
Abstract
This article highlights emerging roles for veterinary pathologists outside of traditional functions and in line with the translational research (TR) approach. Veterinary pathologists offer unique and valuable expertise toward addressing particular TR and associated translational pharmacology questions, identifying gaps and risks in biomarker and pathology strategies, and advancing TR team decision making. Veterinary pathologists’ attributes that are integral to the TR approach include (i) well-developed understanding of comparative physiology, pathology, and disease; (ii) extensive experience in interpretation and integration of complex data sets on whole-body responses and utilizing this for deciphering pathogenesis and translating events between laboratory species and man; (iii) proficiency in recognizing differences in disease end points among individuals, animal species and strains, and assessing correlations between these differences and other investigative (including biomarker) findings; and (iv) strong background in a wide spectrum of research technologies that can address pathomechanistic questions and biomarker needs. Some of the more evident roles in which veterinary pathologists can offer their greatest contributions to address questions and strategies of TR and biomarker integration will be emphasized.
Introduction
Drug discovery and development are evolving in a number of unprecedented ways. These include broadening the scope of combination therapies and expanding into therapeutic platforms with unique means and routes of administration; novel engineered proteins; targeted “polypharmacology”; and gene, cell, epigenetic, next generation nucleic acid, and microbiome therapies. Drug development strategies are also evolving to be increasingly driven by a translational research (TR) approach for improving and accelerating efficacy and safety evaluation. TR focuses on the integration of knowledge from basic science, patient-oriented, and population-based research as means of improving human health (Rubio et al. 2010; Woolf 2008). While TR, translational medicine, and translational science are often used synonymously, TR as used in this article is the ability to translate basic science discovery and patient learnings into new clinical therapies. The TR approach as applied to drug discovery and development highlights translational pharmacology questions to specifically and measurably track therapeutic effects along mechanistic pathways, from molecular to whole-body responses. The TR approach also encompasses objective methods to stratify disease profiles within an indication and identify specific factors of patient susceptibility and response in a “precision” medicine method to treatment. Characterization of disease heterogeneity and susceptibility/response factors supports better target– and patient–treatment connection.
A universal concept of the TR approach is to utilize biomarker end points to assess patient susceptibility factors, pharmacologic target engagement, direct target effects and downstream responses, as well as overall efficacy and safety in the selected population. Biomarker selection is hypothesis driven to test these translational questions and may include targeted “omic” and imaging end points, as well as microscopic and immunology-based measures, sometimes with uncommon biologic matrices and/or kinetics. Ensuring the appropriate markers requires informed scientific input from a broad spectrum of discovery to clinical disciplines. Optimizing translational biomarker selection and application also commonly requires robust preclinical testing and, for some markers, may be most reliably achieved by dedicated in vivo preclinical method development and biological evaluation. The translation between in vitro, ex vivo, and in vivo models challenges our understanding of these systems and their biological differences in target, pathways, and downstream effects. Interpretation of both preclinical and clinical biomarker data also requires understanding and calibration of the impact of confounding secondary factors (e.g., nutrition, stress, inflammation, hormonal cycling, comorbidity, and disease behavior). However, the expected benefits of robust translational biomarker measures and knowledgeable interpretation include early project risk identification, go-no-go decisions on drug candidates, shortened development timelines, and potentially, improved benefit/risk profile.
The ultimate goal of the TR approach is to generate more predictable outcomes and optimize efficacy while limiting side effects for the indicated patient population. The challenge is that it also requires integrated expertise in very diverse sciences, including clinical medicine, disease pathology/pathologic mechanisms, pertinent model systems, biomarker identification, molecular biology, and pharmacology. The approach is also most successful when this multidisciplinary team comes together early in the process to “translate” in timely progression between the preclinical with clinical sciences.
Veterinary pathologists have unique expertise in some of the critical aspects of this translational method, with a multidiscipline, multispecies comparative pathology, and medical research background. In serving on a TR (or “translational medicine”) team, veterinary pathologists take on a role that extends outside of traditional preclinical toxicologic assessment, target validation, efficacy model characterization, or safety biomarker discovery/development. This article is intended to identify and exemplify areas in TR and especially translational pharmacology where pathologists can make particularly important contributions.
Translational Pharmacology
A key component of the TR approach is translational pharmacology. Translational pharmacology bridges traditional pharmacology assessments (e.g., circulating blood levels, scaling for human dose prediction, and nonmechanistic in vivo modeling) with mechanistic translational end points in linking drug exposure at the relevant site to drug–target interactions. These mechanistic end points drive direct, testable links between results (and relevance) of preclinical studies and models with clinical efficacy and safety (figure 1).
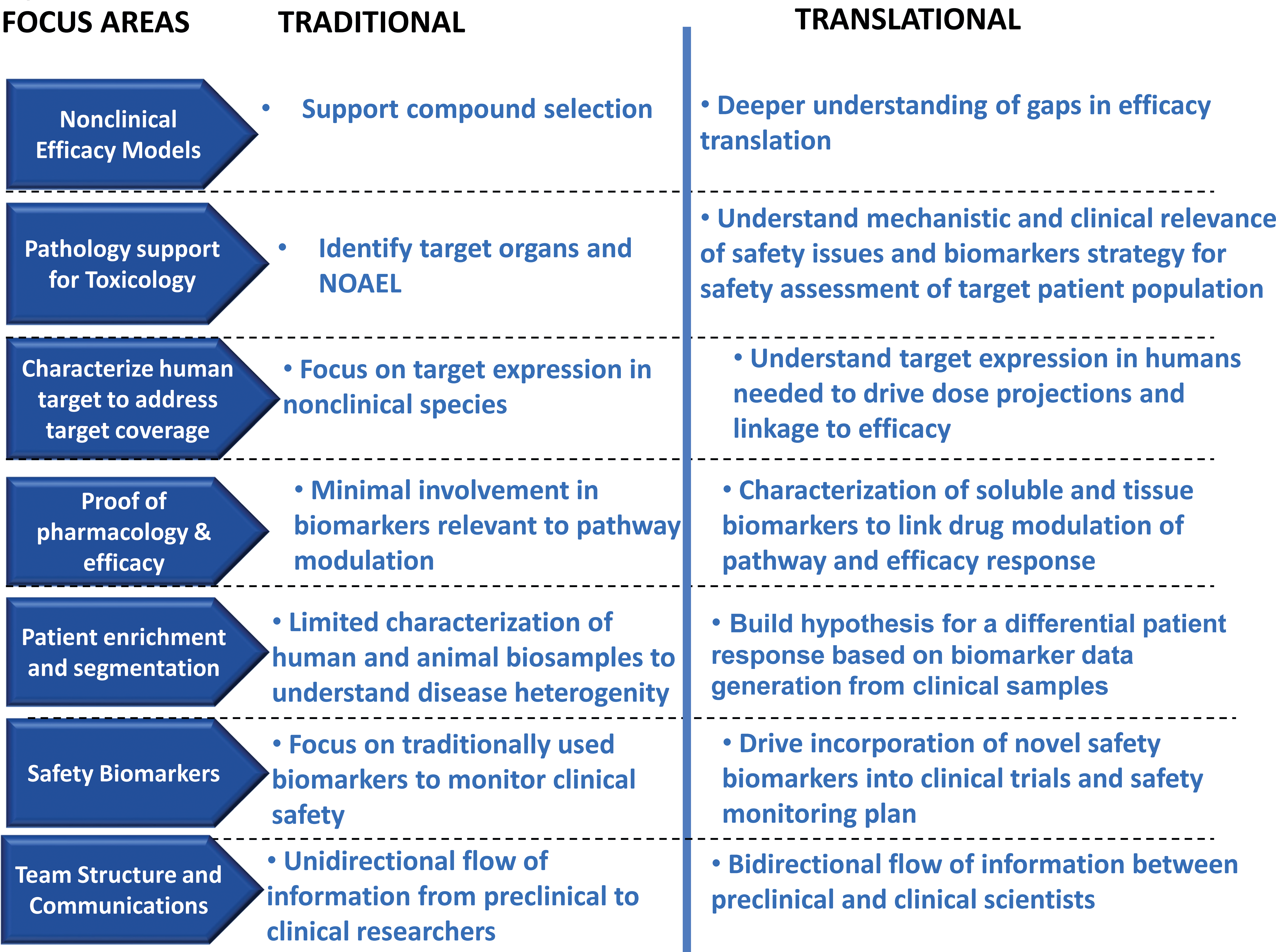
Current and future state of toxicologic pathologist’s impact on translational pharmacology. While there are several focus areas where toxicologic pathologists can impact drug discovery and development, translational pharmacology questions to characterize human target, testing proof of pharmacology and efficacy, and patient enrichment are important. With appropriate training and background, toxicologic pathologists are positioned to impact multiple areas beyond current traditional roles.
Translational pharmacology end points include target selectivity and engagement: whether (and how to determine if) the therapeutic is reaching and then interacting with (e.g., activating/inhibiting) the intended target, what dosing regimen results in the expected measurable molecular or cellular pharmacodynamic response, if/how dosing is linked with the anticipated biological (e.g., organ system or whole body) response, and if the effects correlate with measurable disease mitigation and/or clinical benefit. Translational pharmacology also encompasses physiologically based modeling and comparative target expression and turnover to extrapolate preclinical data to clinical dose and schedule and assess clinical safety (especially when safety is a continuum of pharmacology often referred to as exaggerated pharmacology). Translational pharmacology further includes measures of the disease heterogeneity to support patient-enrichment strategies. Translational pharmacology particularly changes the dynamic within drug development away from unidirectional flow of information (preclinical to clinical) toward multidirectional flow of knowledge between disciplines to address the end point questions for each therapeutic (Murray et al. 2010; Markou et al. 2009; Morgan et al. 2011; Enna and Williams 2009; Anderson and Kodukula 2014; Mullane, Winquist, and Williams 2014).
Because translational pharmacology is focused on understanding connections between molecular mechanisms, cellular response and systems physiology, as well as to on- and off-target biological effects, pathology, and safety risks, and the “translation” of such across species, veterinary pathologists are particularly positioned to contribute (figure 1). In the following sections, key goals and questions presented in a translational pharmacology approach are further described, along with some of the beneficial contributions that veterinary pathologists can offer toward each.
Does the Therapeutic Reach and Engage the Intended Pharmacologic Target(s)?
To be effective, a therapeutic must reach its target—whether receptor, ligand, enzyme, ion channel, nucleic acid, lipid, or selective molecular domain. Early in development, this target site is commonly modeled using in silico and in vitro systems. Whereas clinically, drug concentrations in peripheral blood or other accessible body fluid or cells must suffice to estimate target exposure. Hence, linking these determinations typically requires in vivo relevant animal model(s) to assess exposure at specific target tissues and cell types and correlate end point measures with those of clinical matrices to establish human dosing regimen. For example, correlation of blood and cerebrospinal fluid (CSF) with extracted brain (retina, spinal cord, etc.) drug levels in an animal model may be important to assess the clinical relationship between target and accessible fluid levels.
With new therapeutic modalities, many of which are biologics, determining target exposure in animal models is typically more complex. This is only partly because plasma concentrations are less apt to be in equilibrium with tissue or target site distribution. With protein-binding moieties, comparisons between humans and nonclinical species for target-binding affinity, kinetics, and stoichiometry (stereological density and strength of binding) and target-mediated elimination are also often needed. Preclinical-to-clinical translation of exposure is further complicated with certain targets, therapeutics, and/or mechanisms of action and may require consideration of species differences in therapeutic translocation, metabolism, interaction with membrane transporters, metabolic regulators or other cellular proteins, and impact of immunogenicity. Properties of tested tissues and matrices (e.g., solid organs, plasma, urine, saliva, and bronchoalveolar lavage [BAL] fluid) to assess target exposure and binding can also show important species variation that requires results translation. The findings of these studies provide important information for exposure–response, projected human dose, and translation of both pharmacologic and safety findings between species.
Toward these determinations, veterinary pathologists can input on appropriate strategy and technology for selection and collection of target site(s) and matrices to evaluate for drug engagement, which may incorporate species differences in physiology, anatomy, or target cell location/type(s). Pathologists can also interact on the pros and cons of investigating tissue or cell epitope expression and drug binding, including utility of direct labeling in tissues in vitro or ex vivo in dosed animals (e.g., by immunohistochemistry [IHC] and flow cytometry). Pathologists may bring attention to relevant animal models for pharmacokinetic (PK) testing of alternative matrices (e.g., CSF, vitreous, synovial fluid, lymph, and lacrimal fluid) and species differences in collection method, timing, and sample processing for greatest clinical relevance of target exposure. Pathologists may also contribute toward integrating target exposure data with morphologic and biomarker (including protein and gene expression profiling) findings. Overall, pathologists may show team value in posing important questions to ensure accurate and appropriate translation and integration of cross-species target exposure data.
Does the Therapeutic Induce the Expected Measurable Molecular or Cellular Pharmacodynamic Response(s)? (“Proof of Mechanism” [PoM])
Demonstration of pharmacodynamic (PD) response on a molecular or cellular level with acceptable dose–response relationship links the therapeutic to a temporal and specific effect that supports, but does not go as far as to assess, the desired overall biologic response or efficacy of the treatment. PoM evaluations are designed to determine whether the compound interacts appropriately and predictably with the target to cause the expected local or direct molecular or cellular effect. The PoM response may also inform on early clinical dosing regimen and efficacy and/or toxicity end points within the expected useful dose range. Some examples of PoM end points include concentration of a translated protein of a targeted messenger RNA (mRNA), density of a specific cell type or subcellular component that expresses a targeted epitope, or decreased product directly downstream of an inhibited pathway (e.g., thromboxane A2 metabolites with cyclooxygenase 1 antagonists, phosphorylated intracellular proteins that transduce extracellular signals and activate downstreams as intracellular effectors with transforming growth factor beta receptor kinase inhibitors). Some common PoM end points with oncology drugs include measuring protein (e.g., kinase) phosphorylation with pathway inhibitors or other targeted effects such as markers of proliferation, apoptosis, cell cycle regulation, or epigenetic changes of a tumor cell population. A PoM end point can also involve morphologically and/or functionally identifying a specific responding tissue or cell type.
PoM generally begins with relevant nonclinical models and may be evaluated in healthy or diseased clinical subjects, especially in early phase trials. Nonclinical PoM testing typically utilizes an in vivo model, although in vitro systems may be adequate if relevant and the results provide the needed confidence for corresponding end points in clinical protocols. Assessment of PoM response can be confounded over time, especially in vivo, due to secondary (including adaptive) changes, target modulation, and other temporally dynamic changes. Hence, some PoM effects may be most reliably assessed only early in a study or during a specific window in the course of treatment. Mechanistic end points also need to have sufficient analytical and biological dynamic range to ensure the association between exposure and response. Another common challenge is ensuring appropriate methodology for comparable preclinical–clinical PoM evaluation. These and other aspects in the science and strategy of designing and evaluating pharmacodynamically predictable PoM are critical to success of a program. If a specific drug candidate is found not to have adequate mechanism of action (via PoM determination) early enough, the team can rapidly switch to a more effective candidate.
Veterinary pathologists have backgrounds particularly suited to contribute to decisions and interpretations with PoM end points. The end points may be derived from findings in the indicated human disease or disease models where the mechanism is part of the pathology or in healthy subjects where the mechanism is an aspect of normal physiology. Selection and interpretation of such PoM end points require understanding of the models, prototypes, or representations, associated cellular-to-tissue functional integrations, disease pathogenesis, and physiologic time course. The veterinary pathologists’ background in these areas offers meaningful input on biological sample collection and evaluation for PoM biomarkers in both animal studies and clinical trials. Pathologists can be integral to understanding the variability of the PoM biomarker response (biological, technical, and tissue specific); differentiating a mechanistic response from secondary, adaptive, or toxicologic effects; and considerations of such when designing sample collection strategy for clinical protocols. Pathologists have the background to also investigate novel PoMs as well as help optimize test tissue/matrix(ces), testing methodology, results interpretation, and translation to clinical application. Veterinary pathologists’ involvement in PoM investigations has been one of their most evident roles in the TR approach, contributing to early identification, selection, and accurate translational interpretation of these markers for therapeutic clinical testing.
Is the Therapeutic Linked with Anticipated Clinical Response? (“Proof of Concept” [PoC])
PoC refers to demonstrating adequate (even if just preliminary) evidence of intended, measurable, and specific functional clinical response on a tissue/organ or system level without overriding safety risk. This ultimately requires evidence from actual species intended (e.g., human patients or relevant human volunteers). PoC is the guiding objective of translational pharmacology even in the preclinical phase. Well-designed and accurately translated preclinical studies inclusive of acceptable PoM and PK/PD determinations establish the basis for effective dosing and demonstration of PoC and safety in later stage patient trials. Thus, PoC planning begins early and is considered not an end goal but a targeted focal point for launching later phase evaluations.
Ideally, PoC end points are a logical follow-up to PoM and can be supported with preclinical models to verify adequacy and utility (technically and biologically). PoC end points should be specific to the targeted clinical effect with sufficient dynamic range to show a correlating dose–response. End points also need to be adequately tested, dynamic, and robust for determining PoC in the limited subject pool with early phase trials. End points such as classical surrogate markers (e.g., blood cholesterol, hemoglobin A1c, fever, and peripheral blood pressure) may be too nonspecific alone for early PoC (Enna and Williams 2009). Others, such as soluble biomarkers in accessible matrices (e.g., neurotransmitters or Aβ fragments in CSF and leukocyte proportions in BAL), may not reliably reflect functional tissue levels or true clinical or relative dynamic response (Benilova, Karran, and De Strooper 2012; Mullane and Williams 2012; Mullane, Winquist, and Williams 2014; Cummings and Zhong 2014). These limitations can often be determined or better understood through preclinical investigation via animal models. Some end points may require advanced technical feasibility, atypical matrices or novel functional, biological, or dose–response testing which may also be particularly well assessed or developed in animal models. The spectrum of potential PoC measures along with scientific, technical, and regulatory considerations in an early phase clinical trial is perceptibly broad and a prime reason for the emphasis of a multidiscipline team approach.
Some common types of PoC end points include quantification of downstream disease mediators, such as pertinent synovial cytokines with an antiinflammatory osteoarthritis therapeutic (Romme et al. 2014), measurable shifts in character or cells in tissue biopsies, such as infiltrating lymphocyte subtypes with immune-oncology therapy, dynamic morphologic (e.g., dysplasia) or functional (e.g., nociception) target tissue changes; and concentration of disease-linked specific products or metabolites (e.g., circulating B-lipoproteins with Apolipoprotein C or lipoprotein lipase effectors). There are also well-established PoC end points such as forced expiratory volume 1 following asthma allergen challenge, circulating viral load with antivirals, glomerular filtration rate for monitoring renal transplant perfusion, and radioimaging for bone density and left ventricular ejection fraction for cardiomodulatory treatment assessments (Bar-Zohar et al. 2008; Stevenson et al. 2003). Ability to detect reversal of changes following cessation of therapy may also be an important need in selecting and timing of PoC end points. In general, these and most PoC end points are effectively explored and/or demonstrated in the animal model(s).
Veterinary pathologists can offer valuable input to teams on PoC decisions, including for end points outside of histologic or blood biomarkers, because of their understanding not only in whole-body systems biology and disease pathogenesis but also in comparative physiology of such. Pathologists also comprehend the impact of disease, disease time course and adaption on PK/PD end points (e.g., influence of inflammation on drug metabolism or renal or hepatic functional impairment on drug exposure), and biomarker values (e.g., acute vs. chronic disease response); factors important in determining selection and application of PoC end points. Many pathologists are also experienced in interpreting data from transgenic and disease models, which may be important in PoC modeling and end point derivation. Veterinary pathologists can further contribute directly to PoC end point verification through assessment of banked human samples for linking disease targets with response end points. Veterinary pathologists’ input on PoC biomarkers is further facilitated by their ability to readily bridge communications between nonclinical and clinical scientists in the process of PoC decision making.
Do Therapeutic Effects Correlate with Measurable Disease Mitigation and/or Clinical Benefit? (“Proof of Efficacy” [PoE])
Does the therapeutic show adequate evidence of clinically meaningful benefit on disease progression or symptoms? This is the natural next question to PoC, although formal PoE is typically not determined until a larger number of patients have been evaluated and under more proscriptive trial conditions, study designs, and weighting of data (e.g., phase IIB or III; Sargent and Taylor 2009; Sargent 2010; Food and Drug Administration [FDA] Guidance for Industry Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products 1998; ICH E9 1998). Primary efficacy end points are typically direct objective clinical observations, response rates, or recognized surrogates (http://www.medpagetoday.com/PublicHealthPolicy/FDAGeneral/48244). Hence, primary PoE evaluations may offer less opportunity for adaptive study designs and mechanism-based learnings. However, secondary well-established PD, PoC, and safety end points may also be important to include, and analysis of response with these in the larger patient populations can be highly informative. Late development strategy may further include separate studies, study arms, or add-in testing of a broader spectrum of patient subtypes, related indications, or expanded drug combinations to assess clinical efficacy. This is especially the case when both the therapeutic mechanism of action and disease pathophysiology are understood well enough to explore such applications.
Results of ongoing phases IIB–III studies can remarkably enhance the understanding of clinical disease and patient response variability among discovery and early development researchers. Results may further refine or correct presumptions on appropriate target subpopulations, biomarker behavior, or associations with comedications or companion treatments. Nonclinical team members including veterinary pathologists can also provide useful input in PoE studies on hypotheses setting, inclusion/exclusion criteria, and testing to support a distinct patient therapeutic response and/or incorporation of secondary safety markers and monitoring plan based on earlier phase biomarker data.
Veterinary pathologists may undertake another important role in PoE trials in investigating unexpected issues that can occur partly as an outcome of the larger and more diverse clinical test populations. The combination of signaling pathway bioinformatics with more reliable sources of banked human and animal disease tissues available within most institutions and cellular- and molecular-level marker analyses offer valuable means for pathologists to investigate such issues. The use of these resources and background knowledge of disease pathogenesis and animal models enables veterinary pathologists to take lead roles in such research. Additionally, the regulatory acceptance of these issue investigations and secondary end points relies partly on the rigor of their science, and methods utilized that include diligent control of variables in assay analytical validations, sample collection, processing and storage, and reporting. It can be an important advantage for veterinary pathologists to provide input on these variables in advance of testing and in regulatory reporting and responses to ensure such rigor is captured with study procedures and documents.
Enriching for the Patient Population(s) Most Likely to Have the Intended Therapeutic Response (Patient-enrichment Strategies)
What are the variables and their sources within a target patient population that affect efficacy and safety in drug response? Enrichment strategies have the potential to enable preselection of patients who have relatively greater chances of responding beneficially to and/or present less safety risk from the therapeutic. This can decrease the size of the clinical trials and more rapidly and decidedly identify extent of therapeutic benefit. In effect, this accelerates early development decision making and limits the study population at risk of side effects. Trial subject enrichment is sometimes referred to as “precision medicine.” Subgroups may be selected by patient phenotype, genotype, clinical symptoms, history, pathology, disease phase or stage, and biomarker data pertinent to the disease and target (often an integration of more than one criteria, “cluster analysis” or “systems biology” approach is used). This same information is also used to understand the target relative to disease pathogenesis and course.
Examples of enrichment in clinical trials can be readily found in recent literature and other public resources. Enrichment strategies are customary for some indications such as oncology, anti-infectives, congestive heart disease, and serious neurologic conditions such as schizophrenia and epilepsy (FDA 2012; Matthews et al. 2014). Information on enrichment strategies can also be found in scientific literature for prevalent conditions such as osteoarthritis (Tonge, Pearson, and Jones 2014), inflammatory bowel disease (Biancheri et al. 2013; Laifenfeld et al. 2012), rheumatoid arthritis (Curtis et al. 2012; Drubin et al. 2012), osteoporosis (Moon and Harvey 2014), atrial fibrillation (Hijazi et al. 2014), stroke, multiple sclerosis (Matthews et al. 2014), psoriasis, and many others. Perhaps the best known successes of clinical trial–enrichment strategy are with molecular markers for cancer subtypes (e.g., estrogen receptor detection by IHC in breast cancers to select responders to tamoxifen; human epidermal growth factor receptor 2 [HER2] gene expression for anti-HER2 treatment such as trastuzumab [Herceptin]; and breakpoint cluster region-ABL(Abelson murine leukemia viral homolog) gene-positive tyrosine kinase genotype to identify chronic myelogenous leukemia [CML] patients likely to respond to imatinib). Beneficial clinical trial enrichment has also been based on safety, such as excluding patients with a uridine diphosphate–glucuronosyltransferase 1A1 variant that identifies increased risk for serious side effects with irinotecan; and nonresponders with oncology therapeutics such as CML patients with tumor T315i variant associated with resistance to all tyrosine kinase inhibitors except ponatinib and colon cancer k-ras mutations predictive of resistance to anti–epidermal growth factor receptor therapies (Haznedaroglu 2014).
Importantly, despite heterogenous etiology and/or pathophysiology, a majority of targeted marketed therapies currently lack sufficient evidence of adequately discriminating genetically based end points. This includes common high-medical-need disorders such as asthma (Holgate 2013; Woodruff et al. 2009; Bhakta and Woodruff 2011), depression (Merlo-Pich et al. 2010), atherosclerosis, major subtypes of Alzheimer’s disease (Kohannim et al. 2013; Lorenzi et al. 2010), and autism spectrum disorder (Hollander et al. 2004). Enrichment strategy with such conditions relies on a compilation of variably weighted clinical, historical, epidemiological, environmental, pathophysiological, and increasingly, limited genotypic and quantifiable biomarker end points. Enrichment of patients most likely to respond to an exploratory treatment based on mostly nongenotypic end points stresses consideration of mechanistic biomarkers relevant to the therapeutic target. For example, patients who have persistent prominence of a targeted cytokine or associated cell type and related or downstream mediators in the affected tissue (or accessible biofluid) are hypothetically more likely to respond than those with lower levels of these markers. An example given in the FDA guidance on enrichment strategies (2012) is that patients with high-renin hypertension are more likely to respond in studies of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers than the general hypertension population.
Selected enrichment end points are best identified early in discovery/development, are minimally invasive, provide real-time assessment, and are at least partly method tested and characterized in relation to the disease and response (in vivo or in vitro) prior to clinical trial application. Method development and/or characterization may be addressed in preclinical models or clinical samples where biomarker heterogeneity exists. Identifying enrichment markers may be aided by a “systems biology” approach with analysis of a multitude of genes involved in the pathway of a target, or genetic profiling, as well as mRNA or epigenetic analyses. These same profiling methods can be applied to select animal models to aid human disease–enrichment strategies, especially when objective and consistent clinical evaluation is impractical. For example, genes involved in peripheral pain perception are expected to be common to multiple species, and although stratifying pain perception can be highly subjective clinically, studying rodent models of specific pain phenotypes has enabled identification of genomic variants that link with cognate human genes that are risk factors (and potential markers) for pain susceptibility (Dib-Hijj and Waxman 2014). Preclinical identification also enables early clinical trial exploration and understanding of the behavior of selected enrichment biomarkers. In later phase clinical trials, biomarker validations and cutoff points become more stringent, complicating incorporation into nonadaptive study designs and randomization schemes (Friedlin et al. 2013; Drucker and Krapfenbauer 2013). There are a number of other factors (e.g., patient, disease, logistical, practical, and economic) to consider for rationale, utility, and pursuit of adding target patient enrichment to a particular development program (Trusheim, Berndt, and Douglas 2007; Sumithra, Mandrekar, and Sargent 2009; Friedlin and Korn 2014; Friedlin et al. 2010; Anderson et al. 2014). The decision and strategy to include enrichment markers is thus a complex process and best approached by a team with the collective knowledge to appropriately identify the pros and cons of disease categorization for exploratory clinical testing.
Veterinary pathologists can contribute in these decisions on whether, when, and how to utilize end points for patient-enrichment testing. Pathologists have understanding of heterogeneity in both the pathogenesis and progression of disease. This background can be important in understanding the extent and persistence of some types of disease heterogeneity, and the reliability of a marker to discriminate between patients over time. Pathologists also can offer input on the selection of optimal test matrix relevant to the target tissue and disease response if the enrichment biomarker is expected to be a soluble or tissue marker. Pathologists can further aid in characterizing soluble and tissue biomarkers and evaluating the strength of correlations with the target disease or response in animal models and/or with banked human specimens. These biomarker assessments can lead to identifying new discriminating end points and improve processes such as sample collection, handling, and testing methods. Veterinary pathologists can also contribute to integrating results of all end points evaluated to formulate the marker set which suits the desired criteria and to ensure safety markers are a consideration for patient enrichment.
Biomarker Selection Strategy for Translational Pharmacology
Biomarkers are utilized to establish, support, simplify, and/or quantify each of the translational and clinical pharmacology decision points above and may aid in market approval. A notable distinction of translational pharmacology markers compared with conventional clinical parameters measured in development studies is that the former are generally test article, target, and/or disease specific. They rarely have broad application or general utility as with routine clinical chemistry, hematology, and commercial diagnostic assays. Some general categorical examples are biomarkers (protein, gene, or mRNA) in tissue or fluids of downstream apoptotic pathway mediators; various growth factor family members; neurohormones; less known mediators of inflammation (Kahles, Findeisen, and Bruemmer 2014; Tektonidou and Ward 2010; Liu et al. 2010; Kurt et al. 2015); tumor epitopes; and markers of fibrosis, cellular stress responses, protein or DNA acetylation/methylation, and lipid peroxidation. Specialized imaging for end points such as tissue fibrosis, edema, and active inflammation in whole animals or organs are other examples.
Translational pharmacology biomarkers serve as specific indicators of drug’s pharmacologic effects in relationship to the target. One marker may be used to detect or quantify a drug’s engagement with (or proximity to) the target, while others are intended to measure a desired target response (or PoM) or downstream clinically relevant effect (or PoC). Pharmacodynamic markers (often PoM end points) can also aid selection of clinical dosing regimen and understanding drug resistance mechanisms. Additional biomarkers may be essential for characterizing (and potentially stratifying) patient subpopulations providing competitive advantage and safety and efficacy. Biomarkers can also be selected to overlap these purposes. (Notably, some literature also classifies biomarkers used for translational pharmacology as “predictive,” “diagnostic,” “prognostic,” etc. This is useful, although the terminology is not yet well defined.)
Decisions on which biomarkers to use for dose selection or PoM, PoC end points, patient selection, and so on can be critical to the developmental success of a therapeutic (or entire drug platform). Additional biomarker needs may arise during development and with health authority communications or change in competitive field. Early evaluation of a biomarker’s behavior in association with the targeted disease and therapeutic administration in animal models and with human samples offers greater confidence in the generated biomarker data from clinical protocols. These evaluations can also show whether there is need to segregate testing by gender, stage of disease, and so on. Early testing may also show the need for expanded end point measures (e.g., a larger “panel” of biomarkers) to enhance sensitivity, specificity, or both. There are many other considerations with biomarker and assay development for translational pharmacology that are well reviewed in current literature (e.g., Drucker and Krapfenbauer 2013; Waerner, Urthaler, and Krapfenbauer 2011; Tektonidou and Ward 2011).
There is clear need for diverse discipline involvement in optimizing biomarker decisions, development, and evaluation in translational pharmacology. Pathologists, both human and veterinary, offer expertise to contribute to these needs, especially in having broad understanding of disease pathogenesis and extrapolation of molecular and cellular events to complex tissue, organ, and whole-body responses. In this way, pathologists are situated to help identify gaps in biomarker selection and testing protocols for assessing disease progression versus resolution. Many veterinary pathologists also have expertise in technologies suited to address biomarker analyses and/or in biologically qualifying biomarker performance in animal models with the ability to correlate with available human specimens. Additionally, pathologists can aid biomarker decisions through objective review of exploratory test data relative to other study findings and individual subject variables to ensure novel markers meet expectations in practice and provide information not available through more common measured end points (Tektonidou and Ward 2010).
Expectations and Needs for Adapting to the Translational Medicine/Pharmacology Approach
The expertise of veterinary pathologists can have unique value in TR decision making; yet, pathologists’ participation on TR (or translational medicine) teams has been inconsistent across the industry. This may be simply due to the lack of precedent. Pathologists’ participation on these teams is also often without mentorship; thus, it may be beneficial to note some overarching expectations and needed contributions with this role to further support awareness and interest.
TR teams are predominantly focused on clinical development, although members tend to be from diverse scientific areas with variable clinical experience. This broad expertise together with compelling purposeful goals enables the team to “go where no man has gone before” in discussing novel development strategies. Thus, there is great need for members that have both strong collaborative and investigative drive. Translational medicine teams also motivate members to think strategically about clinical trial study design with integration of PD end points throughout the development. Team understanding and appropriate synthesis of preclinical data to address these needs are important to optimize clinical plans and protocols. Expanding technologies for molecular and biomarker testing further challenges teams to stay up to date on these tools and their effective use to address targeted translational medicine questions. Synthesis of large volumes of data, including the abundance of bioinformatics data, is also increasingly important within translational medicine teams; understanding the variability, strengths, and weaknesses of such “Big Data” in association with biological and clinical events aids productive collaboration with bioinformaticists and computational biologists. The increasing access to this wide array of technologies and information requires teams to tactically prioritize energy and resources on identifying key risks and issues—and the methods and strategies that best address such.
Pathologists are an asset to TR teams most overtly if they are effective in articulating integrated preclinical, and translational biomarker findings and discussing their specific clinical relevance (or lack of relevance) to the teams’ broad audience with leaning toward clinical terminology. Pathologists understand therapeutic mechanisms, disease pathogenesis, confounding comorbidity and comedication, disease stages and phases, epidemiology, and standard of care that are important in translational strategies. These same variables are chief considerations for clinical study designs and biomarker selection, as well. Hence, veterinary pathologists can improve their effectiveness by developing familiarity with both routine and adaptive clinical study designs, general logistics related to clinical biomarker testing (e.g., complexities with multiple test site and multicountry trials), and regulatory testing requirements (e.g., good clinical practice [GCP]). Awareness of the spectrum (and limitations) of testing methodologies and platforms available for a clinical project and ability to innovate and integrate across platforms to address key toxicopathology, PD, and stratification biomarker needs are also beneficial. Notably, one of the greatest contributions that pathologists may provide to translational medicine teams is not in supplying ideas and answers but in applying their expertise to voice the key questions that drive a project forward.
Summary
The TR approach in drug discovery and development has expanded opportunities and roles for veterinary pathologists. This is particularly evident in translational pharmacology which incorporates biomarker end points intended to transcend discovery through human trials to understand relationships between therapeutic exposure and pharmacodynamic response and intended pharmacology and disease outcome. Translational pharmacology further supports biomarker-based enrichment designs for patient populations who may best respond to a specific therapeutic. Success in these approaches is reinforced when strategic planning on study designs and biomarker selection begins early in the preclinical phase and is adapted with information gained through exploratory testing. Translational biomarker strategy for clinical testing can then be predicated on results of early evaluations for feasibility, relevance to exposure, mechanism, disease and/or therapeutic response, and understanding cross-species biology. Success of a translational medicine approach is also predicated on garnering and capitalizing on scientific expertise from diverse translationally relevant disciplines. Understanding these aspects of TR help illuminate why veterinary pathologists are particularly well suited to participate on teams addressing the challenges inherent to the approach.
The roles of veterinary pathologists in TR extend beyond cross-species translation and biomarker needs. Veterinary pathologists have the background and experience to recognize and explore gaps in understanding pathogenic mechanisms and individual variability in a disease or target indication as well as identifying and preventing the common biases in translational study hypotheses, and issues in design or sample timing and management that impact investigative results or interpretation. They are also perceptive to the clinical relevance and development impact of safety, including “exaggerated pharmacology” and off-target effects in the intended patient population.
The roles for veterinary pathologists in translational medicine are often outside of traditional pathology responsibilities and functions; hence, they are still slow to be recognized. Filling these roles will require not only industry recognition of the advantages but also concerted training within the profession and impetus by veterinary pathologists to find and capitalize on TR opportunities.
Footnotes
*
This is an opinion article submitted to the Regulatory Forum and does not constitute an official position of the Society of Toxicologic Pathology or the journal Toxicologic Pathology. The views expressed in this article are those of the authors and do not necessarily represent the policies, positions, or opinions of their respective agencies and organizations. The Regulatory Forum is designed to stimulate broad discussion of topics relevant to regulatory issues in Toxicologic Pathology. Readers of Toxicologic Pathology are encouraged to send their thoughts on these articles or ideas for new topics to
Acknowledgments
The authors acknowledge the STP Advocacy Committee members for recognizing this topic as a current gap of importance to the STP membership. The authors also thank Page Bouchard, Vito Sasseville, Dale Morris, Robert Mauthe, Dan Morton, and Lindsay Tomlinson for their review and invaluable comments on early versions of this article.
Author Contributions
Authors contributed to conception or design (SR and DW); data acquisition, analysis, or interpretation (SR). All authors drafted the manuscript; critically revised the manuscript; gave final approval, and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.