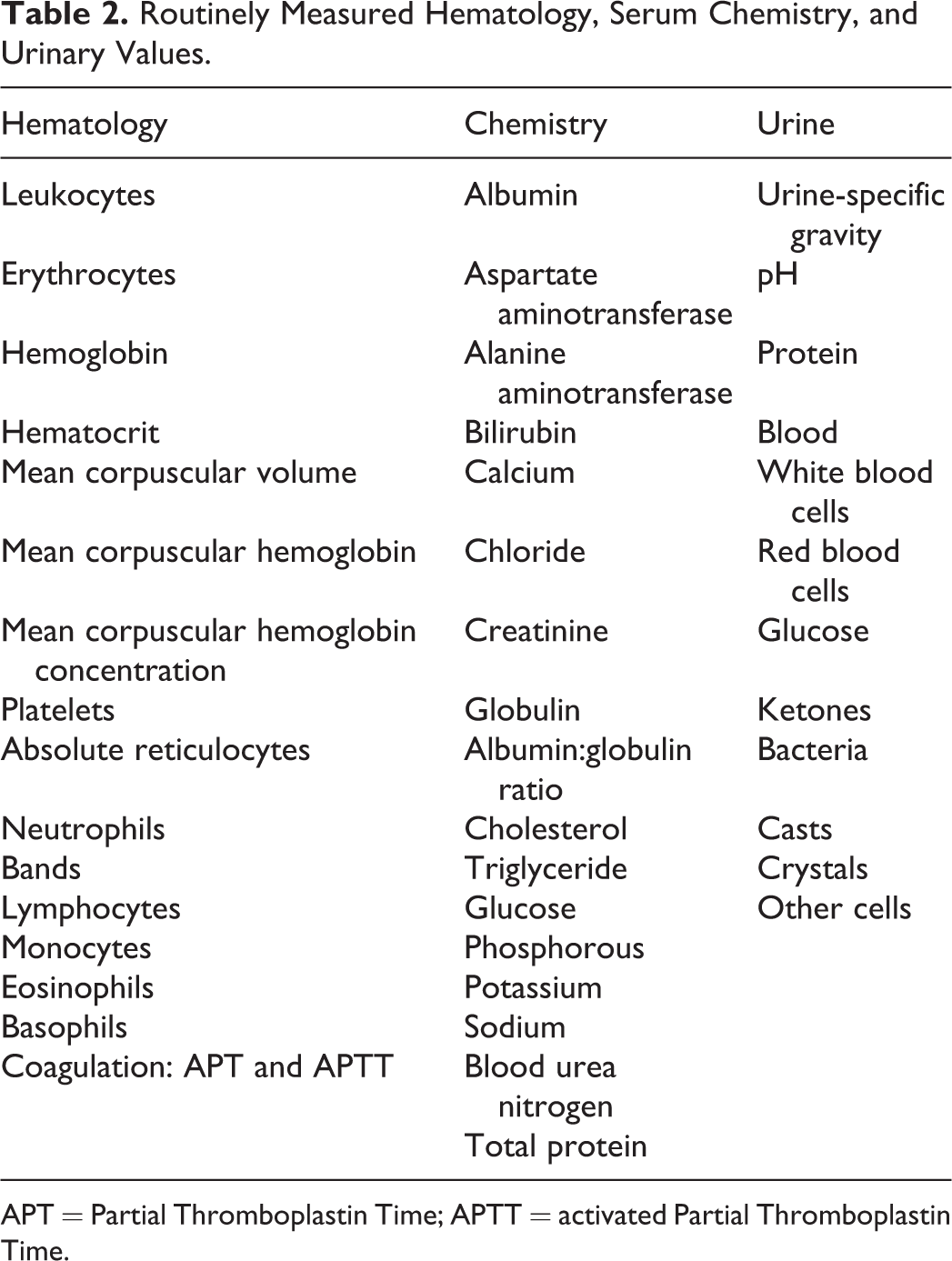
APT = Partial Thromboplastin Time; APTT = activated Partial Thromboplastin Time.
Anatomic Pathology
The thorough evaluation of gross and histologic samples from device-contacting and downstream tissues is at the core of testing for systemic toxicity. Pathologic evaluation provides definitive evidence of organ macro- and microlevel changes, vascular abnormalities, and tissue response to the device and healing process. When interpreting pathologic tissue findings, the characteristics of the animal model and device exposure must be considered. Specifically, anatomic and physiologic differences between the animal model and intended human clinical application (e.g., skin and tissue thickness, vascular supply, organ dimensions, and metabolic capacity) are critical to the extrapolation of animal study data to device use in humans. In this sense, it is logical to evaluate for systemic toxicity in an animal model that most closely replicates or approximates the intended clinical scenario. In many cases, device sizing, anatomic targets, dosing of a drug component if present, and approaches for device implantation or deployment are best suited to a large animal model. The use of a large animal model with bilaterally placed devices—both test or one test and one control—can further reduce required animal numbers while providing clinically relevant conditions for as-intended or worst-case testing. While specific pathology protocols differ between individual contract or academic sites, an acceptable protocol should include histologic assessment of key tissues including heart, liver, kidneys, spleen, lungs, brain, skin, muscle, and others as applicable (adrenals, reproductive organs, long bones, and bone marrow). If other gross abnormalities or clinicopathologic findings suggestive of specific organ dysfunction are documented, then these organs should be sectioned and assessed histologically.
Chronic Implant Testing: ISO 10993-6: 2016
The local tissue response to a chronically implanted device is also of interest. In general, chronic tissue responses are evaluated in tests longer than 3 months as in muscle and connective tissue a steady state tends to be seen in the cell population between 9 and 12 weeks (International Standards Organization 2016). The standard describes implantation in subcutaneous, muscle, bone, and brain tissue but states that the test sample shall be implanted into the tissues most relevant to the intended clinical use of the material (International Standards Organization 2016). Semiquantitative and quantitative scoring systems have been described for evaluation of local biological effects, including capsule formation, inflammation, presence of different types of leukocytes, and degradation of the test material (International Standards Organization 2016). One of the most common models currently in use is the rabbit muscle implantation test, which is used to evaluate for muscular and perimuscular tissue reactivity to the implant. While this can be appropriate for testing a device intended for muscular implantation in a clinical setting, it is likely unreasonable, for example, to similarly test a neurovascular stent. In such instances, a large animal model with a clinically relevant device implantation site would provide superior data regarding the reaction of the tissues that would ultimately be exposed to the device. Furthermore, a clinically relevant large animal model provides the opportunity to establish worst-case conditions at the intended implant site.
In traditional small animal testing models as well as a large animal model, terminal histopathology provides definitive information regarding tissue biological response. In certain circumstances, the large animal model can also facilitate additional in vivo data regarding local tissue changes. Specifically, noninvasive imaging and physical examination findings are commonly performed cage side in awake large animals and may also supplement terminal histopathology findings by providing a context for the timing and evolution of underlying tissue reaction and changes throughout a chronic study period. For example, ultrasound can be used to noninvasively evaluate the appearance of device-contacting tissues or vessels at interim time points sparing sacrificial time points at subacute and subchronic time frames. Davidson (2009), for example, reported specific evaluative criteria for the evaluation of reproductive tissues in the dog. Since dogs are a controversial research species and since the male reproductive tract of the dog is one of the few reasonable models for human male reproductive devices, such a strategy of longitudinal awake examination under ultrasound is not only extremely helpful, it can also provide noninvasive tissue sampling by guided ultrasound fine needle aspirate or Tru-Cut biopsy, but the findings may be put in context of other systemic responses obtained for clinical pathology by phlebotomy (complete blood count/serum chemistry). The aforementioned dedicated SOAP-based veterinary evaluation of large animal subjects includes observation and direct palpation of implantation sites (if accessible).
Thus, SPME commonly employed in large animal veterinary settings seems overall superior to those typically utilized in rodent test labs to satisfy requirements of ISO 10993-11 and should be strongly considered to enhance animal and human welfare.
In Vivo Thrombogenicity Testing: ISO 10993-4: 2017
Thrombogenicity testing is generally required as part of overall hemocompatibility testing when devices are in contact with the blood. Conventional canine testing has been the subject of recent national attention and dialogue due to inherent problems including its lack of clinical relevance, humane concerns, and frequently confounding findings (U.S. FDA 2014). The SPME in alternative testing to the traditional dog in vivo model was generally encouraged in this forum and since then several other in vivo testing strategies have been developed and explored. Since 2014, some ex vivo flow loop models have also been discussed where relevant and may be useful and acceptable to FDA (Grove 2017; Slee and Alferiev 2014).
There are many factors that can influence the thrombogenicity of a device tested in vivo, including species, subject positioning, anticoagulation regimen, implantation technique, device design and coating characteristics, and implantation location (International Standards Organization 2017a). Depending on the nature of the device and its intended clinical use, thrombus formation can occur on an acute (procedural or periprocedural) and/or chronic timescale. It is critically important that, as much as possible, the thrombogenicity testing be performed in a model that closely approximates its clinical use and that if multiple componentry such as delivery systems for short-term intravascular use and chronic intravascular implants receives adequate evaluation consistent with FDA’s interest in understanding each component’s contribution to any thrombogenicity.
For this reason, a clinically relevant large animal model is ideal, however, the acceptance criteria and end points should be determined consistent with the overall design controls and risk mitigation plans. Successful strategies involve a combination of techniques similar to the traditional canine in vivo test including digital photographs of the device before and after use and scoring paradigms for regional and downstream tissues. It is recommended that the preclinical team confer with the study pathologist and veterinary surgeons, develop a priori acceptance criteria and end points, and seek opinion from FDA in the form of a presubmission.
For most intravascular devices, an essential component of thrombogenicity testing is that the study subjects receive an appropriate anticoagulation regimen that matches or closely approximates the intended clinical regimen. Monitoring of coagulation parameters (activated clotting time, activated partial thromboplastin time, etc.), hemogram, and clinical pathology parameters before, during, and throughout the in-life period following the index treatment procedure provides confirmation of anticoagulation maintenance and absence of systemic signs of thrombogenesis. In particular models, additional hematologic testing for coagulation and fibrin formation (e.g., Enzyme-linked immunoassay (ELISA) tests for Thrombin antithrombin Thrombin (TAT) and Fibrinopeptide A (FPA)), platelet activation markers (e.g., beta-Thromboglobulin (βTG)), or blood coagulation efficiency (thromboelastography) might be useful (International Standards Organization 2017). At the index procedure, any temporarily blood-contacting device such as a delivery catheter should be visually inspected and documented by visual (photographic) and written means for presence or absence of thrombus immediately following use. During the in-life study phase, SOAP-based veterinary evaluation should be routinely performed to document clinical signs that could be related to thromboembolism (e.g., neurologic signs, lameness, dyspnea, and edema/vascular congestion). Noninvasive vascular imaging techniques such as ultrasound, echocardiography, fluoroscopic angiography, and CT-angiography can be performed to provide interim assessments of vessel and device patency, thrombus formation, and blood flow characteristics. Terminal necropsy should be performed with careful preservation and thorough evaluation of device-containing tissues, downstream thromboembolic target organs (lungs, kidneys, brain, etc.), and any other tissues with gross abnormalities or implicated by abnormal clinical signs. Under this approach, utilizing a clinically relevant large animal model, animal usage is limited, and clinically useful data are maximized.
Conclusions
The FDA has recently published key guidance documents emphasizing efficiency of medical device testing and best practices for animal studies (Table 3). Common to these are suggestions for ways to replace or reduce the overall animal numbers while simultaneously enhancing the periodicity and intensity of information reported for any particular animal model. Thorough interrogation of medical device safety and the principles of 3R in animal use are well aligned to produce robust, reliable data that ultimately benefit human health. Large animal studies that utilize clinically relevant SPME as well as objective, problem-oriented veterinary medical care provide comprehensive information encompassing device performance, acute and in-life device effects, and macro- and microlevel device-organ interactions. Data derived from well-designed large animal studies can potentially be leveraged to replace traditional small animal models for safety tests such as systemic toxicity, chronic implantation, and in vivo thrombogenicity. FDA/CDRH has shown a willingness to consider such leveraged testing and a dedication to finding ways to reduce overall animal burden. Continued cooperation between industry, academic, and regulatory parties to uphold and implement 3R principles will lead to streamlined safety testing, reduced animal use and improved welfare, and an overall reduction in economic resources needed to prove a medical device safe for use in humans.
Formative Public Law and Food and Drug Administration (FDA) Guidance Documents for Provision of Animal Reduction and Replacement Rationales.
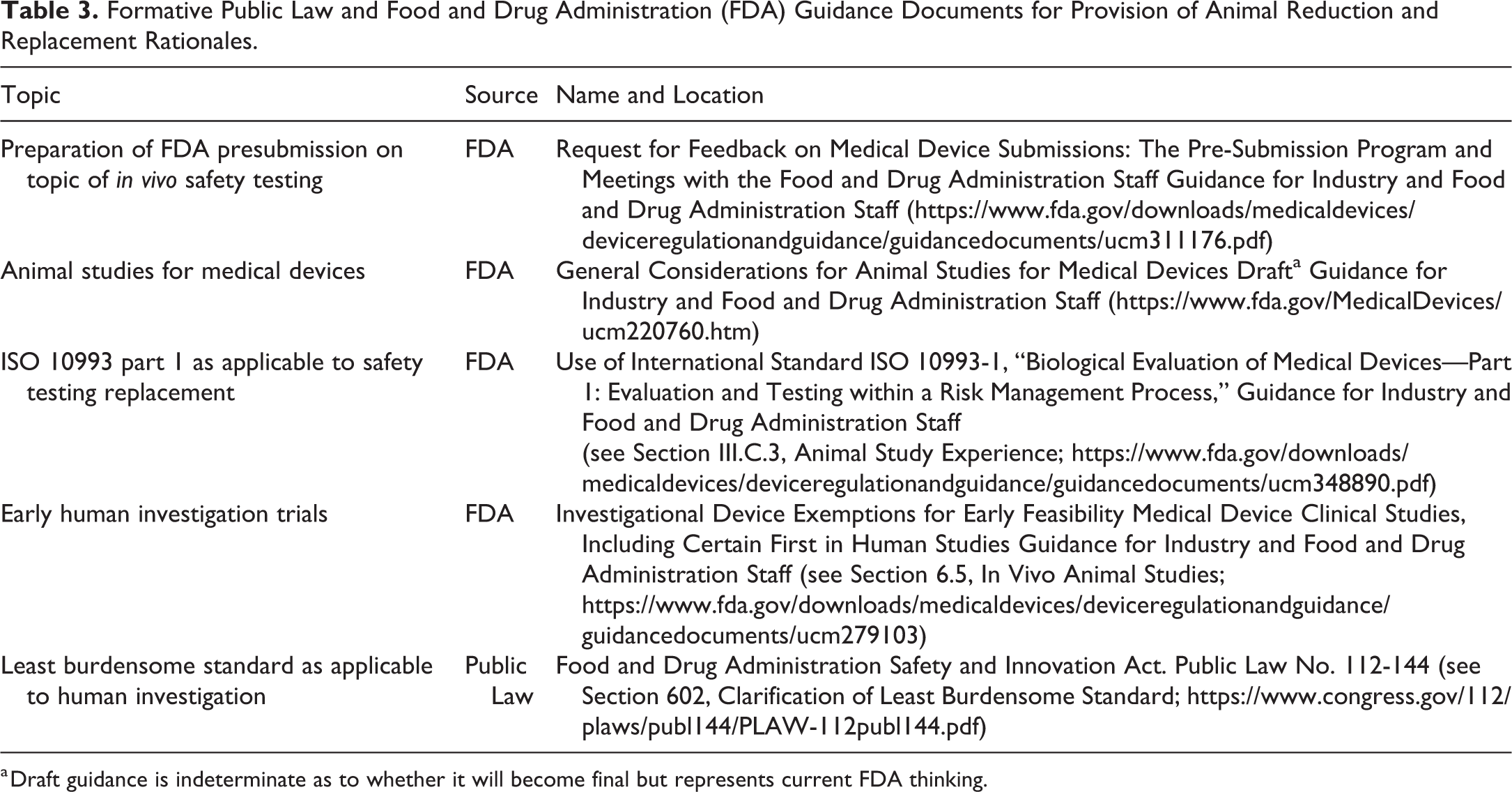
a Draft guidance is indeterminate as to whether it will become final but represents current FDA thinking.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.