
Abstract
Background and Aims:
Rats are resistant to acetaminophen (APAP) hepatotoxicity. In this study, we evaluated whether by augmentation of the hepatic oxidative stress, through the induction of hepatic iron overload (IO), it will be feasible to overcome the resistance of rats to the toxic effects of APAP.
Method:
Rats with no or increased hepatic IO.
Results:
Providing iron by diet induced hepatocellular IO, while parenteral iron administration induced combined hepatocellular and sinusoidal cell IO. APAP administration to rats with no IO caused an increase in hepatic oxidative stress and a decrease in the hepatic antioxidative markers but no hepatic cell damage. APAP administration to rats with hepatocellular IO further amplified the hepatic oxidative stress but induced only hepatocyte feathery degeneration without any increase in serum aminotransaminases. APAP administration to rats with combined hepatocellular and sinusoidal cell IO caused an unexpected decrease in hepatic oxidative stress and increase in the hepatic antioxidative markers and no hepatic cell damage. No hepatic expression of activated c-jun-N-terminal kinase was detected in any of the rats.
Conclusions:
The hepatic distribution of iron may affect its oxidative/antioxidative milieu. Augmentation of hepatic oxidative stress did not increase the rats’ vulnerability to APAP.
Keywords
Acetaminophen (APAP) is the most widely used analgesic-antipyretic. APAP is remarkably safe when taken at a therapeutic dose; APAP overdose has become the most common cause of acute toxic liver failure (Lee 2013; Jaeschke 2015).
During the metabolism of APAP in toxic doses, a reactive intermediate toxic metabolite, N-acetyl-p-benzoquinone imine (NAPQI), is formed. NAPQI depletes hepatic glutathione and binds to cellular proteins including mitochondrial proteins. These events are associated with an increase in the formation of mitochondrial reactive oxygen species (ROS) and peroxynitrite. As a consequence of hepatic glutathione depletion, the detoxification of ROS and nitrogen species is severely impaired (Jaeschke 2015; Xie et al. 2014). These events lead to phosphorylation of c-jun-N-terminal kinase (C-JNK) and its transformation into phospho-JNK (P-JNK). P-JNK then translocates to the mitochondria and amplifies the oxidant stress and peroxynitrite formation, mitochondrial DNA damage, mitochondrial membrane permeability transition (MPT) pore opening with collapse of mitochondrial membrane potential, cessation of adenosine triphosphate synthesis, and hepatocyte cell death (Jaeschke 2015; Xie et al. 2014).
Iron has a role in the pathogenesis of APAP hepatotoxicity. Studies in primary mouse hepatocytes (PMHs) revealed that during exposure to a toxic dose of APAP, ferrous iron is released into the cytosol from ruptured lysosomes. This form of free cytosolic iron initiates MPT pore opening and a sequence of events that lead to hepatic cell death (Uchiyama et al. 2008; Kon et al. 2010).
To date, the data regarding the effect of iron overload (IO) on acute liver injury from hepatotoxic agents are scarce and inconsistent (Moon, Richie, and Isom 2010; Moon et al. 2011). Studies performed in PMHs revealed that addition of an iron donor to PMHs that were treated with APAP potentiated the APAP toxicity, while the administration of an iron chelator (deferoxamine) protected the cells from APAP toxicity (Moon, Richie, and Isom 2010). Different findings were reported during similar manipulations in a whole animal model. Induction of both hepatic parenchymal and non-parenchymal cell IO did not result in potentiating of APAP toxicity (Moon et al. 2011). We recently demonstrated that thioacetamide administration to rats with IO exacerbated the extent of liver injury only in the rats with hepatic parenchymal IO. In the rats with both hepatic parenchymal and non-parenchymal cell IO, the extent of liver injury from thioacetamide did not differ from that observed in the rats without IO (Ackerman et al. 2015).
Rats in comparison to mice are relatively resistant to the hepatotoxic effects of APAP (McGill et al. 2012). This originates from dissimilar effects of APAP overdose in these rodent species: compared to mice, rats have a delay in glutathione depletion, an absence of JNK activation, and reduced mitochondrial oxidative stress and mitochondrial dysfunction (McGill et al. 2012).
In the present study, we examined the possibility that by augmentation of the hepatic oxidative stress, by means of increasing the hepatic iron concentration (HIC), it will be feasible to overcome the resistance of rats to APAP hepatotoxicity.
Method
Experimental Design
Thirty-two male Sprague-Dawley rats (Harlan Laboratories Ltd., Jerusalem, Israel) weighing 140 to 220 grams (5–7 weeks old), were studied. Rats were housed in regular cages situated in an animal room at 22°C, with a 14/10-hr light/dark cycle. This research project was approved by the ethical committee of the Hadassah-Hebrew University Medical School (Ethical approval number MD-83.03-4). All animal studies were conducted according to the regulations for the use and care of experimental animals. Rats were randomly divided into the following groups: Rats with “normal” HIC without any additional manipulation (control group, n = 4). Rats with “normal” HIC that were subjected to APAP overdose (APAP group, n = 13 Rats that were first subjected to orally induced IO and later to APAP overdose (Fe·Po·APAP group, n = 5). Rats that were first subjected to subcutaneously induced IO and later to APAP overdose (Fe·Sc·APAP group, n = 10).
All rats were maintained on standard rat chow diet (SRCD; pellets #19520; Koffolk, Tel Aviv, Israel) and were given tap water to drink ad libitum. The composition of SRCD is reported elsewhere (Ackerman et al. 2015).
Orally Induced IO
For six consecutive weeks, rats included in the Fe·Po·APAP group were fed ground SRCD mixed with 2.5% (by weight) of carbonyl iron powder (Sigma Chemical Co., Saint Louis, MO, catalogue number-C 3518; Galleano, Aimo, and Puntarulo 2002). Rats were given tap water to drink ad libitum. At the end of the IO period, these rats were subjected to APAP overdose. This form of IO is known to increase oxidative stress markers (Galleano, Aimo, and Puntarulo 2002).
Subcutaneous Induced IO
For six consecutive weeks, rats included in the Fe·Sc·APAP group received weekly subcutaneous injection of 200 mg/kg of iron dextran (ferric hydroxide dextran complex, 100 mg/ml, Sigma Chemical Co., Saint Louis, MO; Wood et al. 2008). Four days after the last iron dextran injection, the rats from both groups were subjected to APAP overdose. This form of IO is known to increase HIC and oxidative stress markers (Galleano, Aimo, and Puntarulo 2002; Ackerman et al. 2015).
APAP Overdose
APAP was administered to rats from APAP, Fe·Po·APAP, and Fe·Sc·APAP groups, six weeks after inclusion into the study. APAP (1g/kg of body weight) was administered into the gastric cavity after a 24-hr period of fasting but with free access to water. APAP was administered as a liquid solution (100 mg/ml, CTS Chemical industries Ltd., Kiryat Malachi, Israel). This dose of APAP is known to produce in rats a form of acute, nonfulminate liver injury (McGill et al. 2012).
Twenty-four hours after APAP overdose administration, while the rats were under ketamine anesthesia (90 mg/kg intramuscularly), blood was withdrawn from inferior vena cava, centrifuged, aliquoted, and frozen. Rats were then sacrificed, and their livers were harvested. A portion of liver tissue was taken for histological evaluation, while the remaining liver tissue was snap-frozen and stored at −80°C until required. The effects of APAP overdose were studied 24 hr after APAP administration, since it was reported that after such a time period, no recovery of hepatic glutathione occurred in such rats (McGill et al. 2012).
Plasma Biochemistry Parameters
Plasma biochemical parameters including albumin, liver enzymes, and kidney function tests were measured by the dry chemistry method (Vitros 950-5.1 analyzer, Johnson & Johnson Clinical Diagnostics, Rochester, NY).
Determination of Hepatic Nonheme Iron Concentration
Hepatic nonheme iron concentration was measured by colorimetric assay described by Torrance and Bothwell (1968). Briefly, 0.1-g liver tissues were digested in 2 ml acidic solution for 24 hr at 70°C. Digested samples were incubated with chromogen reagent, and the absorbance at 535 nm was measured using spectrophotometer.
Plasma APAP Levels
Plasma APAP levels were measured by an enzymatic method (Cobas Integra 800, Roche Diagnostics GmbH, Manheim, Germany). Results were expressed as µg/ml.
Hepatic Histology
Hepatic histological specimen from each animal was stained with hematoxylin and eosin for the evaluation of hepatic cell necrosis and with Perl’s Prussian blue for the detection of iron. In the present study, no lobular inflammation was observed within the histological slides, and the type of necrosis found was only of feathery degeneration type (Moon et al. 2011; Nayak et al. 1996). Feathery degeneration was scored by points of increment, according to the severity of the finding as follows: 0 = none, 1 = up to 33% of the lobule, 2 = up to 66% of the lobule, and 3 if the feathery degeneration was >66% of the lobule. Damage with intermediate severity between points was graded accordingly. Hepatic iron deposition was assessed separately within the parenchymal and non-parenchymal cells using the method described by Deugnier et al. (1993). By this scoring system, the maximal points that can be gained for cellular iron deposition is 36 for hepatocyte cells and 12 for sinusoidal cells.
Determination of Hepatic Lipid Peroxidation
Assessment of hepatic lipid peroxidation products was estimated spectrophotometrically, using thiobarbituric acid assay (Yagi 1987). The pink chromogen produced by the reaction of thiobarbituric acid with malondialdehyde, a secondary product of lipid peroxidation, was estimated. Results were expressed as the concentration of thiobarbituric acid reactive substances (TBARS).
Determination of Hepatic Glutathione and Hepatic Antioxidant Enzymes
Hepatic antioxidant enzymes were measured in the rat liver cytosolic fraction. Paraoxonase activity was measured using phenylacetate as substrate (Gan et al. 1991). Glutathione peroxidase activity was determined using the method described by Beutler (1975). Glutathione reductase (GSSG-R) activity was determined using the method of Carlberg and Mannervik (1985). Results of enzyme activity were reported as mmol/min/g liver.
Assessment of hepatic glutathione content was measured by a kinetic chemical assay as described previously by Akerboom and Sies (1981). By this method, the sums of both forms of glutathione (reduced and oxidized) were determined. Since both forms of glutathione were measured, the results of this method were expressed as “glutathione equivalents.” It should be noted that the results of this reaction depend on the GSSG concentration of oxidized glutathione and the activity of GSSG-R in the assay (Akerboom and Sies 1981).
Determination of Hepatic α-Tocopherol
Rat liver extract was prepared as previously described by Folch, Lees, and Sloane Stanley (1957). Determination of α-tocopherol, in the lipid extract, was based on the reduction of ferric ions to ferrous ions by α-tocopherol and the formation of red-colored complex with 2.2′-dipyridyl (Baker et al. 1980).
Immunohistochemistry and Image Analysis
Formalin-fixed, paraffin-embedded hepatic tissues were sectioned at 3 µm. Deparaffinized, rehydrated sections were subjected to the following procedures sequentially: heat-induced epitope retrieval in Citrate buffer and blocking by 3% peroxide and Background Buster (Innovex Biosciences, Richmond, CA).
The following antibodies were applied overnight at 4°C: Rabbit anti-nitrotyrosine (NITT; Millipore Corporation, Billerica, MA), diluted 1:500; Rabbit anti-copper–zinc–superoxide dismutase 1 (SOD1; Proteintech, Chicago, IL), diluted 1:100; Mouse monoclonal anti-metallothionein (MT; that cross-reacts with MT1 and MT2; Dako, Denmark), diluted 1:100; Rabbit anti-cytochrome P4502E1 (CYP2E1; Boster Biotech, Pleasanton, CA), diluted 1:200.
Antibodies for immunohistochemistry stains of either C-JNK (Mouse anti C-JNK that cross-reacts with JNK1, JNK2, and JNK3, Santa Cruz Biotechnology, Dallas, TX) or P-JNK (Mouse anti P-JNK, Santa Cruz Biotechnology, Dallas, TX) were applied in 2 dilutions (either 1:20 or 1:100), by 2 modes of incubation (either overnight at 4°C or for 2 hr at 37°C), by 2 modes of retrieval (Decloaker chamber either at 120°C or 97°C), and by using 2 retrieval solutions (either Citrate or EDTA). Antibodies were detected with ZytoChem Plus (HRP) Polymer anti-Mouse/Rabbit/Rat (Zytomed, Berlin, Germany) followed by diaminobenzidine tetrahydrochloride (DAB Chromogen System [Signet], Covance, London, England), according to standard procedures. Each staining procedure included negative controls. Sections were counterstained lightly with Mayer’s hematoxylin and mounted in Eukitt.
Protein expression (for NITT, SOD1, MT, CYP2E1, P-JNK, and C-JNK) was assessed by measuring the optic intensity of hepatic cell immunopositivity (in pixels), in 3 fields from 3 hepatic lobules, in the hepatic tissue located around the central veins (nine fields for each animal). Image analysis was carried out using modified approach in ImageJ software. The histological evaluation of the liver sections was done in blinded fashion.
Hepatic Cell Apoptosis
Apoptosis of hepatocytes was assessed through terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) immunohistochemistry. For TUNEL assay, apoptotic cells were identified in formalin-fixed, paraffin-embedded liver tissue sections using the in situ cell death detection kit POD (Roche Diagnostics, Corp., Indianapolis, IN), according to the manufacturer’s protocol, using a fluorescence microscope. Apoptosis quantification was performed by counting TUNEL-positive cells (magnification 400×). A total of 10 fields per liver were analyzed, and the mean value ± standard error of the mean (SEM) was determined. (Matot et al. 2008).
Statistical Evaluation
Comparisons between groups were performed with one-way analysis of variance (ANOVA) with Tukey–Kramer multiple comparisons test, or when needed, the Kruskal–Wallis tests (nonparametric ANOVA). The p value ≤.05 was considered statistically significant.
Results
Effects of APAP Overdose
Histological Grading of Acetaminophen Induced Liver Injury and Iron Staining Scores.
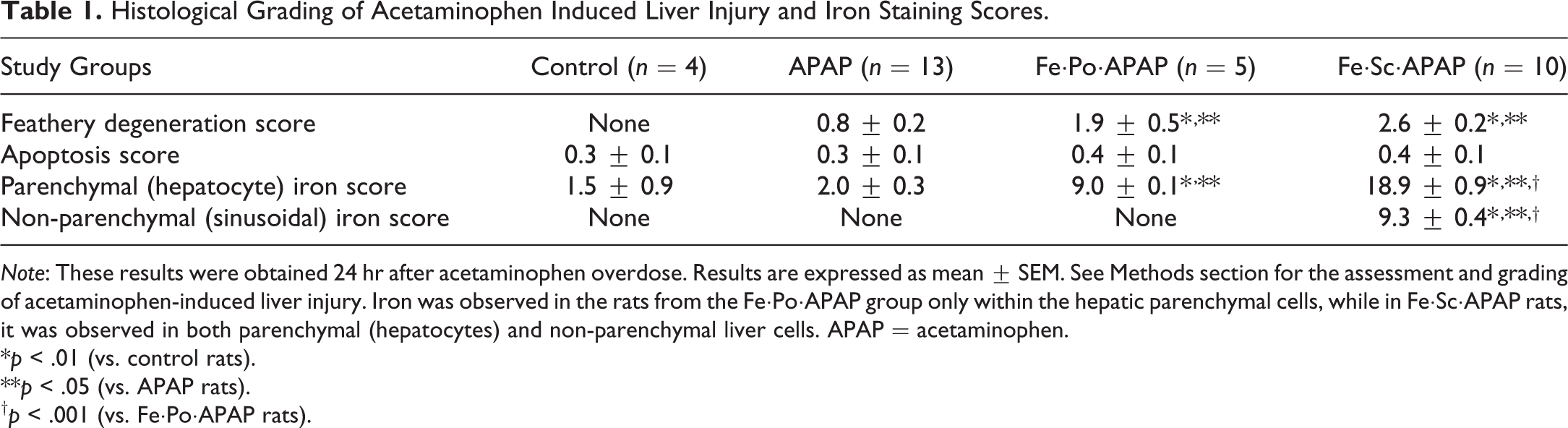
Note: These results were obtained 24 hr after acetaminophen overdose. Results are expressed as mean ± SEM. See Methods section for the assessment and grading of acetaminophen-induced liver injury. Iron was observed in the rats from the Fe·Po·APAP group only within the hepatic parenchymal cells, while in Fe·Sc·APAP rats, it was observed in both parenchymal (hepatocytes) and non-parenchymal liver cells. APAP = acetaminophen.
*p < .01 (vs. control rats).
**p < .05 (vs. APAP rats).
† p < .001 (vs. Fe·Po·APAP rats).
Changes in liver histology, function, and liver enzymes:
Changes in oxidative/antioxidative milieu: An increase in TBARS concentration (Table 3) and NITT expression (Figure 1A, B, and E). A decrease in α-tocopherol concentrations and paraoxonase activity (Table 3). An increase in MT expression (Figure 2A, B, and E
End of Study Weight and Plasma Biochemistry Parameters, Plasma Acetaminophen, and Hepatic Iron Concentration (HIC).
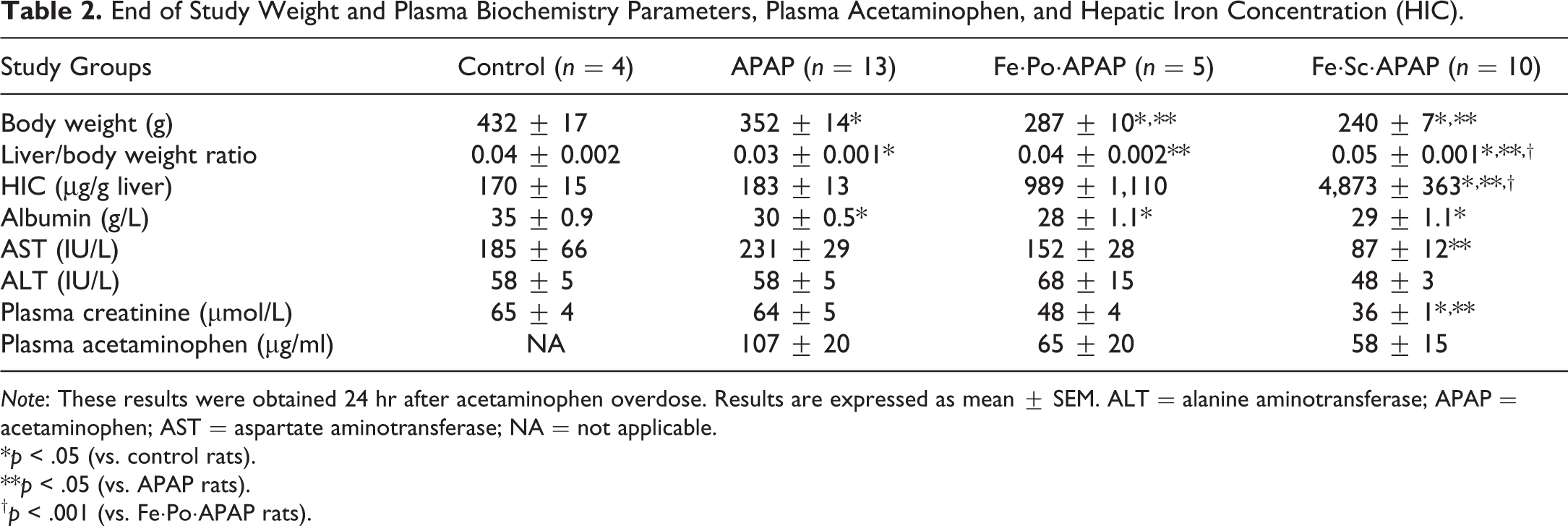
Note: These results were obtained 24 hr after acetaminophen overdose. Results are expressed as mean ± SEM. ALT = alanine aminotransferase; APAP = acetaminophen; AST = aspartate aminotransferase; NA = not applicable.
*p < .05 (vs. control rats).
**p < .05 (vs. APAP rats).
† p < .001 (vs. Fe·Po·APAP rats).
Hepatic Content of Oxidative and Antioxidative Substances and Activity of Antioxidative Enzymes.
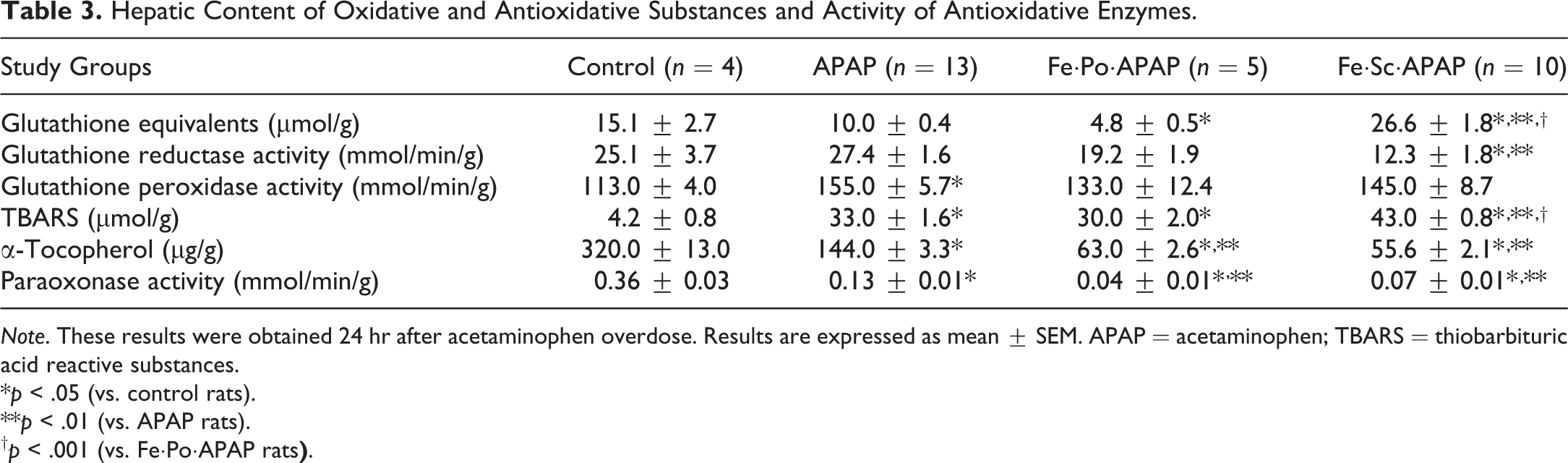
Note. These results were obtained 24 hr after acetaminophen overdose. Results are expressed as mean ± SEM. APAP = acetaminophen; TBARS = thiobarbituric acid reactive substances.
*p < .05 (vs. control rats).
**p < .01 (vs. APAP rats).
†
p < .001 (vs. Fe·Po·APAP rats
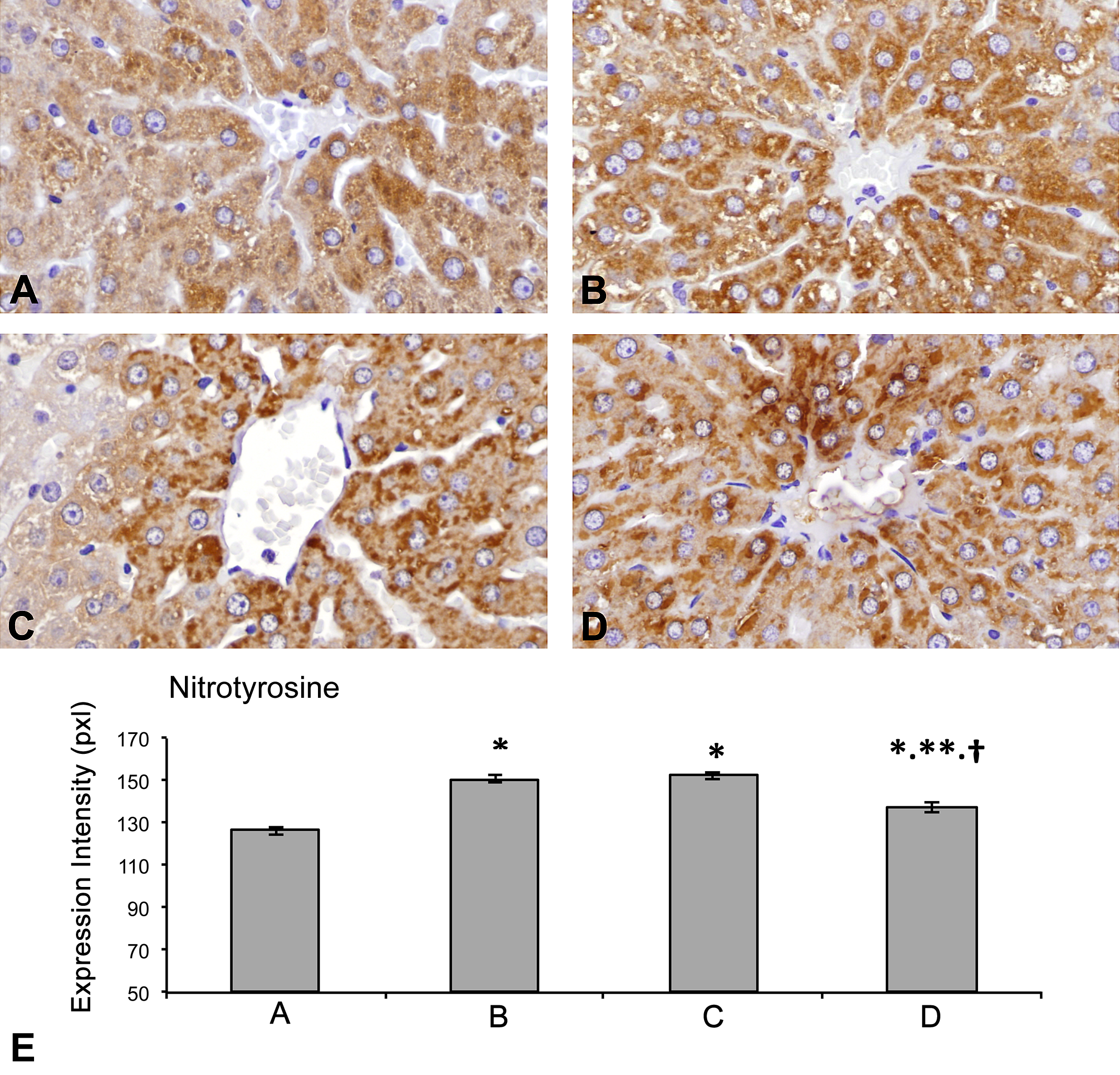
(A–E) Hepatic nitrotyrosine (NITT) expression (by immunohistochemistry). Compared to the control rats (A), rats from the 3 rat groups that received APAP (APAP [B], Fe·Po·APAP [C], Fe·Sc·APAP [D]) exhibited an increase in the intensity of NITT expression (in pixels) in the perivenular hepatocytes. The lowest degree of increase in the intensity of NITT expression was observed in the Fe·Sc·APAP rats. (*p < .01, vs. A; **p < .01, vs. B; †p < .01, vs. C [E]).
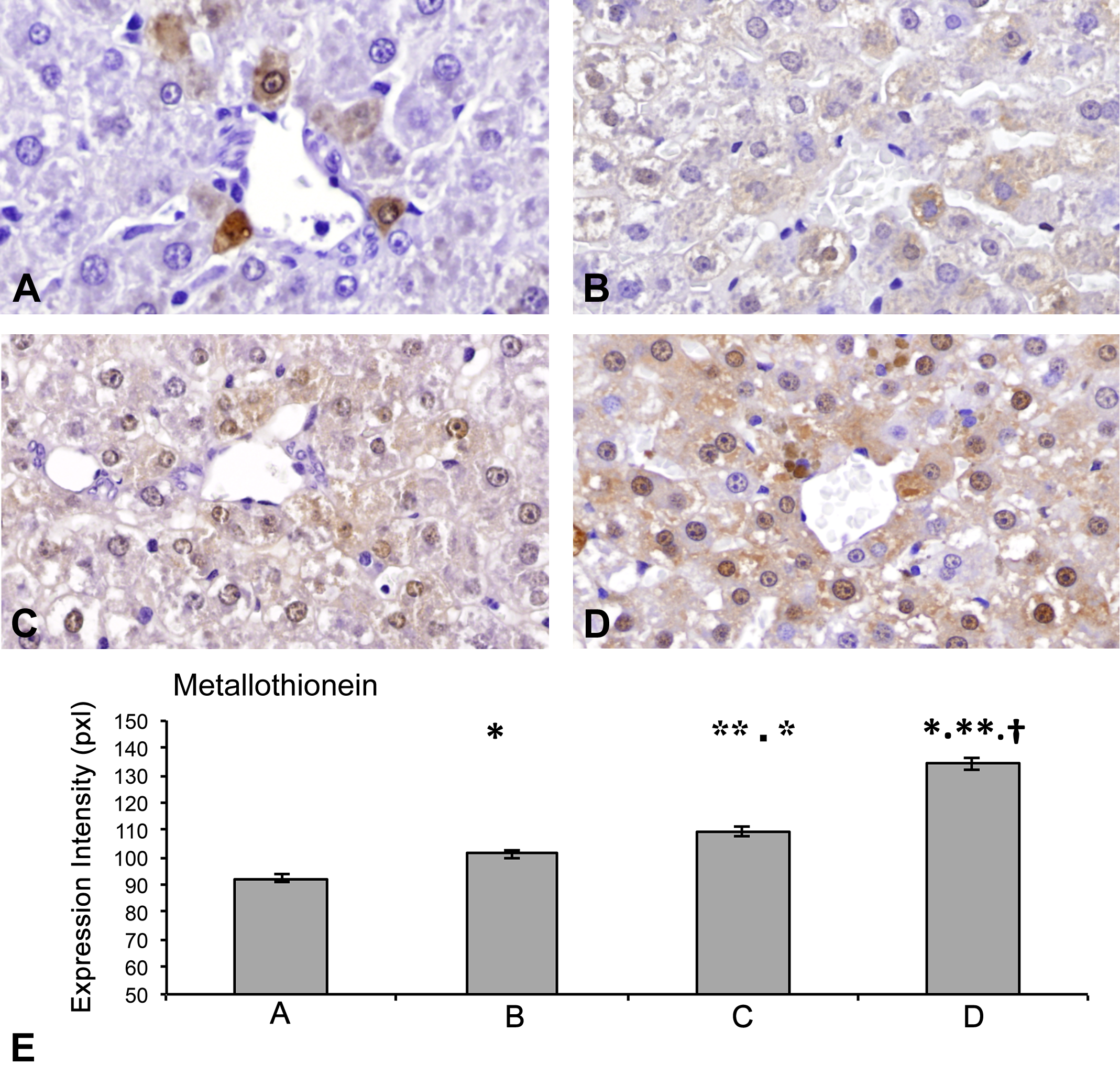
(A–E) Hepatic metallothionein (MT) expression (by immunohistochemistry). Compared to the control rats (A), rats from the 3 groups that received APAP (APAP [B], Fe·Po·APAP [C], Fe·Sc·APAP [D]) exhibited an increase in the intensity of MT expression (in pixels) in the perivenular hepatocytes. The increase in MT expression was proportional to the degree of hepatic iron concentration (HIC). The highest expression of MT was observed in the Fe·Sc·APAP rats. (*p < .01, vs. A; **p < .01, vs. B; †p < .01, vs. C [E]).
Effects of Orally Induced IO and APAP Overdose
The estimated total dose of carbonyl iron powder that was ingested by each Fe·Po·APAP rat during the study period was 17.0 g (4788 mg of elemental Fe). The hepatic iron deposits in these rats were observed mainly in the periportal hepatocytes. No sinusoidal cell iron deposits were observed (Table 1 and Figure 3A and B).
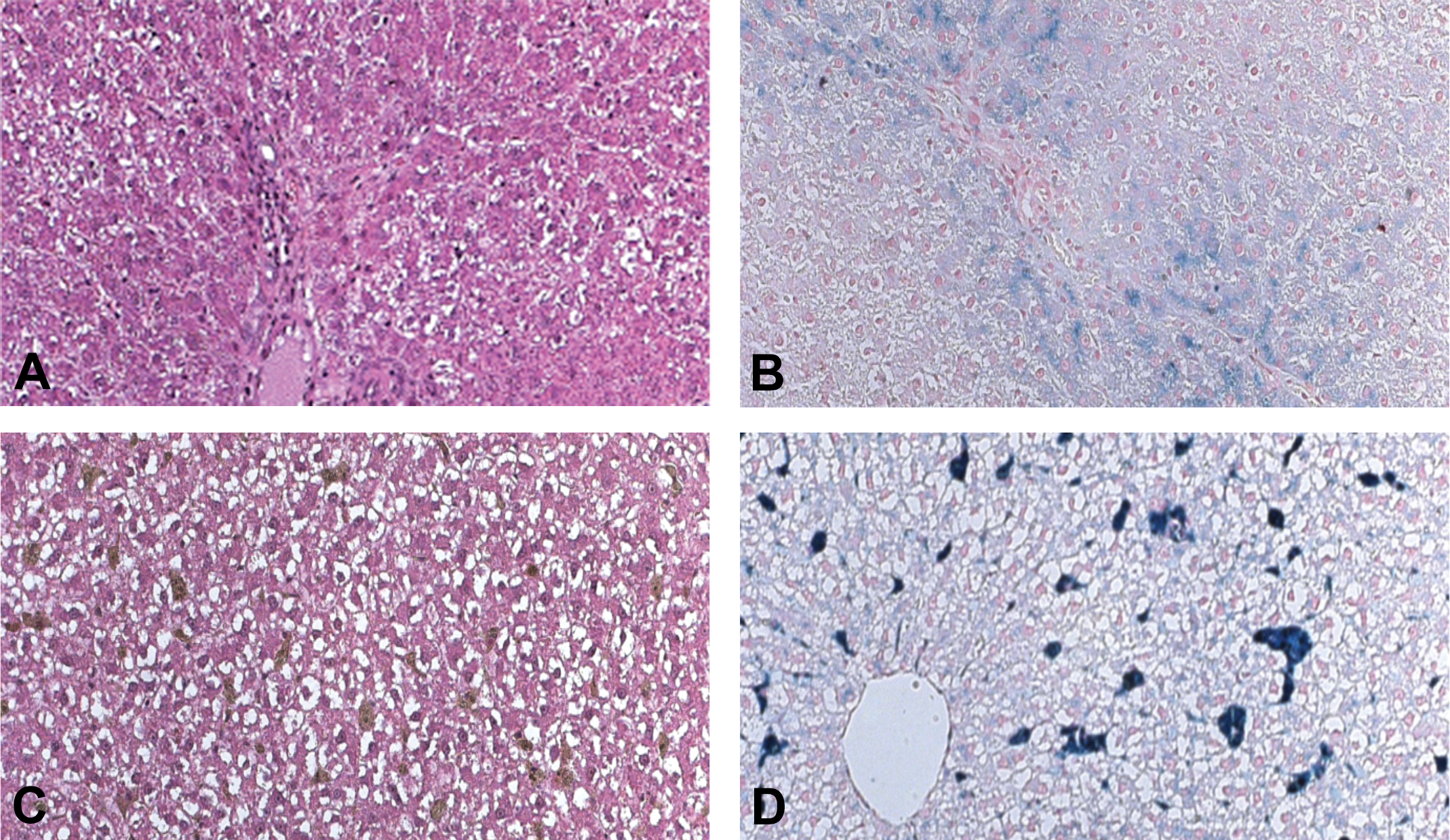
(A–D) Representative hematoxylin and eosin and Perls’ Prussian blue stains performed on livers from an Fe·Po·APAP rat (A and B, respectively) and from an Fe·Sc·APAP rat (C and D, respectively). Iron accumulated in the periportal hepatocytes in the Fe·Po·APAP rats and within both the periportal hepatocytes and sinusoidal cells in the Fe·Sc·APAP rats. An increase in the extent of hepatocyte feathery degeneration was observed in the rats from both of these groups.
Compared to the APAP rats, the following were observed in the Fe·Po·APAP rats:
Changes in liver histology, function, and liver enzymes:
Changes in oxidative/antioxidative milieu: A decrease in α-tocopherol concentrations and paraoxonase activity (Table 3). Increase in MT expression (Figure 2B, C, and E).
Effects of Subcutaneously Induced IO and APAP Overdose
The mean (±SEM) calculated total volume of iron dextran that was injected (on 6 sessions) to each rat from the Fe·Sc·APAP group was 2.2 ± 0.05 ml (110 mg of elemental Fe). The iron deposits in these rats were observed in both hepatocyte and sinusoidal cells (Table 1 and Figure 3C and D).
Compared to the APAP rats, the following were observed in the Fe·Sc·APAP rats:
Changes in liver histology, function, and liver enzymes:
Changes in oxidative/antioxidative milieu An increase in both TBARS and “glutathione equivalents” concentrations but a decrease in α-tocopherol concentrations and in paraoxonase activity (Table 3). A decrease in NITT expression (Figure 1B, D, and E). An increase in the expression of MT (Figure 2B, D, and E) and SOD1 (Figure 4B, D, and E).
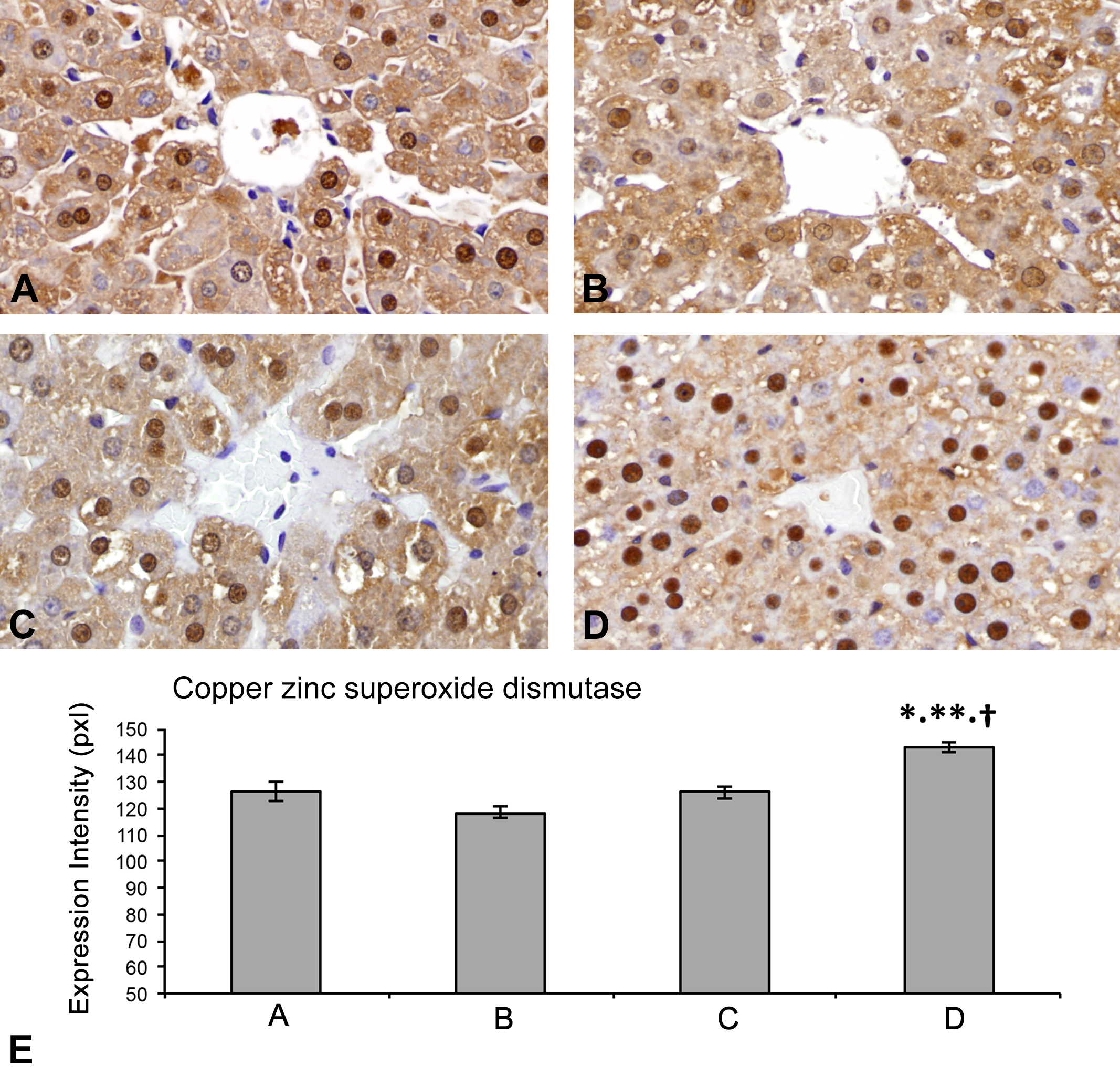
(A–E) Hepatic copper–zinc–superoxide dismutase (SOD1) expression (by immunohistochemistry). Compared to the control (A), APAP (B), and Fe·Po·APAP rats (C), the Fe·Sc·APAP rats (D) exhibited an increase in SOD1 expression (in pixels) in the perivenular hepatocytes. (*p < .01, vs. A; **p < .01, vs. B; †p < .01, vs. C [E]).
Hepatic Expression of CYP2E1
An increase in the immunohistochemical CYP2E1 hepatic expression was observed in all rat groups that received APAP overdose (Figure 5A to E).
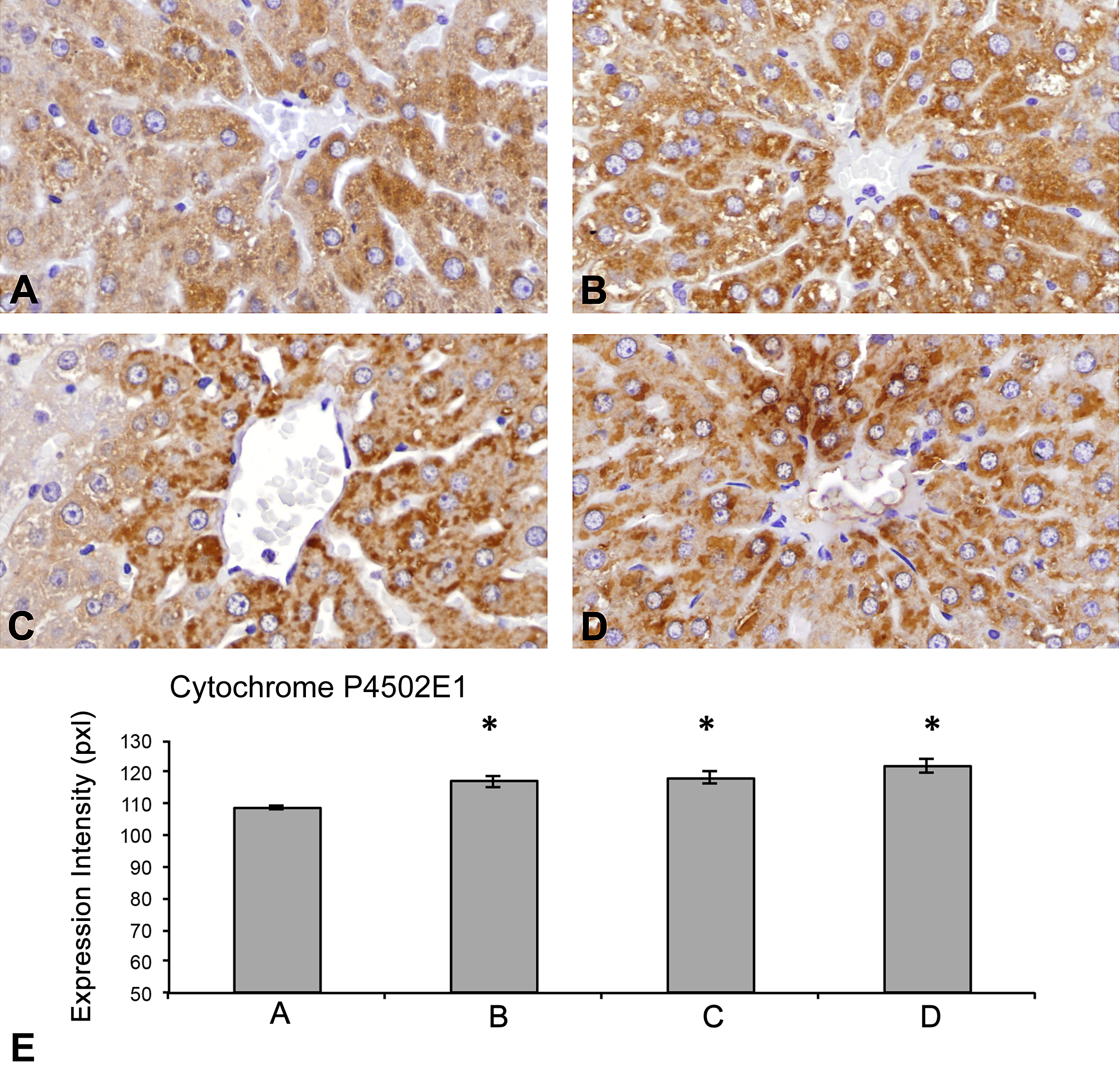
(A–E) Hepatic cytochrome P4502E1 (CYP2E1) expression (by immunohistochemistry). Compared to the control rats (A), rats from the 3 groups that received APAP (APAP [B], Fe·Po·APAP [C], and Fe·Sc·APAP [D]) exhibited an increase in the intensity of CYP2E1 expression (in pixels) in the perivenular hepatocytes. The increase in CYP2E1 expression was not proportional to the degree of HIC. (*p < .01, vs. A [E]). HIC = hepatic iron concentration.
APAP Plasma Levels
None of the manipulations to increase the HIC had any effect on the APAP plasma levels measured 24 hr after APAP overdose (Table 2).
Hepatic Expression of JNK
Immunohistochemical hepatocyte expression of C-JNK and P-JNK were not detected in any of the rat groups from the present study.
Mouse livers, taken from healthy mice that were not involved in this study, revealed positive stain (within hepatocytes) only for P-JNK but not for C-JNK (data nor shown).
Hepatic Cell Apoptosis
The administration of APAP overdose and the increase in HIC had no effect on the hepatocyte apoptosis score in any of the rat groups (Table 1).
Discussion
In the present study, no significant liver damage was observed in any of the APAP rats after exposure to APAP overdose. The inability to produce liver damage in these rats, despite the increase in hepatic oxidative stress and NITT expression, was probably due to the absence of hepatic JNK activation in the Sprague-Dawley rats that were used for this study (McGill et al. 2012).
In the present study, manipulations to increase the HIC in two groups of rats were performed. Increase in HIC is known to augment hepatic oxidative stress (Galaris and Pantopoulos 2008; Jomova and Valko 2011). Thus, it was expected that the increase in HIC would increase the rats’ vulnerability to APAP overdose despite the absence of activated JNK.
As expected, APAP administration to the Fe·Po·APAP and Fe·Sc·APAP rats was associated with an increase in the hepatic feathery degeneration, which is reported to be a typical feature of APAP toxicity (Moon et al. 2011). In addition, changes in several components of the hepatic oxidative/antioxidative milieu were observed in these rat groups. However, despite these findings, no increase in serum aminotransferases was observed. Moreover, unexpected findings were observed in the Fe·Sc·APAP rats. These included a decrease in AST levels, an increase in hepatic “glutathione equivalents” content, an increase in hepatic expression of the antioxidative substances (SOD and MT), a decrease in hepatic NITT expression, and an increase in TBARS concentration.
The causes for the above described changes in several components of the hepatic oxidative/antioxidative milieu in the Fe·Sc·APAP rats such as the increase in hepatic “glutathione equivalents” content, increase in the hepatic SOD and MT expression, and the decrease in the hepatic NITT expression are not clear. However, such findings were previously reported in experimental animals with IO (Moon et al. 2011; Brown et al. 2003; Zhang et al. 2012; Brown et al. 2007). Two speculative mechanisms may be responsible for the above described findings and to our inability to produce hepatic damage from APAP overdose in the Fe·Sc·APAP rats: either Kupffer cell inactivation or hepatic preconditioning.
Kupffer Cell Inactivation
Differences in the cellular localization of hepatic iron deposits were reported to play a significant role in the exacerbation of acute or chronic liver damage in both human subjects and experimental animals (Ackerman et al. 2015).
As mentioned earlier, the hepatic iron accumulation in the Fe·Sc·APAP rats was found to occur both in the parenchymal and in the non-parenchymal (including Kupffer) cells. Proliferation and activation of Kupffer cells has been observed early in various models of liver disease. These cells produce a wide range of cytokines that may contribute to hepatic injury, inflammation, and fibrosis (Olynyk and Clarke 2001).
Given the data presented in the previous sections, it is hypothesized that in the Fe·Sc·APAP rats, the iron overloading within the Kupffer cells impaired their own function, and thus prevented the augmentation of liver injury that was expected to occur from the exposure of these rats to the combination of APAP overdose, IO, and increased lipid peroxidation (Ackerman et al. 2015; Olynyk and Clarke 2001). A similar phenomenon, with lessening of hepatic damage from either APAP (in mice) or thioacetamide (in rats) with combined parenchymal and non-parenchymal hepatic cell IO, was previously described (Moon et al. 2011; Ackerman et al. 2015). Inactivation of Kupffer cells and hepatic macrophages, by treatment with either gadolinium chloride or dextran sulfate, was associated with decreased degree of hepatic damage from APAP overdose (Michael et al. 1999; Ju et al. 2002; Choi et al. 2015).
Hepatic Preconditioning
Hepatic preconditioning refers to the development of increased tolerance to various insults. This tolerance may be induced by indirect induction of low level of stress that triggers cellular defense mechanisms against a subsequent stronger insult (Selzner et al. 2003; Bahde and Spiegel 2010). An example for such an event was described by Galleano and colleagues (2011). These researches induced a state of IO in rats. This was made by parenteral iron dextran administration during a 10-day course. On the first and the second day after concluding the iron overloading, markers of increased hepatic oxidative stress were detected. Three days after concluding the iron overloading, while the markers hepatic oxidative stress normalized, the iron overloaded rats and rats from a control groups were subjected to ischemia–reperfusion (IR) injury. While the rats from the control groups developed, as a consequence of the IR injury, markers of severe liver injury, the iron overload rats exhibited lesser degree of hepatic injury, lower serum levels of tumor necrosis factor-α, higher hepatic glutathione content, and higher expression of hepatic haptoglobin (an acute phase protein; Galleano et al. 2011). Similar responses to iron preconditioning that protected the rats’ livers from IR injury, by augmentation of antioxidant and anti-inflammatory responses, were also reported after the administration of L-3, 3’, 5-triidothyronine (T3). Administration of T3 to the rats was associated with increased hepatic mitochondrial oxygen consumption and with increase in hepatic ROS production. ROS generation within the Kupffer cells triggered the activation of various transcription factors, which unregulated the expression of Kupffer cell cytokines such as tumor necrosis factor-α, interleukin-1, and interleukin-6. Interaction of these cytokines with specific receptors within the hepatocytes triggered the expression of various antioxidant enzymes, antiapoptotic and acute phase proteins. In addition, these reactions promoted hepatocytes and Kupffer cell hyperplasia. The combination of all of the abovementioned events provided protection to the liver of rats that were subjected first to T3 administration and later to IR injury (Videla 2010).
During the metabolism of APAP in toxic doses, a reactive intermediate hepatotoxic metabolite (NAPQI) is formed by the cytochrome P450 system. The enzyme that is most active in the biotransformation of APAP to NAPQI is CYP2E1 (Bessems and Vermeulen 2001). Upregulation or downregulation of CYP2E1 may either enhance or decrease the extent of APAP hepatotoxicity (Moon et al. 2011; Stål et al. 1996; Martin-Murphy et al. 2013; Du et al. 2015). In the present study, an increase in the CYP2E1 hepatic expression was observed in the three rat groups that received toxic doses of APAP. However, the CYP2E1 hepatic expression in all these rat groups was similar and did not correlate with the HIC of these rats. Others have reported that increase in HIC may have either no role (in mice) or may cause upregulation of hepatic CYP2E1 levels (in rats) or CYP2E1activity (in thalassemic patients; Moon et al. 2011; Stål et al. 1996; Somparn et al. 2007).
In summary, male Sprague-Dawley rats were insensitive to toxic doses of APAP. This probably occurred due to the absence of hepatic JNK activation. Parenchymal hepatic cell IO augmented the hepatic oxidative stress but did not increase the rats’ vulnerability to APAP. Combined parenchymal and non-parenchymal hepatic cell IO was associated with an unexpected increase in hepatic antioxidative markers. This phenomenon may indicate that the hepatic oxidative/antioxidative milieu may be affected by the cellular distribution of hepatic iron.
Footnotes
Author Contributions
All authors (ZA, GS, GL, MG, OP, and MG) contributed to conception or design; data acquisition, analysis, or interpretation; drafting the manuscript; and critically revising the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The authors declared no potential, real or perceived conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by internal funds from the authors’ institutions.