
Abstract
Simvastatin, a competitive inhibitor of HMG-CoA reductase widely used in the treatment and prevention of hyperlipidemia-related diseases, has recently been associated to in vitro anticancer stem cell (CSC) actions. However, these effects have not been confirmed in vivo. To assess in vivo anti-CSC effects of simvastatin, female Sprague-Dawley rats with 7,12-dimethyl-benz(a)anthracene (DMBA)–induced mammary cancer and control animals were treated for 14 days with either simvastatin (20 or 40 mg/kg/day) or soybean oil (N = 60). Tumors and normal breast tissues were removed for pathologic examination and immunodetection of CSC markers. At 40 mg/kg/day, simvastatin significantly reduced tumor growth and the expression of most CSC markers. The reduction in tumor growth (80%) could not be explained solely by the decrease in CSCs, since the latter accounted for less than 10% of the neoplasia (differentiated cancer cells were also affected). Stem cells in normal, nonneoplastic breast tissues were not affected by simvastatin. Simvastatin was also associated with a significant decrease in proliferative activity but no increase in cell death. In conclusion, this is the first study to confirm simvastatin anti-CSC actions in vivo, further demonstrating that this effect is specific for neoplastic cells, but not restricted to CSCs, and most likely due to inhibition of cell proliferation.
Introduction
Mammary glands have the ability to reshape themselves during cycles of pregnancy, lactation, and involution throughout the lifetime, and this is generally attributed to adult stem cells (SCs) that lie within the tissue (Villadsen et al. 2007; Woodward et al. 2005). Mammary SCs have 3 main functions: to give rise to tissue constituents of mammary glands during embryogenesis, to allow cyclic tissue plasticity as mentioned previously, and, less frequently, to serve as a cellular reserve for self-limited repair after tissue damage (Woodward et al. 2005). SCs can only perform these important functions because they bear 2 fundamental properties: the ability of self-renewal (through symmetrical or asymmetrical mitosis) and that of giving rise to different cell lines (through asymmetrical mitosis; Al-Haji and Clarke 2004; Klonisch et al. 2008; Soltanian and Matin 2011). In breast tissue, adult SCs can originate 3 distinct cell subtypes: the luminal epithelial cell (located at the inner lining of ductal structures), the myoepithelial cell (which has contractile capacity), and the basal cells (Li and Rosen 2005). The distinction between the latter subtypes is a difficult one, as they present overlapping morphology, immunophenotype, and topology (both share the outer layer of ducts, i.e., the interface with stroma). Some authors have suggested that adult mammary SC are actually a subgroup of basal cells (Velasco-Velázquez et al. 2012; Woodward et al. 2005). Recent lines of evidence have also linked SCs to pathological phenomena such as inflammatory and neoplastic processes, particularly in the genesis, maintenance, and progression of malignant tumors (Gökmen-Polar, Nakshatri, and Badve 2011; Klonisch et al. 2008).
Breast cancer is the most common malignancy and the leading cause of death by neoplasia among women worldwide. The onset of breast cancer seems to occur as a result of deregulation of multiple intracellular processes that are cumulative and persistent throughout their lifetime, since an exponential increase in the incidence of breast cancer parallels aging. According to the classical model of carcinogenesis, these mutations can occur in practically any cell, regardless of its stage of maturation. In a recent innovative paradigm, however, it is said that the main targets of those genetic mutations leading to oncogenic transformation are stem and progenitor cells. This hypothesis is currently known as the cancer stem cell hypothesis (Charafe-Jauffret et al. 2009; Kakarala and Wicha 2009; Woodward et al. 2005).
Whether or not SCs are the true origin of most neoplastic processes is still a topic of debate. Nonetheless, recent studies have suggested that in the majority of neoplasms—including solid tumors—a hierarchy of cells in various stages of maturation seems to be driven by cells with phenotypic and functional features of SCs (Al-Haji and Clarke 2004; Kakarala and Wicha 2009). As a result, these cells have come to be designated as cancer stem-like cells (CSCs). Although CSCs may represent a small portion of a given neoplasia, they have been linked in numerous studies to therapeutic resistance (including the main treatment modalities: chemotherapy, radiotherapy, and immunotherapy [Klonisch et al. 2008; Park et al. 2010; Soltanian and Matin 2011]). From a immunophenotypic viewpoint, CSCs are characterized by specific molecular expression profiles (Klonisch et al. 2008), mostly defined by the presence or absence of certain surface molecules such as CD133, CD34, CD24, and CD44 (Velasco-Velázquez et al. 2012). Such immunophenotypic profile may vary across different neoplastic entities, tumor areas, cell lineages, and patients, suggesting the coexistence of distinct subclasses of CSCs within any given neoplasm (Klonisch et al. 2008).
Following intensive investigation dedicated to the development of antitumoral drugs in the past 4 decades, great attention has been given to inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, also known as statins (Sassano and Platanias 2008). Being primarily antidyslipidemic drugs, statins have traditionally been applied to the treatment and prevention of cardiovascular diseases (the leading cause of death in many countries). Interestingly, in the past decade, there have been several lines of experimental evidence suggesting that statins could have some anticancer effects in vitro (Gauthaman, Manasi, and Bongso 2009) and in vivo (Kubatka, Zihlavnikova, Kajo, et al. 2011; Kubatka, Zihlavnikova, Solar, et al. 2011). For example, Kubatka et al. showed evidence of cancer chemoprevention with the use of simvastatin (at 180 mg/kg) in Sprague-Dawley rats submitted to the mammary carcinogenic agent N-methyl-N-nitrosourea (NMU). These properties could be related to specific effects on cell cycle and growth, ultimately resulting in tumor cell apoptosis (Kubatka, Zihlavnikova, Kajo, et al. 2011; Kubatka, Zihlavnikova, Solar, et al. 2011). Nonetheless, even more recent data have suggested that this effect could include actions on cancer CSCs (Gauthaman, Manasi, and Bongso 2009). In 2009, Gauthaman, Manasi, and Bongso demonstrated in an unprecedented manner that lipophilic statins have in vitro inhibitory effect on specific embryonic SC with karyotypic alterations and neoplastic cells of a breast cell line with CSC phenotype, while not affecting the growth of normal breast SC. Of notice, in studies characterizing the antineoplastic effects of statins, there was no report of the degree of susceptibility of specific subpopulations of CSCs to the action of statins in vivo.
Thus, this study aimed to characterize simvastatin effects on tumor growth and on the expression of CSC-like markers in an in vivo model of breast carcinogenesis. In order to do so, we selected one of the most studied models of breast cancer, the 7,12-dimethylbenz(a)anthracene (DMBA) model, which is characterized by high induction efficiency (100% of treated animals develop a malignant tumor, 13 weeks after single administration of the carcinogen) and specificity for breast neoplasms, when applied to virgin female Sprague-Dawley rats (Barros et al. 2004; Welsch 1985).
Materials and Methods
Animals and Tumor Induction
Sixty virgin female Sprague-Dawley rats from the Multidisciplinary Center for Biological Investigation at the Laboratory for Animal Science of the State University of Campinas (CEMIB-Unicamp), aged 40 days through 45 days (200–250 g), were used. They were housed in a temperature- and humidity-controlled facility in the Department of Pharmacology-Unicamp with a 12 hr light–dark cycle for 10 days and then subjected to the chemical induction of mammary carcinoma. The rats were kept in plastic cages (5 animals per cage) and fed with ration for rodents (Nuvital®, São Paulo, Brazil) and water ad libitum. Malignant breast neoplasms were induced by a single dose of DMBA at a concentration of 100 mg/kg diluted in 1 ml of soybean oil and given intragastrically by gavage (a modification of Barros et al. 2004 protocol). All procedures were approved by the local ethics committee (CEUA-Unicamp; protocol number: 2335-1), in accordance with National guidelines (established by the Brazilian Society of Laboratory Animal Science [SBCAL], former College for Animal Experimentation [COBEA]) and NIH standards.
Treatment Protocol
The animals were randomly divided into 6 groups. Three of them were comprised by noninduced animals treated for 14 days with either 1 ml of soybean oil by gavage (Control Group [CG], n = 10) or with simvastatin (at the dose of 20 or 40 mg/kg/day in 1 ml of soybean oil; henceforth, designated as simvastatin-20 [S20] and simvastatin-40 groups [S40], n = 10/group). The remaining groups were composed of induced (tumor-bearing) animals either treated with 1 ml of soybean oil by gavage (untreated DMBA Group [DMBA], n = 10) or with simvastatin (at the dose of 20 or 40 mg/kg/day in 1 ml of soybean oil; DMBA/simvastatin-20 [DMBA+S20] and DMBA/simvastatin-40 groups [DMBA+S40] respectively, n = 10/group), continuously for 14 days. All treatment protocols were started as soon as the tumors reached 0.5 cm3. Throughout the protocol, the animals were monitored daily by an experienced veterinarian for tumor growth (number and size of developing tumors), weight changes, overall clinical status, as well as for the detection of common clinical syndromes associated with the developing neoplasms. The volume of each individual neoplasm was calculated following the equation V = (a × b × c) × π/6 (where a, b and c represent the greater dimensions of the tumors).
Tumor Specimens and Postmortem Examinations
At the end of each experimental protocol, the rats were euthanized by deep anesthesia with isoflurane and cervical dislocation. All mammary glands (regardless of tumor status) were excised and macroscopically examined. Once again, the volume of each individual neoplasm was calculated following the equation stated previously. All animals were submitted to complete necropsy (by either a veterinary [P.C.S.] or a human pathologist [A.A.S.]) in order to assess metastatic disease and other comorbidities (e.g., toxic effects caused by pharmacological treatments). Pathology assessment was performed at both macroscopic and microscopic levels, with special attention to brain, liver, lung, heart, kidney, bone marrow, and skeletal muscle. Slices of 3- to 5-mm thick of each specimen were fixed by immersion in 10% buffered formalin for 24 hr and submitted to automated histological processing for paraffin embedding.
Histological Staining
Sections of 4- to 5-µm thick were obtained from each paraffin block containing a representative slice of the largest tumor, normal breast tissue, or any other tissue previously sampled at the postmortem exam. These sections were stained with hematoxylin-eosin (H&E stain) or submitted to immunostaining for CD133, CD34, CD24, and CD44 (CSC markers), as well as Ki67 (cell proliferation marker).
Briefly, the 4-μm thick sections were placed on silanized slides, deparaffinized in xylene and rehydrated. Endogenous peroxidase activity was quenched with 3% hydrogen peroxide. Antigen retrieval was performed by heating slides in citrate buffer (10 mm, pH 6.0) at 95°C for 30 min. The primary antibodies used were rabbit anti-CD133 (polyclonal, cat#PAB12663, Abnova, Taiwan; diluted at 1:250), goat anti-CD34 (polyclonal, cat#sc7045, Santa Cruz, USA; diluted at 1:50), rabbit anti-CD24 (polyclonal, cat#251181, ABBIOTEC, USA; diluted at 1:200), rabbit anti-CD44 (polyclonal, cat#Ab65829, ABCAM, USA; diluted 1:200), or rabbit anti-Ki67 (polyclonal, cat#Ab15580 ABCAM, USA). Antigen–antibody reaction was revealed using LSAB + System—HRP (DakoCytomation, Carpinteria, CA, USA). In addition, the sections were counterstained with Mayer’s hematoxylin and mounted on glass slides with Entellan resin (Merck, Dermstad, Germany). Negative controls were included in each batch and consisted in the omission of the primary antibody. Positive controls included specific tissues of rat origin that were known to consistently react to the primary antibodies used (according to the suppliers’ instructions).
Morphologic Analysis and Quantitation of Immunomarkers
Digitization of histologic slides was carried out automatically using a whole slide scanner (ScanScope XT, Aperio). Six randomly selected images, each representing one medium power field (=200× magnification), were manually retrieved from hotspots of positivity within the virtual slides using ImageScope software (Aperio Technologies Inc.) and further analyzed using the ImageJ software (National Institutes of Health [NIH]). Using the multipoint selection tool, positive and negative cells within each image were counted in order to determine the mean frequency of positive cells per medium power field, in each case. Histological classification and grading of the lesions were performed on H&E slides by 2 experienced breast pathologists (A.A.S. and N.G.M.S.), using the latest World Health Organization (Lakhani et al. 2012) criteria and the Nottingham grading system for breast tumors. The diagnosis of malignancy was based on the criteria provided by Russo and Russo (2000), which includes (1) rapid growth and skin ulceration in a short period of time, (2) soft fleshy appearance (with or without necrosis), (3) loss of normal architecture with varying pleomorphism and layering or formation of papillae, (4) a monotonous cell population (intermediate cells, predominating over dark or myoepithelial cells), (5) varying response of the host (such as fibrosis or inflammatory infiltrates), (6) increased nucleocytoplasmic ratio and nuclear atypia (round to oval nuclei, with smooth contour, leptochromatic appearance, and 1 or 2 prominent nucleoli), (7) increased mitotic activity, and (8) microscopic signs of invasiveness (neoplastic cells infiltrating surrounding structures such as muscle, dermis, and fat). Morphologic evaluation also included an estimation of tumor necrosis (average percentage of necrosis) in 4 low magnification fields (=40× magnification), and determination of the mitotic index (number of mitotic figures) in 10 high power fields (=400× magnification). This method was validated in a random sample representing 10% of the total number of analyzed images by 2 experienced observers, A.L.R. and A.A.S. (blinded to the intervention/origin of the images). In order to assess the correlation between the 2 independent observers (and thus, the objectivity and reproducibility of the countings), we performed Pearson’s correlation coefficient (Pearson’s r). In this validation procedure, Pearson’s r was greater than .8 for all quantitative variables (p < .0001).
Statistical Analysis
Differences between groups were analyzed using Student’s t test, analysis of variance (ANOVA; followed by Bonferroni post hoc test), or Fisher’s exact test, according to the specific context, as stated in figure legends. Correlation between quantitative variables was further analyzed by Pearson’s r. A p value of less than .05 was considered statistically significant. All tests were performed in GraphPad Prism 5.0 software for Windows.
Results
In this study, a single dose of DMBA (adjusted to animal weight) was able to successfully induce malignant breast tumors in all of the animals submitted to the protocol. The average number of neoplasms per animal was 2.47, the mean size was 3.60 cm3, and, in general, these tumors were located bilaterally. The diagnosis of malignancy was mainly based on the finding of locally aggressive behavior and cytoarchitectural atypia. By the time the experimental protocol had been initiated, virtually all neoplasms presented macroscopic signs of soft tissue invasion, frequently associated with necrosis, skin ulceration and, occasionally, the animals presented early stage signs of consumptive syndrome (discrete weight loss and decreased appetite). Most animals (72%) had at least 1 palpable tumor by the 8th week posttreatment with DMBA.
Neither metastatic disease nor second primary neoplasms were associated with the use of DMBA. DMBA tumors were characterized by confluent nodules of varying size and shape, usually soft and friable, unencapsulated, microlobulated, with central necrotic foci and frequent hemorrhagic areas. None of the animals developed overt morphological signs of toxicity, classically reported with the use of simvastatin (such as, myotoxicity, hepatotoxicity, and renal failure).
Figure 1 illustrates tumor growth during experimental protocols. As can be seen in this figure, simvastatin, at the dose of 40 mg/kg (but not at 20 mg/kg) significantly inhibits the expected tumor growth, particularly from the 12th day on. Figure 2 further confirms the effect of 40 mg/kg simvastatin, showing an average decrease of 80% in tumor increment for simvastatin treated animals, when compared to untreated ones. Also, as depicted in Figure 3, simvastatin treatment at 40 mg/kg results in a significant decrease in the total number of tumors per animal. Of notice, no significant differences were found between control (untreated) animals and those treated with simvastatin at 20 mg/kg.
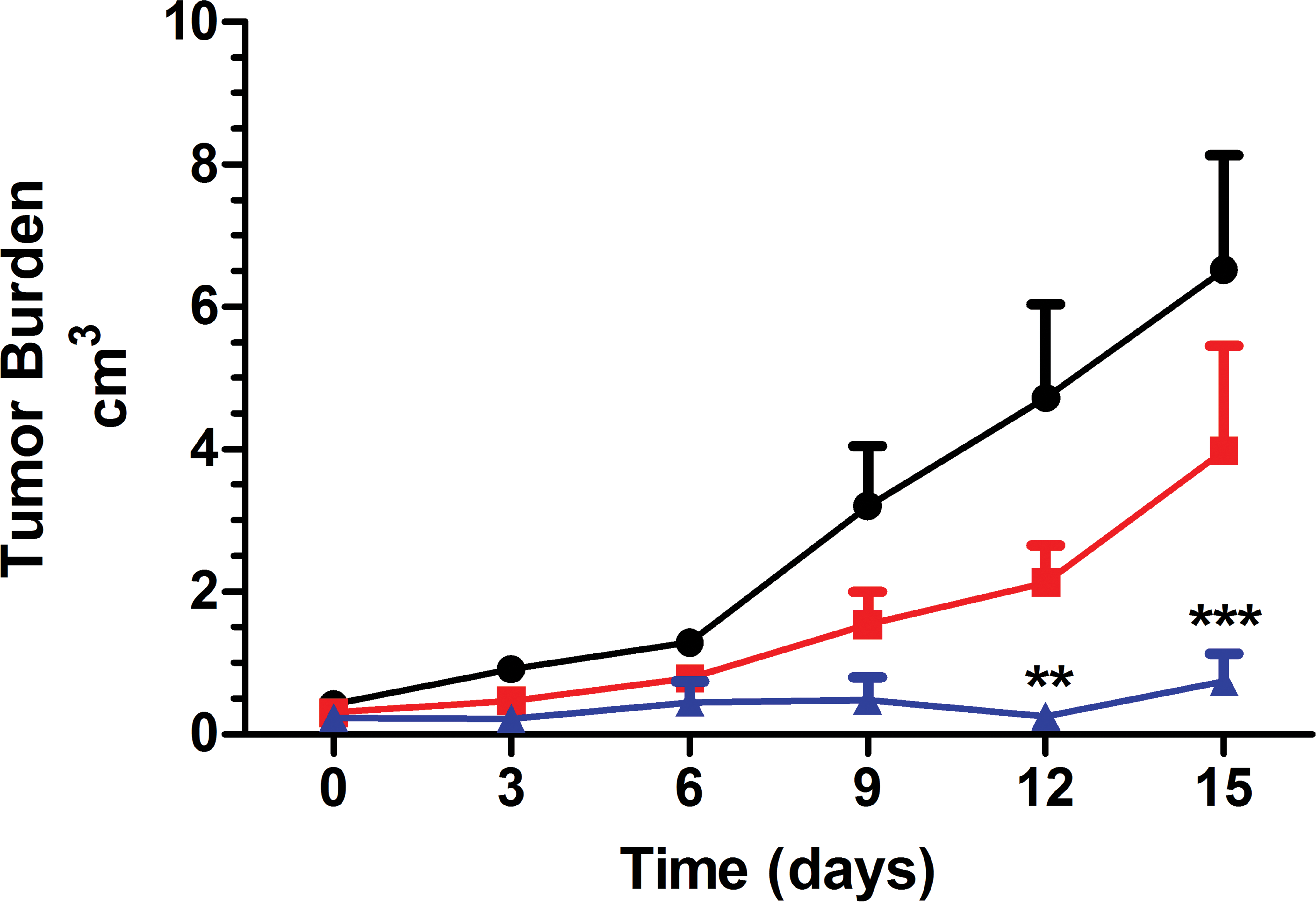
Progression of tumor burden (sum of individual tumor volumes within each animal) during experimental protocol. This graph demonstrates a significant inhibition in tumor growth among animals treated with simvastatin at the dose of 40 mg/kg (p = .0034 at 12 days and p = .00027 at 15 days). The groups were as follows: (-•-) DMBA; (-▪-) DMBA+S20; (-▴-) DMBA+S40. The results represent mean (M) ± standard error of the mean (SEM); **p < .01 and ***p < .001, when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni post hoc test).
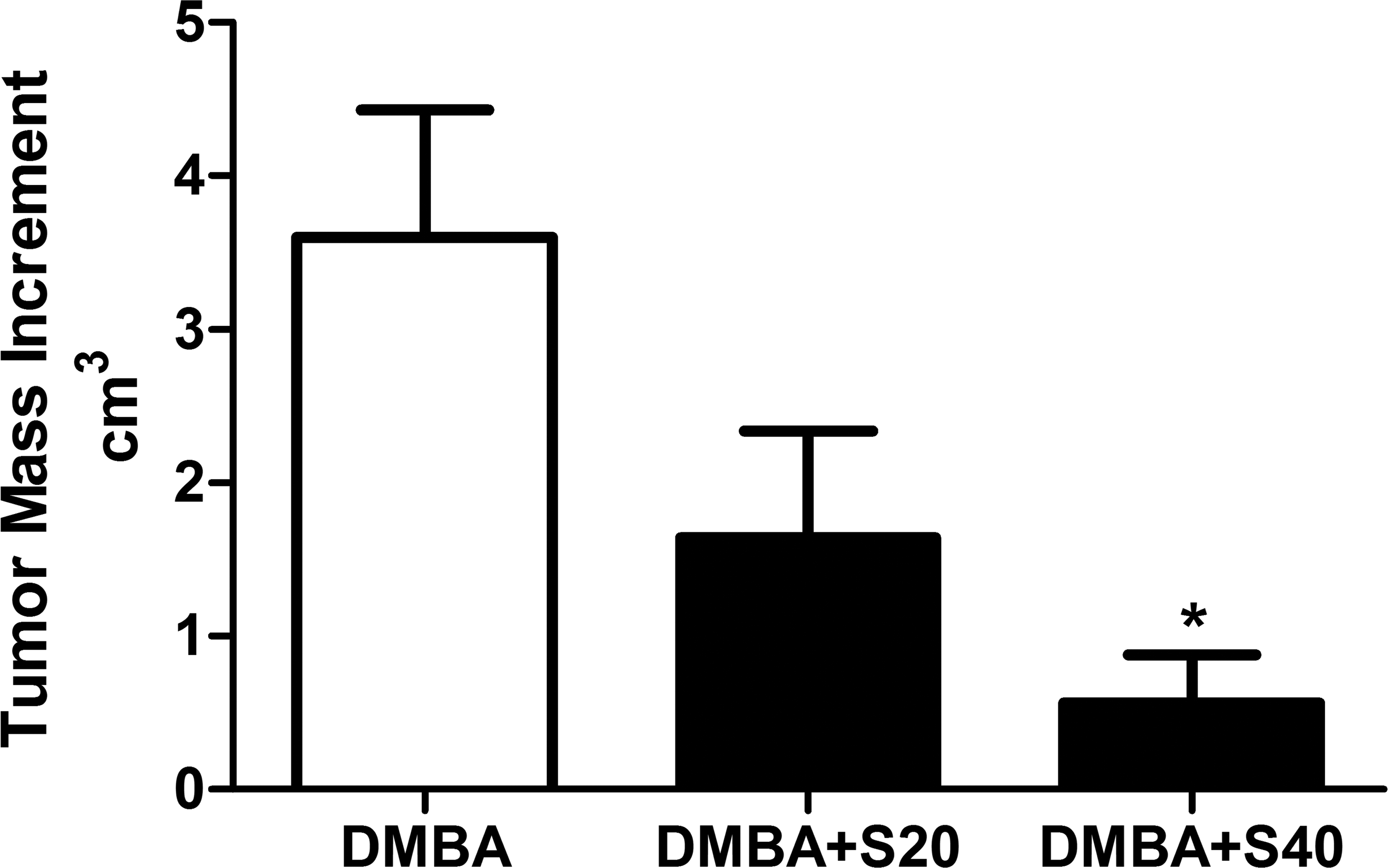
Tumor mass increment (TMI = Final tumor burden − Initial tumor burden, expressed in cm3). Mean tumor increment at the end of treatment demonstrates a significant decrease in tumor growth among animals treated with simvastatin at the dose of 40 mg/kg (p = .0240). Of notice, this decrease is on average greater than 80% when compared to untreated animals (DMBA group). The results represent the mean (M) ± SEM of the observations. TMI% decrease was obtained by comparing paired DMBA and DMBA+S40 animals and calculated as follows: ([TMI from DMBA animal n − TMI from a paired DMBA+S40 animal n ]/TMI from DMBA animal n ) × 100%; the 80% decrease not shown on graph represents the mean of all decrement results obtained by pairing. *p < .05 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
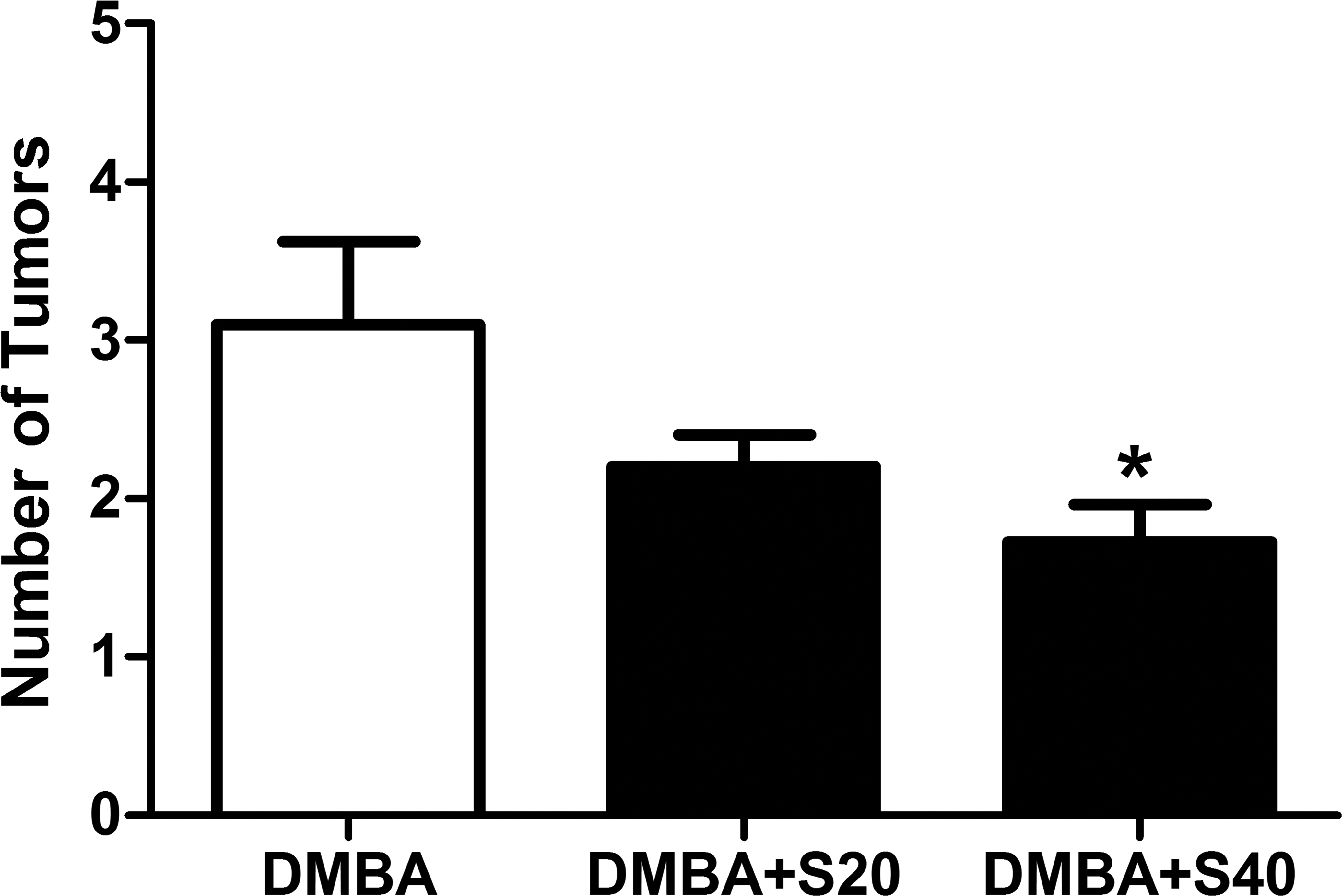
Number of tumors per animal at the end of experimental protocol. As shown, use of simvastatin at 40 mg/kg results in a significant decrease in total number of neoplasms at the end of treatment (p = .0113). The results represent the mean (M) ± SEM of the observations. *p < .05 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
The majority of DMBA-induced tumors were represented by a mixture of histologic types, such as invasive ductal carcinoma, histologically malignant phyllodes tumor and invasive papillary carcinoma. Of notice, in mixed tumors, the main component (i.e., the most extensively represented histologic subtype) was that of an invasive ductal carcinoma. Simvastatin seems to increase the relative percentage of the invasive ductal carcinoma subcomponent (from 82% to 90%, on average); however, differences between experimental and CGs did not reach statistical significance (Figure 4).
Mean percentage of invasive ductal carcinoma component and histological subtypes observed in this sample. Mean percentage of invasive ductal carcinoma component (invasive ductal carcinoma of no special type) seems to be increased in DMBA+S animals (i.e., treatment with simvastatin, regardless of dose) in comparison to DMBA rats (untreated tumors) (A); however, such difference does not reach statistical significance. Photomicrographs illustrating the main histological subtypes observed: invasive ductal carcinoma (B), malignant phyllodes tumor (C), and invasive papillary carcinoma (D). H&E.
The distribution of induced tumors according to histologic grading is presented in Table 1. As can be seen, a higher percentage of well-differentiated tumors (low grade tumors) was observed among DMBA+S animals in comparison to untreated animals (p = .0312). In addition, no cases of poorly differentiated tumors were observed among simvastatin groups.
Distribution of induced tumors according to the final histologic grade, as determined by the Nottingham Score System (Lakhani et al. 2012).

Note: p = .0312 (Fisher’s exact test). “DMBA” refers to control animals (n = 10), while DMBA+S refers to the experimental groups combined (i.e., animals treated with simvastatin at the dose of 20 mg/kg [DMBA+S20] or 40 mg/kg [DMBA+S40]; n = 20).
Figures 5 to 7 present the results concerning tumor necrosis mapping, mitotic count and Ki67 index, respectively. In the current protocol, tumor necrosis was less than 10%, regardless of treatment protocol. Of notice, both proliferative indices (mitotic and Ki67 counts) were significantly decreased by treatment with high-dose simvastatin.
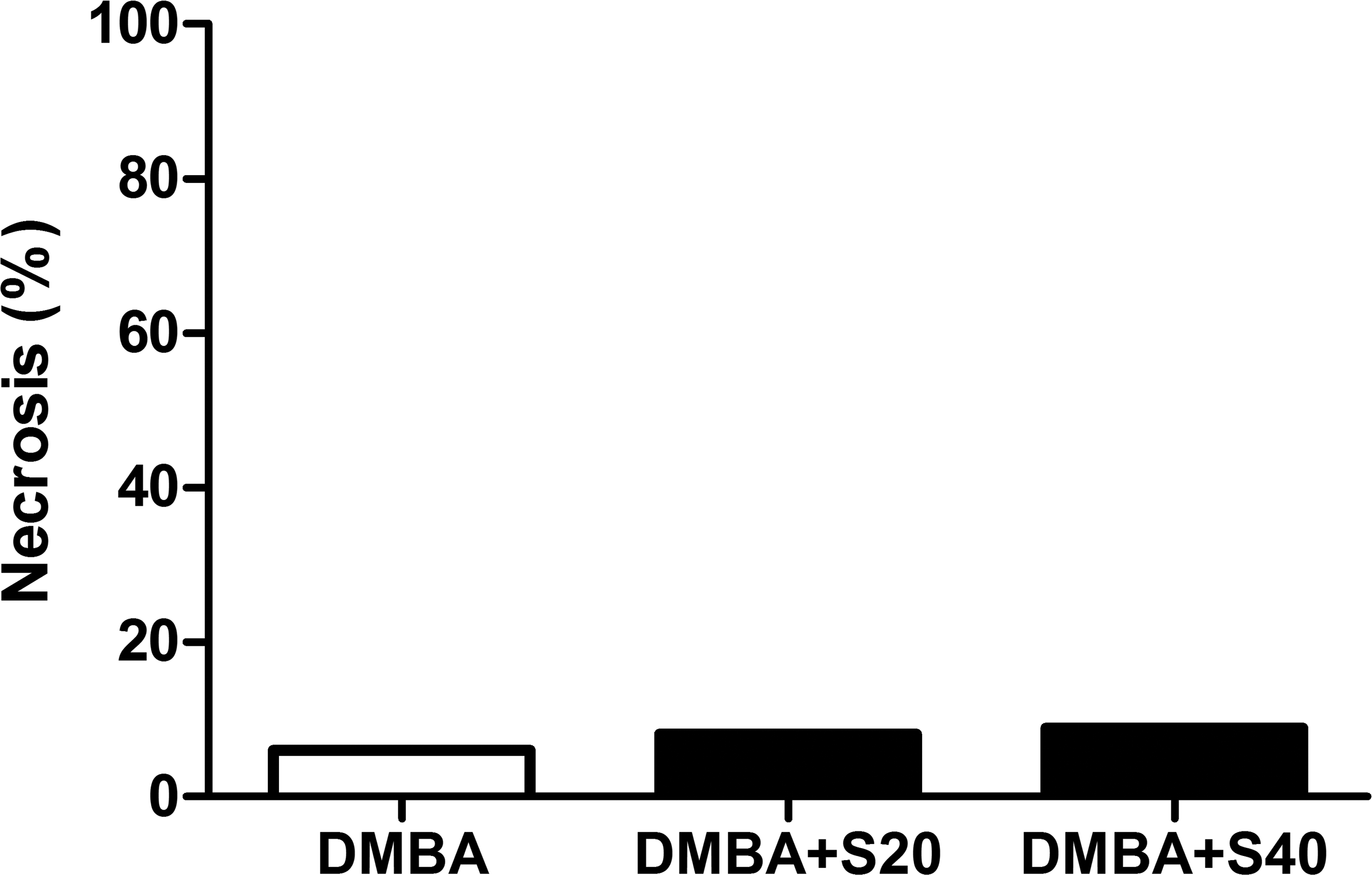
Mean percentage of necrosis. Mean percentage of necrotic area observed in DMBA, DMBA+S20, and DMBA+S40 female Sprague-Dawley rats. No significant differences were seen between groups (p = .6530; analysis of variance [ANOVA]).
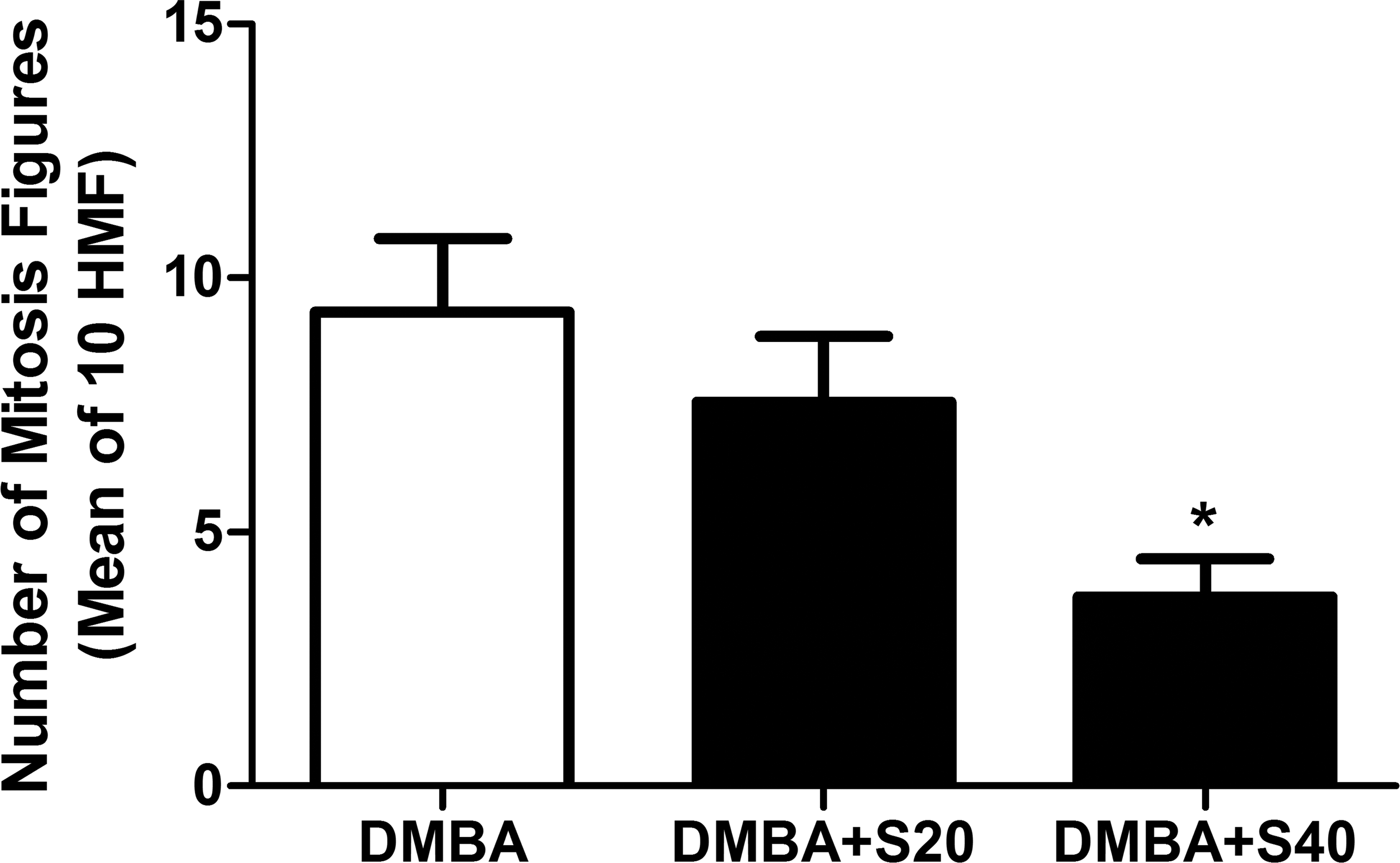
Number of mitotic figures. Number of mitosis in 10 high power fields observed in DMBA, DMBA+S20, and DMBA+S40 groups. The results represent the mean ± SEM of the observations. p = .0333. *p < .05 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
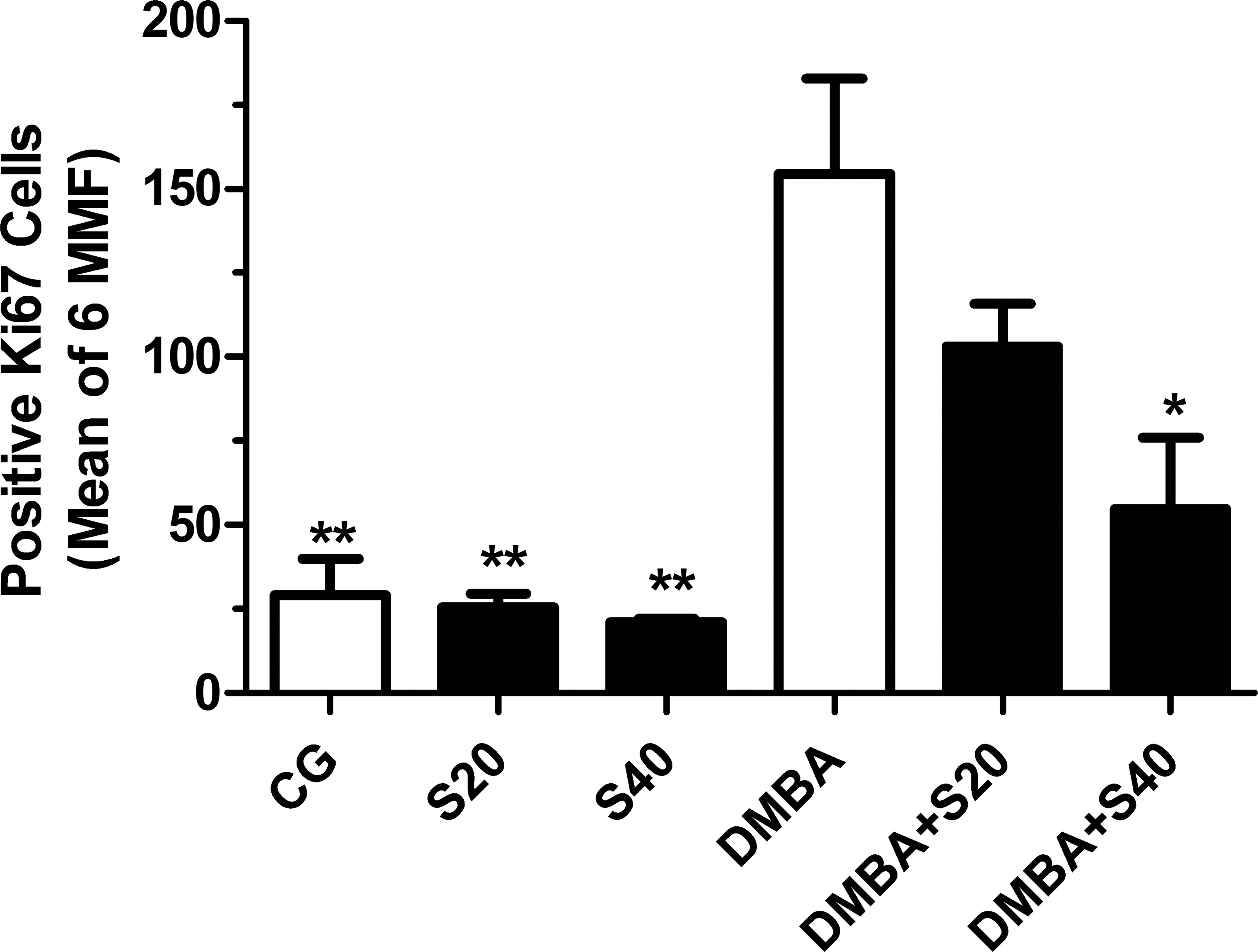
Proliferation index by Ki67. Number of Ki67+ cells in CG, S20, S40, DMBA, DMBA+S20, and DMBA+S40 groups. The results represent the mean ± SEM of the measures. *p < .05 and **p < .01 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
The results for stem cell-like markers (CD133, CD34, CD24, and CD44) are presented in Figures 8 through 12. As shown, the frequency of cells expressing progenitor/stem cell markers was significantly elevated in DMBA tumors when compared to normal breast tissue. In normal breast tissue samples, the expression of progenitor/stem cell markers (number of positive cells) was not significantly affected by simvastatin, regardless of dosage (there was no significant difference between treated and untreated normal breast tissue, in terms of stem cell markers expression). On the other hand, treatment of tumor-bearing rats with high-dose simvastatin (40 mg/Kg) was associated with a significant decrease in the frequency of CD133+ (62.2 cells/image), CD24+ (19.9 cells/image), and CD44+ cells (8.7 cells/image), in comparison to untreated DMBA tumor-bearing rats (CD133 = 131.65 cells/image; CD24 = 51.61 cells/image; CD44 = 19.68 cells/image) and low-dose groups (CD133 = 89.22 cells/image; CD24 = 33.95 cells/image; CD44 = 10.1 cells/image). Treatment with simvastatin, in either dose regimen, did not affect CD34+ tumor cell counting.
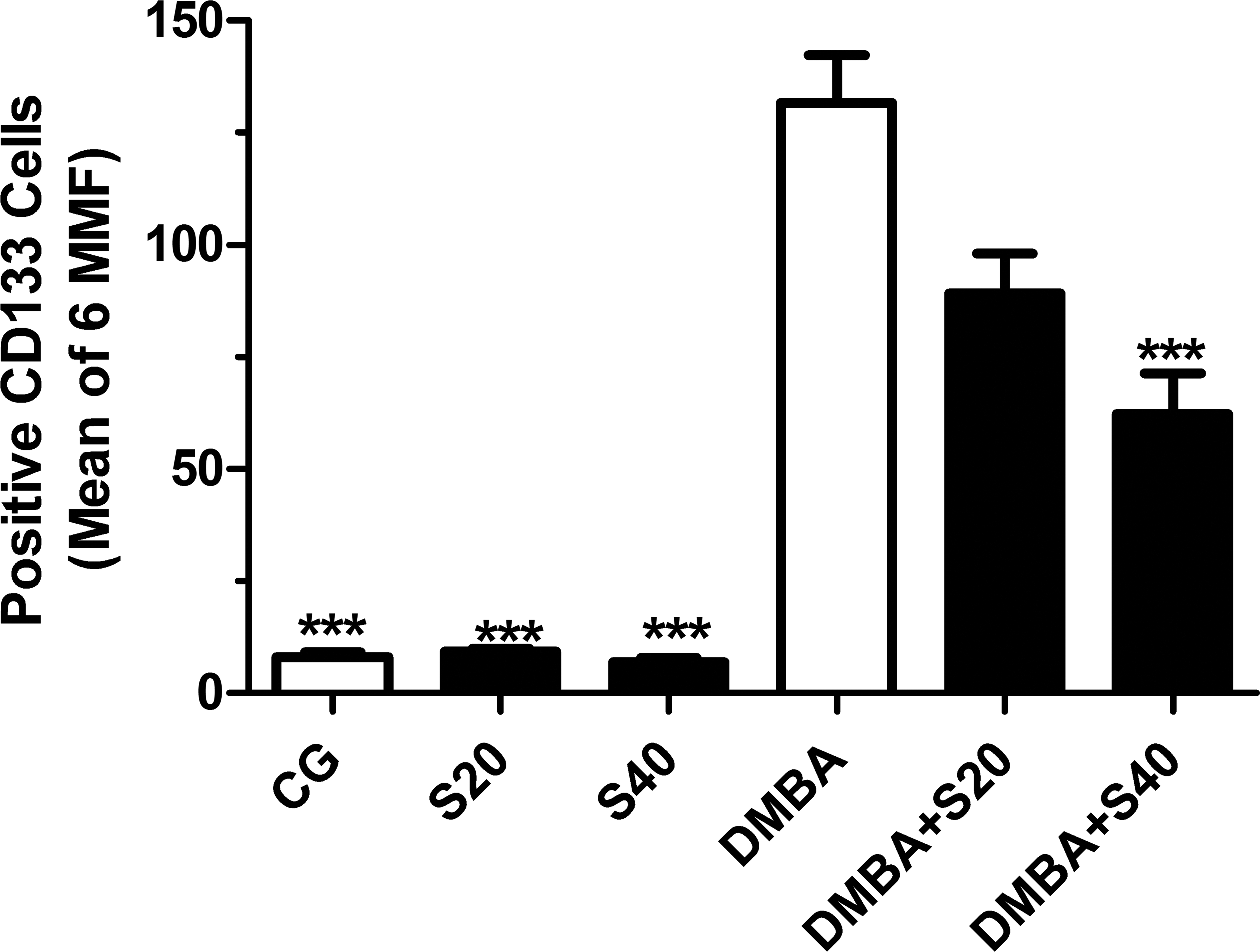
Frequency of CD133+ cells. Number of CD133+ cells in CG, S20, S40, DMBA, DMBA+S20, and DMBA+S40 groups. The results represent the mean ± SEM of the measures. ***p < .001 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
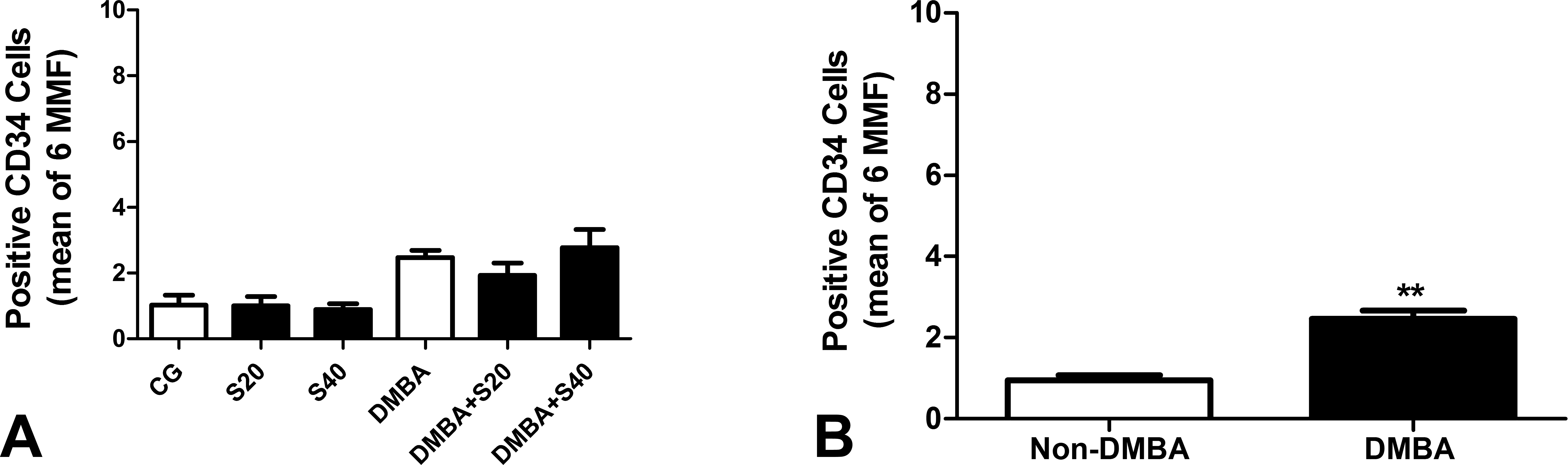
Frequency of CD34+ cells. Number of CD34+ cells in CG, S20, S40, DMBA, DMBA+S20, and DMBA+S40 groups (n = 10/group). The results represent the mean ± SEM of the measures. In (A), there was no significant difference between groups when one compares all six of them (analysis of variance [ANOVA]). In (B), there is a small but significant (**p < .01) difference in terms of CD34 positivity when non-DMBA animals are grouped and compared to DMBA-exposed animals altogether (Student’s t test).
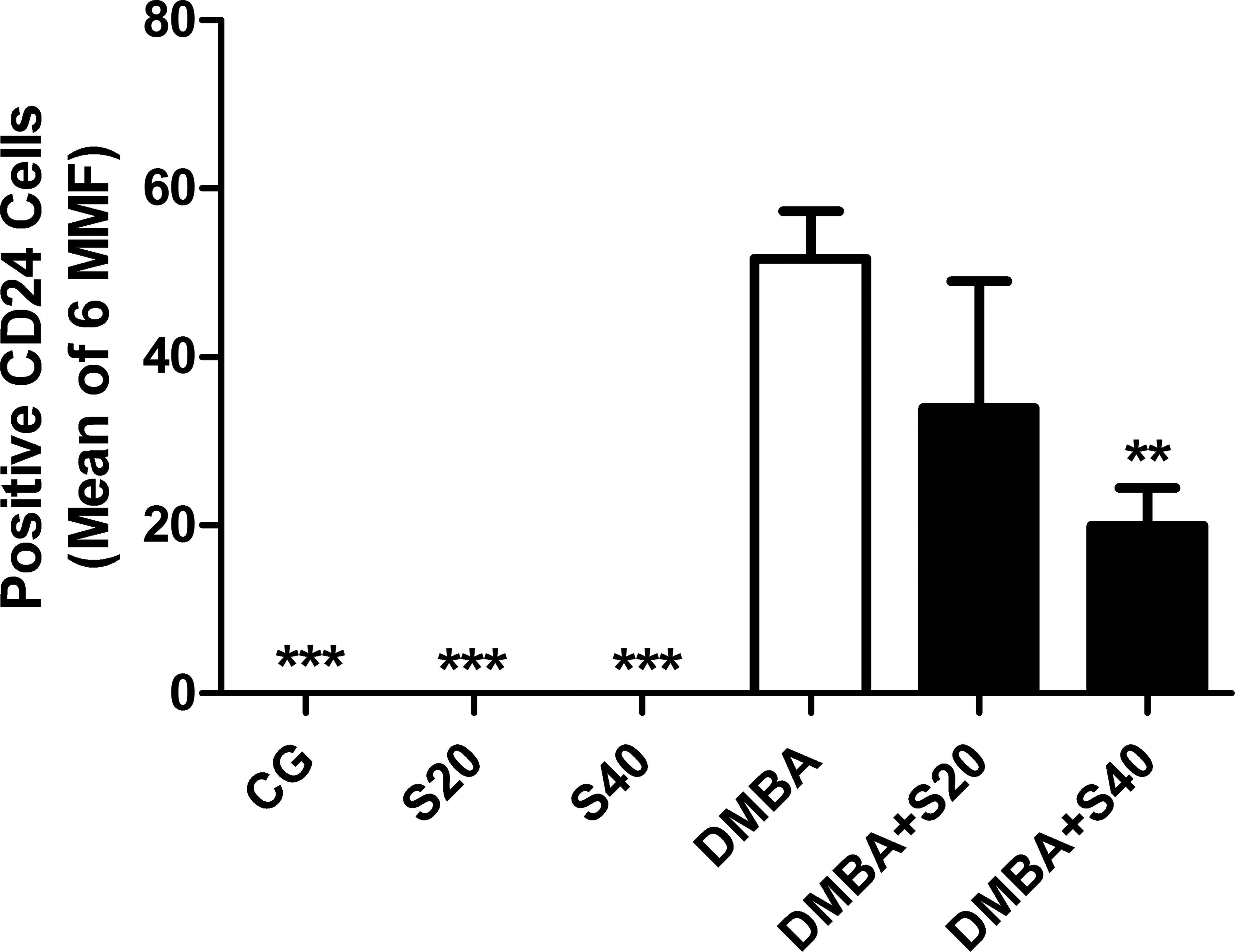
Frequency of CD24+ cells. Number of CD24+ cells in CG, S20, S40, DMBA, DMBA+S20, and DMBA+S40 groups. The results represent the mean ± SEM of the measures. **p < .01, ***p < .001 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
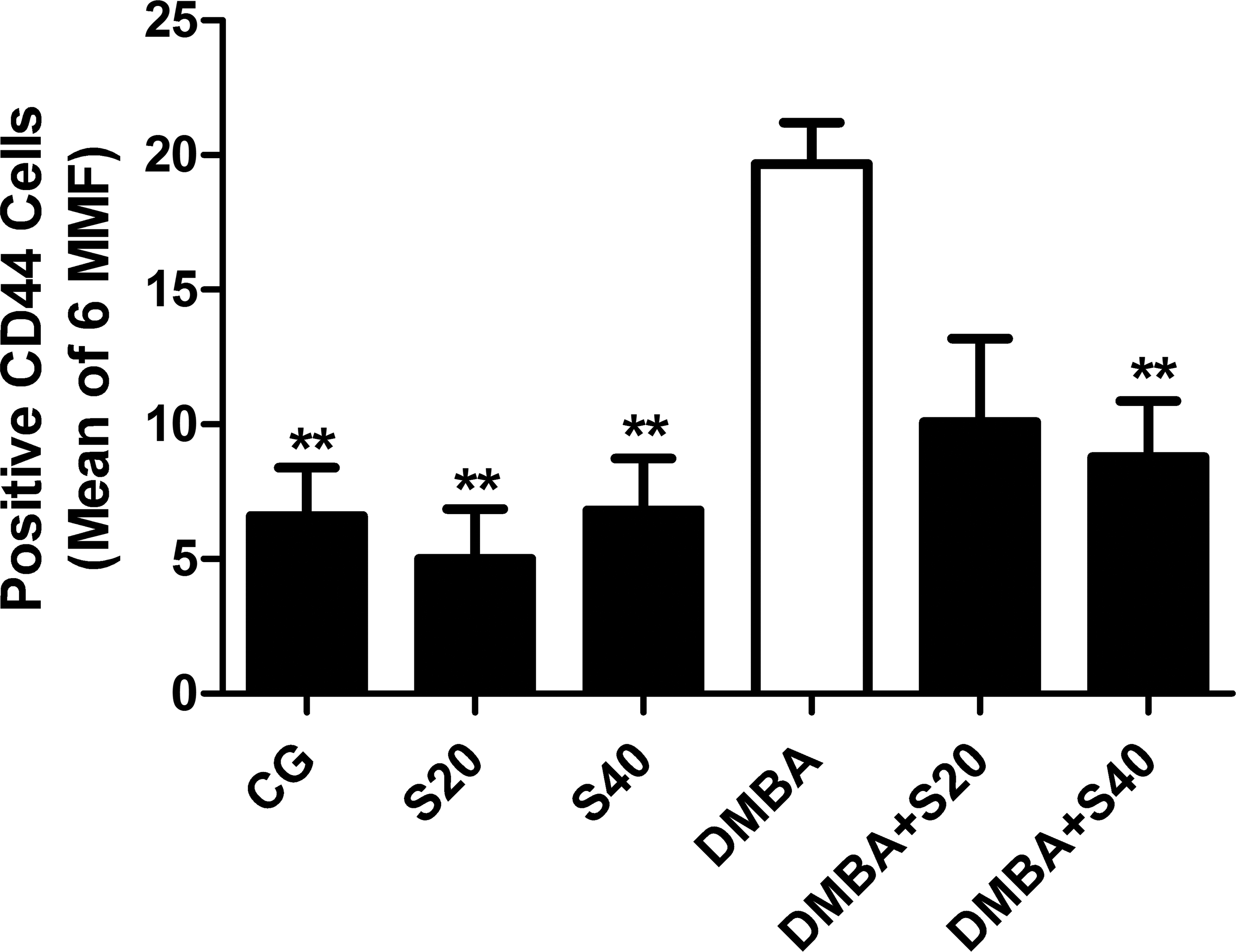
Frequency of CD44+ cells. Number of CD44+ cells in CG, S20, S40, DMBA, DMBA+S20, and DMBA+S40 groups. The results represent the mean ± SEM of the measures. **p < .01 when compared to DMBA group (analysis of variance [ANOVA] followed by Bonferroni test).
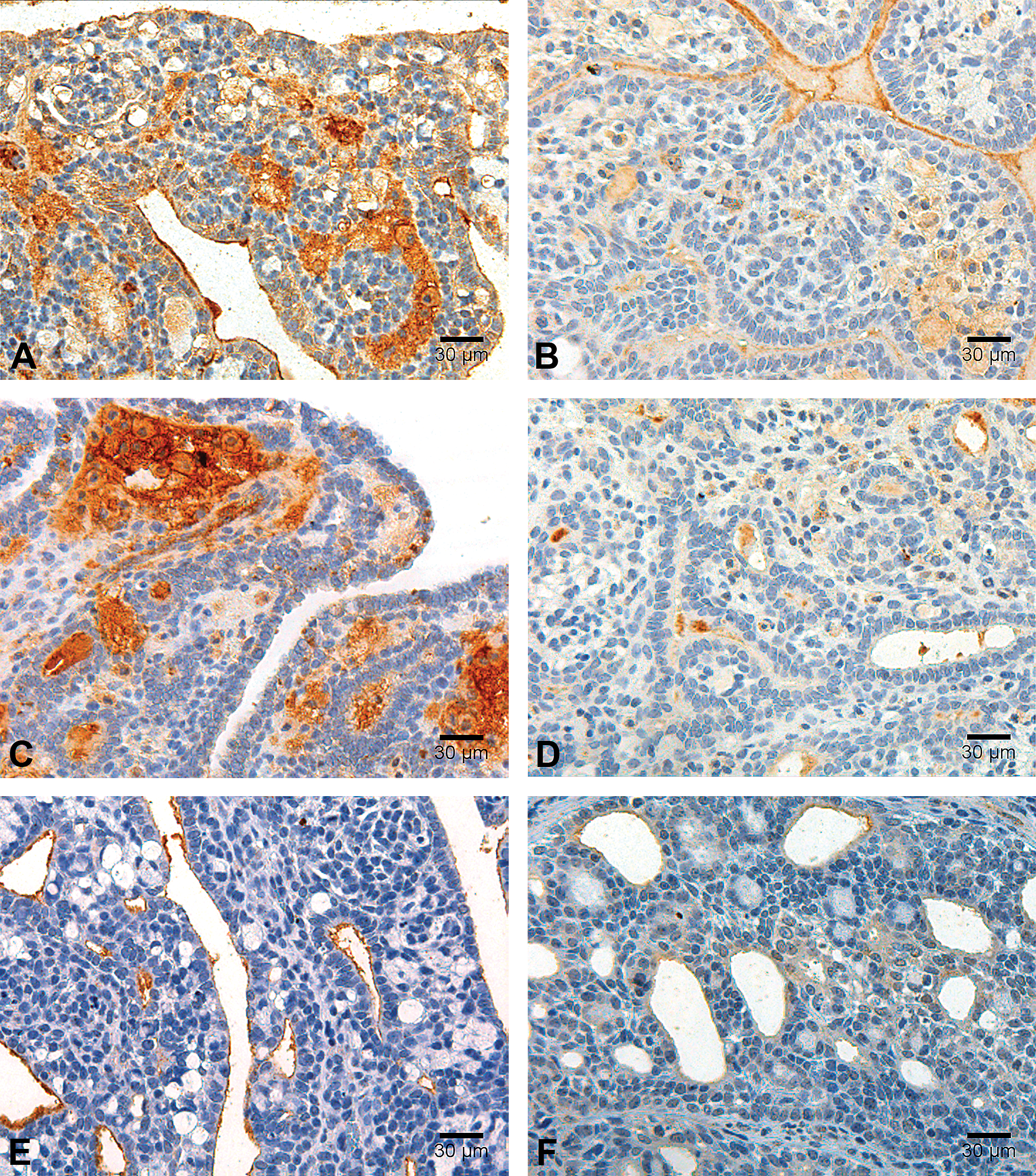
Immunoexpression of cancer stem-cell markers. Photomicrographs illustrating immunoexpression of CD24 (A and B), CD44 (C and D), and CD133 (E and F) among treated (right side column: B, D, and F) and nontreated tumors (left side column: a, c, and e). Immunoperoxidase, scale bar = 30 µm.
The relationship between tested biological variables (percentage of invasive ductal carcinoma component, histological grade, percentage of necrosis, and mitotic index) and the mean frequency of positivity for different stem-cell marker cells is shown in Table 2 and Figures 13 and 14. Briefly, in the present model, only CD133 was significantly correlated to the biological variables herein considered. CD133 was positively correlated to total tumor volume and inversely correlated to the percentage of invasive ductal carcinoma.
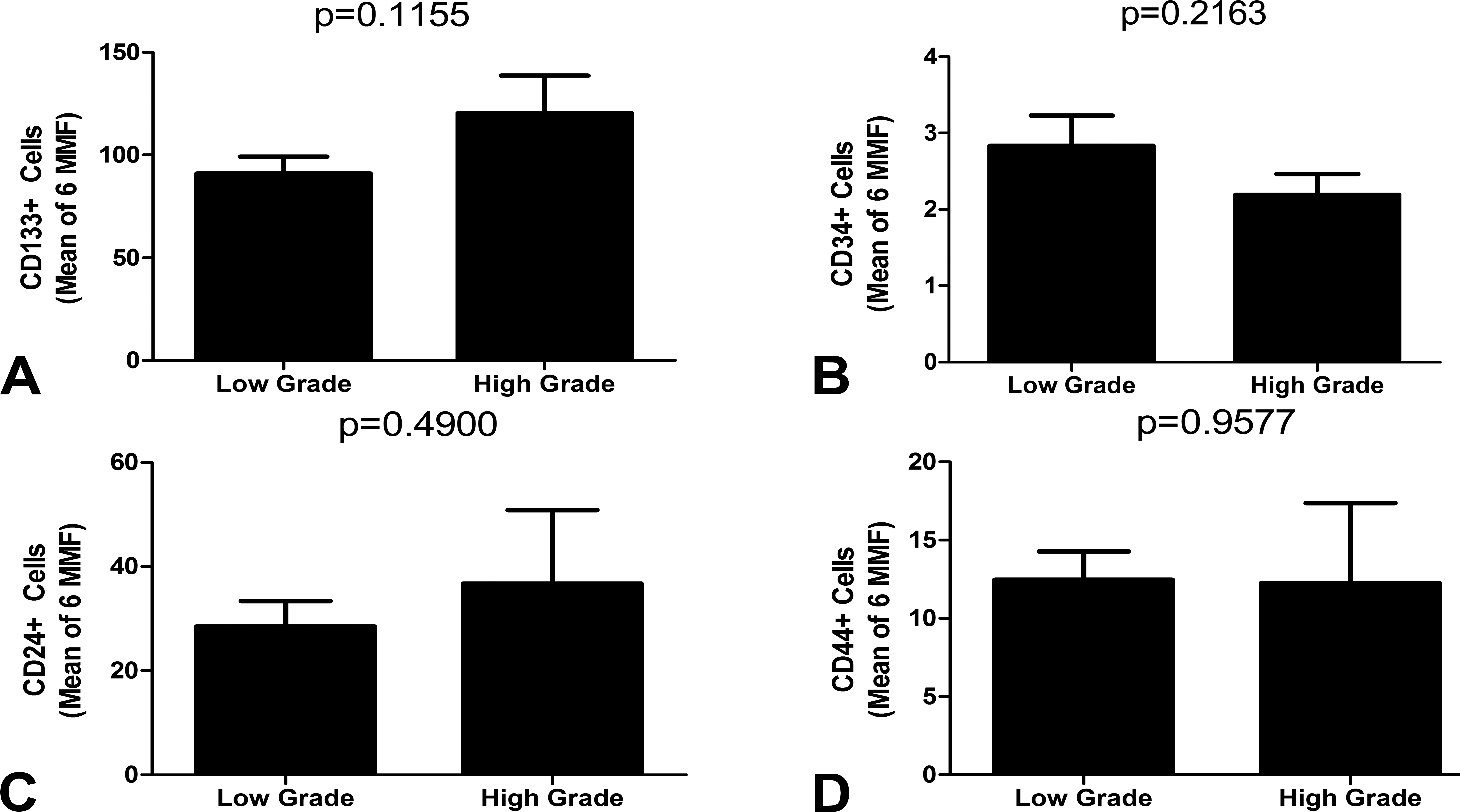
Number of positive cells and final histological grade. The relationship between number of cells expressing CD133 (A), CD34 (B), CD24 (C), and CD44 (D) and the final histological grade (by the Nottingham scoring system) in groups of tumor bearing female Sprague-Dawley rats. The results represent the mean ± SEM of the measures.
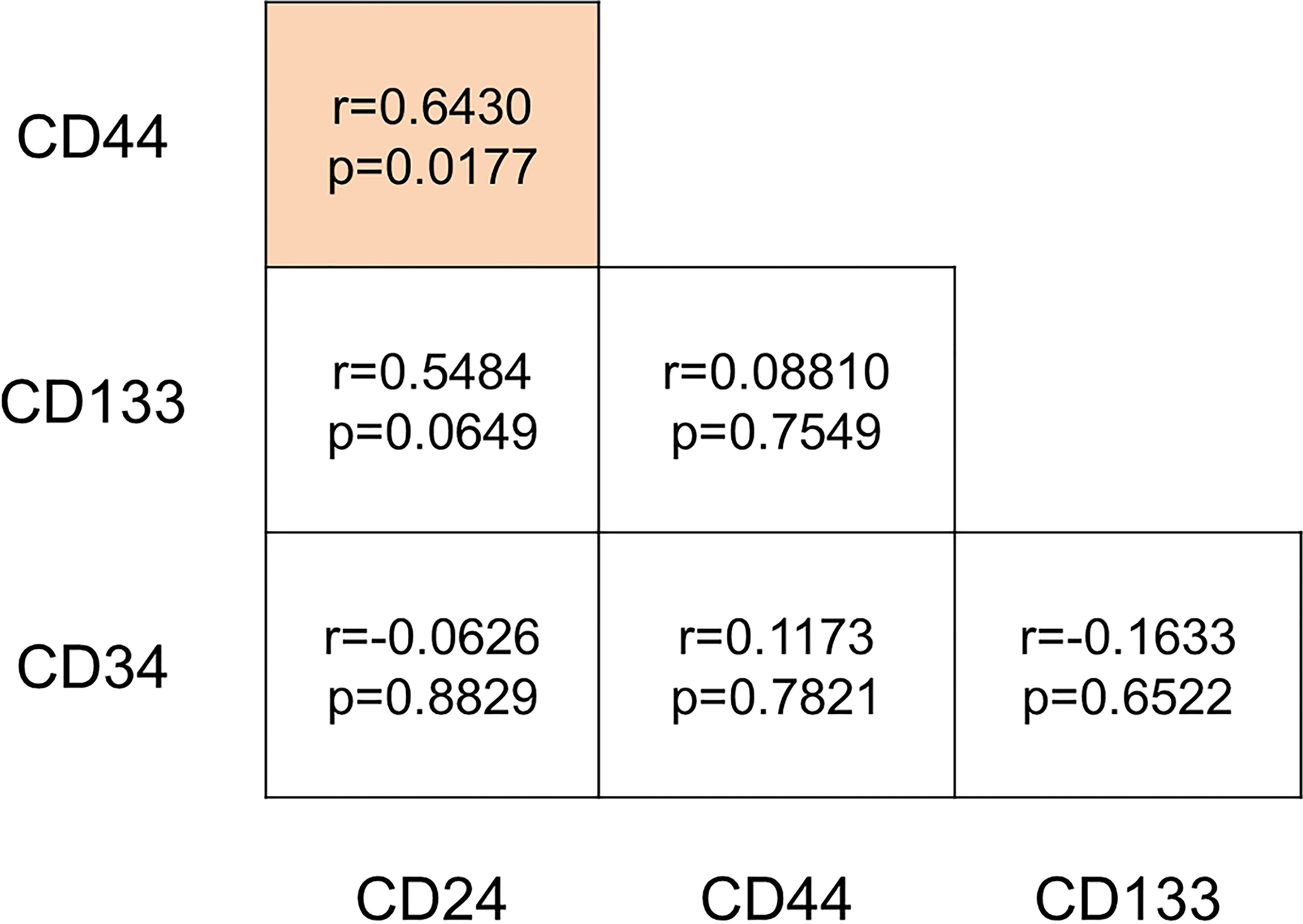
Pairwise correlation between cancer stem cell markers. As can be seen, only CD44 and CD24 were significantly and positively correlated (although the degree of correlation is moderate to low). The analysis includes all experimental groups (N = 60).
Correlation between number of positive cells and various biological variables (tumor burden, percentage of invasive ductal carcinoma component, percentage of necrosis, and mitotic index).

Note: CSC = cancer stem cell.
As for the association between SC markers, a moderate and yet significant correlation r = .6430, p = .0177) was found between CD24 and CD44.
Discussion
Data provided by our experiments clearly demonstrated an increase of CD133, CD34, CD24, and CD44 biomarkers in DMBA-induced breast tumors, when compared to control mammary tissue. This finding supports the adequacy of this protocol as a model for pathophysiology and drug testing studies focused on CSCs, progenitor cells, and CSC-like cells. DMBA is one of the best characterized models in breast cancer research, and this is the first time that this particular model was applied to the study CSCs as potential pharmacological targets.
It is important to mention that the range of dosage used in this study was established in accordance with the literature (Butterweck et al. 2009) and preliminary studies performed by the authors (data not shown). In addition, as evidence for the safety of the drug, one should notice that CD133+, CD34+, CD24+, and CD44+ cells of normal breast tissue were not significantly affected in any of the subjects with the chronic treatment with simvastatin.
Few studies have discussed the in vitro antineoplastic effects of statins, particularly of the lipophilic type (Gabrys et al. 2008; Kang, Kim, and Moon 2009; Kochuparambil et al. 2011; Wu et al. 2009). Two of the most recent and influential studies agree in that simvastatin had significant antineoplastic effects on breast cancer models. Kubatka et al. (2011) have reported a significant reduction in the number of animals developing breast cancer when a prophylactic treatment with simvastatin or atorvastatin is applied. Similarly, Lubet et al. (2009) have described a significant reduction in mammary carcinogenesis among rats treated with atorvastatin or lovastatin. In accordance with these studies, our experiments demonstrate that chronic treatment with simvastatin can prevent both an increase in tumor volume and in the number of tumors per animal. Nevertheless, it is important to emphasize that all previous studies have dealt with the concept of chemoprevention (i.e., simvastatin was given before and during chemical induction of breast neoplasia). In other words, these studies show that simvastatin has a potential role as a primary chemoprophylactic agent. In this study, however, simvastatin was given after the malignant tumors had been established. As a consequence, our results suggest for the first time that simvastatin may also be used as a true chemotherapeutic agent (either in adjuvant or in neoadjuvant protocols).
In brief, we demonstrate herein that a chronic treatment with simvastatin can not only inhibit tumor growth/regeneration (in a dose-dependent manner), but may also modulate neoplastic morphologic heterogeneity, 2 of the most consistent features of cancer stem cell role in malignant neoplasms. Taken alone, these findings are already highly indicative of an anti-CSC effect, but they are not the only evidence provided by this study. This suspicion was further reinforced when we found that simvastatin was able to significantly decrease the immunoexpression of cancer stem cell markers at the highest tested dose. This reduction in immunoexpression was documented for all SC markers, with the exception of CD34. Hence, taken altogether, these data demonstrate for the first time in an in vivo model of breast carcinogenesis the inhibitory of simvastatin over CSCs.
It is important to notice that simvastatin seems to inhibit cancerous but not normal cells expressing SC markers. Among cancer cells, however, this inhibitory effect does not seem to favor specifically either CSCs or more differentiated neoplastic cells: the lack of tumor increment in treated animals cannot be explained by a simple reduction in CSCs since they do not account for more than 10% of the neoplastic cell population within the tumors examined in this study. In addition, finding that CSCs constitute a minority subpopulation in most malignant tumors is not an exclusive feature of this study or of DMBA model, but a common finding in studies that have shaped CSC theory (Meacham and Morrison 2013). From a clinical perspective, the evidence that simvastatin negatively affects both differentiated neoplastic cell and CSCs further increases its potential role in the pharmacological treatment of breast cancer. It shows that simvastatin may not only reduce the cellular bulk of the tumor (often constituted by more differentiated cells) but also the small subpopulation of treatment resistant cells known as CSCs.
Concerning putative mechanisms for simvastatin action, the reduction in tumor increment is likely due to a direct effect on cell proliferation rather than cell death since simvastatin significantly reduced mitotic and Ki67 indexes while not altering the percentage of necrosis. This hypothesis awaits, however, further confirmatory studies addressing other forms of cell death (e.g., apoptosis). These experiments are currently being executed in our laboratories.
Most of the analyzed antineoplastic/anti-CSC effects were more clearly demonstrated at the highest dose tested, thus suggesting they are dose-dependent. The same dose was able to reduce most of the cells expressing SC markers, such as CD133, CD24, and CD44. CD133, also called prominin-1, is a 5 transmembrane domain molecule known to be expressed not only in the cytoplasm but also on the cell membranes (Fan et al. 2011; Immervoll et al. 2008; Qun et al. 2009). CD133 expression is commonly described as a biomarker for a subgroup of breast stem/progenitor cells (Keysar and Jimeno 2010; Qun et al. 2009; Wright et al. 2008), although its function has not yet been fully established (Wright et al. 2008). Positive CD133 cells have the capacity of self-renewal, by means of asymmetric division (Wright et al. 2008). Several studies have reported that the levels of CD133 in various tumors are negatively correlated with patient survival, tumor stage, and tumor size (Fan et al. 2011; Qun et al. 2009). Our results show that the number of CD133+ cells is positively correlated to tumor volume and inversely correlated to the percentage of the ductal component. These correlations may indicate that, in this model, CD133 could have a role in tumor proliferation and cell differentiation, that is, favoring tumor growth and histologic heterogeneity.
Of notice, none of the other markers followed CD133 behavior, thus confirming the idea that these SC associated molecules may actually identify different types of subpopulations of CSC, and further suggesting that the CD133+ subpopulation could be more relevant for these experimental tumors than the other CSCs, at least from a pathophysiological point of view. Nevertheless, from a pharmacological point of view, as far as simvastatin is concerned, this hypothesis may actually be irrelevant once this drug seems to equally affect the expression of the majority of CSC markers herein tested. So regardless of the relative importance of the different CSC subtypes detected in this model, high-dose simvastatin seems to cover them all. This observation is strategically very important in therapeutical terms. It anticipates that the emergence of a resistant clone (i.e., derived from a resistant type of CSC) with the chronic use of simvastatin is less likely. It should be noticed, however, that when using simvastatin as an antineoplastic agent, a potential source of therapeutic resistance could be represented by CD34+CSCs, since these cells have not been significantly affected in number by simvastatin, in this study.
The mechanisms involved in the antiproliferative and anti-CSC actions of simvastatin are not fully understood. Several lines of evidence indicate that an inhibitory effect on the production of isoprenoids may be a major event in these antineoplastic actions (Gauthaman, Manasi, and Bongso 2009; Lee et al 2001). By inhibiting HMG-CoA reductase activity, simvastatin is capable of blocking not only cholesterol synthesis, but also the production of isoprenoids, such as farnesyl and geranylgeranyl. Isoprenylation, which is the transfer of a farnesyl or geranylgeranyl moiety to proteins, is an important step in the attachment of small GTPases (such as Ras and Rho) to the plasma membrane and their subsequent actions in signal transduction pathways relevant to cancer, such as the phosphoinositide 3-kinase/Akt and Ras/MEK/ERK pathways (Steelman et al. 2008). Perhaps the most convincing data supporting this theory are recent observations associating downstream inhibition of isoprenoids by statins and decreased cell growth in squamous cell carcinoma (Dimitroulakos et al. 2001), prostate cancer (Moyad and Merrick 2005), breast cancer (Gauthaman, Manasi, and Bongso 2009), and acute myeloid leukemia (van der Weide et al. 2012). It should be noticed, however, that most of these studies are based on in vitro data. Therefore, the exact mechanisms involved in the anti-CSC effects of simvastatin await in vivo validation and further development on the specific intracellular signaling pathways that are affected by the lack of isoprenoids.
In summary, this is the first study to confirm the inhibitory effect of simvastatin over CSCs in vivo. Although this effect is specific for neoplastic cells (simvastatin does not affect normal SCs), it is not specific for CSC, which may be considered an actual advantage from the therapeutical point of view (i.e., the drug is active both on CSCs and more mature cancer cells). Finally, this effect, which is probably more related to cell proliferation inhibition than to the promotion of cell death, does not seem to favor any specific subtype of CSC, which means a lesser chance for the emergence of CSC-derived resistant clones and consequently for the development of therapeutical failure.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo [Fapesp; #2010/10703-0], Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundo de Apoio ao Ensino à Pesquisa e à Extensão (FAEPEX).