
Abstract
Cell-based therapies have the potential to treat a diversity of disease conditions, many representing significant and long-standing unmet medical needs. Certain properties of cell-based therapies, such as differentiation potential and proliferative potential, present safety concerns uniquely distinct from those of small molecule drugs and other macromolecule biologics. These cellular products carry risks associated with localized host tissue response, long-term persistence, ectopic tissue formation, differentiation to undesirable cell and tissue types, uncontrollable biodistribution, tumorigenicity, and immunogenicity. Such risks are generally evaluated in preclinical animal model studies as part of a comprehensive safety program prior to administration in humans. However, safety assessment for these products can be challenging because of inconsistent approaches to product characterization, inadequately defined product parameters that anticipate adverse events, and the lack of standardized approaches in evaluating in vivo host responses. In this symposium, we introduced cell-based therapies as an emerging product class to the Society of Toxicologic Pathology (STP) and highlighted key challenges for consideration during product biosafety evaluation.
Keywords
Introduction
The search for bioactive agents capable of modulating disease and catalyzing the body’s inherent ability to repair itself has evolved from the identification, isolation, and application of biologically derived or chemically synthesized small molecules and protein-based biologics to the conceptual recognition that the cell itself may be regarded as an active biological ingredient (ABI). Mechanistically operating in part through action-at-a-distance paracrine or endocrine signaling pathways, the cell in its capacity as a therapeutic agent may serve to recruit and mobilize native (i.e., host-derived) stem and progenitor cell populations. Additional medicinal functions catalyzed by the cell may include a response to existing growth factors by migration and proliferation, or release of growth factors and exosomes that promote angiogenesis and neurogenesis, and modulation of inflammatory, fibrotic, and apoptotic cascades that generally function to interfere with the onset of disease while promoting self-repair and regeneration. Furthermore, the cell as medicinal agent may itself contribute directly to regeneration and repair of native tissues and organs by niche-specific differentiation along defined developmental lineages. These are typically regulated by contextual signaling cues derived from the surrounding tissue or organ parenchyma (reviewed by Basu and Ludlow 2010, 2011, 2012).
Critical Quality Attributes of Cell-based Therapy Product Candidates
Current regulatory paradigms for the characterization of medicinal products are tailored principally toward small molecule chemical entities and biopharmaceuticals such as natural or recombinant sourced proteins. Current emphasis in therapeutic product research pipelines continues its evolution away from consideration of the cell as a mere manufacturing platform and toward a realization of the cell itself as the manufactured product. It is therefore imperative that frameworks for process and manufacturing controls, quality and supply chain management, evaluation of toxicologic/pathologic profiles, and therapeutic bioactivity through preclinical animal studies and subsequent clinical trials are sufficiently flexible to accommodate the emerging class of novel cell-based therapies (CBTs) that were unimaginable during the inception of these frameworks. Alternatively, existing policies and regulatory infrastructure will require reformulation to remain relevant. This will permit the appropriate governmental agencies to effectively deliver on their mission to facilitate approval of safe and effective novel therapeutic products without needlessly burdensome oversight of the nascent CBT industry.
Current U.S. Food and Drug Administration (FDA) practice is to evaluate CBTs using a tiered, risk-based, case-by-case approach based on product attributes. FDA expectations for analytic definition of CBTs identify 4 principal critical quality attributes (CQA): (i) identity, (ii) purity, (iii) safety, and (iv) potency. (www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm). Evaluation of identity is required to establish the presence of bioactive cellular components and parameters of manufacturing control. Measures of purity confirm the absence of undesirable elements, including cellular contaminants and processing chemicals. Safety demonstrates the absence of bio-burden (contaminating bacteria, fungi, protozoa, viruses, etc.) and an acceptable adverse risk/benefit outcome. Finally, potency represents an index of the product’s therapeutically relevant biological activities. Recent efforts by industry working groups have provided standardized guideline criteria for the definition of CBT product potency (Guthrie et al. 2013). Here, we examine the current state of the CBT field globally and attempt to provide clarification as to how these novel products may be evaluated for biosafety. Toward this end, a broad summary of the state of the Regenerative Medicine (RM) and CBT fields is presented in Figures 1 to 3.
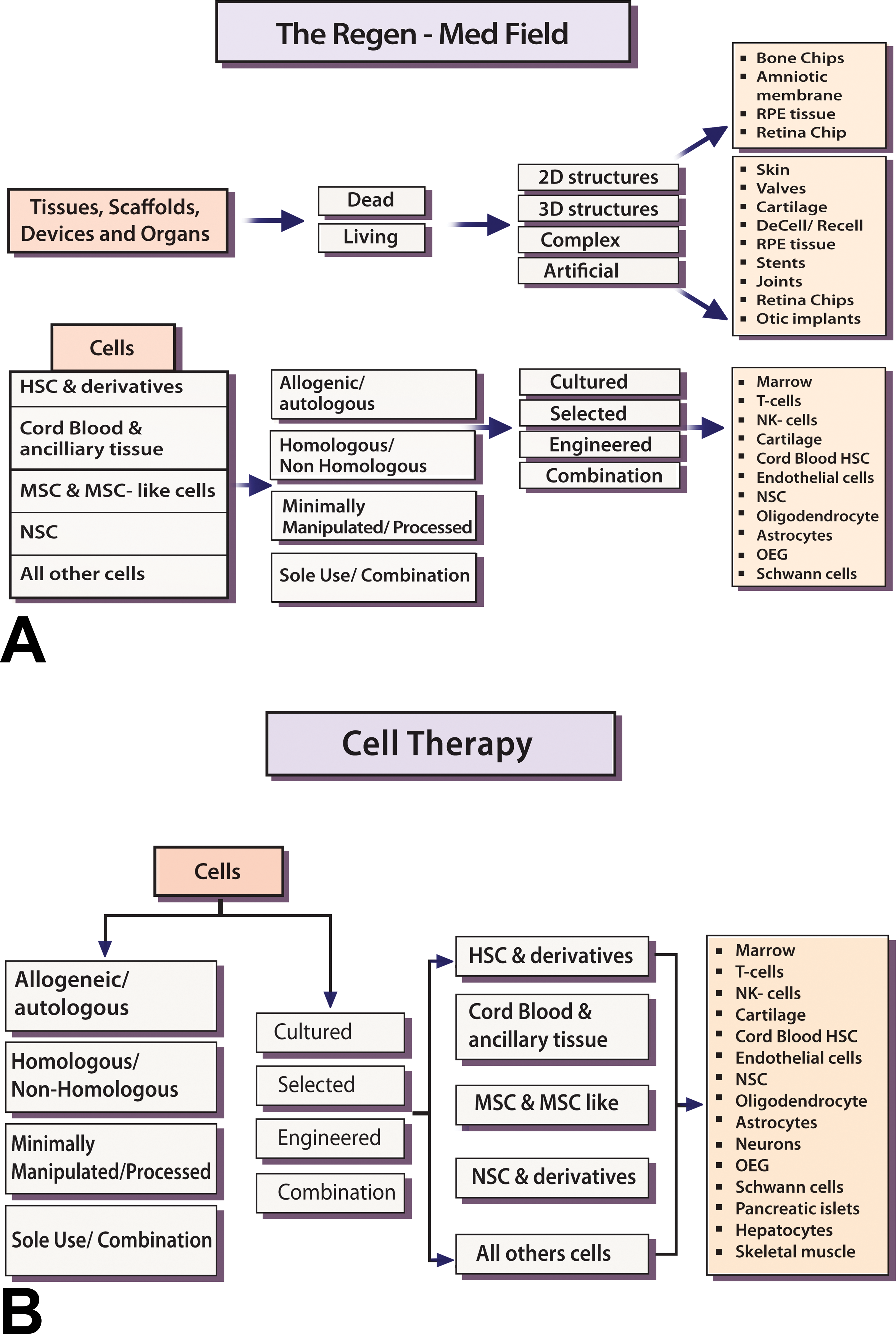
(A) Overview of the field of regenerative medicine. (B) Overview of cell therapy.
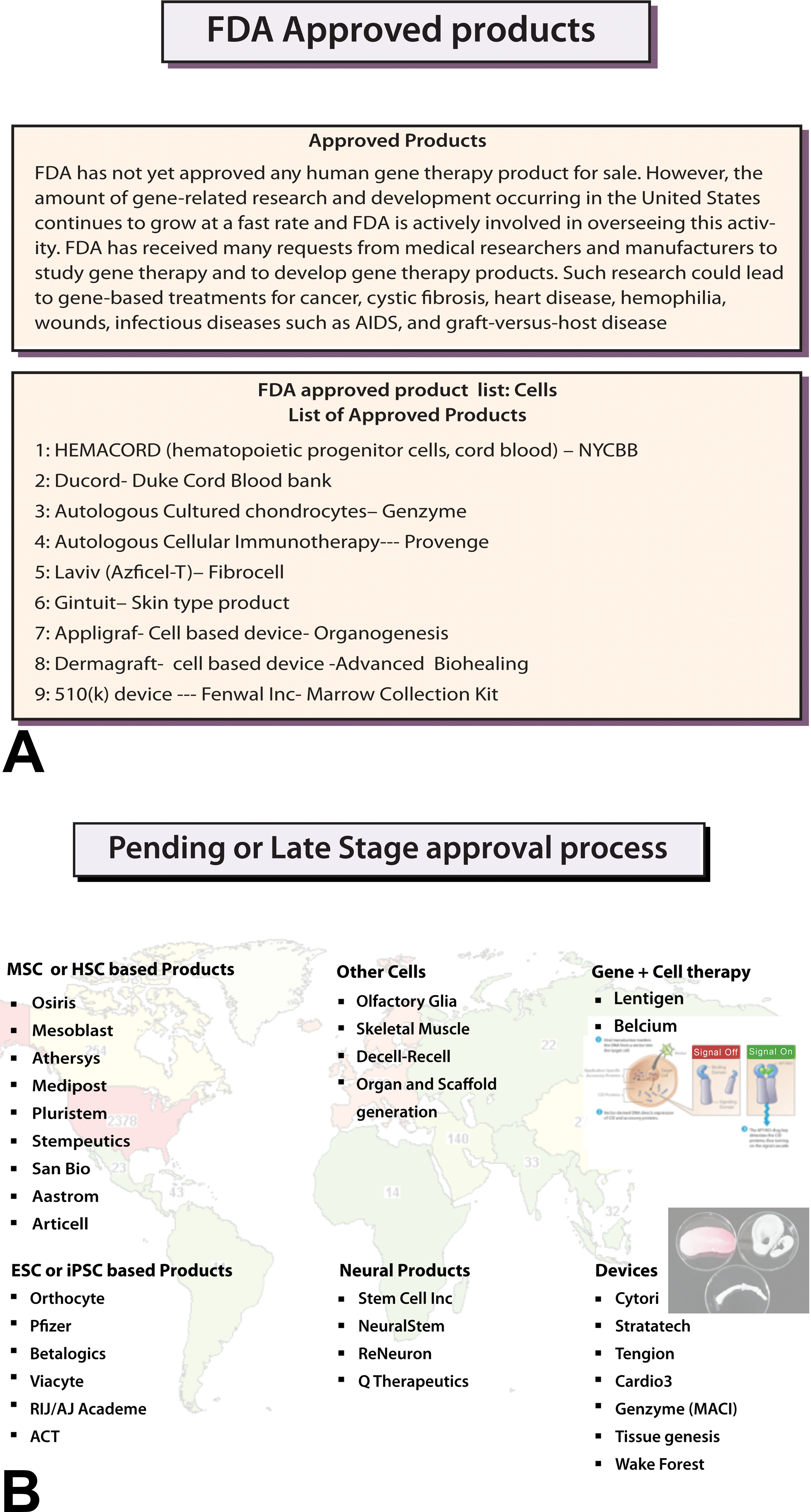
(A) Food and Drug Administration (FDA)-approved cell-based therapeutics. (B) Cell-based therapeutics currently pending or in late stage approval process.
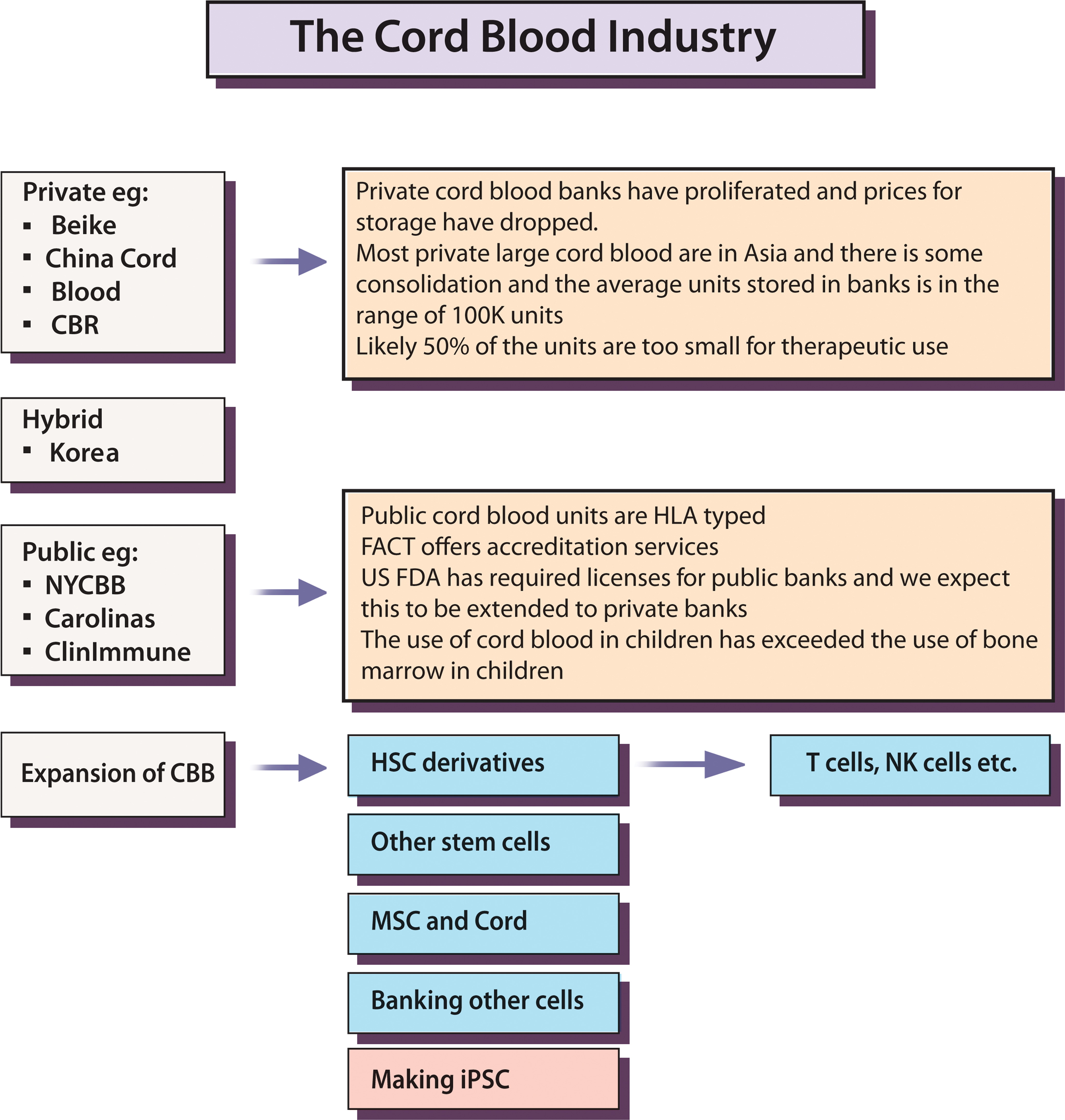
Overview of the cord blood industry.
Cosmeceuticals Provide a Regulatory Shortcut for RM Product Candidates
Regulatory requirements for the promotion and marketing of cosmeceuticals are relatively straightforward and consistent across international boundaries. With approval in the cosmeceutical and plastic surgery markets, many surgical clinics have looked at extension into related fields, based on the operating premise that application of autologously sourced, minimally processed cells would be classified as well-established surgical procedures and could therefore also be used for therapy. Cell types that fit this criteria include adipose-derived stem cells (ADSCs), mesenchymal stem cells (MSCs), immune modulators such as T cells, natural killer cells (NKCs), and other blood derivatives. In each case, the modus operandi is straightforward: cells are harvested and processed as needed to remove blood and other contaminants or to enrich for a specific cellular subpopulation. Additionally, expansion of the cell population by culturing in a sterile environment for a time period of up to 28 days postharvest may be required. Such processing may be performed at a clinic with an attached good manufacturing practice (GMP) processing site, similar to a bone marrow hematopoietic stem cell (HSC) processing site, and several hospitals and clinics have begun to offer such cell-based therapeutic services. These include application of CD34+ cells, ADSC or autologous MSC for the treatment of nonhealing ulcers, critical limb ischemia, or congestive cardiac failure (Caulfield and McGuire 2012). Likewise, immunotherapy for cancer has become popular in Japan and China where autologously sourced T cells, NKCs, and dendritic cells are harvested, expanded using a standard protocol and reinfused back into the patient as a possible salvage or “no other option” therapy, all of which is based on a protocol pioneered by Rosenberg and colleagues (1988, 2008). Case studies have been reported in reputable journals, and clinical trials have been performed in small numbers of patients; based on the results, hospitals and clinics have begun to offer these services as part of a growing medical tourism industry in Asia.
Other groups have begun to use autologous adipose- or marrow-derived cells for the treatment of osteoarthritis, rheumatoid arthritis, and a variety of similar autoimmune disorders (e.g., www.sinostemcells.com). More aggressive clinics have begun to offer services based on research protocols in animals or completely unproven methodologies, leveraging the assertion that their cell manufacturing experience and surgical expertise negate the need for regulatory approval. Such therapies include the use of MSC and ADSC to treat spinal cord injury, stroke, multiple sclerosis, graft-versus-host disease, and an associated plethora of otherwise untreatable or chronic diseases. Anecdotal reports abound, and as these cases are performed on a clinical-service-for-fee basis, follow-up has been limited. Data that are typically collected in clinical trials or research investigations have not been forthcoming. Physicians and surgeons have argued that CBT protocols are surgical procedures that fall within their area of expertise and because regulatory authorities do not govern the practice of medicine, such governance should be left to their respective medical associations. As governments and regulatory authorities have largely agreed, this situation has engendered a multitude of groups that offer CBT services, plus the spawning of companies that provide ancillary services to collect, process, and store cells. However, excesses in the field have led to changes in regulations. As a result, since 2007, the extent of such activity has diminished significantly (www.manhattan-institute.org/html/lpr_17.htm).
Although similar studies have been performed in the United States and Europe, cosmeceutical services cannot be offered in the United States as approved therapy because the FDA has intervened to define CBT as experimental products, successfully arguing that such products were indeed manufactured/processed and were therefore subject to regulatory approval. These regulations were introduced relatively recently (within the past 5 years) and the few clinics offering these services have been forced to cease this practice (Sipp and Turner 2012; Carpenter and Couture 2010). A number of clinics have appealed these regulations, arguing that FDA had overstepped its jurisdiction in regulating the practice of medicine. However, recent rulings by the federal courts have sided with FDA (www.manhattan-institute.org/html/lpr_17.htm). Such approval was not required in most other countries until relatively recently (within the past 5 years), when Chinese authorities defined most CBT as experimental with requirements to get approvals for the same. This has led to a virtual freeze in CBT development activity until clarity emerges regarding exactly which CBT product candidates may be approved. Current regulations in Japan, Korea, and Australia allow for such unregulated therapies, but they are being actively evaluated by the relevant authorities to ensure the continued safety of such activities (Ikegaya 2012).
Overall, the unregulated RM industry is in a state of flux worldwide. Despite this uncertainty, the cell-based medicine industry continues to grow. However, recent court rulings in the United States (within the past 5 years), the publicized closure of clinics in Germany and Switzerland, and the ban of unproven CBT in mainland China (Cyranoski 2012), have had a chilling effect on this rapid expansion. Such clinics typically have not conducted any clinical trials or published their data in any peer-reviewed journals. Local governments and institutional review boards may not have the expertise to rigorously review treatment outcomes for safety and efficacy (Spits 2012). Nevertheless, companies that provide the tools and reagents for this space continue to expand, and there is ongoing approval of devices that allow the practice of this type of medicine. South Korea, Japan, Malaysia, and Thailand appear to be the most active in this area, with Singapore, Indonesia, and India being somewhat less active (Arcidiacono, Blair, and Benton 2012). Hospitals and private physician groups have established personalized medicine clinics that provide a variety of cells for a range of applications. Such clinics are legal but not well regulated and are often prone to overreaching claims and undefined safety risks. However, other centers may be quite reputable and provide important clinical services. These clinics are largely self-funded and, given their rapid spread, are likely to be profitable. Hospitals have also begun to set up stem cell processing laboratories that provide clinical grade cells processed on-site by experienced and trained personnel for marrow, orthopedic, and immunotherapy applications. Device companies have partnered with hospitals to fund trials and develop customized devices for these procedures.
Umbilical Cord Banking
The banking of umbilical cord cells for personal use has become a significant global initiative, with more than 150 cord blood banks (CBBs) worldwide that contain an estimated aggregate of over half a million samples (Zhou, Chang, and Rao 2012; M. Rao, Ahrlund-Richter, and Kaufman 2012; Allison 2012). This private collection of banked samples dwarfs that of the public banks in the United States, United Kingdom, and Europe. Few public banks exist in Asia. This has translated to a rapidly developing medical field and the provision of over 15,000 cord blood transplants worldwide through 2009. In the United States, more than one half of all stem cell transplants from unrelated donors in children now use cord blood (www.nationalcordbloodprogram.org). The uptake from private CBBs is far more limited and perhaps less than a thousand (authors estimate) have been utilized.
However, cell banks market the collection and storage of umbilical cord blood (UCB) to expectant parents who are highly motivated to store UCB for possible therapeutic needs in the future. Private banks charge fees for processing samples and subsequent storage. Additionally, private CBBs spend significant amounts of money educating the public about hematopoietic cell therapy (HCT) and establishing relationships with clinics to ensure the integrity of the supply chain. Private CBBs have grown rapidly, although as might be expected, collections are restricted to the more affluent segments of society. Many of the largest CBBs are now in Asia, with the greatest concentration in China, Hong Kong, and Taiwan. In the absence of public/government support of public banking, these Asian CBBs have developed a model of private/public banking that has allowed otherwise discarded cell lines to become available for potential therapeutic use. This subscription model with a storage fee has become a viable biorepository template for processing and storage of other cell types. As overall global economies have suffered within the past 5 years, the rate of growth of CBBs has also slowed, and there has been significant consolidation in that industry (blogs.nature.com/theniche/2008/03/umbilical_cord_blood_companies.html).
As a result, this has led companies to explore alternative methods of generating revenue within their overall framework of expertise, that is, the collection, processing, and storage of tissues. Some companies have begun to offer archived umbilical cords as a source of MSC-like cells, whereas others process and store MSC. Still other entities have begun looking at placental-sourced CBT. Public CBBs in the United States have seen a change in regulations with the introduction of a licensure requirement. Several of these banks have now obtained the requisite licensure (www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Blood/UCM187144.pdf) and perhaps this signals a further tightening of regulations by FDA to ensure safe and effective CBT products are available to consumers.
New regulatory requirements will undoubtedly lead to cost increases, and with public banks functioning at a deficit, it is likely that there will be pressure on them to develop additional sources of revenue. Indeed, some of the more ambitious cord blood companies, which include public CBBs, have demonstrated that induced pluripotent stem cell (iPSC) can be manufactured from small amounts of cord blood, suggesting that this route may help deliver on the promise of personalized medicine (Meng et al. 2012; M. Rao, Ahrlund-Richter, and Kaufman 2012). Since cells are human leukocyte antigen (HLA) typed, it is also possible that a hybrid model akin to a bone marrow registry could be developed in which licensed public CBBs provide clinically compliant iPSC or iPSC-derived products. Overall, the demand for the core services of private CBBs has declined, and support for public CBBs continues to be limited. However, the established infrastructure and potential for breakthrough technologies offers these banks an opportunity to remain relevant and even thrive as the RM field continues to evolve rapidly (see also Figure 3).
Regulated Therapies and Product Approvals
The largest number of approved CBT products involves adult cells that require limited processing. These include fibroblasts, allogeneic MSC, derivatives of HSC such as dendritic cells, T cells, and NKC. These join the ranks of previously approved cartilage-related and dermal products. Although quantities are currently very small and approvals are country specific, it is important to note that the present number of CBT is larger than the number of approved gene therapy products, although recent successes with genetically engineered CBT for hematological disorders within the past 5 years suggest that a wave of such products may be in the CBT pipeline. Another very active niche field is the combination product strategy, in which a scaffold or device is combined with autologous cells. This model is built on the well-established expertise of device manufacturers coupled with a clear path to regulatory approval of devices. Combination products may be regulated as devices or biologicals. Orthopedic and skin substitute products (e.g., Dermagraft®, Carticel®, etc.) are the best examples of this area of the RM industry.
The rapid pace with which new CBT applications were being evaluated suffered a major setback with the establishment of new regulatory guidelines by various countries; as a consequence, clinics providing such services were forced to stop. Under such regulations, all “manufactured” cells would have to be evaluated though a formal regulatory process to ensure safe and effective product development and would not be classified as the “practice of medicine.” Guidelines approved in the United States with small variations were rapidly adopted by many countries including India, China, and those of the European Union. Draft guidelines intended to achieve similar oversight are in preparation in Taiwan, South Korea, and Singapore (Kellathur and Lou 2012). As a result, it became evident that some entities that initiated development of CBT candidates lacked the expertise and the infrastructure to convert their systems to FDA-approved, cGMP-compliant manufacturing processes. Therefore, new participants need to enter the market to supply such services and provide the investment required to obtain necessary regulatory approvals. The pharmaceutical industry in particular has been reluctant to enter the cell therapy field (Liras 2010); therefore, the single most important source of expertise in dealing with the FDA and related agencies is not an active participant. Other traditional sources of investment such as angel investors, venture funds, and initial public offering (IPO) markets have been similarly limited (M. S. Rao 2011; M. Rao 2013), leaving support mainly to universities, hospitals, and governments.
In Asia, governments have been less generous investors than in the West, and because of this, it has typically been hospitals and small biotechnology companies that tend to enter the market. In doing so, they have faced major financial challenges. Venture capitalists have not historically been able to model returns on CBT candidates as these therapies have undefined development timelines and unknown market potential. Additionally, the pricing power for such therapies is far smaller in Asia, although the Affordable Care Act may have a similar dampening effect in the United States. Consequently, many companies have had to look to other models of financing. This has come in large part from private investors and from hospitals, which see CBT as a natural extension of their efforts to provide needed clinical services to the patient community. Led by their private investors, cell therapy companies have chosen to try and tap the market with a reverse merger strategy (where a private company merges with a public shell company as an alternative, less costly route to go public without an IPO) or to generate additional revenue by selling nonregulated products (see previously, e.g., cosmeceuticals). The South Korean market is perhaps the most active in approving CBT products and the South Korean FDA has taken the lead in product approvals (www.mfds.go.kr/eng/index.do).
MSC-based Products Form by Far the Largest Segment of the Regulated Market
MSC-based cell therapy is by far the largest segment of the current RM market (Figure 4). MSC and MSC-like cells can be harvested relatively easily from a wide variety of sources. Cells can be obtained in sufficient numbers to treat hundreds of patients, and the growth of these cells in the scale required is relatively straightforward with few complex steps. MSC also appear versatile in that they can provide trophic support to many tissues and organs and are able to target and engraft to injury sites to form bone, cartilage, and connective tissue elements (Caplan and Correa 2011). Furthermore, bone marrow–derived mesenchymal cells and likely other MSC-like cells can enhance the engraftment, survival, and expansion of HSC (Battiwalla and Hematti 2009). More recently, it has been shown that MSCs have strong immune-modulatory activity and may be able to both suppress and enhance the immune response as required for cancer therapy (Serakinci, Fahrioglu, and Christensen 2014). MSC products derived from either bone marrow or placenta are available as approved products for certain clinical indications (Table 1). The first MSC-based products have been targeted for their immune modulation activity, but the utility of MSC in treating kidney, liver, and central nervous system (CNS) disorders is being explored actively, and clinical trials using MSC are the largest fraction of clinical trials being undertaken in the stem cell field (www.clinicaltrials.gov). There are possibly 3 major drivers for the current interest in the therapeutic potential of MSC: (1) the Department of Defense (DOD) in the United States, which has funded several high risk–high reward clinical trials, (2) the immune-modulatory effect of MSC, and (3) the possibility that MSC can be derived from iPSC and may be engineered using newer genome editing technologies (Somoza and Rubio 2012). More recently, novel uses of MSC have been considered, including the application of MSC for creating organ scaffolds and the treatment of nervous system and ocular disorders (Marion and Mao 2006; Carvalho et al. 2011).
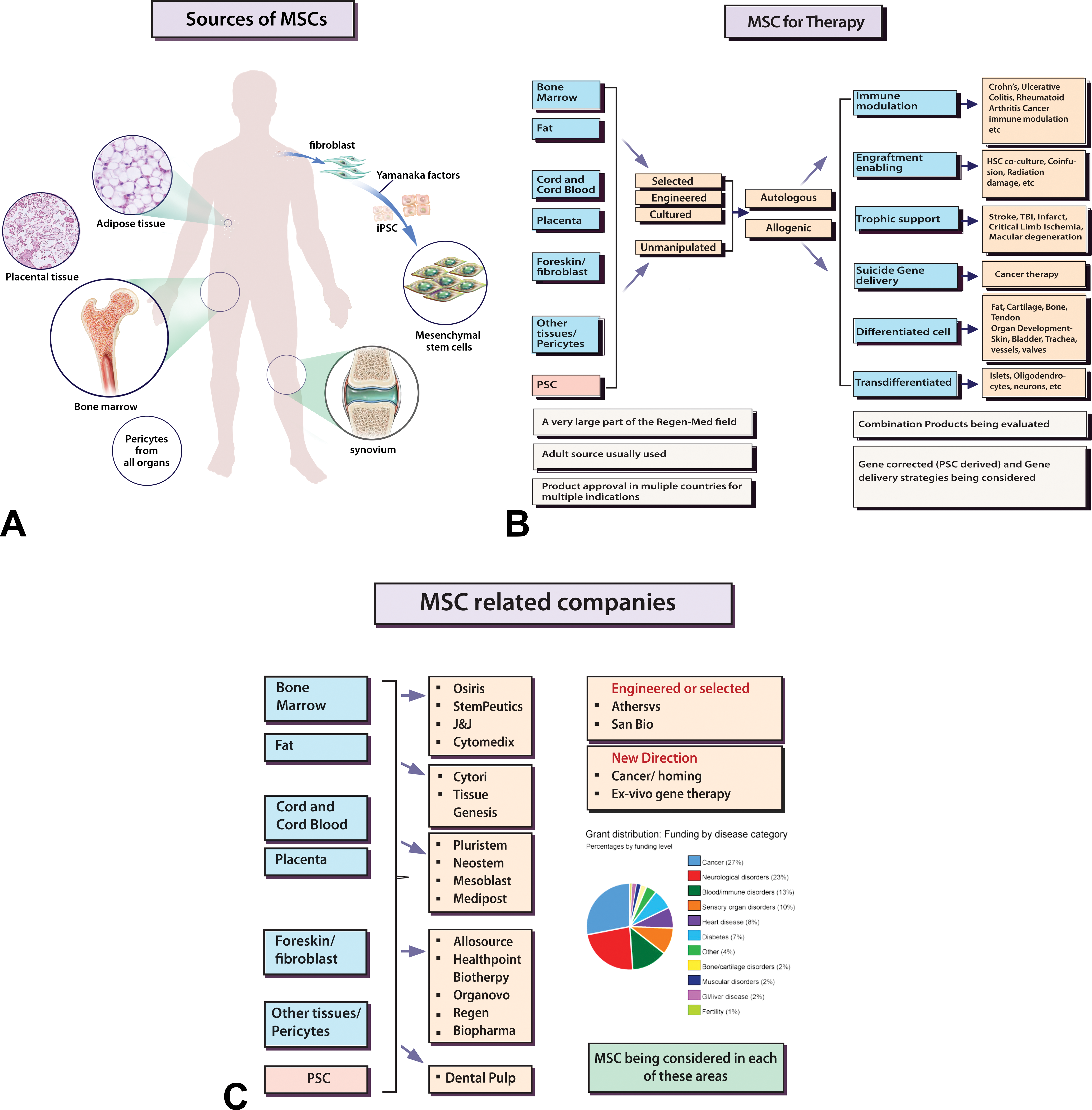
(A) Sources of mesenchymal stem cell (MSC). (B) Overview of applications of MSC for cell-based therapies. (C) Overview of biotechnology companies developing MSC and MSC-like therapies.
Summary of MSC and MSC-like products in development.
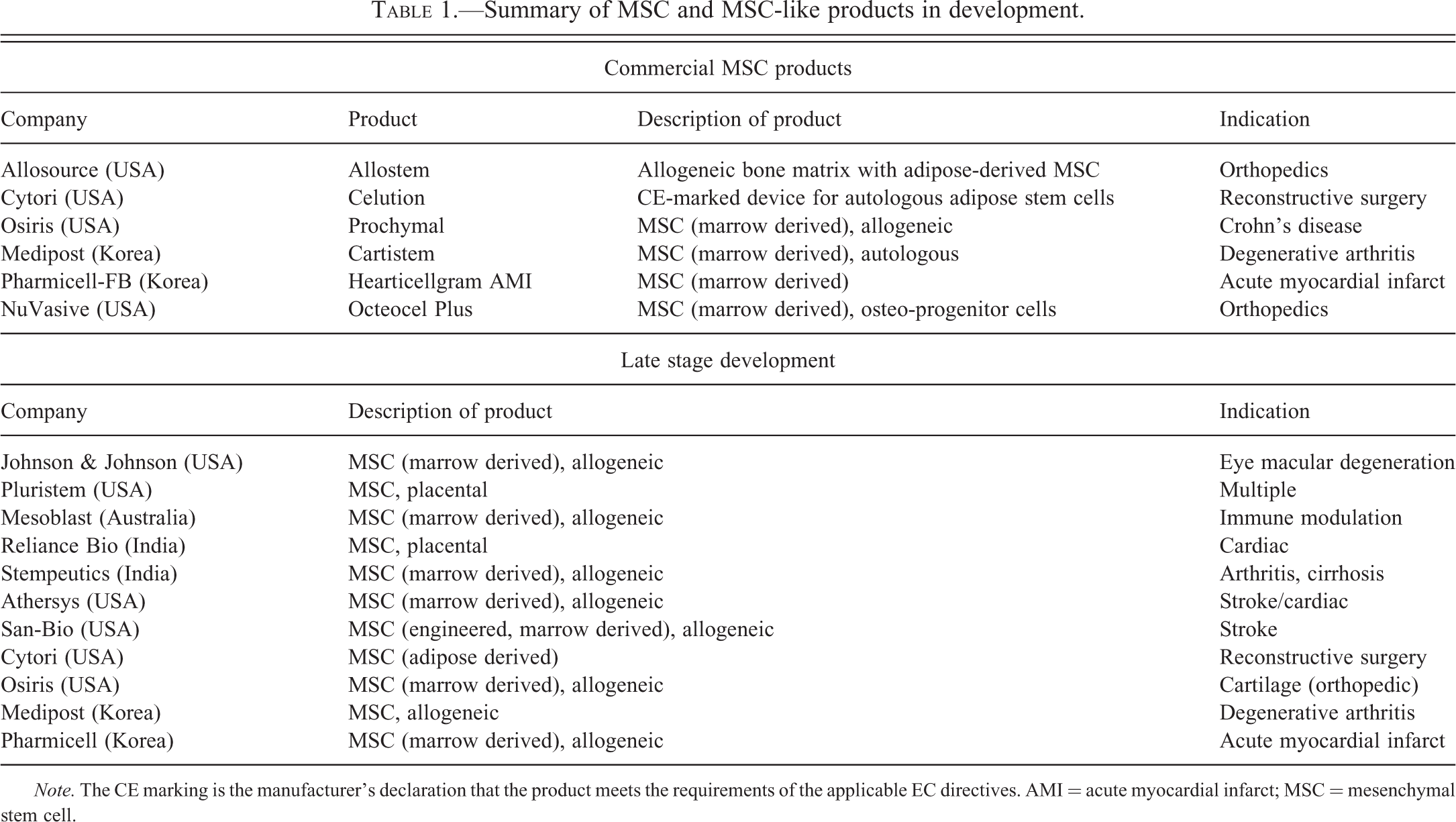
Note. The CE marking is the manufacturer’s declaration that the product meets the requirements of the applicable EC directives. AMI = acute myocardial infarct; MSC = mesenchymal stem cell.
HSC and Neural Stem Cell Subfields Remain Active
Applications of bone marrow– and hematopoietic-derived stem cells have not expanded at the same pace as other cell types and remain largely a physician- and hospital-based therapeutic practice. Marrow registries exist worldwide and a well-established process for collection of either marrow or peripheral blood–derived HSC is currently in place. Cells are harvested and transplanted in autologous or related donor transplants and, in a smaller number of cases, in unrelated donors (Anderlini et al. 2001). Increasing the application of HSC by expanding cells in culture, combining them with other products, or sorting the cells to optimize engraftment remain active areas of research. Additional uses of cells to treat autoimmune disorders or to engineer them via lentivirus, adenovirus, or retrovirus technologies have been evaluated, as has their potential for transdifferentiation. Within the past 5 years, investigators have begun to evaluate blood and marrow for the presence of other types of stem cells or differentiated cell populations that when expanded may represent suitable cells for therapy. These could include hemangioblasts, endothelial cells, macrophages, T cells, NKC, and so on. Many of these activities build on the expertise of the well-established marrow transplant infrastructure and remain the domain of investigator initiated studies. Few companies are active in this field. Other companies have attempted to produce nonnucleated cells such as red blood cells and platelets in bulk by using immortalizing strategies to augment the enormous expansion potential of stem cells. Companies such as Megakaryon, for example, have begun experimenting with such a technology for the manufacture of platelets (www.megakaryon.com; see Figure 5A).
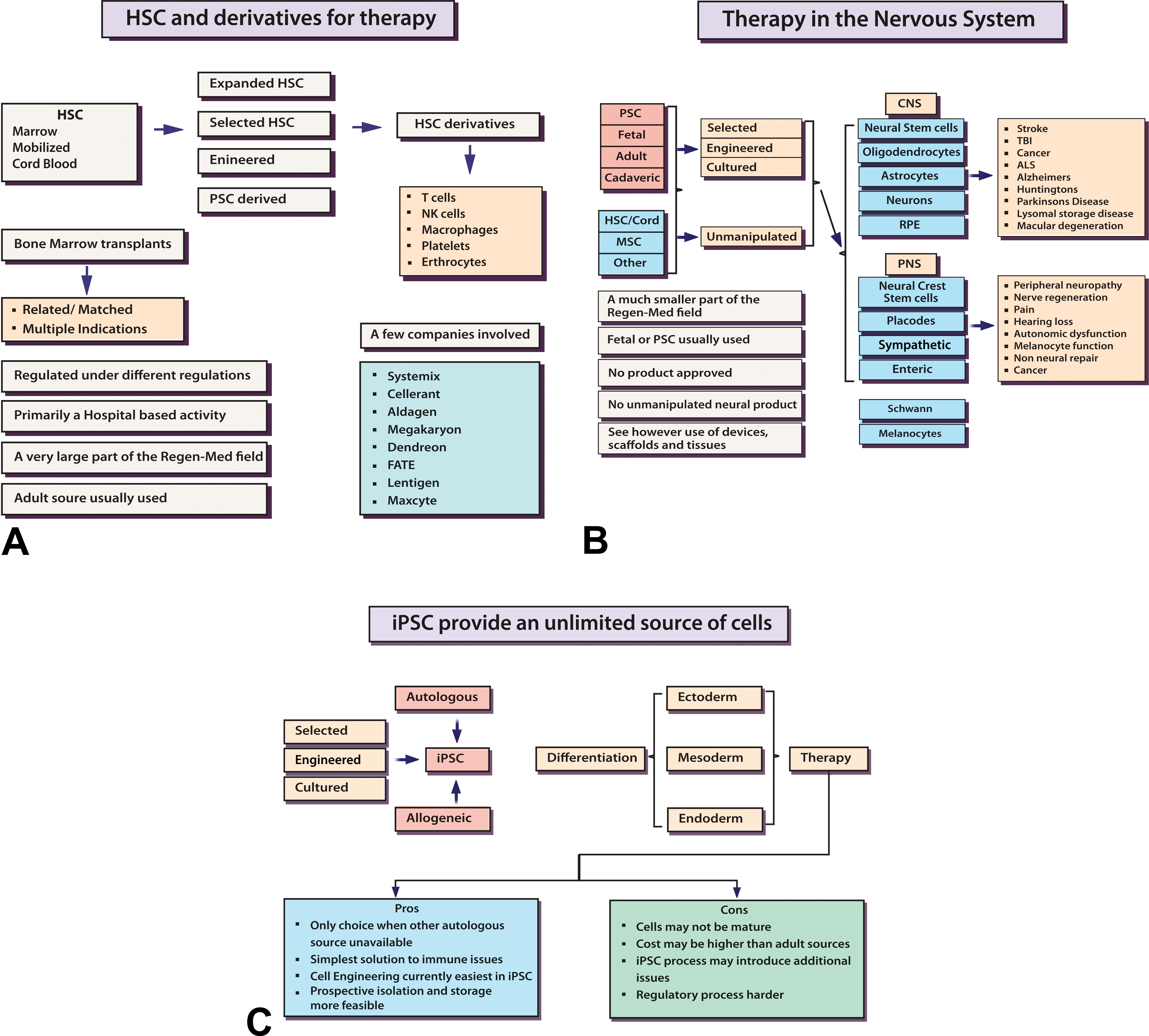
(A) Overview of hematopoietic stem cell (HSC) products in development. (B) Overview of therapeutic approaches for the nervous system: neural stem cell (NSC). (C) Overview of induced pluripotent stem cell (iPSC).
The neural stem cell (NSC) and neural derivative field are comparatively smaller than the HSC field. NSCs are used primarily as allogeneic therapies and have been developed by small companies that own proprietary technologies involved with cell manufacturing or composition of matter patents that concern specific cell types. Examples of such commercial entities include Stem Cells Inc. and Neural Stem Biopharmaceuticals. The indications considered are numerous, although no commercially approved product is currently available (Chiu and Rao 2011). It is important to note that MSC-based companies have now begun exploring the use of MSC and MSC-like cells for the treatment of neurological disorders (Chiu and Rao 2011; Kim and de Vellis 2009). Early results seem promising, at least based on the case reports provided and on the preliminary results from several phase I and phase II trials that are currently underway (see Figure 5B).
The Rest of the RM Market Includes Several Other Cell Types
In addition to MSC, HSC, NSC, UCB, and their derivatives, investigators have considered the application of several other cell products. These include the autologous cell model pioneered by Genzyme (Cambridge, MA) that uses cartilage cells for knee joint repair. Several other companies have obtained approval for competing products and this field remains an active and evolving niche activity. Fibrocell (Exton, PA) uses autologous fibroblasts to aid in wrinkle and anti-scar therapy. Other efforts include the use of dental pulp, hair cell regeneration, skin grafts, and so on. While many of these are cellular products and correctly classified as RM, they are in most cases not derived from stem cells or progenitor cells and thus represent somatic cell therapies. However, with the availability of embryonic stem cell (ESC) and iPSC which can differentiate into many of these cell types, it is entirely possible that in the future, these cell products may be derived from such stem cell populations (Georgieva and Love 2010; see Figure 5C). It is important to note while this represents a small section of the market, this segment has by far the largest number of approved products and perhaps is the most profitable of the current RM-related activities.
Combination Products, Devices, and 3-D Structures Represent the Next Phase of RM
Manufacturers who make implantable devices have recognized the need for combination products, and their development has been an active ongoing activity (Zorlutuna, Vrana, and Khademhosseini 2013; Takagi et al. 2012). Cellular products include skin, heart valves, urinary bladders, blood vessels, and other simple 3-D structures. Additional effort has been expended to develop synthetic scaffolds covered with cells for structures such as the retina and cornea, and for applications such as bone grafts or tracheal and esophageal transplants. Organ regeneration technologies aim to restore the original structure and functionality of a diseased organ. Typically, healing responses in mammals are characterized by fibrosis and scar tissue formation, rather than marked regeneration. Tubular organ regeneration involves a specific, temporal sequence of cellular infiltration, vasculogenesis, neurogenesis, and the differentiation of mucosal, stromal, and parenchymal laminar tissue architectures (reviewed by Basu and Ludlow 2010, 2011). Strategies for organ and tissue regeneration must therefore achieve the dual objectives of triggering a true regenerative response while ameliorating any tendency toward fibrotic repair. The strategy first pioneered for regeneration of the urinary bladder (Basu and Ludlow 2010, 2011) may serve as a foundational platform for the regeneration of other tubular organs. To this end, Tengion, Inc. (Winston-Salem, NC) is actively pursuing commercial development of hollow organ regeneration technologies. This activity represents an important focus in the RM field and the expertise of such bioengineering is widespread. This field lies in a regulatory gray zone, and it is likely that regulations will differ among countries unless efforts at harmonization (currently in their infancy) are successful (Sanzenbacher et al. 2007). To this end, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) has worked to create harmonized guidelines for product efficacy, safety, and quality between Japan, Europe, and the United States (www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000035.jsp&mid=WC0b01ac0580027645).
Induced Pluripotent Stem Cells
The field of RM underwent a seismic upheaval when Dr. Yamanka and colleagues showed that virtually every adult cell in the human body can be modified by a technically simple process to become a pluripotent stem cell. This process, which was first discovered in Japan, was rapidly replicated worldwide with many resulting important advances (Takahashi, Okita et al. 2007; Takahashi, Tanabe et al. 2007; Malik and Rao 2013). Because of the potential for this new technology to provide innovative solutions to intractable health problems, many governments have targeted iPSC CBT as an important focus for resource allocation. These proposals range from establishment of iPSC banks and development of disease-specific cell panels for biopharmaceutical drug screening, to combining pluripotent stem cell technology with genome modification strategies to effect a “cure in a dish.” This latter process involves transplantation of the modified cells back in the patient to effect a clinical cure (Tan et al. 2012; Capecchi 1989; Thomas and Capecchi 1987). Most recently, iPSC-based therapy for macular degeneration using autologous retinal pigment epithelium is slated for clinical trials in Japan as early as 2015 (stemcellstm.alphamedpress.org/site/misc/News159.xhtml).
Preclinical Safety Evaluation of Cell-based Therapies: The Role of Toxicologic Pathologists
CBT products have the potential to treat a diverse array of medical conditions, many of which have unmet therapeutic needs. Properties of stem cells, such as their potential for differentiation and proliferation, pose safety concerns that are unique from those of small molecule drugs and other macromolecule biologics. These cellular products carry risks associated with localized host tissue response, long-term persistence, ectopic tissue formation, differentiation to undesired cell and tissue types, off-target distribution, tumorigenicity, and immunogenicity. Such risks are generally evaluated in preclinical studies as part of a comprehensive preclinical safety program prior to administration in humans. However, safety assessment for these products can be challenging because of inadequately defined host tissue responses and the lack of standardized approaches for evaluating in vivo host responses. A continuing education session on “Scientific and Regulatory Considerations in the Safety Evaluation of Stem Cell-Derived Therapies in Preclinical Studies” was organized and held at the Society of Toxicologic Pathology 33rd Annual Symposium, Washington, DC, June 2014, to introduce this product class to the toxicologic pathology community. It also provided a forum for discussion of the scientific and the regulatory considerations concerning improvements in the evaluation of host responses to stem cell-derived therapies.
Cell therapy and RM is an emerging and diverse technology that aims to regenerate diseased tissues. In this symposium review, we have provided a comprehensive overview of CBT industry and technologies (Figures 1 to 3). Although the CBT industry has advanced from a handful of products to commercialization, this is an emerging technology and many products have not been able to achieve registration. Reasons for product failure have been complex and multifactorial, including the challenge of rebuilding diseased tissues and organs. Several international organizations have been active in advancing CBT including Tissue Engineering International and Regenerative Medicine Society (TERMIS), International Society for Stem Cell Research (ISSCR), and International Society for Stem Cell Therapy (ISCT). Governmental agencies including National Institutes of Health (NIH) and FDA have focused resources on identifying barriers and advancing CBT technologies and products. A number of major initiatives have been put forward by NIH to advance CBT technologies, with emphasis on the assessment of potential safety risks as a key focus. Mechanisms of developmental toxicity, genetic toxicity, and tissue-specific toxicity, including cardiac, liver, and neurologic toxicity, are CBT areas in significant need of predictive toxicology.
Stem cell–derived products represent a more complex entity than small molecule drugs. Their function is expected to persist in a dynamic microenvironment that may influence the product behavior depending on intra- and extracellular signals. Consequently, safety evaluation of CBT products extends beyond the well-defined metabolic pathways of drugs. Current methods, lexicons, and diagnostic criteria used in toxicological pathology are not always adequate for evaluating RM technologies. Additional studies are necessary to identify cellular changes and identify the presence of cellular components of CBT products. Such special evaluations include immunohistochemistry for the detection of infused cells and for the evaluation of cellular proliferation and stages of differentiation and in situ hybridization for localization of target gene expression to specific cell types. Considerable value can be placed on selection of the appropriate animal species and strains (for rodents) and the choice of healthy animals versus animal models of disease. Improved animal models may be required to evaluate both the safety and the efficacy of RM products. Manufacturing processes, product composition, and delivery methods are generally considered when evaluating the safety of CBT products. Because of the unique dynamic features of CBT products, investigational new drug (IND) applications are generally reviewed in a case-by-case approach. Several other unique approaches are being considered. FDA/Center for Biologics Evaluation and Research (CBER)/Office of Cellular, Tissue, and Gene Therapies (OCTGT) are engaging sponsors in the pre-pre-IND discussion phase during the early stages of CBT product development in order to facilitate the translation of CBT products and to support the “Reduce, Refine, Replace” (3Rs) initiative while concurrently evaluating the safety and efficacy of CBT products.
Most CBT products begin evaluation by the administration of product to patients with disease, so the first clinical studies usually evaluate both safety and pharmacodynamics. This clinical testing paradigm requires that animal models provide insight into both the safety and pharmacology of the CBT being evaluated. Key to FDA’s consideration of biosafety are the potential for CBT tumorigenicity, immunogenicity, infectious disease propagation, and genetic modification. In formulating their position, the Society of Toxicologic Pathology (STP) may consider the diversity of CBT products, expansion in the numbers of IND applications reviewed by FDA, and several risks and potential mitigation strategies designed to address these risks. Although CBT products are ideally anticipated to proliferate and differentiate into the desired cell type, FDA places greatest concern on the potential for undifferentiated stem cells to become neoplastic. The safety evaluation of CBT for carcinogenicity is challenging, considering that stem cells are ideally required to proliferate and differentiate, that stem cells are principally defined by their ability to form teratomas, and the fact that conventional rodent carcinogenicity bioassays may be inadequate to evaluate such risk. Two key safety aspects of CBT products include delivery toxicity and histological efficacy. Since CBT products are administered by a parenteral route, there are significant translational challenges in establishing the proper dose. Classical methods of dosing pharmaceuticals are not necessarily relevant for CBT, especially as the dose may change after implantation as a result of cellular expansion or elimination. Unique morphological alterations observed with some CBT products, such as the formation of epithelial lined cysts, serve to highlight the need to understand the actual risk imposed by the unique histological changes that might be induced by CBT.
Overall, the symposium highlighted the need for the STP to consider biosafety evaluation of CBT as a strategic imperative in the future. The symposium emphasized the need for new toxicological assays for CBT products. Several speakers highlighted this as a potential strategic direction for STP and identified the need for enhanced characterization of animal models of disease that provide a robust clinical translation. Translational aspects of identified risks are particularly relevant since many CBT are evaluated in patients during the first clinical trial. The importance of in vivo assays for assessment of dynamic effects of CBT was also considered. These include early changes that may be proliferative in nature, but actually represent a regenerative response rather than an indication of safety risk. Another potential strategic direction for STP is the development of standardized nomenclature that covers novel observations for CBT products (e.g., teratoma) and indicators of histological efficacy (e.g., blastema). Each of these could represent a regenerative or biosafety signal depending on the time postimplantation and the animal model being used. A significant challenge highlighted was the need to understand the tumorigenicity and immunotoxicity risks that may be associated with CBT products.
Footnotes
Acknowledgment
The authors would like to thank Mercedes Serabian, Alexander Bailey, Julia Baker, and Jane Lebkowski for their participation in the CE Session on “Scientific and Regulatory Considerations in the Safety Evaluation of Stem Cell-Derived Therapies in Preclinical Studies” held at the Society of Toxicologic Pathology 33rd Annual Symposium, Washington, DC, June 2014.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.