
Abstract
Toxic tendinopathy is a rare but reproducible complication in humans, given agents of four drug classes: aromatase inhibitors, fluoroquinolone antibiotics, glucocorticoids (long-term regimens), and statins. Toxic tendinopathy in humans has been linked less consistently to treatment with anabolic steroids, antiretroviral agents (mainly protease inhibitors), metalloproteinase inhibitors (MMPI), and isotretinoin. Classic drug-induced tendinopathies appear as “tendinosis” (i.e., progressive tendon degeneration without inflammation), although cases associated with aromatase inhibitors exhibit mainly tenosynovitis. Any tendon may be affected, but fluoroquinolones, glucocorticoids, and statins most frequently affect large load-bearing tendons in the lower limb, especially the calcaneal (“Achilles”) tendon—which ruptures in approximately 30 to 40% of cases. The time to symptom onset ranges from days (fluoroquinolones) to weeks, months, or even years. The pathogenesis is incompletely understood, but proposed mechanisms include apoptosis of tenoblasts and tenocytes, deficient tenocyte function (leading to abnormal extracellular matrix maintenance and repair as well as disrupted intercellular signaling), and structural disintegration (via a combination of increased expression of lytic enzymes, lessened cholesterol content in cell membranes, and neoangiogenesis within highly ordered tendon tissue). Nonclinical safety assessment of therapeutic candidates in these drug classes should incorporate tendon routinely as a protocol-specified tissue for pathology evaluation.
Toxic tendinopathy associated with drug administration is an underappreciated outcome in human patients. This minireview summarizes our current understanding of toxic tendinopathy and proposes reasonable means for incorporating routine evaluation of tendons into nonclinical safety assessment strategies.
Normal tendon structure is conserved throughout the body and across vertebrate species. At the macroscopic level, tendons form white, rounded, smooth, and slick bridges that attach skeletal muscles to bones. At the microscopic level (Józsa and Kannus 1997; Kirchgesner et al. 2014), tendon bodies are formed of elongated collagen fibers (with collagen type I comprising about 70–80% of the dry tendon weight) and fewer elastic fibers assembled into fibrils, which in turn are organized in elongated parallel bundles (“fascicles”) except for a few obliquely arranged fibers at the tendon ends. Groups of fascicles are reinforced by thin fibrous septa and enveloped by a thicker fibrous capsule. Highly mobile tendons reside in synovium-lined tendon sheaths and are lubricated by synovial fluid. Within the tendon, about 90% of cells are modified fibroblasts designated as tenoblasts (highly active) and tenocytes (less metabolically active), which together generate collagen and elastin fibers as well as the extracellular matrix (e.g., chiefly glycosaminoglycans [GAGs] and proteoglycans, which sustain elasticity due to their highly hydrophilic nature) as well as secreted proteins (e.g., enzymes, signaling molecules). The remaining 10% of tendon cells are chondrocytes, which are found at tendon–bone junctions (called “entheses” [singular: enthesis]), as well as elements of small-caliber blood and lymphatic vessels and also nerves scattered sparingly among the fascicles. Vessels and nerves enter at the tendon–muscle junction, the tendon–bone junction, or via the synovial membrane, but many regions of the tendon (especially the middle [or “body”]) are poorly vascularized. Tendon color, cellularity, and size vary by age (Cooper and Valentine 2016) and sex (Sarver et al. 2017).
Tendon restoration under normal conditions involves a stepwise process of repair and remodeling (Kirchgesner et al. 2014; Cooper and Valentine 2016). During the initial vascular phase, acute damage leads to neoangiogenesis in response to extravasated erythrocytes and fibrin culminating in local accumulation of vascular growth factors. After a few days, the proliferative phase yields multiplication of tenoblasts in the tendon and nearby fibrous capsules, after which the new cells fabricate collagen fibers and extracellular matrix components (especially GAGs). The remodeling phase begins approximately 6 weeks later and features the normalization of collagen and GAG generation by tenoblasts and tenocytes along with reorientation of the new fibrils along the longitudinal axis (as defined by lines of force experienced when the tendon is loaded). The maturation phase starts after another 4 weeks (i.e., about 8 weeks following initial injury), at which time the metabolic capacities of tenocytes return to normal and the vascular bed regresses. The repaired tissue is of lesser quality than the original tendon because the new fibrous tissue beds are less organized and contain substantially more collagen type III—which is less elastic than collagen type I—relative to the original tendon (Cooper and Valentine 2016).
Toxic tendinopathy may be recognized by the presence of stereotypical macroscopic and microscopic changes associated with chronic degeneration (designated “tendinosis”), which generally develops in the absence of notable inflammation (or “tendinitis”). At the macroscopic level, affected tendons may be discolored (usually tan or brown), rough, and irregularly thickened. Viewed microscopically, degeneration is characterized by fibril disorganization, distortion and sometimes apoptosis of tenoblasts and tenocytes, altered extracellular matrix composition (found using special histologic stains), and neovascularization (Figure 1; Kirchgesner et al. 2014). In chronic conditions, the tendon may exhibit fibrocartilaginous metaplasia and/or mineralization, especially near entheses (lesions of which are designated as “enthesopathy”). These changes are common end-stage changes and are not specific to any particular etiology. However, spontaneous tendinosis as a background finding, such as occurs in aged individuals, is of modest degree (e.g., tan or brown discoloration macroscopically, minimal tendinosis and cartilage metaplasia microscopically) relative to that seen in cases of toxicant-induced tendon degeneration (Cooper and Valentine 2016).
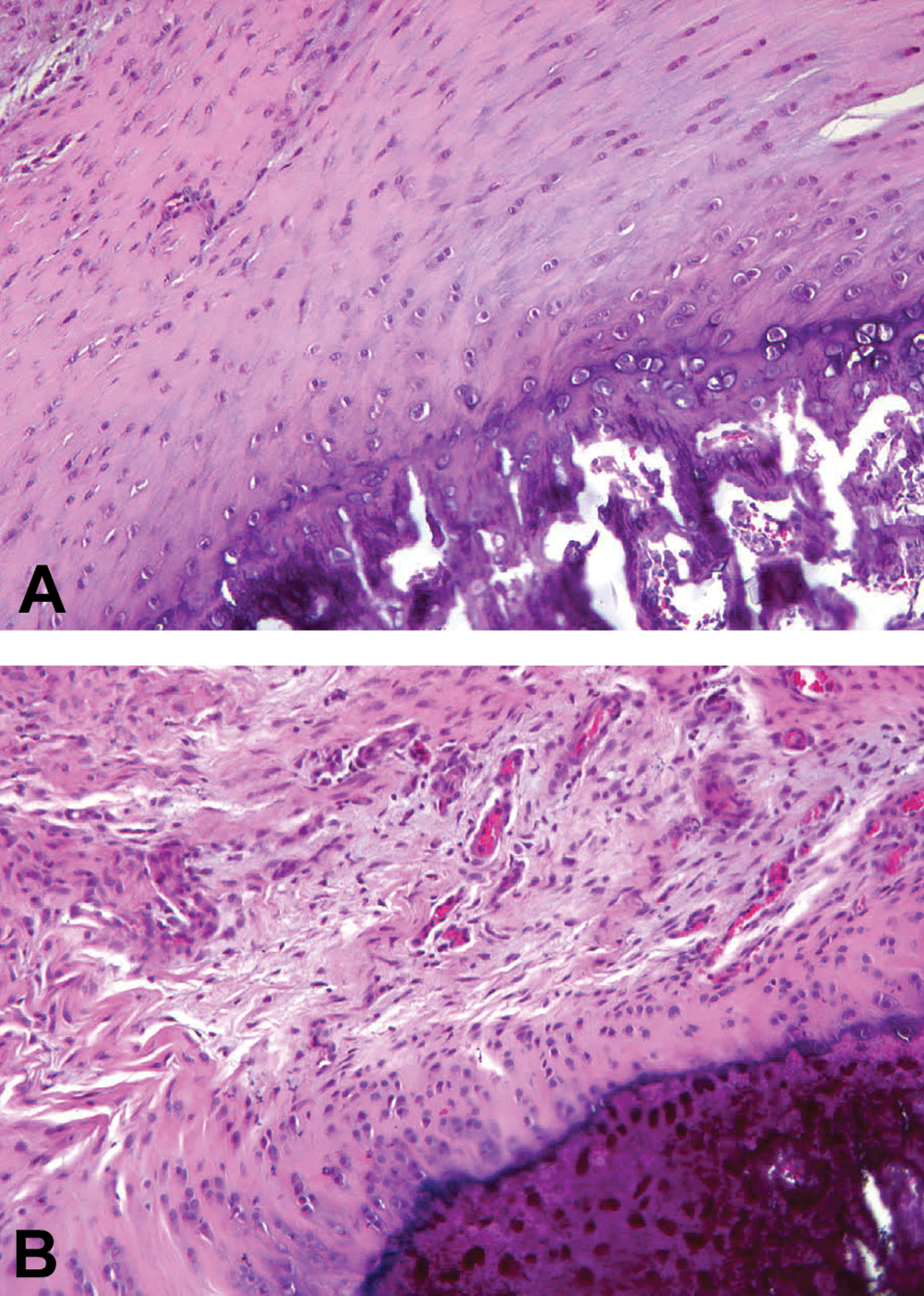
Chemically induced tendinopathy in an adult Sprague-Dawley rat. In control rats (A), the tendon is characterized by short rows of oval to spindle tenocyte nuclei interspersed among regularly spaced collagen fibers. In contrast, tendons of rats that received a single intratendinous injection of collagenase (B) exhibited marked disorganization of collagen fibers associated with many new capillaries. Location: Enthesis of the calcaneal (“Achilles”) tendon. Stain: Hematoxylin and eosin stain; original objective magnification, 50×. Source: Images adapted from Kirchgesner et al. (2014), by courtesy of the author (Professor Benjamin Dallaudière, Centre d’Imagerie Ostéo-Articulaire, Clinique du Sport de Bordeaux, Bordeaux, France) and the publisher (Elsevier Masson SAS; all rights reserved).
Toxic tendinopathy has been linked conclusively to treatment with four chemical classes (Kirchgesner et al. 2014; Knobloch 2016). The best established iatrogenic tendinopathies are correlated with administration of fluoroquinolone antibiotics or glucocorticoids. Tendon toxicity caused by fluoroquinolone antibiotics appears to occur most consistently with ciprofloxacin but also arises with other class members, regardless of the route of administration and dose. Tendinopathy develops as an acute event—within days, and sometimes following a single dose—and has an estimated incidence of 0.14 to 2%. The calcaneal (Achilles) tendon is involved in 90% of human cases, and nearly half of affected individuals experience bilateral damage; degenerative changes are centered in the tendon body rather than at the entheses, leading to rupture in approximately 40% of cases in as little as 2 weeks after onset. Toxic tendinopathy that follows long-term glucocorticoid therapy is a known outcome of uncertain incidence, often appearing in patients after 3 or more months of oral or inhalational dosing, especially in the presence of predisposing autoimmune diseases of connective tissue (e.g., rheumatoid arthritis, systemic lupus erythematosus). Primary sites of degeneration are large lower limb tendons (especially the Achilles tendon), leading to rupture (usually unilateral) several years after initiation of therapy. Toxic tendinopathy associated with statins is a dose-independent class effect that arises approximately 8 to 10 months after exposure, with an estimated incidence of 2%. The Achilles tendon is affected in just over 50% of cases, usually as a unilateral lesion, and about a third of cases result in spontaneous tendon rupture. Tendinopathy may recur if statin treatment is reinitiated. Finally, aromatase inhibitors have been linked to toxic tendinopathy, though the clinical presentation seems to differ from that of the other three classes. Aromatase inhibitors have been linked to tenosynovitis rather than tendinosis and tend to affect tendons in the hands and wrists rather than the lower limbs. The incidence is unclear but is high (almost 50%) in some reports.
Toxic tendinopathies also have been reported following exposure to several other drugs, though the evidence implicating these agents is less consistent (Kirchgesner et al. 2014). Anabolic steroids alter the biomechanical properties of tendons and have been connected to ruptured large tendons in upper and lower limbs of weight lifters. Therapy with isotretinoin has been tied to tendinopathy and also enthesopathy (which features osteophyte formation, especially along the vertebral column entheses). Antiretroviral agents, and especially protease inhibitors, have been linked to tenosynovitis and capsulitis in various joints. Peritendinous injection of local anesthetics has been shown to alter regional tenocyte function and gene expression (Honda et al. 2016). Treatment with metalloproteinase inhibitors (MMPI) has been associated with tendon pain (“musculoskeletal syndrome”) and proximal tendinitis in human patients (Groves et al. 2002; Miller et al. 2004). Histopathologic changes seen in MMPI-treated animals are dominated by exuberant fibroblast proliferation in tendons of Beagle dogs (Westwood et al. 2009) and extracapsular ligaments and joint capsules of rats (Renkiewicz et al. 2003).
Several attributes have been connected to toxic tendinopathies in human patients or animal models (Kirchgesner et al. 2014). Principal risk factors include advanced age (suggesting that senescent tenocytes have reduced maintenance abilities), obesity and physical exertion (indicating high loads and sudden shifts in axial stress as key players), preexisting disease (e.g., autoimmune diseases of connective tissues, renal failure [since the kidney is the main excretory route for quinolones]), and cotherapy with two drugs that are known to induce toxic tendinopathy. The predilection for the tendon body, which is the least vascularized region, as the usual site of rupture implies that ischemia also plays a role, perhaps by thwarting repair of degenerate tendon tissue. Finally, many cellular and biochemical mechanisms are reported to launch and sustain toxic tendinopathies (as discussed below and in Table 1; Molloy et al. 2006; Dirks and Warden 2011; Kirchgesner et al. 2014).
Mechanisms Linked to Induction or Progression of Toxic Tendinopathy.
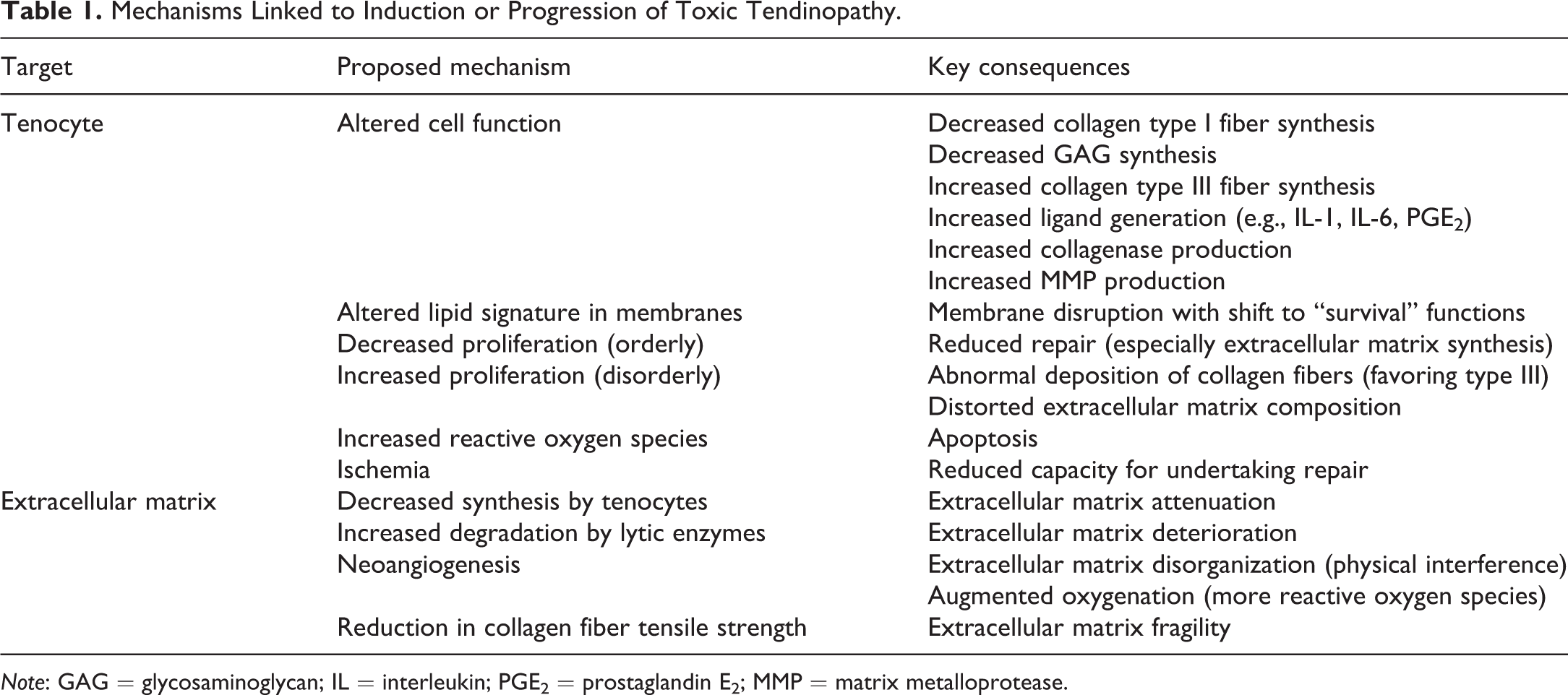
Note: GAG = glycosaminoglycan; IL = interleukin; PGE2 = prostaglandin E2; MMP = matrix metalloprotease.
One key mechanism encountered in degenerating tendons is aberrant tenocyte function. The altered cells express this change in multiple fashions, the chief of which are less proliferation, diminished production of collagen type I (in favor of the less elastic collagen type III), and decreased synthesis of GAGs. Tenocyte apoptosis is thought to result in part from the local action of reactive oxygen species, generation of which may be higher in normally avascular areas of the tendon due to neoangiogenesis leading to improved oxygen delivery to the damaged tissue. Another proposed driver of tenocyte apoptosis is glutamate-mediated excitotoxicity. In particular, upregulated expression of multiple receptors and accessory proteins associated with glutamate signaling has been observed in cells within degenerate tendons (Molloy et al. 2006).
A second main mechanism is postulated to be altered intercellular signaling among tenocytes. Several different pathways have been implicated. For example, increased tendon loading ex vivo has been linked to amplified generation of proinflammatory cytokines, such as interleukins (IL)-1 and IL-6, as well as elevated expression of prostaglandin-endoperoxide synthase-2 (also designated as cyclooxygenase-2), leading to augmented production of prostaglandin E2 (Dirks and Warden 2011; Kirchgesner et al. 2014). These data suggest that the microenvironment of tendons may be naturally primed to mount an immune response. Similarly, expression of many genes related to immune system function is altered in acutely damaged tendons of rats (Molloy et al. 2006). Thus, despite the scarcity of leukocytes in most cases of toxic tendinopathy, inflammation as a cause of tendon degeneration following drug exposure cannot be discounted as a possible contributing mechanism.
Such intercellular mediators invoke a third toxic mechanism: increased lytic enzyme activity (Figure 1; Dirks and Warden 2011). Fluoroquinolones and statins boost matrix metalloprotease activity while glucocorticoids have been shown to enhance collagenase activity, thus yielding greater degradation of extracellular matrix. In contrast, platelet-derived growth factor (PDGF) delivered by neovessels promotes the differentiation of tenoblasts into tenocytes leading to subsequent local overproduction of collagen, unregulated accrual of which alters the extracellular matrix composition (in favor of collagen type III) and organization (Cooper and Valentine 2016).
A fourth putative mechanism of toxic tendinopathy is lipid dysregulation in tenocytes. Statins, which act as 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are proposed to function in this regard by modifying the cholesterol content of tenocyte plasma membranes. The affected cells redirect their machinery to cell survival processes and downregulate their attempts to maintain the structure of the tendon fibrils and extracellular matrix.
A final primary mechanism is purported to be blighted tendon repair. Long-term glucocorticoid exposure diminishes the ability of tenocytes to refurbish foci of tendon microtrauma. Furthermore, glucocorticoids reduce the tensile strength in the remaining collagen fibers. Over time, these effects lead to buildup of weakened tendon tissue.
Given the severity of toxic tendinopathies in affected patients, improved techniques for nonclinical safety testing to predict the potential for drug-induced tendon injury would be a welcome addition to the risk assessment paradigm. Many animal models of toxic tendinopathy have been developed, including intratendinous injection, peritendinous injection, and systemic administration of chemicals and enzymes (Dirks and Warden 2011). Tendon stress might be boosted by surgical introduction of a structural defect, exercise-related overuse, or even disuse (Dirks and Warden 2011; Hast, Zuskov, and Soslowsky 2014). Collectively, such models have utility in investigating mechanisms and validating targets of drug-induced tendon toxicity but typically would not be used for safety assessment. For at least some tendon toxicants, humans and animals develop comparable functional abnormalities and pathologic abnormalities (Groves et al. 2002; Renkiewicz et al. 2003; Miller et al. 2004; Westwood et al. 2009).
Instead, nonclinical safety testing typically will employ conventional rodent (mouse and rat) and nonrodent (canine, nonhuman primate, and rabbit) species. The potential for induction of toxic tendinopathy may be explored in standard screening bioassays by including 1 or more major tendons (Hast, Zuskov, and Soslowsky 2014), usually the calcaneal (Achilles) tendon and/or the supraspinatus tendon (1 of the 4 tendons that form the “rotator cuff”) in the battery of protocol-specified tissues. Both tendon proper and an enthesis should be assessed since the lesion character and severity may vary in different parts of the tendon. Standard evaluation of tendon morphology can rest on macroscopic evaluation at necropsy and microscopic analysis of routinely processed specimens (immersion fixation in neutral buffered 10% formalin, paraffin embedding, and sectioning to produce 4- to 8-μm-thick hematoxylin and eosin [H&E]-stained sections), though decalcification may be necessary if an enthesis is included in the sample. Tendons generally should be examined in both longitudinal and transverse orientations, but if a single facet will be viewed, the longitudinal plane typically provides a better means of judging fibril and fascicle organization as well as tenocyte numbers and characteristics. Scoring schemes may combine diverse features including nuclear and cytoplasmic morphology, cell density and organization, fibril orientation, extracellular matrix appearance, and vascularity (Maffulli et al. 2008). If warranted, special methods may be employed to further characterize the nature of test article–related findings in tendons (e.g., polarized light to examine fibril orientation in H&E-stained sections, Alcian Blue and/or periodic acid-Schiff staining to assess extracellular matrix integrity, and immunohistochemical detection of specific markers such as collagen types I and III, elastin, and tenascin). Inclusion of tendon in nonclinical studies thus would require minor increases in cost and time (to collect, process, and analyze 1 more tissue sample) and no expenditure for specialized analytical equipment or processes.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.