
Abstract
Bedaquiline (BDQ) is an antibiotic to treat pulmonary multidrug-resistant tuberculosis (MDR-TB). Studies up to 39 weeks were conducted orally in dogs to assess the toxicity and pharmacokinetics of BDQ and its N-desmethyl metabolite (D-BDQ). Phospholipidosis (PLD) seen in the monocytic phagocytic system was considered an adaptive change. Skeletal muscle, heart, stomach, liver, and pancreas toxicities with D-BDQ as the main contributor were associated with a less-than-dose-proportional increase in plasma exposure and an overproportional tissue uptake of BDQ and D-BDQ at high-dose levels. Tissue concentrations of BDQ and D-BDQ slowly decreased after lowering the dose, contributing to the recovery of the pathological findings. Treatment was better tolerated at mid-dose levels, characterized by a dose-proportional increase in plasma and tissue exposures. Treatment at a low dose, reaching exposures approximating therapeutic exposures, was without adverse effects and not associated with PLD. There was no evidence of delayed toxicities after treatment cessation. Intermittent dosing was better tolerated at high doses. Since MDR-TB patients are dosed within the linear plasma exposure range and plasma levels of BDQ and D-BDQ are similar or lower than in dogs, PLD and adverse findings related to tissue accumulation that occurred at high doses in dogs are unlikely to occur in humans.
Keywords
Bedaquiline (BDQ, also known as SIRTURO™, TMC207, R207910) is a diarylquinoline drug with a unique mechanism of action, targeting the mycobacterial adenosine 5′-triphosphate synthase (Andries et al. 2005). BDQ obtained accelerated approval by the United States Food and Drug Administration in December 2012 for use as part of combination therapy in adults (≥18 years) with pulmonary multidrug-resistant tuberculosis (MDR-TB) and has since been approved in over 40 countries, many of them high-burden countries representing approximately 80% of the burden of disease. In a randomized, double-blind, placebo-controlled phase 2b trial, 24 weeks of oral treatment with BDQ (400 mg once [qd] daily for 2 weeks, followed by 200 mg 3 times a week [tiw] for 22 weeks) in combination with a preferred background regimen, resulted in faster culture conversion and significantly more culture conversions at 120 weeks, as compared with placebo (Diacon et al. 2014). BDQ administered as 400 mg qd for 2 weeks and 200 mg dosed tiw up to an additional 22 weeks was generally safe and well tolerated in adults as part of combination therapy of pulmonary TB due to MDR Mycobacterium tuberculosis.
BDQ is a lipophilic and basic compound (calculated partition coefficient [cLog P] 7.25, dissociation constant [pKa] 8.77; the pKa was determined for an analogous structure due to low solubility) and contains a cationic amphiphilic structure (Figure 1). BDQ is primarily metabolized into N-desmethyl bedaquiline (D-BDQ) in human and nonhuman species. D-BDQ is formed by demethylation of BDQ and, as such, has the characteristics of a cationic amphiphilic drug (CAD) with high log P (cLog P 6.59 and pKa 9.97), like BDQ. D-BDQ is 5-fold less active in vitro than BDQ (Andries et al. 2005; Rustomjee et al. 2008; Rouan et al. 2012). Compounds with the characteristics of CADs, such as BDQ and D-BDQ, bind to intracellular phospholipids, resulting in reversible drug accumulation (drug–phospholipid complexes arranged in the so-called lysosomal lamellar bodies) in tissues, generally referred to as phospholipidosis (PLD; Reasor, Hastings, and Ulrich 2006; Hanumegowda et al. 2010). The toxicological significance of PLD is unclear. There may be functional changes in cells and tissues where lamellar bodies are present; however, a causal linkage to PLD has not been clearly made (Lullmann, Lullmann-Rauch, and Wassermann 1975; Reasor, Hastings, and Ulrich 2006). The prevailing theory is that drug-induced PLD is an adaptive response by the host in response to the presence of a drug and is not a toxic condition. Each drug must be evaluated separately regarding indication, time required for development, species susceptibility, organs affected, and reversibility (FDA Advisory Committee meeting material 2010).
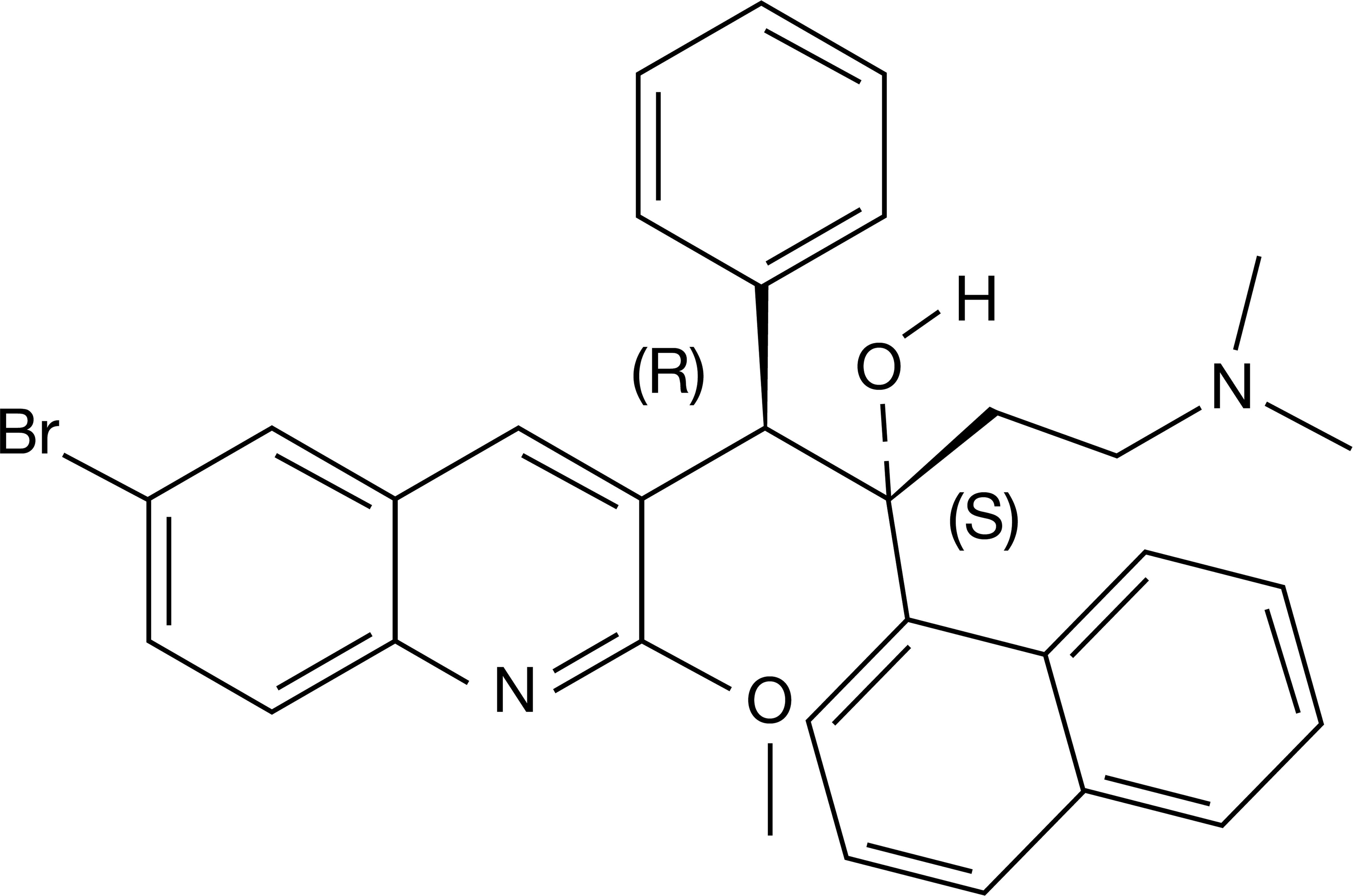
Molecular structure of bedaquiline as the free base form. From Andries et al. (2005). Reprinted with permission from American Association for the Advancement of Science.
Both BDQ and D-BDQ were shown to induce PLD in vitro with D-BDQ exhibiting a greater phospholipidogenic potential than BDQ (Mesens et al. 2010). In vivo, BDQ-induced PLD in drug-treated animals during nonclinical studies, mainly in cells of the monocytic phagocytic system (MPS). All species tested (mouse, rat, and dog) showed drug-related increases in pigment-laden and/or foamy macrophages in a variety of tissues. Across species, BDQ-induced PLD became apparent after administration of high doses for short duration, as well as after prolonged administration of lower doses and subsequent increases in plasma and tissue concentrations of the drug, as a consequence of the long terminal elimination half-life (t 1/2, term) of BDQ and D-BDQ. The observed PLD in macrophages and lymphoid tissues had no functional consequences as evidenced by normal macrophage function in a rat host resistance assay and a normal T-cell dependent antibody response (Janssen, data on file).
Here, we describe the dose- and time-dependency of the toxicity and pharmacokinetic profiles of BDQ and D-BDQ after long-term (i.e., 26 or 39 weeks) oral administration in dogs.
Materials and Methods
Test Compound
BDQ ((1R,2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4-(dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol) (8-[(2-ethyl-6-methylbenzyl)amino]-2,3-dimethylimidazo[1,2-a]pyridine-6-carboxamide) was synthesized by Janssen. The molecular structure of BDQ as the free base form is shown in Figure 1. BDQ, as free base or fumarate salt, was formulated as an aqueous solution containing 20% or 40% hydroxypropyl-β-cyclodextrin (HP-β-CD) and was administered to dogs by oral gavage at a dosing volume of 2.5 (26-week study no. 1) or 5 ml/kg (26-week study no. 2 and a 39-week study).
Animals
Male and female beagle dogs from Marshal Farms, United States (26-week study no. 1), Marshal Farms, Italy (26-week study no. 2), or Marshal BioResources, United States (39-week study) were used. The dogs were housed in custom designed dog pens under standard conditions of temperature, relative humidity, ventilation, and illumination. The dogs were acclimatized to laboratory conditions for at least 2 weeks before study start. Animals were provided tap water ad libitum and a daily ration of a dog laboratory diet based on their body weight. Approval from the animal research ethics committee had been obtained for using these animals in preclinical safety studies. All animal experiments were performed in an Association for Assessment and Accreditation of Laboratory Animal Care–accredited lab in accordance with the European and Belgian guidelines and with the principles of euthanasia as stated in the Report of the American Veterinary Medical Association Panel.
Experimental Design
An overview of the experimental design for the 3 long-term studies performed in beagle dogs is presented in Table 1. In each study, vehicle control dogs were given the vehicle solution (20% or 40% HP-β-CD) via oral gavage.
Overview of Experimental Designs for Dog Long-Term Toxicity Studies.
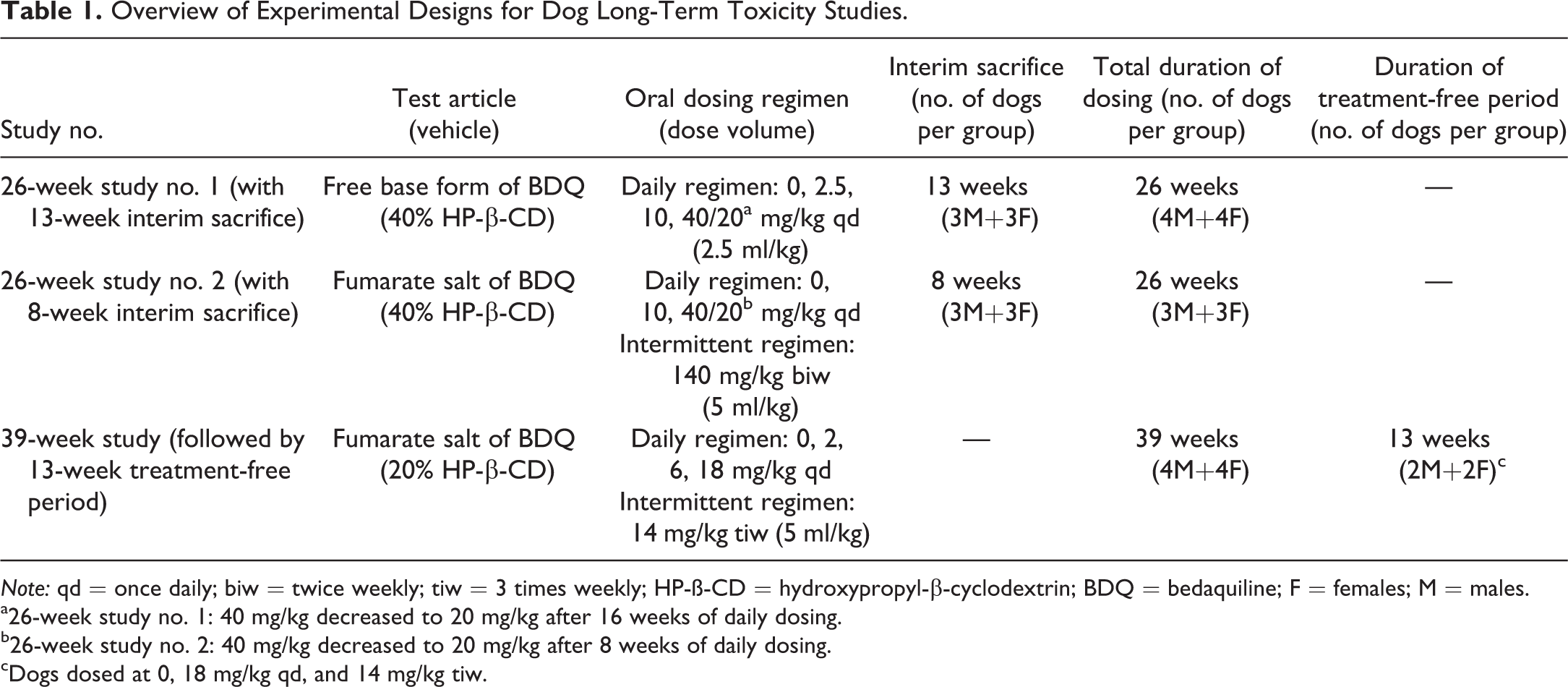
Note: qd = once daily; biw = twice weekly; tiw = 3 times weekly; HP-ß-CD = hydroxypropyl-β-cyclodextrin; BDQ = bedaquiline; F = females; M = males.
a26-week study no. 1: 40 mg/kg decreased to 20 mg/kg after 16 weeks of daily dosing.
b26-week study no. 2: 40 mg/kg decreased to 20 mg/kg after 8 weeks of daily dosing.
cDogs dosed at 0, 18 mg/kg qd, and 14 mg/kg tiw.
In 26-week study no. 1 (with 13-week interim sacrifice), 7 males and 7 females, aged approximately 7 months at the start of the study (respective body weight ranges: 6.4 to 9.5 kg and 7.0 to 8.8 kg), were included at each dose level. The dose levels were 2.5, 10, and 40 mg/kg BDQ qd. Three males and 3 females at each dose level were sacrificed ad interim after 13 weeks of daily dosing. The high-dose level was reduced to 20 mg/kg qd after 16 weeks of daily dosing due to poor condition of the dogs.
A subsequent 26-week study no. 2 (with 8-week interim sacrifice) was conducted to better characterize the toxicities seen in the first 26-week study and to assess biomarkers related to these toxicities; with this purpose, the high-dose level of the first study was included again. In 26-week study no. 2, groups of 6 males and 6 females, aged 5.5 to 6.5 months at the start of the study (respective body weight ranges: 6.2 to 8.6 kg and 4.0 to 6.4 kg) were given BDQ at a daily dose level of 10 or 40 mg/kg (base form equivalent) and the high-dose level was reduced to 20 mg/kg qd (base form equivalent) after 8 weeks of daily dosing due to progressive deterioration of the clinical condition of the dogs. An intermittent dose group was included in this study with dogs receiving 140 mg/kg BDQ twice weekly (biw). Three males and 3 females from each group were sacrificed ad interim after 8 weeks.
In a 39-week study (followed by a 13-week treatment-free period), 4 males and 4 females, aged approximately 7 to 8 months at the start of the study (respective body weight ranges: 7.4 to 9.5 kg and 5.5 to 7.7 kg), were included at each dose level. The dose levels were 2, 6, and 18 mg/kg qd (base form equivalent) and 14 mg/kg tiw. An additional 2 males and 2 females were assigned to the groups receiving vehicle, qd high dose, and tiw dose for assessment of reversibility of findings after 13 weeks without dosing.
In-life Observations and Measurements
Clinical signs and body weight were monitored throughout the studies. Electrocardiography (ECG) measurements and blood sampling for clinical chemistry and safety biomarkers analyses were carried out as follows in the 3 studies.
In the 26-week study no. 1 (with 13-week interim sacrifice), ECG examinations and blood sampling for routine clinical chemistry analyses including creatine kinase (CK) were performed prior to the start of dosing and after 13 and 26 weeks of daily dosing.
In the subsequent 26-week study no. 2 (with 8-week interim sacrifice), ECG measurements and routine clinical chemistry were carried out before the first dose and after 4, 8, 13, and 26 weeks of dosing. In addition, biomarkers indicative of cardiac and muscle injury (cardiac troponin I, CK, and myoglobin), pancreatic lesion (amylase, lipase, and trypsin-like immunoreactivity), and gastric lesion (gastrin-17) were measured monthly. Light microscopic evaluation for peripheral vacuolated cells was performed monthly on Wright-stained blood smears.
In the 39-week study (followed by a 13-week treatment-free period), ECG measurements and clinical chemistry evaluations were undertaken predose; after 13, 26, and 39 weeks; and again at the end of the 13-week treatment-free period, and included evaluation of cardiac troponin I, CK, and myoglobin, in addition to the routine clinical chemistry parameters.
Cardiac troponin I was measured using a canine-specific enzyme immunoassay (26-week study no. 2—Life Diagnostics, Inc., West Chester, PA) or using Abbott Test Kit on an Abbott Architect CI8200 Clinical Chemistry Analyzer (39-week study). A canine-specific enzyme immunoassay was used for the quantitative determination of myoglobin (26-week study no. 2—Life Diagnostics, Inc.; 39-week study—Kamiya Biomedical Company, Seattle, WA).
Pathology
Full necropsy was performed after 8 weeks (interim necropsy in 26-week study no. 2), 13 weeks (interim necropsy in 26-week study no. 1), 26 weeks, or 39 weeks of dosing, and following the 13 weeks treatment-free period in the 39-week study. Animals were anaesthetized with sodium pentobarbital or thiopental sodium and sacrificed by exsanguination. For each animal, a full range of organ and tissue samples were taken and fixed in neutral buffered 10% formalin. Tissue samples were paraffin embedded, sectioned, and stained with hematoxylin and eosin for examination by light microscopy. For electron microscopy, buffy coats (from peripheral white blood cells) and samples of gall bladder, kidney, liver, lung, pancreas, skeletal muscle (quadriceps), spleen, and stomach were fixed in 3% glutaraldehyde in potassium phosphate buffer 0.09M 1.4% sucrose (26-week study no. 2) and after 1 to 2 weeks transferred to 10% buffered formalin. Ultrathin sections were prepared for examination with a Philips CM100 electron microscope.
Toxicokinetic Assessment, Including Bioanalysis
Blood sampling was performed after the first dose (39-week study), after 4 weeks of dosing (26-week studies), in week 13 (39-week study), and at the end of treatment before interim and terminal sacrifice in all animals at the following time points: predose, 0.5, 1, 2, 4, 8, and 24 hr after dosing in the qd regimen, additionally 48 hr after dosing in the tiw regimen (39-week study), and predose, 0.5, 2, 8, 24, 72, 72.5, 74, 80, 96, 144, and 168 hr after the first weekly dose in the biw regimen (26-week study no. 2). Plasma was separated and stored at −20°C before bioanalysis. BDQ and D-BDQ were quantified in dog plasma by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after protein precipitation using a 2-in-1 assay as in the following description. Aliquots of 50 μl of plasma were spiked with isotope-labeled internal standards, and proteins were precipitated with 2 aliquots of 200 μl of methanol. After centrifugation, the supernatant was analyzed by LC-MS/MS. Chromatography was performed on a 4.6 by 30 mm Polaris C18 5 μm column at a temperature of 40°C. The mobile phase was pumped at a flow rate of 1.5 ml/min and consisted of 25% 0.01M ammonium formate (pH 4) (A)/75% methanol (B). The starting condition of 25% A to 75% B was held for 0.5 min and was followed by a step gradient to 2% A to 98% B, which was held for 3.2 min. Detection was by tandem MS using an API 4000 Sciex instrument with electrospray operating in positive ion mode. The following transitions were used for monitoring BDQ, D-BDQ, and their respective internal standards: BDQ, m/z 555.2 to 58.0 and internal standard, m/z 561.2 to 64.0; D-BDQ, m/z 541.2 to 480.0 and internal standard, m/z 545.2 to 480.0. The range of the assay was 10.0 ng/ml to 5,000 ng/ml.
Plasma concentration–time profiles were subjected to a noncompartmental pharmacokinetic analysis using WinNonLin (v 4.0.1a, Pharsight Corporation, Mountain View, CA; 26-week studies) or PKAA R2.1 software (39-week study). Maximum plasma concentrations (Cmax) and areas under the plasma concentration–time curves (AUCs) were calculated. The AUCs were calculated using the linear up/log down trapezoidal rule.
Tissue samples for determination of the concentration of BDQ and D-BDQ were collected in the 26-week studies at interim and terminal necropsy about 24 hr after administration of the last dose (lung, liver, lymph node, spleen, and thymus in 26-week study no. 1 and lung, liver, lymph node, spleen, thymus, heart, pancreas, muscle, and stomach in 26-week study no. 2). Tissue samples for BDQ and D-BDQ were analyzed by means of the LC-MS/MS method described above, following tissue homogenization. Mean tissue concentrations of BDQ and D-BDQ were calculated as µg/g tissue.
Statistical Analysis
Statistical analyses in the 26-week study no. 1 for body weight, ECG, and serum analysis were performed according to the Dunnett test (homogeneous variances) or the Dunn test (heterologous variances). Statistical analyses were not performed in the 26-week study no. 2; the recorded parameters obtained from the BDQ-dosed males and females were compared with predose values and with those of the vehicle-dosed groups. In the 39-week study, statistical analyses for body weight and serum were performed using Fisher’s F-protected least significant difference method via Student’s t-test (homogeneous variances) or using Kruskal–Wallis nonparametric analysis of variance (ANOVA) and χ2 protection (z-tests; heterogeneous variances).
Results
PLD and Adverse Findings in 26-week Studies after Once Daily Oral Dosing with BDQ
Tables 2 and 3 present the main toxicity findings from the first and second 26-week study, respectively. Toxicities in dogs mainly occurred at high-dose levels (≥10 mg/kg qd), whereas treatment with BDQ at a lower dose (2.5 mg/kg qd) was well tolerated and without adverse effects. Two females in the high-dose group (40 mg/kg qd) were sacrificed in the second half of study no. 1 after 15 and 18 weeks of dosing due to a deteriorating condition. Based on microscopic examination, pronounced subacute pancreatitis associated with slight degeneration/necrosis of the fundic mucosa of the stomach in one animal and myocardial degeneration/necrosis (subendocardial and subepicardial) with slight multifocal pancreatitis in the other animal were considered to be the cause of the poor condition and their subsequent sacrifice. Because of a poor condition and effects on body weight among dogs at the 40 mg/kg qd dose level in both studies, the high-dose level was reduced to 20 mg/kg qd after 16 and 8 weeks of dosing in study no. 1 and no. 2, respectively. Lowering the dose to 20 mg/kg qd stabilized the condition of the animals as manifested by regain of body weight.
Adverse Findings in the 26-week Study No. 1 with 13-week Interim Sacrifice.
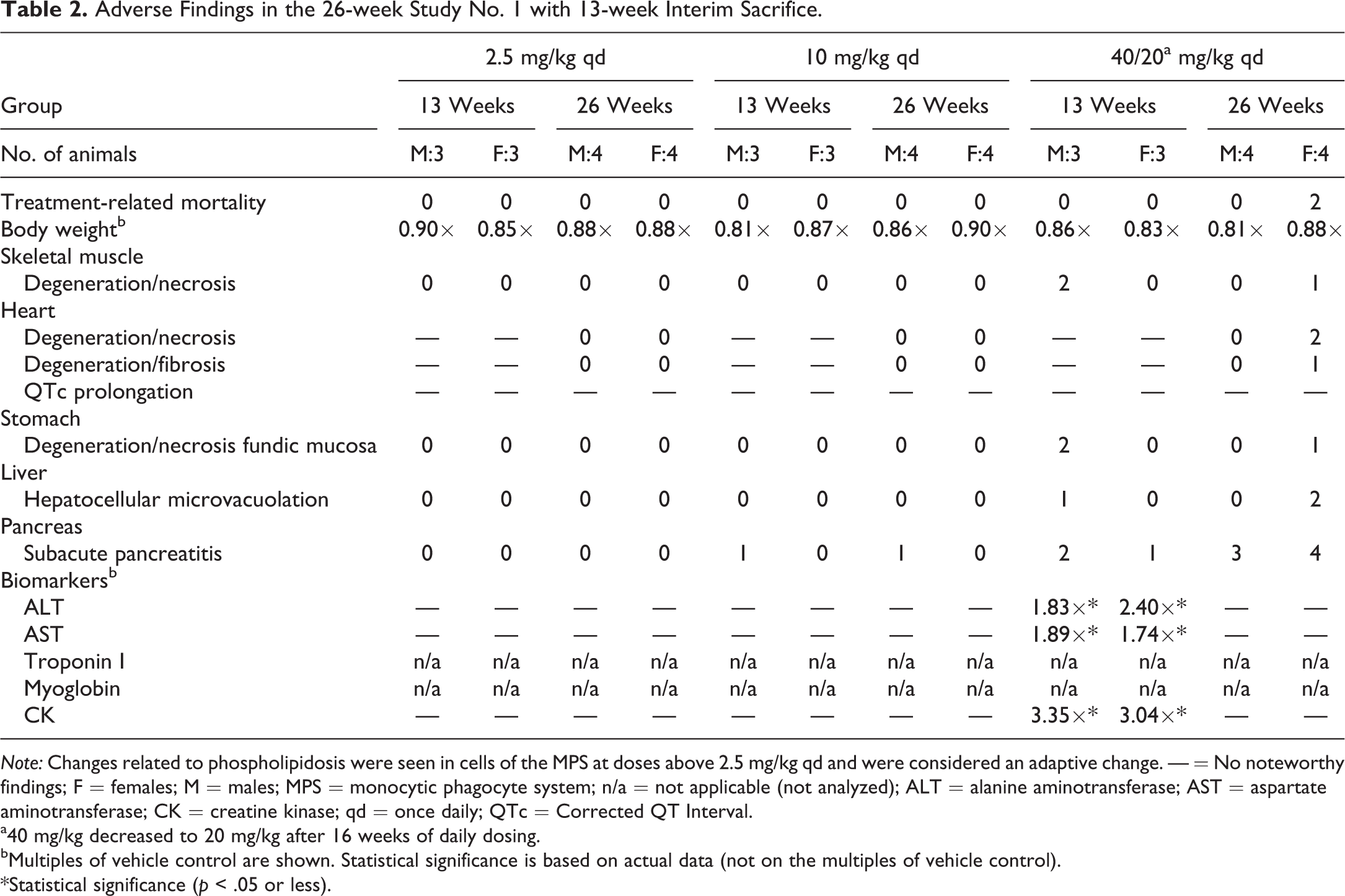
Note: Changes related to phospholipidosis were seen in cells of the MPS at doses above 2.5 mg/kg qd and were considered an adaptive change. — = No noteworthy findings; F = females; M = males; MPS = monocytic phagocyte system; n/a = not applicable (not analyzed); ALT = alanine aminotransferase; AST = aspartate aminotransferase; CK = creatine kinase; qd = once daily; QTc = Corrected QT Interval.
a40 mg/kg decreased to 20 mg/kg after 16 weeks of daily dosing.
bMultiples of vehicle control are shown. Statistical significance is based on actual data (not on the multiples of vehicle control).
*Statistical significance (p < .05 or less).
Adverse Findings in the 26-week Study No. 2 with 8-week Interim Sacrifice.
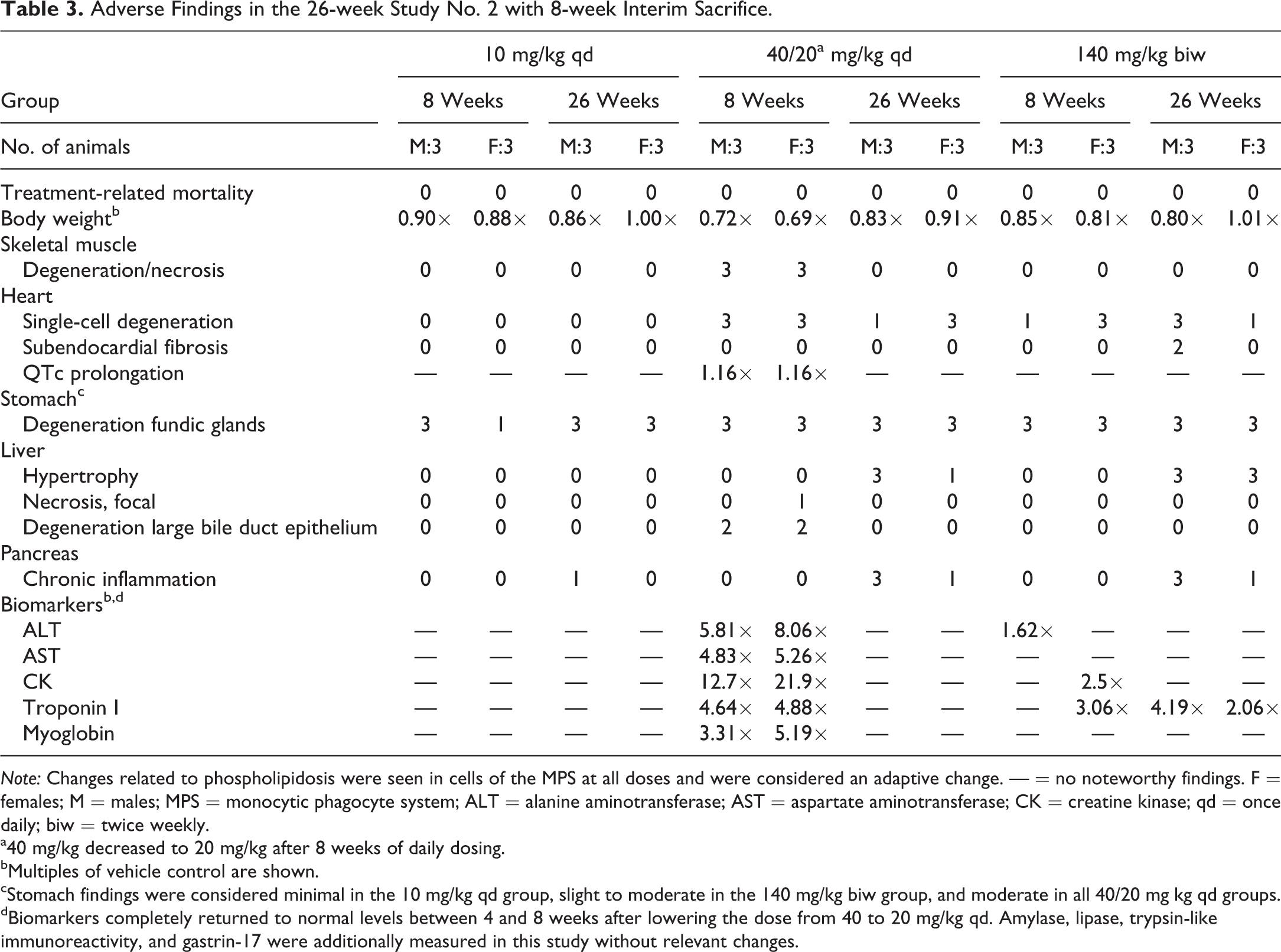
Note: Changes related to phospholipidosis were seen in cells of the MPS at all doses and were considered an adaptive change. — = no noteworthy findings. F = females; M = males; MPS = monocytic phagocyte system; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CK = creatine kinase; qd = once daily; biw = twice weekly.
a40 mg/kg decreased to 20 mg/kg after 8 weeks of daily dosing.
bMultiples of vehicle control are shown.
cStomach findings were considered minimal in the 10 mg/kg qd group, slight to moderate in the 140 mg/kg biw group, and moderate in all 40/20 mg kg qd groups.
dBiomarkers completely returned to normal levels between 4 and 8 weeks after lowering the dose from 40 to 20 mg/kg qd. Amylase, lipase, trypsin-like immunoreactivity, and gastrin-17 were additionally measured in this study without relevant changes.
The most prominent microscopic findings after repeated dosing with BDQ in both studies were the light microscopic and ultrastructural changes related to PLD in cells of the MPS. A higher incidence of pigmented and/or foamy macrophages (see Figure 2) was seen in various tissues and principally in lymphoid organs (spleen, thymus, lymph nodes, and any mucosa associated lymphoid tissue), lungs, liver, stomach, skeletal muscle, pancreas, and/or uterus; in the lungs, this was associated with the presence of granulomas. Although pigmented macrophages were also seen in vehicle control dogs, not exposed to BDQ, the incidence and/or grade was increased in the 10 and 40/20 mg/kg qd dose groups. Foamy macrophages were seen from 10 mg/kg qd onward. Ultrastructural examination by electron microscopy (Figure 3) indicated the presence of myeloid bodies in the examined tissues confirming PLD at doses above 2.5 mg/kg qd. The lamellae of the myeloid bodies were usually concentric but occasionally organized in a laminar fashion. Myeloid bodies were observed in peripheral white blood cells (mainly in lymphocytes and to lesser extent in monocytes) and in macrophages; parenchymal cells; vascular endothelial cells; and/or smooth muscle cells of liver, lung, exocrine pancreas, skeletal muscle, spleen, and stomach (fundus).
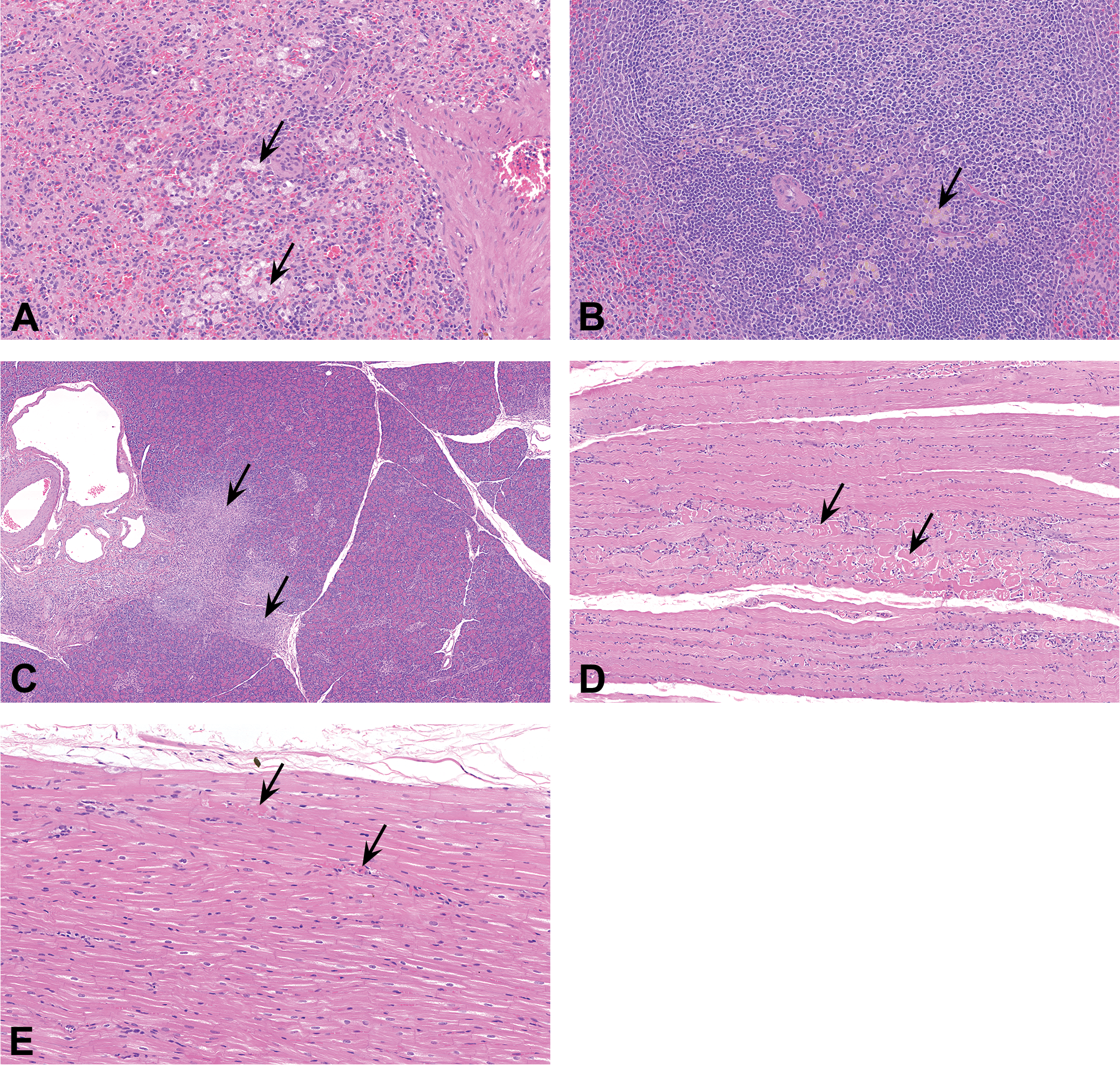
Photomicrographs of phospholipidosis in the spleen and of adverse findings in pancreas, skeletal muscle, and heart from dogs dosed for 8 or 26 weeks with bedaquiline (26-week study no. 2). (A) Swollen, foamy macrophages and (B) pigmented macrophages in spleen at 10 mg/kg once daily (qd) after 26 weeks. (C) Chronic inflammation in pancreas at 40/20 mg/kg qd after 26 weeks of dosing. (D) Necrosis/degeneration in skeletal muscle at 40 mg/kg qd after 8 weeks of dosing. (E) Minimal single-cell degeneration of cardiomyocytes in the subendocardial region of the left ventricle of the heart at 40 mg/kg qd after 8 weeks of dosing.
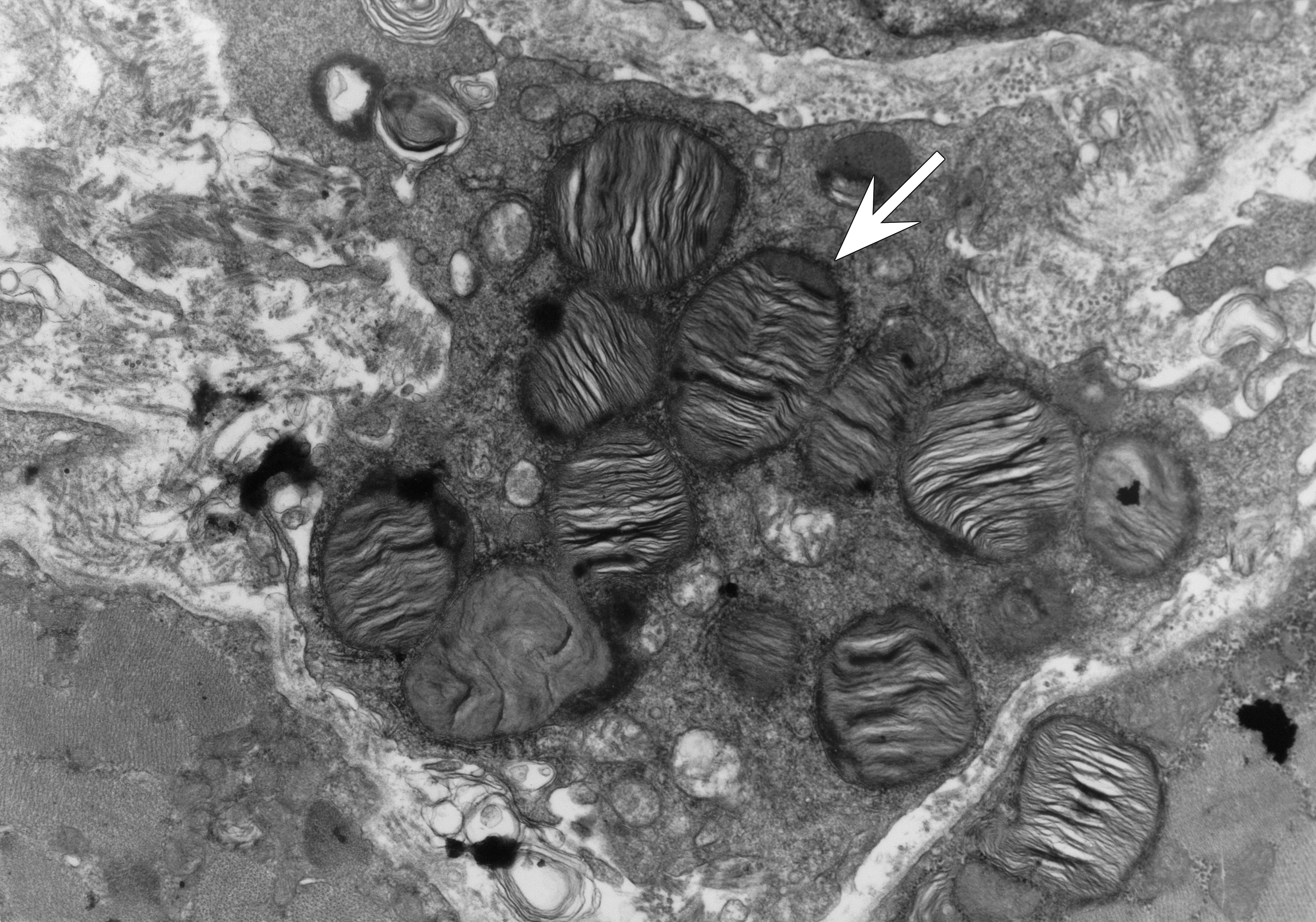
Electron microscopic image of skeletal muscle. Myeloid bodies (arrow) were noted in muscle fibers and macrophages. In this picture, the lamellae are mainly organized in a laminar fashion (in contrast to the predominant concentric organization that was observed).
Other light microscopic changes after repeated dosing with BDQ were mostly confined to animals receiving the high dose of 40 mg/kg qd and appeared to be more pronounced in study no. 2 where, compared to the first study, higher exposure levels were measured, despite the same high-dose level. Dogs dosed with BDQ for 8 weeks at 40 mg/kg qd in study no. 2 showed degenerative and necrotic lesions in skeletal muscle. The skeletal muscle lesions had completely recovered by the end of the 26-week dosing period and after lowering the dose from 40 to 20 mg/kg qd in week 8. No skeletal muscle changes were seen in the study no. 1 when dogs were dosed with BDQ at 40 mg/kg qd for 13 weeks. Also in study no. 2, myocardial degeneration/necrosis was observed after 8 weeks of daily BDQ dosing at 40 mg/kg qd. At the end of the 26-week dosing period and after lowering the dose to 20 mg/kg qd in week 8, histological changes in the myocardium consisted of degeneration of a few single cardiomyocytes with a minimal lymphohistiocytic infiltrate, associated with minimal to slight endocardial fibrosis. In study no. 1, no histological changes in the myocardium were seen in dogs dosed with BDQ at 40 mg/kg qd for 13 weeks, but slight myocardial degeneration/necrosis or myocardial degeneration/fibrosis was observed in the 2 surviving high-dose females at the end of the study and after lowering the dose to 20 mg/kg qd in week 16. These changes were located subendocardially in the left ventricle. Changes in the stomach and pancreas were noted after 8 (study no. 2, only changes in stomach) or 13 weeks (study no. 1) of dosing at 40 mg/kg qd and after 26 weeks of dosing at 40/20 mg/kg qd (both studies). Changes in the stomach consisted of multifocal to diffuse degenerative and atrophic changes in the glandular part of the stomach (fundus) and were noted alongside a mixed inflammatory cell infiltrate associated with the presence of pigmented macrophages. Changes in the pancreas consisted of a focal to multifocal subacute or chronic pancreatitis. Hepatocellular microvacuolation was noted in study no. 1 after 13 and 26 weeks of dosing at 40 and 40/20 mg/kg qd, respectively. In study no. 2, hepatocellular hypertrophy was seen after 26 weeks at 40/20 mg/kg qd. Degenerative changes in hepatocytes (focal necrosis) and large bile duct epithelium were only present after 8 weeks of dosing at the highest dose level of 40 mg/kg qd (study no. 2) and disappeared after lowering the dose.
The histological changes in animals receiving the high dose of 40 mg/kg qd for 8 weeks in study no. 2 were accompanied by changes in several safety biomarkers. Alanine aminotransferase (ALT), aspartate aminotransferase (AST), and CK increased from 4 weeks of dosing and myoglobin concentrations as well as cardiac troponin I after 8 weeks of dosing; these biomarker changes completely returned to normal levels between 4 and 8 weeks after lowering the dose from 40 to 20 mg/kg qd, such that no biomarker changes were seen at the end of the 26-week dosing period. Although no histological muscle lesions were observed in study no. 1, dogs receiving 40 mg/kg qd for 13 weeks showed similar increases in ALT, AST, and CK that disappeared at completion of dosing after 26 weeks and including the dose level reduction to 20 mg/kg qd after 16 weeks. Changes in amylase, lipase, trypsin-like immunoreactivity, or gastrin-17 levels were absent despite histological changes in the pancreas and the stomach. By light microscopy, PLD can possibly be identified by the appearance of cytoplasmic vacuoles in peripheral blood mononuclear cells in whole blood smears; however, phospholipids-containing inclusions resulting from BDQ administration were not clearly observed as vacuoles in blood smears by light microscopy.
ECGs in dogs dosed at 40 mg/kg qd for 8 weeks in study no. 2 showed increased QT intervals (216 msec vs. 173 msec in control dogs; 25% increase) coinciding with decreased heart rates (98 bpm vs. 123 in control dogs). When corrected for heart rate according to Fridericia’s formula, QTc intervals were still prolonged (254 msec vs. 219 in control dogs; 16% increase). QTc intervals normalized within 4 weeks after lowering the dose from 40 to 20 mg/kg qd. No QTc prolongation was seen in the study no. 1 where dogs were dosed with BDQ at 40 mg/kg qd for 13 weeks.
PLD and Adverse Findings in 39-week Study after Once Daily Oral Dosing with BDQ and Reversibility
Key toxicity findings from the 39-week study in dogs are presented in Table 4. The dose level of 2 mg/kg qd was not associated with PLD after 39-weeks of daily dosing. One male and 2 female dogs at the high-dose level of 18 mg/kg qd were sacrificed during the last trimester of the study showing a body weight loss of more than 10% relative to the start of the study; in at least 1 of these dogs, pancreatitis was considered the cause of weight loss. BDQ administration at dose levels of 6 mg/kg qd and above was associated with pigmented or foamy macrophage accumulation in various organs, suggestive of PLD; a low incidence and minimal to moderate atrophy or degeneration of the fundic mucosal glands of the basal region of the stomach (not impacting the general health state of the animals) and chronic pancreatitis (considered adverse) were present. Findings were most pronounced in the 18 mg/kg qd dosed animals. Skeletal muscle and myocardial degeneration as well as ECG findings (QTc prolongation) were absent, and levels of ALT, AST, CK, and myoglobin did not change in the 39-week study; however, cardiac troponin I concentrations were elevated in dogs at 18 mg/kg qd.
Adverse Findings in the 39-week Study Followed by 13-week Treatment-free Period.

Note: Changes related to phospholipidosis were seen in cells of the MPS at doses above 2 mg/kg qd and were considered an adaptive change. — = no noteworthy findings; F = females; M = males; MPS = monocytic phagocyte system; ALT = alanine aminotransferase; AST = aspartate aminotransferase; CK = creatine kinase; qd = once daily; tiw = 3 times weekly.
aMultiples of vehicle control are shown. Statistical significance is based on actual data (not on the multiples of vehicle control).
bActual data (vehicle group data: M: 0.014 µg/L; F: 0.000 µg/L).
*Statistical significance (p < .05 or less).
No delayed toxicities occurred during the 13-week treatment-free period following the 39-week treatment period. Thirteen weeks after cessation of treatment, decreased occurrence and severity, or disappearance of the changes in the MPS, pancreas, and stomach in animals that previously received 18 mg/kg qd suggested at least partial and ongoing recovery. Pigment-laden macrophages were present at reduced grades of severity, and incidences for most affected tissues and foamy macrophages in the spleen, lymph nodes, and lungs had disappeared. Minimal to mild chronic pancreatitis and atrophy or degeneration of the fundic mucosal glands of the basal region of the stomach remained in 1 female animal. Cardiac troponin I had completely returned to normal vehicle control values.
PLD and Adverse Findings after Intermittent Oral Dosing with BDQ
In 26-week study no. 2, a group of dogs was dosed intermittently (140 mg/kg biw) with the same weekly dose (280 mg/kg) as to the group dosed daily (40 mg/kg qd). Intermittent dosing of BDQ was better tolerated than daily dosing despite the same weekly dose (280 mg/kg/week). Twice weekly dosing at 140 mg/kg resulted in findings in the heart, pancreas, and stomach similar to those observed after daily dosing at 40 mg/kg qd, but other changes were limited (PLD) or absent (QT/QTc prolongation, skeletal muscle necrosis, and degenerative changes in liver/large bile ducts). In contrast to the dogs that were dosed at 40 mg/kg qd for 8 weeks, only marginally or slightly elevated levels of ALT, CK, and cardiac troponin I were present in animals dosed intermittently at 140 mg/kg biw, and AST and myoglobin concentrations were in the normal range.
When a lower weekly dose was administered in the 39-week study (42 mg/kg/week), findings after intermittent dosing at 14 mg/kg tiw were similar to those after daily dosing at 6 mg/kg qd.
Plasma and Tissue Concentrations of BDQ and D-BDQ
Administration of a single dose of BDQ resulted in maximum plasma concentrations around 3 hr postdose followed by a slow decline. D-BDQ concentrations slowly increased up to 8 hr postdose and then hardly decreased. Repeated dosing resulted in accumulation of BDQ and D-BDQ (2- to 7-fold [BDQ] and 8- to 21-fold [D-BDQ] higher plasma exposures at the end of the 39-week study vs. after single dosing). For both compounds, the plasma profiles at the end of the studies were flat at all doses, with the exposure parameters of D-BDQ generally being 2-fold lower than those of BDQ (Figure 4). Steady state was reached at 13 weeks or later, and no clear gender effect on plasma exposure was apparent in any study.
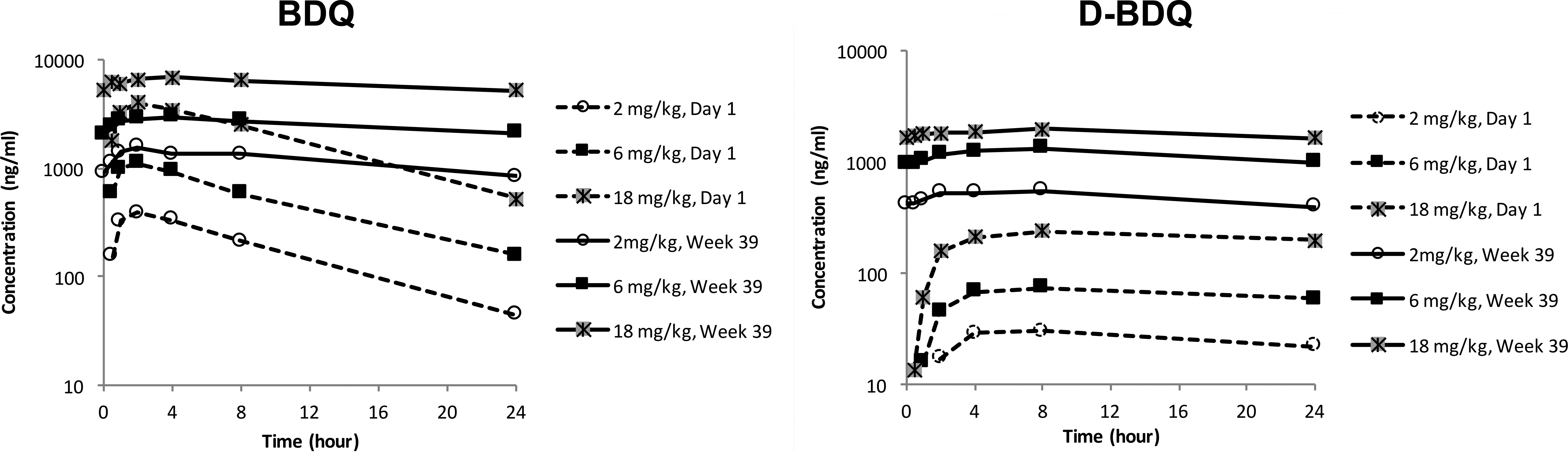
Mean bedaquiline (BDQ) and D-BDQ plasma concentration–time profiles in male dogs upon BDQ once daily dosing (39-week study). Mean 24-hr plasma concentration–time profiles for BDQ and D-BDQ determined in the 39-week study on day 1 (first administration) and at week 39 following once daily administration of BDQ to male dogs at 2 mg/kg (n = 4), 6 mg/kg (n = 4), or 18 mg/kg (n = 6). On day 1, plasma concentrations of BDQ were maximum at around 3 hr postdose and then declined slowly, while D-BDQ concentrations slowly increased up to around 8 hr postdose and then hardly decreased. In week 39, plasma profiles were flat at all doses for BDQ and D-BDQ.
After 4 weeks of daily dosing, BDQ exposure between 2.5 and 10 mg/kg qd increased roughly dose proportionally (males) or somewhat less-than-dose proportionally (females). By contrast, exposure between 10 and 40 mg/kg qd hardly increased or increased up to 2-fold only in both 26-week studies. BDQ and D-BDQ exposure (AUC) was comparable to higher (2- to 3-fold for BDQ) in the second than in the first 26-week study. At the end of 26-week study no. 1, BDQ exposure at 2.5 and 10 mg/kg qd was higher than after 4 weeks and exposure increased less-than-dose proportionally. Following reduction of the top dose from 40 to 20 mg/kg qd (after 16 weeks of dosing in study no. 1 and 8 weeks in study no. 2), BDQ plasma exposure at the end of the treatment was generally comparable or up to 2-fold higher compared to the exposure after 4 weeks, most probably because steady state was not yet reached after 4 weeks.
BDQ exposure increased fairly dose proportionally between 2 and 18 mg/kg qd in the 39-week study. BDQ plasma exposure reached in the 39-week study at 18 mg/kg qd at week 13 and at the end of the treatment was similar to the exposure reached in the 26-week study no. 2 at 40 mg/kg qd after 4 weeks.
At the same weekly dose of BDQ, the systemic exposure to BDQ and D-BDQ (AUC over one week) was generally comparable between the daily and intermittent dosing regimens, that is, at 40 mg/kg qd and 140 mg/kg biw after 4 weeks of dosing in the second 26-week study and at 6 mg/kg qd and 14 mg/kg tiw at the end of the treatment in the 39-week study.
Distribution of BDQ and D-BDQ to most tissues was extensive, with high BDQ tissue concentrations in lung, spleen, lymph nodes, and pancreas (tissue/plasma concentration ratio >30). A lower tissue/plasma ratio was observed in skeletal muscle, and the concentration in the brain was only slightly above that in plasma. D-BDQ concentrations in most tissues were higher than those of BDQ despite D-BDQ plasma concentrations being lower than those of BDQ. Between 2.5 and 10 mg/kg qd, the tissue trough concentrations of both compounds increased almost dose proportionally at the end of the treatment in the 26-week study no. 1 (Figure 5, panel A). By contrast, the trough concentrations increased more than dose proportionally between 10 and 40 mg/kg qd for most tissues at 8 weeks in the 26-week study no. 2 (Figure 5, panel B).
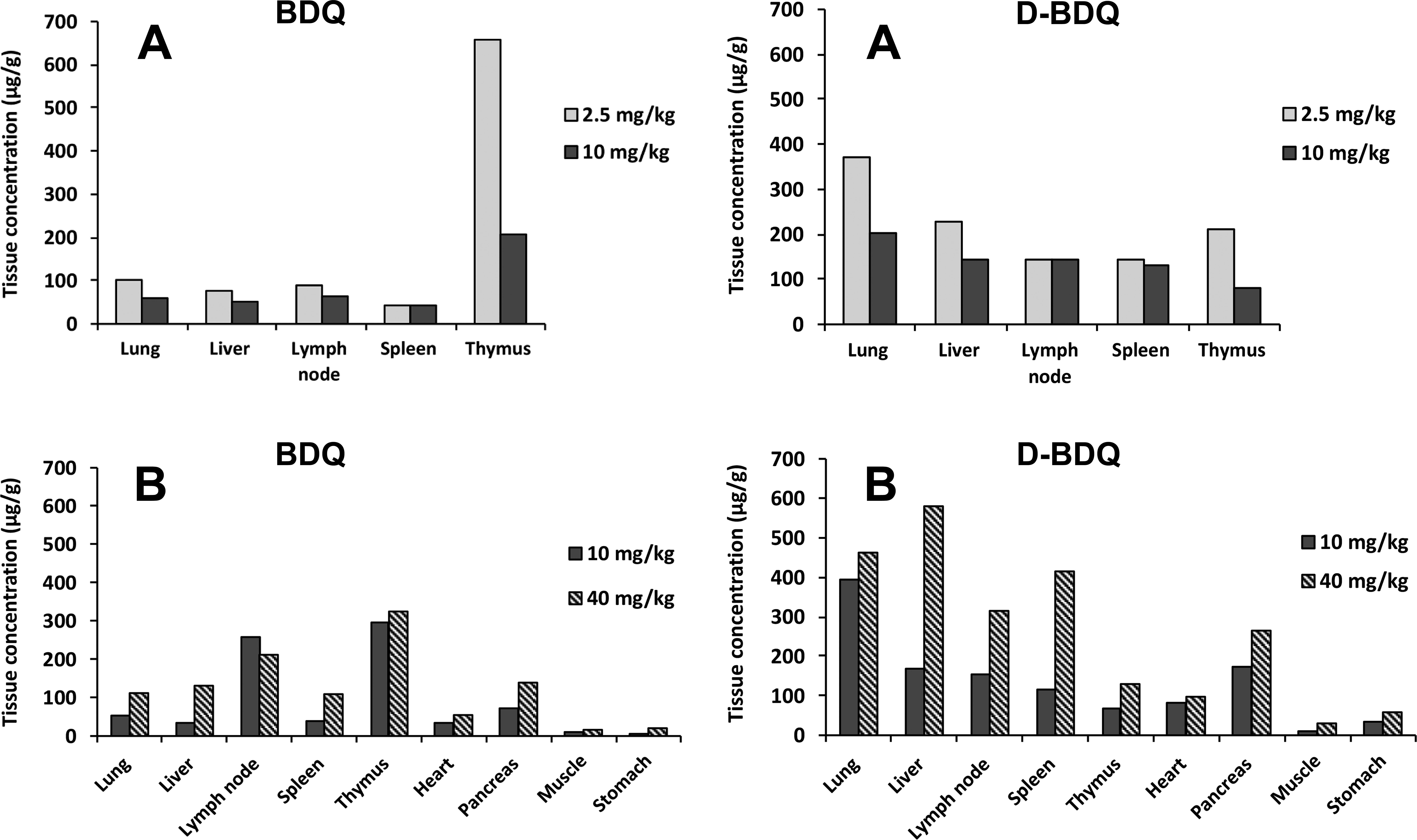
Mean dose normalized trough tissue concentrations of bedaquiline (BDQ) and D-BDQ in male dogs upon BDQ once daily dosing. Mean trough tissue concentrations of BDQ and D-BDQ normalized for a dose of 10 mg/kg following once daily dosing of BDQ to male dogs: (A) at 2.5 mg/kg (n = 4) and 10 mg/kg (n = 4) at the end of the 26-week study no. 1 (at necropsy around 24 hr after administration of the last dose) and (B) at 10 mg/kg (n = 3) and 40 mg/kg (n = 3) after 8 weeks of dosing in the 26-week study no. 2 (at interim kill around 24 hr after administration of the last dose). D-BDQ concentrations in most tissues were higher than those of BDQ. Tissue trough concentrations of both compounds increased almost dose proportionally between 2.5 and 10 mg/kg once daily (qd) at the end of the treatment in the 26-week study no. 1 (A). By contrast, the trough concentrations increased more than dose proportionally between 10 and 40 mg/kg qd for most tissues at 8 weeks in the 26-week study no. 2 (B). The greater than proportional increase was more pronounced with D-BDQ.
The trough tissue concentrations of BDQ and D-BDQ after 8 weeks of dosing at 40 mg/kg qd were compared to those measured after 8 weeks of dosing at 40 mg/kg qd plus 18 weeks of dosing at 20 mg/kg qd in the 26-week study no. 2. Tissue concentrations of BDQ and D-BDQ were 35% to 92% lower after a dose reduction from 40 to 20 mg/kg qd (Figure 6). This effect was more pronounced with D-BDQ.
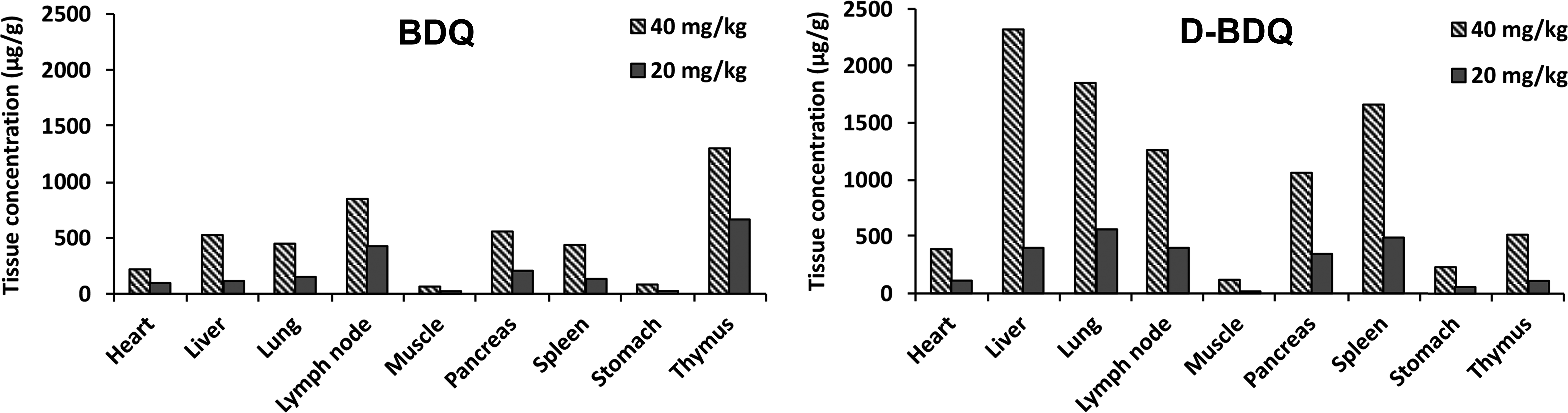
Mean trough tissue concentrations of bedaquiline (BDQ) and D-BDQ in male dogs after BDQ dose reduction from 40 to 20 mg/kg. Mean trough tissue concentrations of BDQ and D-BDQ in the 26-week study no. 2 following once daily dosing of BDQ to male dogs at 40 mg/kg (n = 3) after 8 weeks of dosing (interim kill around 24 hr after administration of the last dose) and at 20 mg/kg (n = 3) after 8 weeks of dosing at 40 mg/kg and 18 weeks of dosing at 20 mg/kg (necropsy around 24 hr after administration of the last dose at the end of the study). Tissue concentrations of BDQ and D-BDQ were much lower after the dose reduction from 40 to 20 mg/kg once daily. This effect was more pronounced with D-BDQ.
Discussion
The structure of BDQ and its primary metabolite D-BDQ is consistent with that of a CAD. CADs are known to distribute extensively to tissues and to be slowly released, mainly as a result of binding to phospholipids and trapping in acidic compartments (lysosomes) in the cells. The drug–phospholipid complexes are arranged in the so-called lysosomal lamellar bodies, visible at the ultrastructural level, a condition generally referred to as PLD (Reasor, Hastings, and Ulrich 2006; Hanumegowda et al. 2010). The dose level of 2 (39-week study) or 2.5 mg/kg qd BDQ (26-week study) was not associated with any changes indicative of PLD in dogs after daily dosing. Light microscopic and ultrastructural changes related to PLD were observed in chronic toxicity studies in dogs at doses above 2 or 2.5 mg/kg qd, mainly in cells of the MPS. Accumulation of pigment-laden and/or foamy macrophages was observed in the lymph nodes, spleen, liver, stomach, skeletal muscle, pancreas, and particularly the lungs. At electron microscopic examination, BDQ-induced PLD was characterized by the prominent presence of intracytoplasmic multilamellar inclusions (myeloid bodies) in the affected tissues. There were no other relevant ultrastructural findings and clearly no mitochondrial morphological changes. The latter was in line with the high in vitro selectivity of the compound toward the mycobacterial ATP synthase (Haagsma et al. 2009). The potential risk for toxicities with CADs is not related to PLD as such (= accumulation of multilamellar lysosomal bodies) but can be attributed to extensive drug accumulation resulting in very high tissue concentrations and/or to interference with lysosomal membranes resulting in leakage of lysosomal proteases (Lullmann, Lullmann-Rauch, and Wassermann 1975; Reasor, Hastings, and Ulrich 2006).
Plasma concentrations of BDQ in dogs increased upon repeated administration and concentration–time profiles became flat, consistent with the slow concentration decline observed after single administration. A t 1/2, term of at least 14 days was expected after single intravenous administration to dogs based on blood sampling up to 11 days after dosing (Janssen, data on file from a single dose intravenous pharmacokinetics study at 1 mg/kg). Trough tissue concentrations of BDQ and D-BDQ provide a good representation of tissue distribution in place of an area under the curve after repeated daily administration since the tissue concentration–time profiles are expected to be flat as observed for the plasma profiles. Distribution of BDQ and D-BDQ to tissues rich in lysosomes was extensive, with tissue/plasma concentration ratios generally above 30, including in the lungs. Whereas plasma concentrations of D-BDQ were generally below those of BDQ with plasma AUC ratios of D-BDQ to BDQ ranging from 0.20 to almost 1, tissue concentrations of D-BDQ were generally higher than those of BDQ. The strong tissue retention of D-BDQ might be due to its basic properties (pKa D-BDQ 9.97 as compared to pKa BDQ 8.77) making it a stronger CAD than BDQ. This is in agreement with its higher potential to induce PLD in an in vitro PLD screening assay (Mesens, Steemans, Hansen, Peters, et al. 2009; Mesens, Steemans, Hansen, Verheyen, et al. 2010). The same in vitro assay also demonstrated D-BDQ to be more cytotoxic than BDQ in the human monocytic cell line THP-1. Thus, because of its higher cytotoxicity and its stronger retention in tissues, D-BDQ is expected to contribute significantly to the toxicity observed in dogs after long-term administration of high doses of BDQ.
Plasma and tissue exposure for BDQ and D-BDQ increased roughly proportionally or somewhat less than proportionally with the dose of BDQ following repeated administration between 2.5 and 10 mg/kg qd. At the high-dose levels administered in dogs (>10 mg/kg qd), a more than dose-proportional increase in tissue concentrations was associated with a less-than-dose-proportional increase in plasma exposure for both compounds, especially for D-BDQ. This suggests that BDQ and D-BDQ were more extensively retained in tissues at doses above 10 mg/kg qd, consistent with more pronounced PLD and accumulation of phospholipids resulting in an increase in available binding sites (Mesens et al. 2012).
Toxicities related to daily BDQ administration were more pronounced in the 26-week studies than in the 39-week study despite similar plasma levels being reached at the end of treatment in the 39-week study. In both 26-week studies an (initial) high dose of 40 mg/kg qd was administered that was associated with an overproportional tissue uptake of BDQ and D-BDQ and a less-than-dose-proportional increase in plasma exposure. Chronic administration of high doses of BDQ (40 mg/kg qd) resulted in pronounced, dose-related toxicities in skeletal muscle, heart, stomach, liver, and pancreas as well as QTc-interval prolongation. In contrast, in the 39-week study a lower top dose of 18 mg/kg qd was administered, which was characterized by a dose proportional to slightly less-than-dose proportional plasma exposure for BDQ and D-BDQ across the dose range up to the high-dose level at the beginning and at the end of treatment. Since tissue concentrations for BDQ and D-BDQ in the first 26-week study increased roughly dose proportionally at low doses between 2.5 and 10 mg/kg qd when plasma concentrations increased with the dose, it can be assumed that tissue concentrations also increased dose proportionally in the 39-week study between 2 and 18 mg/kg qd. Thus, the overproportional loading of BDQ and D-BDQ in tissues seen in the 26-week studies is not expected to have occurred in the 39-week study, which may explain why less organ toxicities were seen in the 39-week study than in the 26-week studies despite similar plasma exposures. Liver, heart, and skeletal muscle were unaffected in the 39-week study. While BDQ is only slowly eliminated from the body and the t 1/2, term is long, biomarker changes (ALT, AST, CK, myoglobin, and cardiac troponin I) had already completely disappeared between 4 and 8 weeks after lowering the top dose from 40 to 20 mg/kg qd in the 26-week study no.2, indicating that biomarkers recover faster than plasma levels decline. The FDA database of PLD-positive approved drugs shows a high association between PLD and QT prolongation; the QTc-interval prolongation seen in our study normalized within 4 weeks after lowering the dose from 40 to 20 mg/kg qd.
Upon termination of CAD administration, PLD is typically reversible with the drug diffusing out of the cells, the phospholipid levels returning to normal, and the ultrastructural changes gradually disappearing (Lullmann, Lullmann-Rauch, and Wassermann 1975; Reasor, Hastings, and Ulrich 2006). In the 39-week dog study, findings of PLD were slowly reversible upon treatment cessation and at least partial recovery was seen at the end of the 13-week treatment-free period. The slow reversibility with BDQ is consistent with the slow elimination of this drug from the body. In the 26-week studies, tissue concentrations of BDQ and D-BDQ were much lower (35% to 92%) at the end of the study, that is, following 8 or 13 weeks of dosing at 40 mg/kg qd followed by 16 or 13 weeks of dosing at 20 mg/kg qd, than after 8 weeks of dosing (interim time point) at 40 mg/kg qd, whereas corresponding plasma concentrations were generally not reduced following reduction of the top dose. This indicates that the amounts of BDQ and D-BDQ that had accumulated into tissues at mid or high doses were slowly but consistently released from the tissues into the peripheral blood after treatment cessation or lowering the dose. Release of BDQ from tissues after cessation of high-dose administration was also noted in other studies; BDQ treatment for 4 weeks in dogs (160 mg/kg qd) clearly showed a similar decline (49% to 87% reduction) of plasma and tissue concentrations of BDQ during a 4-week treatment-free period (Janssen, data on file from 4-week study followed by a 4-week treatment-free period, not discussed in this article).
Drugs with long t 1/2, term like BDQ allow intermittent dosing regimens. In dogs, biw administration of BDQ (280 mg/kg/week) was better tolerated than a daily administration of a same total weekly dose despite comparable plasma exposure in terms of overall AUC over 1 week. PLD changes were limited and no QT/QTc prolongation, skeletal muscle necrosis, or degenerative changes in liver/large bile ducts were seen. The AUC was previously shown to be the main driver of the pharmacokinetic and pharmacodynamic response in a murine model of tuberculosis, supporting intermittent administration of BDQ, in agreement with its slow release from tissues (Rouan et al. 2012). The same benefit was not seen at low doses. Three times weekly administration of BDQ (42 mg/kg/week) resulted in similar findings than those seen after daily dosing of a same total weekly dose.
BDQ also induces PLD in mice and rats (Janssen, data on file from repeated dose toxicity studies up to 13 weeks and 26 weeks, respectively). The D-BDQ plasma level in rats and dogs is close to those measured in human, with the plasma exposure of D-BDQ relative to BDQ being approximately 2-fold lower in rats and dogs and 3- to 4-fold lower in humans (van Heeswijk, Dannemann, and Hoetelmans 2014). On the other hand, in mice, D-BDQ is higher than BDQ in plasma and is therefore not representative of the human situation. Furthermore, the dog is considered the most important species for safety assessment because it was found to be the most sensitive one covering all preclinical target tissues.
The approved treatment regimen in MDR-TB patients is 400 mg qd for 2 weeks, followed by 200 mg tiw for 22 weeks. BDQ displayed a dose-proportional increase in exposure following multiple dose administration up to a dose of at least 400 mg given qd to healthy volunteers for 2 weeks (van Heeswijk, Dannemann, and Hoetelmans 2014). Therefore, no tissue overproportional loading of BDQ or D-BDQ is expected in patients as observed in dogs at high doses. Furthermore, intermittent administration of BDQ at 200 mg tiw, following an initial 2 weeks of qd dosing at 400 mg, should limit the potential for induction of PLD. At the approved dosing regimen in MDR-TB patients, the plasma exposure of BDQ and D-BDQ (AUC) amounted to 22 and 6 μg·h/ml, respectively, after 8 weeks of treatment (i.e., 400 mg BDQ qd for 2 weeks followed by 200 mg tiw for 6 weeks), and was 14 and 3.6 μg·h/ml, respectively, at the end of the 24-week treatment period (i.e., 400 mg BDQ qd for 2 weeks followed by 200 mg tiw for 22 weeks). The plasma exposure of BDQ and D-BDQ in MDR-TB patients after 24 weeks of BDQ administration was similar or lower than that in dogs receiving a dose of 2 mg/kg qd for 39 weeks corresponding to a plasma exposure (AUC, males and female dogs combined) of 24.6 and 12.5 μg·h/ml for BDQ and D-BDQ, respectively. PLD in dogs was seen at 3- and 6-fold higher plasma exposures compared to those observed in MDR-TB patients for BDQ and D-BDQ, respectively, and toxicities in skeletal muscle, heart, stomach, liver, and pancreas were only observed at higher plasma exposures compared to those observed in patients. In current clinical monitoring, no sensitive biomarker is available for PLD. However, all toxicities seen in preclinical species are monitored in clinic.
In conclusion, toxicities in dogs mainly occurred at high-dose levels associated with a less-than-dose-proportional increase in plasma exposure but an overproportional tissue uptake of BDQ and D-BDQ. Consistent with the drop in tissue concentrations upon treatment cessation, partial, or ongoing recovery of the pathological findings was observed in dogs, with no evidence of delayed toxicity. Exposures of BDQ in dogs at lower doses (2.5 mg/kg qd for 26 weeks and 2 mg/kg qd for 39 weeks) are consistent with the exposures achieved at the therapeutic dose in man, and were well tolerated, without adverse effects and not associated with PLD.
Footnotes
Author Contributions
The following authors had a significant involvement in the conception and/or design of the study (IS, SDJ, ST, SS, and M-CR), or acquisition and/or analysis and/or interpretation of data (IS, SDJ, AL, GM, TV, AL, and M-CR). All authors were involved in the development of the primary manuscript, have read and approved the final version, agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved and have therefore met the criteria for authorship as established by the International Committee of Medical Journal Editors.
Acknowledgments
The authors thank Dr. Fons Verheyen (Maastricht University, Maastricht, The Netherlands) for electron microscopic examinations, Erik Hansen for support with figures, and Janssen team members, in particular Koen Andries, Nyasha Bakare, Brian Dannemann, Chrispin Kambili, Koné Kaniga, Sophie Lachau-Durand, Karin Rombouts, Hans Smit, and Myriam Theeuwes, for their review and input into this article.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are full-time employees of Janssen and potential stockholders of Johnson and Johnson.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Janssen.