
Abstract
Glucokinase activators (GKAs) are being developed for the treatment of type 2 diabetes. The toxicity of 4 GKAs (PF-04279405, PF-04651887, piragliatin, and PF-04937319) was assessed in mice, rats, dogs, and/or monkeys. GKAs were administered for 2 to 8 weeks. Standard endpoints, glucose, and insulin were assessed. All compounds produced varying degrees of hypoglycemia in all species. Brain neuronal necrosis and/or peripheral neuropathy were observed with most compounds. These findings are consistent with literature reports linking hypoglycemia with nervous system effects. Arteriopathy, mainly of cardiac vessels, was observed at a low frequency in monkey and/or dog. Arteriopathy occurred only at doses that produced severe and prolonged periods of repeated hypoglycemia. Since this lesion occurred in multiple studies with structurally distinct GKAs, these results suggested arteriopathy was related to GKA pharmacology. The morphological characteristics of the arteriopathy were consistent with that produced by experimental catecholamine administration. We hypothesize that the prolonged periods of hypoglycemia resulted in increased local and/or systemic concentrations of catecholamines via a counterregulatory and/or stress-related mechanism. Alternatively, prolonged hypoglycemia may have resulted in endothelial dysfunction leading to arteriopathy. This risk can be managed in human patients in clinical studies by careful glucose monitoring and intervention to avoid prolonged episodes of hypoglycemia.
Keywords
Introduction
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public epidemic affecting over 343 million people worldwide (World Health Organization 2014). This disease is characterized by elevated fasting plasma glucose, insulin resistance, abnormally elevated hepatic glucose production, and reduced glucose-stimulated insulin secretion (Pfefferkorn et al. 2012). The standard of care for T2DM pharmacotherapy is metformin, followed by sulfonylureas, dipeptidyl peptidase-4 inhibitors, and thiazolidenediones as second line oral therapies (Pfefferkorn et al. 2012). As disease progression continues, patients typically require injectable agents such as glucagon-like peptide-1 analogs and insulin (Pfefferkorn et al.). Even with these treatment options, many patients fail to adequately maintain glycemic control placing them at risk for diabetic complications. Thus, there continues to be a need for new therapies with improved efficacy and safety to help patients achieve their treatment goals (Coghlan and Leighton 2008).
Glucokinase activators (GKAs) offer a promising new opportunity for the treatment of T2DM patients. Glucokinase (GK) is the enzyme responsible for the conversion of glucose to glucose-6-phosphate, and it functions as a key regulator of glucose homeostasis. In the liver, GK regulates hepatic glucose utilization and output whereas in the pancreas it functions as a glucostat establishing the threshold for beta-cell glucose stimulated insulin secretion (Cardenas 1995). GK is also found in glucose sensing neurons in the ventromedial hypothalamus where it regulates the counterregulatory response to hypoglycemia (Dunn-Meynell et al. 2002).
Initially, we identified PF-04279405 as a potent (EC50 = 23 nM), selective, and full GK agonist (Figure 1A and Table 1). Since PF-04279405 exhibited metabolic instability in both rat and dog, toxicological evaluations were conducted in species with better metabolic stability (i.e., mouse and cynomolgus monkey). The choice of cynomolgus monkey as the nonrodent large animal species for toxicological evaluation significantly impacted the nonclinical toxicology development strategy for subsequent GKAs. In cynomolgus monkeys, GKAs produced profound and sustained reductions in serum glucose usually in the absence of clinical signs of hypoglycemia. Mild cardiac arteriopathy (fibrinoid necrosis and /or intramural hemorrhage) was observed in high-dose female monkeys administered PF-04279405. At the time this observation was made, it was unclear whether this finding resulted from chemotype-related (i.e., related to the chemical structure) toxicity or from hypoglycemia due to exaggerated pharmacology. To answer this question, additional GKAs with similar and diverse chemical structures (Figure 1B, C, D, and Table 1) were studied in cynomolgus monkeys, beagle dogs, and rats. We show that arterial and cardiac changes are tightly associated with profound hypoglycemia, a phenomenon that is now suspected in humans experiencing iatrogenic hypoglycemia (Chow and Heller 2012). Several pathophysiological factors may be responsible for these arterial and cardiac changes and should be further investigated.
Chemical structures for (A) PF-04279405, (B) PF-04651887, (C) piragliatin, and (D) PF-04937319.
Biochemical activities of glucokinase (GK) activators (GKAs).a

Note. aThe intrinsic potency (EC50) of GKAs for the human, monkey, rat, and dog GK enzyme was determined using purified recombinant enzymes. The EC50 is reported as the concentration of activator that elicits the half-maximum change in glucose Km of glucokinase.
Material and Methods
Animals and Husbandry
CD-1 (Crl:CD-1[ICR]BR) mice (6–8 weeks of age) and Sprague-Dawley rats (Crl:CD®(SD) IGS BR; 10 weeks of age) were obtained from Charles River Breeding Laboratories (Kingston, NY). Beagle dogs (8–12 months of age) were obtained from Marshall Bioresources (North Rose, NY) and cynomolgus monkeys (>3 years of age, 2–6 kg) were obtained from Biomedical Resources Foundation/Charles River Laboratories (Houston, TX). Animals were randomly assigned to treatment groups. The animal room environment was controlled (21 ± 3°C, humidity 50 ± 10%, 12 hr light/dark). Animals received appropriate certified laboratory diets; monkey diets were supplemented with vegetables and/or fruit. The protocols and any amendments or procedures involving the care and use of animals in this study were reviewed and approved by Pfizer Global Research & Development Institutional Animal Care and Use Committee prior to study conduct. The animal care and experimental procedures of this study were conducted in compliance with the US Animal Welfare Act and the ILAR Guide.
Test Substances
PF-04279405, PF-04651887, piragliatin, and PF-04937319 were synthesized (>96% purity) and supplied by Pfizer Worldwide Research & Development as previously described (Pfefferkorn et al. 2011; Sarabu et al. 2012). To enhance its biopharmaceutical properties, PF-04279405 was prepared as a spray-dried suspension using hydroxypropyl methyl cellulose acetate succinate (25.9% active moiety). The spray-dried material was suspended in 0.5% methylcellulose. Piragliatin was prepared as a suspension in 0.5% methylcellulose. PF-04651887 was prepared as a suspension in 1.25% hydroxypropyl cellulose and 0.5% docusate sodium. PF-04937319 was prepared as a nanoparticle suspension in 1.25% hydroxypropyl cellulose and 0.5% docusate sodium. Control animals received a matching vehicle. Dose volumes were 10 ml/kg for rats and mice and 5 ml/kg for dogs and monkeys. All compounds were administered by oral gavage once daily except as noted.
Experimental Design, Observations, and Measurements
Observations in all studies included clinical signs, body weights, food consumption, standard hematology, coagulation, clinical chemistry, and urinalysis, toxicokinetics, electrocardiogram (monkey and dog), complete necropsy, organ weights, and microscopic tissue examination. Studies designated as exploratory toxicity studies utilized smaller numbers of animals and an abbreviated tissue collection and evaluation list relative to full toxicity studies.
PF-04279405
The nonclinical safety was assessed in mouse and monkey because PF-04279405 was metabolically stable in these species.
Mouse study
Male and female CD-1 mice (10/sex/dose) received once daily oral gavage doses of PF-04279405 at 3, 30, or 300 mg/kg for 1 month. Toxicokinetic parameters were assessed (18/sex/group for controls and 24/sex/group for treated mice) on days 1 and 30 (0.5, 2, 5, and 24 hr postdose) in satellite mice.
Monkey study
Male and female cynomolgus monkeys (3/sex/dose) received once daily oral gavage doses of PF-04279405 at 1, 3, or 10 mg/kg for 1 month. Toxicokinetic parameters were assessed on days 1 and 30 (1, 4, 7, and 24 hr postdose). In addition, time courses for plasma glucose and insulin were evaluated during weeks 2 and 4 at the same time points used for the toxicokinetic assessments.
PF-04651887
The nonclinical safety was evaluated in rats, dogs, and monkeys because PF-04651887 was metabolically stable in these species thus enabling comparison of the pharmacodyanmic and toxicologic profile of this GKA across all 3 species.
Rat study
In this exploratory toxicity study, male and female Sprague-Dawley rats (5/sex/dose) received once daily oral gavage doses of PF-04651887 at 5, 50, or 500 mg/kg for 14 days. Toxicokinetic parameters were assessed on days 1 and 14 (0.5, 2, 8, and 24 hr postdose). Additional observations included plasma glucose and insulin time courses at the same time points as the toxicokinetic assessments.
Monkey study
In this exploratory toxicity study, cynomolgus monkeys (2 females/dose) received once daily oral gavage doses of PF-04651887 at 3, 30, or 300/150 mg/kg (dose lowered on day 25 due to adverse clinical signs) for 1 month. Female monkeys were selected because, in previous studies, cardiac arteriopathy was noted only in female monkeys administered PF-04279405. Toxicokinetic parameters were assessed on days 1 and 27 (0.5, 1, 4, 7, and 24 hr postdose). Additional observations included plasma glucose and insulin time courses at the same time points as the toxicokinetic assessments.
Dog study
In this exploratory toxicity study, beagle dogs (3 females/dose) received once daily oral gavage doses of PF-04651887 at 5, 50, or 500 mg/kg for 1 month. Toxicokinetic parameters were assessed on days 1, 7, and 24 (0.5, 1, 4, 7, and 24 hr postdose). Additional observations included plasma glucose and insulin time. The neurological examinations were conducted pretreatment and on day 22 and consisted of the following measurements: (1) Mental status—overall evaluation of higher brain functions, (2) Gait—overall evaluation of strength and coordination, (3) Postural reactions—localized evaluation of strength and coordination, (4) Cranial nerves—evaluation of brainstem, and Spinal reflexes—segmental spinal cord evaluation.
Piragliatin
Piragliatin is another structurally distinct GKA that was discontinued from development following the phase 2 program due to an unfavorable risk–benefit balance (Sarabu et al. 2012). Due to its relatively advanced clinical development status prior to discontinuation, it was considered to be a good comparator to evaluate in a monkey toxicity study.
In this exploratory toxicity study, piragliatin was administered to groups of 3 females monkeys at 4, 8, and 16 mg/kg for 5 (4 and 16 mg/kg) or 8 (vehicle control and 8 mg/kg) weeks. The goal of the study was to determine whether severe and prolonged hypoglycemia with a structurally distinct GKA could result in arteriopathy similar to that seen with PF-04279405 and PF-04651887. To achieve this level of hypoglycemia, mid study dose-level adjustments were made to the mid-dose group based on interim blood glucose data. Thus, the dose level for animals in the 8 mg/kg/day group was increased to 32 mg/kg/day (16 mg/kg/day twice a day [BID]) on day 29 and increased again to 48 mg/kg/day (24 mg/kg/day BID) on day 41. Toxicokinetic parameters were assessed on days 1, 7, and 24 (0.5, 1, 4, 7, and 24 hr postdose). Serum troponin I was assessed at termination and plasma glucose and insulin time courses were determined at the same time points as the toxicokinetic assessments.
PF-04937319
Rat study
Male and female Sprague-Dawley rats (10/sex/dose toxicity assessment, 4/sex/dose toxicokinetic and glucose time course assessment) received once daily oral gavage doses of PF-04937319 at 10 (females), 50 (males and females), 250 (males), or 1,000 (males and females) mg/kg for 1 month. Different doses were administered to male and female rats to match exposure across gender. Toxicokinetic parameters were assessed on days 1 and 28 (1, 3, 7, and 24 hr postdose). In addition, plasma glucose time course was assessed at the same time points as the toxicokinetic assessments.
Dog study
Male and female beagle dogs (3/sex/dose) received once daily oral gavage doses of PF-04937319 at 5, 50, or 500 mg/kg for 1 month. Toxicokinetic parameters were assessed on days 1 and 30 (1, 4, 7, and 24 hr postdose). In addition, time courses for plasma glucose and insulin were evaluated during weeks 2 and 4 at the same time points as the toxicokinetic assessments.
Clinical Pathology Assessments
Blood samples for hematology, serum chemistry, and plasma glucose determinations were collected by jugular venipuncture from main study animals prior to necropsy. Rats were bled in a fasted state following CO2:O2 anesthesia. Standard hematology parameters were assessed using an Advia 120 automated analyzer (Siemens Diagnostics, Tarrytown, NY). Blood samples taken from the vena cava at necropsy were used to determine prothrombin and activated partial thromboplastin times that were measured using the STA Compact Automated Coagulation System (Stago) analyzer (Diagnostica Stago, Parsippany, NJ). Standard serum clinical chemistry assessments were measured on the Hitachi chemistry analyzer (Roche Diagnostics, Indianapolis, IN) and serum troponin I was measured with the Bayer Advia Centaur analyzer (Siemens Diagnostics, Tarrytown, NY). Rat glucose concentrations presented in Figure 2a and b were measured with the Analox Instruments Fast Glucose Analyzer (Lunenburg, MA). Dog and monkey insulin were measured with the Advia Centaur analyzer (Siemens Diagnostics, Tarrytown, NY) whereas rat insulin was measured by an ELISA method (Molecular Devices, Sunnyvale, CA).
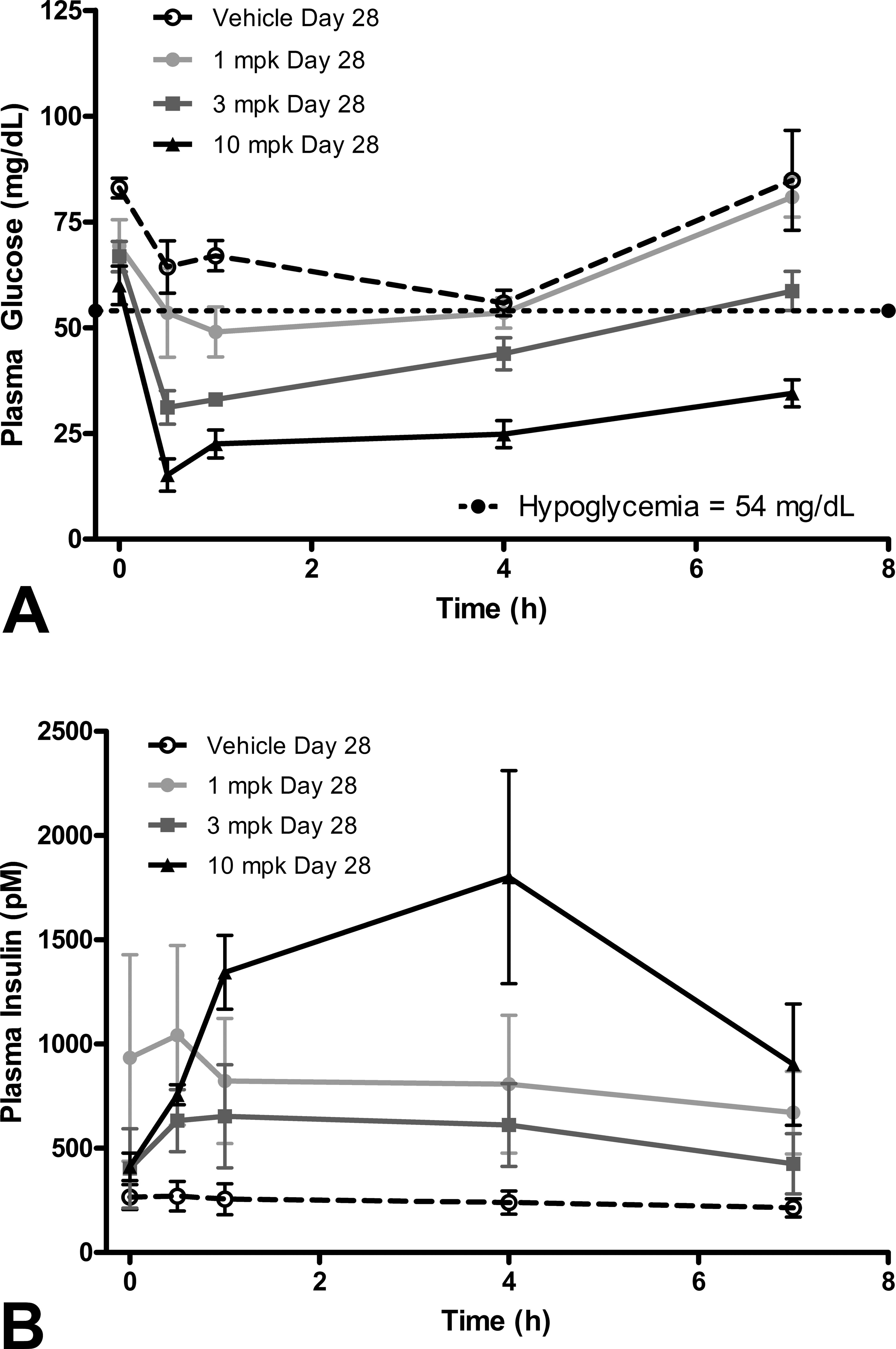
The time course for plasma (A) glucose and (B) insulin in monkeys administered PF-04279405 for 1 month. Each point represents the mean ± standard deviation (N = 3/sex/group, male and female data combined).
Toxicokinetics
Plasma drug concentrations were assayed using HPLC/MS. Toxicokinetic calculations were performed in WinNonlinTM (version 3.2, released June 11, 2001, Pharsight Corporation, St. Louis, MO) and mean data were used in all toxicokinetic analyses. The area under the mean serum concentration–time curve (Area Under the Curve [AUC]0–24 hr) was estimated using the linear trapezoid approximation. C max was defined as the maximum mean serum concentration observed and T max was defined as the time at which mean C max was first observed.
Pathology
Mice and rats were euthanized by isoflurane gas anesthesia followed by exsanguinations. Dogs and monkeys were euthanized by dosing with barbiturate intravenously followed by exsanguinations. Animals were fasted overnight prior to necropsy. Complete necropsies were performed and selected tissues were weighed. For standard studies, a complete set of tissues was collected whereas for exploratory studies an abbreviated set of tissues was collected. Representative samples of collected organs were fixed in 10% buffered formalin except eye (3% glutaraldehyde), optic nerve (3% glutaraldehyde and 10% buffered formalin), and testis and epididymis (Modified Davidson’s). All tissue except larynx and joint were sectioned and stained with hematoxylin and eosin. Tissues were evaluated microscopically. A second pathologist performed a peer review evaluation.
Statistical Analysis
Treated group means were compared with the control group mean. Dunnett’s multiple comparison procedure (Dunnett 1955, 1964) was used if a preliminary Bartlett’s test (Sokal and Rohlf 1969) or F test for homogeneity of variance was not significant at the α = .05 level. If there was significant variance heterogeneity, the Cochran–Cox modified t-test (Cochran and Cox 1957) was used for comparison between treated and control group means. Statistical significance of the comparisons was indicated at both the α = .05 and .01 levels. Tests were two-tailed.
Results
PF-04279405 (Full Agonist)
PF-04279405 was administered to mice at 3, 30, or 300 mg/kg/day for 1 month. There were no adverse treatment-related clinical signs or changes in body weight, food consumption, hematology, or clinical chemistry parameters. Systemic exposure increased with increasing dose (Table 2). Microscopic findings were limited to minimal to mild testicular degeneration at 300 mg/kg, and a very minor increase in the incidence and severity of ellipsoids (digestion chambers), within the sciatic nerve, was noted in females treated at 3 and 300 mg/kg/day (Table 2). Similar sciatic nerve findings have been associated with hypoglycemia (Horvath et al. 2013), particularly in female mice (Tabata 2000).
Summary of systemic plasma exposure values and key findings from PF-04279405, PF-04651887, and PF-04937319 rodent studies.a
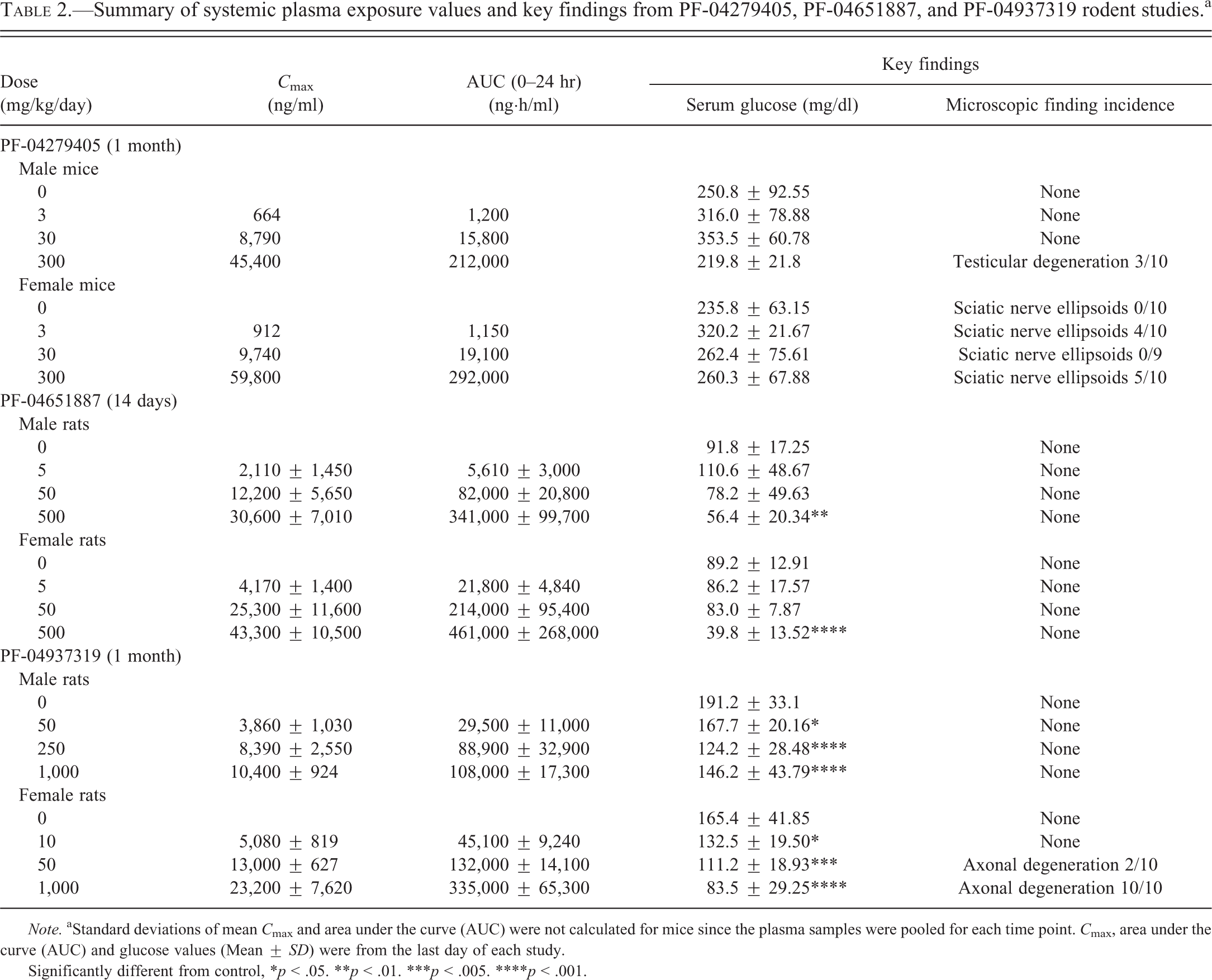
Note. aStandard deviations of mean C max and area under the curve (AUC) were not calculated for mice since the plasma samples were pooled for each time point. C max, area under the curve (AUC) and glucose values (Mean ± SD) were from the last day of each study.
Significantly different from control, *p < .05. **p < .01. ***p < .005. ****p < .001.
Monkeys received 1, 3, or 10 mg/kg/day for 1 month. There were no treatment-related clinical signs or changes in body weight, food consumption, and electrocardiograms. Systemic exposure increased with increasing dose (Table 3). There were no adverse effects on hematology, coagulation, and clinical chemistry parameters. Plasma glucose and insulin time course data on day 28 are included in Figure 2a and b. Glucose was decreased in a dose- and time-dependent manner in all treated animals. During week 4, group mean glucose values dropped to nadirs of 49.0, 28.7, and 15.2 mg/dl at 1, 3, and 10 mg/kg, respectively, at 0.5 to 1 hr following administration of PF-04279405 (Figure 2a). During weeks 2 and 4, predose mean insulin concentrations tended to be increased in PF-04279405-treated animals relative to controls (week 2 mean control 256 pM and PF-04279405-treated group means ranged from 261 to 736 pM; week 4 mean control 266 pM and PF-04279405-treated group means ranged from 404 to 934 pM). Following administration of PF-04279405, insulin was typically increased in a dose- and time-dependent manner in all treated animals. During week 4, group mean insulin values peaked at 1,042, 654, and 1,801 pM at 1, 3, and 10 mg/kg, respectively, while the maximum control value was 270 pM (Figure 2b).
Summary of systemic plasma exposure values and key findings from PF-04279405, PF-04651887, piragliatin, and PF-04937319 monkey and dog studies.a
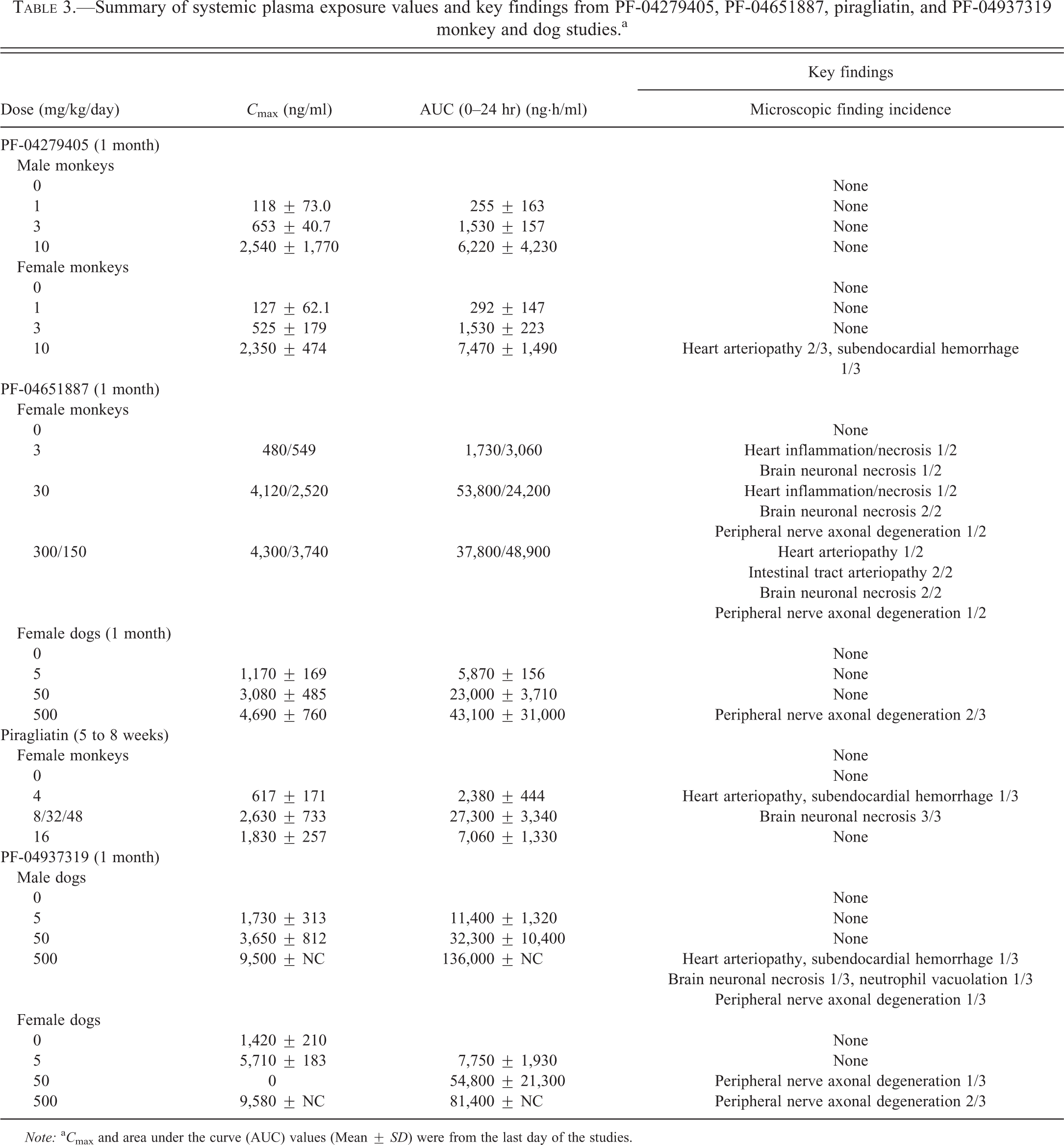
Note: a C max and area under the curve (AUC) values (Mean ± SD) were from the last day of the studies.
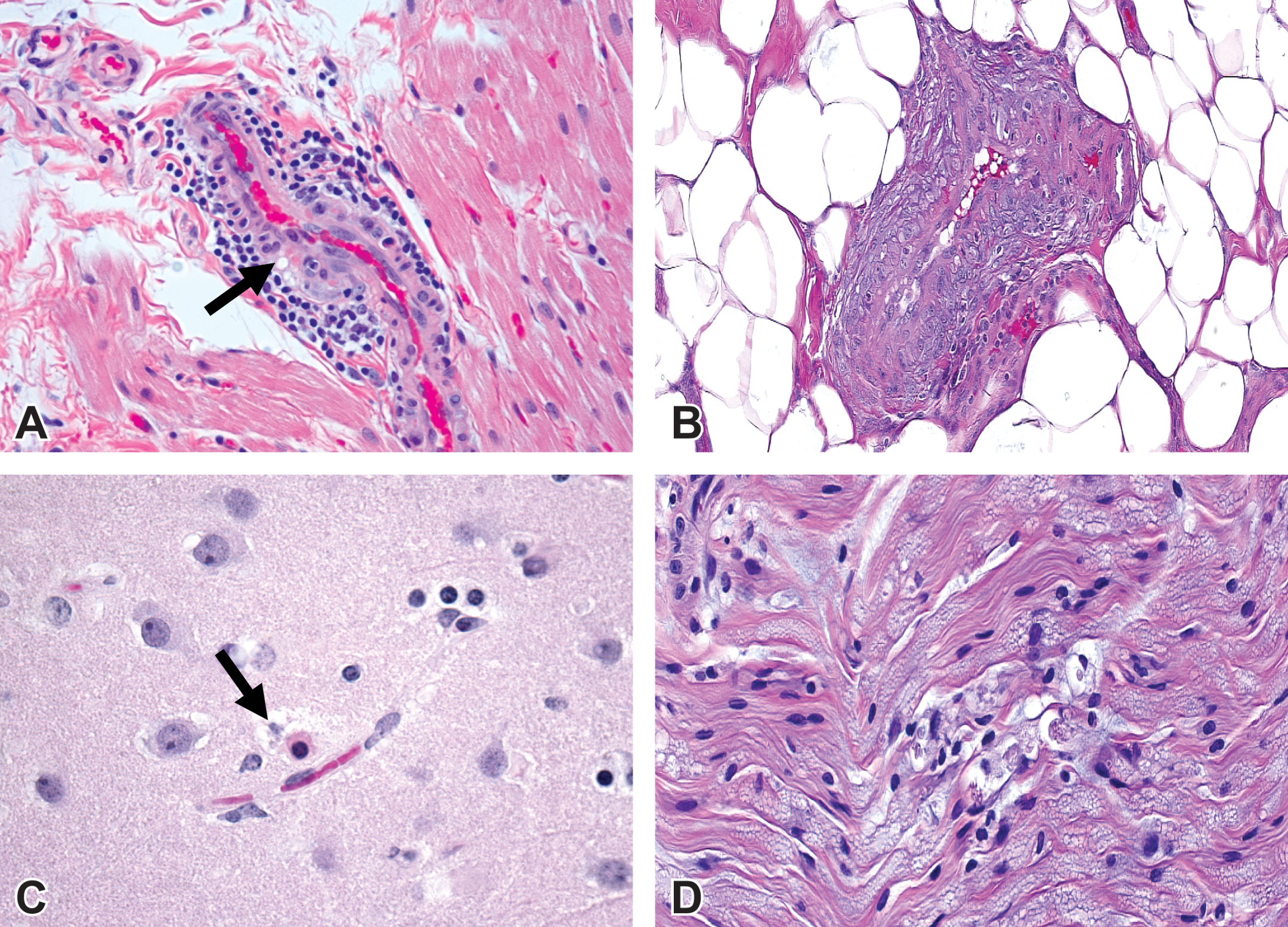
Microscopic findings included cardiac arteriopathy in 2 of the 3 females at 10 mg/kg. The arteriopathy was characterized by medial smooth muscle cell necrosis and intramural hemorrhage, in single, small-gauge intramural coronary arteries in the right ventricle (Figure 3a and b). A focal, subendocardial hemorrhage (Figure 3c) was observed in the left ventricular papillary muscle of 1 female at 10 mg/kg.
Representative photomicrographs (hemotoxylin and eosin) of the heart from a female monkey administered 10 mg/kg/day PF-04279405. (A and B) Right ventricle showing fibrinoid necrosis of medial smooth muscle and intramural hemorrhage in single, small gauge intramural coronary arteries of the right ventricle. (C) Left ventricle showing subacute subendocardial hemorrhage of the papillary muscle.
PF-04561887 (Partial Agonist)
The nonclinical safety of PF-04651887 was initially evaluated in a 14-day exploratory rat toxicity study at doses of 5, 50, and 500 mg/kg/day. Systemic exposure increased with increasing dose (Table 2). There were no treatment-related effects on clinical signs, body weight, or food consumption. Treatment-related reductions in serum glucose were observed in both sexes at 500 mg/kg (Table 2, 0.61× control in males and 0.44× control in females at 24 hr postdose). The glucose changes were moderate in males and marked in females, where values close to 30 mg/dl were recorded. There were no treatment-related histopathology findings.
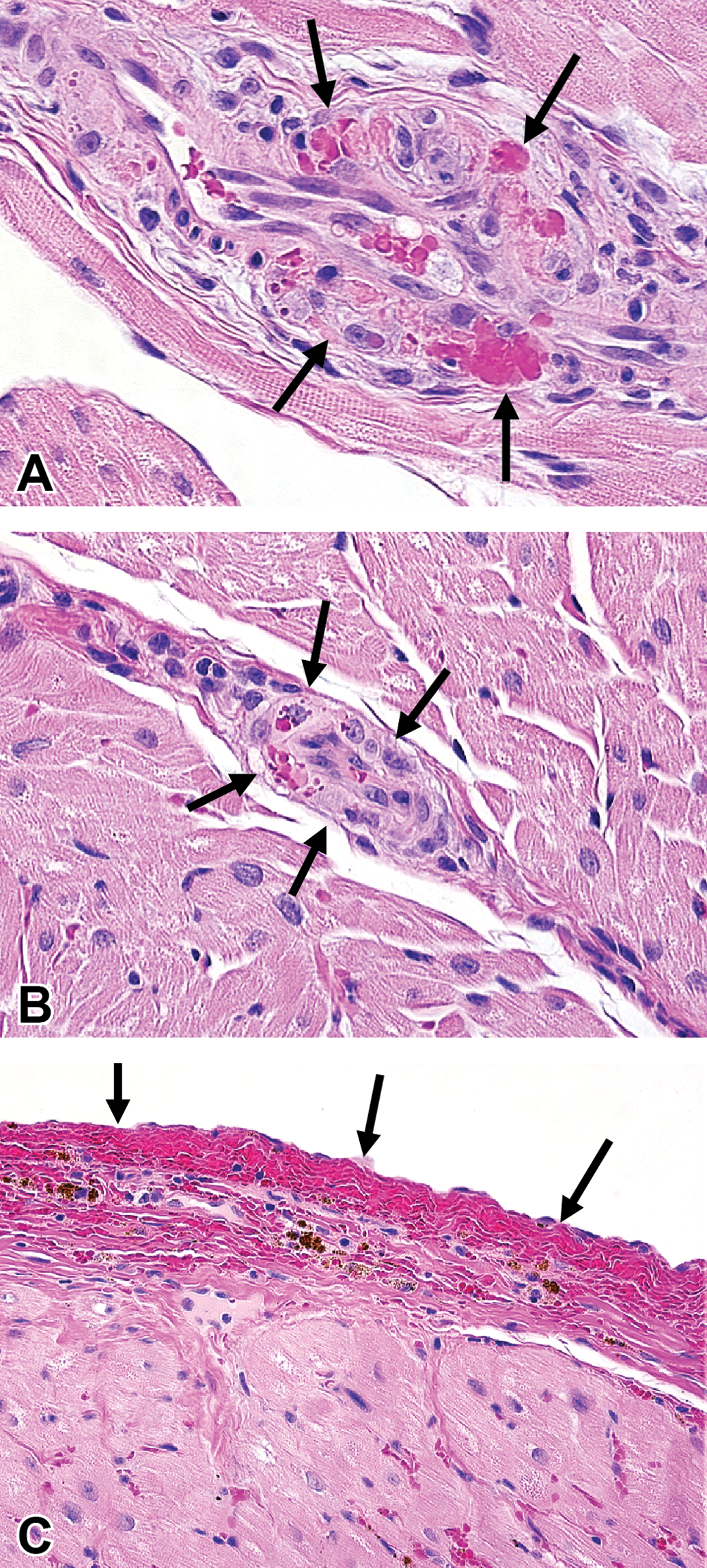
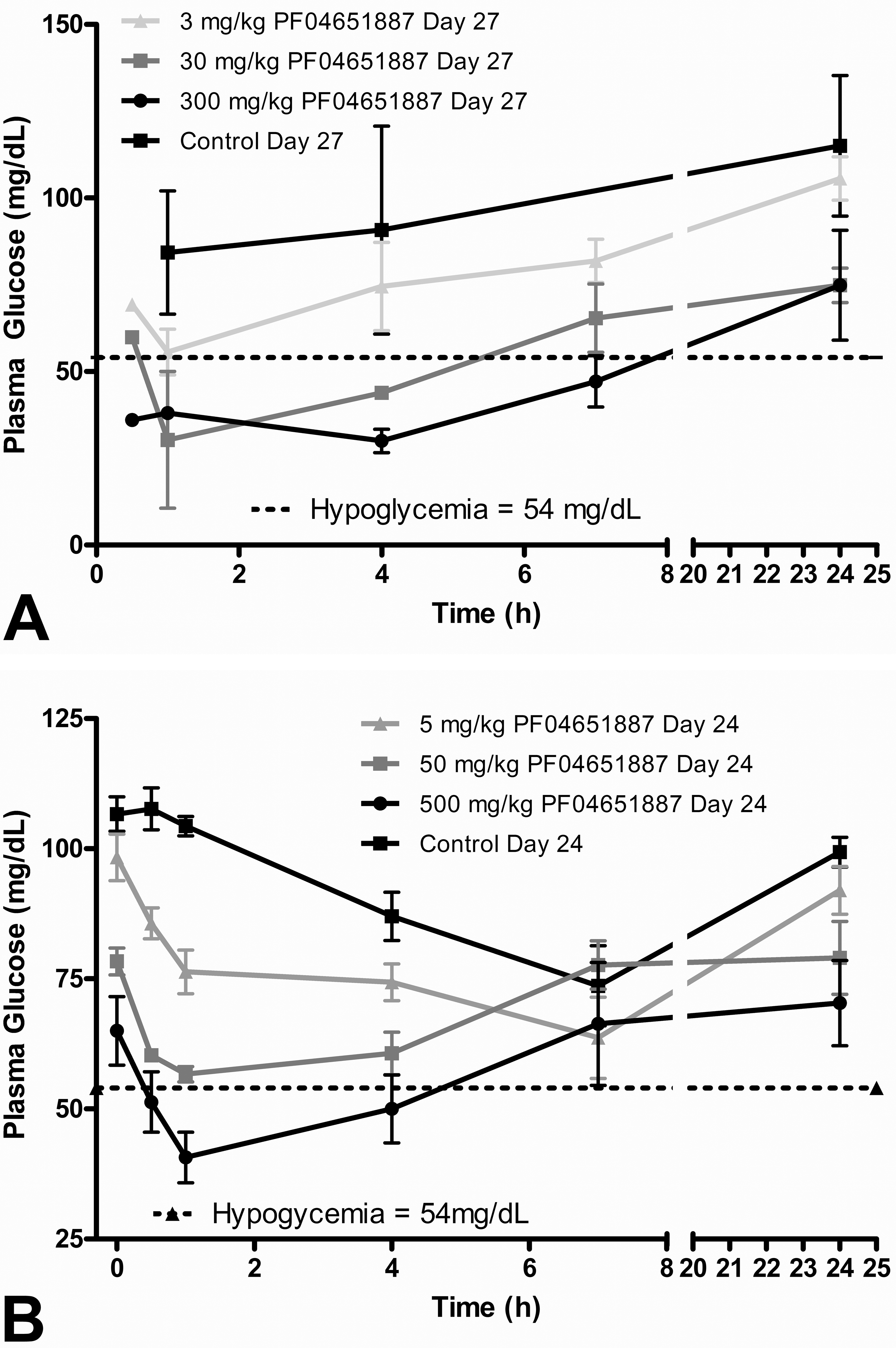
PF-04651887 was also evaluated in a 1-month exploratory monkey toxicity study at doses of 3, 30, and 300/150 mg/kg/day. There were no treatment-related effects at the 3 and 30 mg/kg doses. Systemic exposure increased with increasing dose (Table 3). At 300 mg/kg, treatment-related clinical signs included decreased activity and generalized tremor on days 23 and 24, respectively. These clinical signs resulted in a reduction of the high dose from 300 to 150 mg/kg from day 25 to the end of the study. Decreases in plasma glucose were noted at 0.5, 1, 4, and 7 hr postdose and returned to normal values 24 hr post dose on day 27 (Figure 4a). At 300/150 mg/kg/day, insulin levels were elevated 4 to 12× control at 4 and 7 hr post dose on days 1 and 27. Treatment-related microscopic findings occurred in the myocardium (3 and 30 mg/kg), intramural coronary arteries (Figure 5a, 300/150 mg/kg), arterioles of the intestinal submucosa (Figure 5b, 300/150 mg/kg), brain (Figure 5c, 3, 30, and 300/150 mg/kg), and peripheral nerves (Figure 5d, 30, and 300/150 mg/kg). Myocardial arteriopathy affecting the intramural myocardial (coronary) vasculature was present in 1 monkey of the 300/150 mg/kg group. Inflammation and necrosis of the myocardium were present in 2 monkeys, one each from the 3 and 30 mg/kg groups, respectively. The myocardial arteriopathy consisted of fibrinoid necrosis with inflammation in a single arteriole at the atrioventricular junction and subtle evidence of fibrinoid material within the media of several other arterioles in the left and right ventricles. The myocardial inflammation and necrosis involved the apex and papillary muscles of the left ventricle and was characterized by cardiomyofiber lysis, the presence of occasional neutrophils, and a few macrophages. Although qualitatively similar changes of focal myocardial necrosis and inflammation are sometimes found as incidental findings in untreated monkeys, the severity of lesions in these animals exceeds that which could be attributed to spontaneous background change. The intestinal arteriopathy involved the jejunum, ileum, cecum, and colon. The cecal and colonic arteriopathy was characterized by prominent submucosal arterioles associated with proliferating tunica adventitia, pericytes and macrophages, swollen and disrupted tunica media smooth muscle cells undergoing degeneration, and enlarged endothelial cells. In the brain, there were very few to more numerous scattered necrotic neurons, observed at 3, 30, and 300/150 mg/kg, which were generally localized within the temporal cortex, thalamus, and basal nuclei (caudate nuclei and globus pallidus). The peripheral (sciatic) nerve axon degeneration, observed at 30 and 300/150 mg/kg, was characterized by the presence of multiple ellipsoids (digestion chambers) with occasional infiltration by macrophages and proliferation of Schwann cells.
The time course for plasma glucose in (A) monkeys or dogs (B) administered PF-04651887 for 1 month. Each point represents the mean ± standard deviation (N = 2 females monkeys/group or 3 female dogs/group).
Representative photomicrographs (hemotoxylin and eosin) from monkeys administered 300/150 mg/kg/day PF-04651887. (A) Left ventricle at the atrioventricular junction of the heart: Arteriopathy with inflammation associated with mixed mononuclear leukocytes. (B) Cecum: Submucosal arteriopathy. (C) Brain: Neuronal necrosis. (D) Peripheral nerve: Axonal degeneration.
Additionally, PF-04651887 was evaluated in female beagle dogs at 5, 50, or 500 mg/kg/day for 1 month. Treatment-related clinical signs were limited to soft/watery and/or discolored feces at 50 and 500 mg/kg/day. There were no treatment-related changes in body weight, food intake, neurological examinations, hematology, coagulation, or electrocardiogram parameters. Systemic exposure increased with increasing dose (Table 3). A dose-dependent decrease in plasma glucose was noted (Figure 4b). An increase in plasma insulin concentrations was observed in PF-04651887 treated dogs at 500 mg/kg/day, but the values were quite variable. Minimal to mild axonal degeneration in the peripheral (sciatic) nerve was observed in 2/3 dogs at 500 mg/kg/day. The finding was characterized by the presence of multiple ellipsoids (digestion chambers) with an occasional infiltration by macrophages and proliferation of Schwann cells. This lesion is considered to be secondary to hypoglycemia in the affected dogs (Sidenius and Jackson 1983).
Piragliatin (Full Agonist)
Female cynomolgus monkeys were administered piragliatin at doses of 4, 8, or 16 mg/kg/day for 5 or 8 weeks. Systemic exposure increased with increasing dose (Table 3). Based on interim plasma glucose data, the dose level for animals in the 8 mg/kg/day dose group was increased to 32 mg/kg/day (16 mg/kg/day BID) on day 29, and increased again to 48 mg/kg/day (24 mg/kg/day BID) on day 41 in a effort to achieve a similar reduction in glucose as seen with PF-04279405 and PF-04651887. All animals survived the treatment period. There were no changes in body weight, food intake, electrocardiograms, organ weight, or macroscopic observations. Treatment-related clinical signs included decreased activity, ataxia, and hunched posture in 1 animal in the 8/32/48 mg/kg/day dose group on day 56. These observations were likely secondary to decreased plasma glucose noted in that animal, which is consistent with the pharmacology of piragliatin. There were small increases in white blood cell and neutrophil counts in the 8/32/48 mg/kg/day treated animals.
Treatment-related decreases in mean plasma glucose levels were observed falling below the hypoglycemic range within 4-hr post dose in 4 and 16 mg/kg/day animals on day 35 and in 8/32/48 mg/kg/day treated animals on day 56 (Figure 6A and B). Changes relative to pretreatment plasma glucose values for individual animals ranged from 48 to 70%. There was no treatment-related effect on plasma insulin concentration. Treatment-related microscopic findings included arteriopathy in the heart of 1/3 monkeys and necrotic neurons in the gray matter regions of the brain in 3/3 monkeys in the 8/32/48 mg/kg/day treated group. In the 1 affected monkey, there was minimal to mild myocardial arteriopathy which consisted of fibrinoid necrosis and hemorrhage of the intramural vessels of the left ventricle (Figure7A and B). Additionally, subendocardial hemorrhage of the left ventricle was also observed. The randomly scattered necrotic neurons were distributed in the gray matter of the temporal cortex.
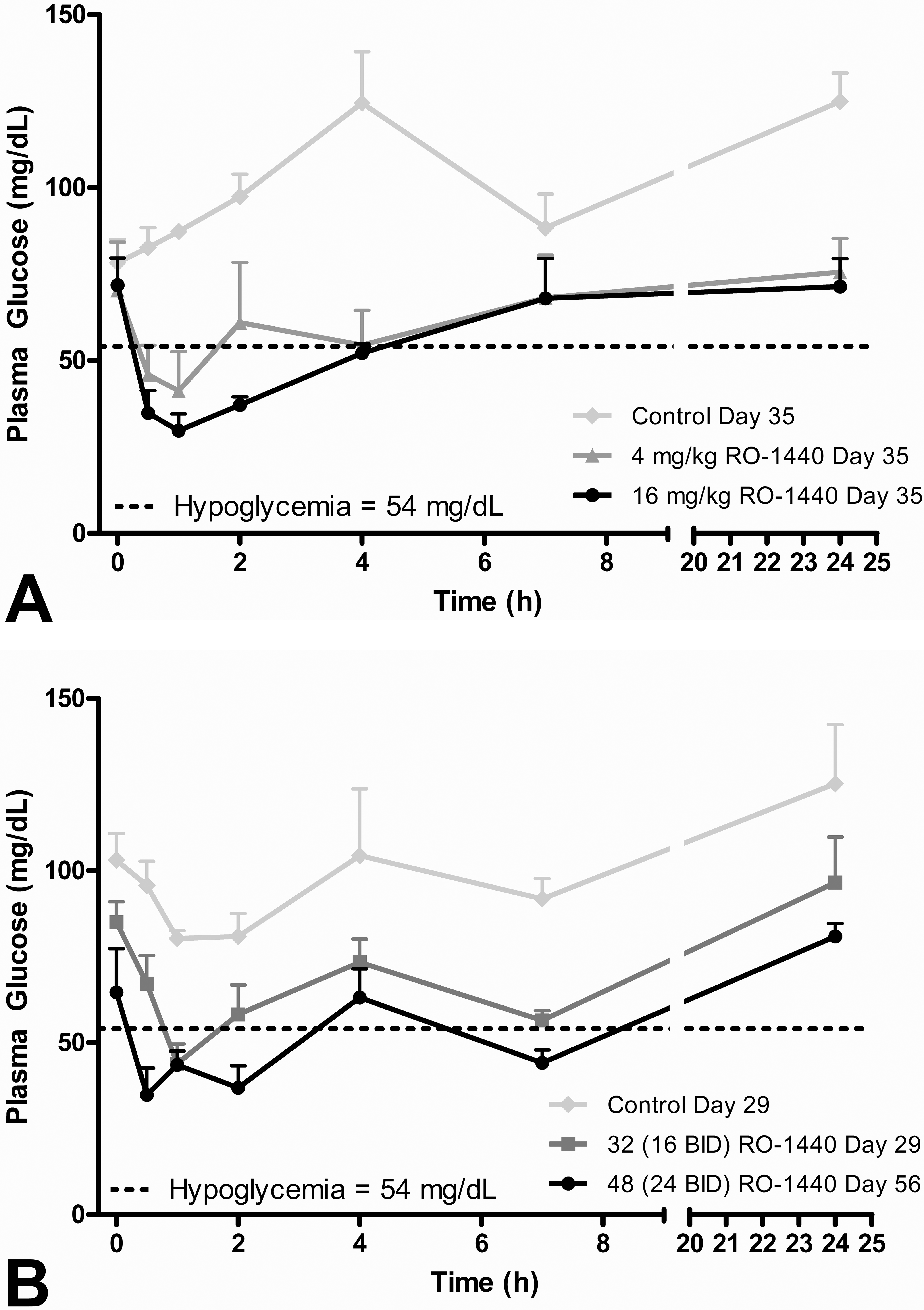
The time course for plasma glucose in monkeys administered piragliatin (A) 4 or 16 mg/kg/day every day (QD; day 35) or (B) 32 (16 BID) mg/kg/day (day 29) or 48 (24 BID) mg/kg/day BID (day 56). Each point represents the mean ± standard deviation (N = 3 females/group). BID = twice a day.
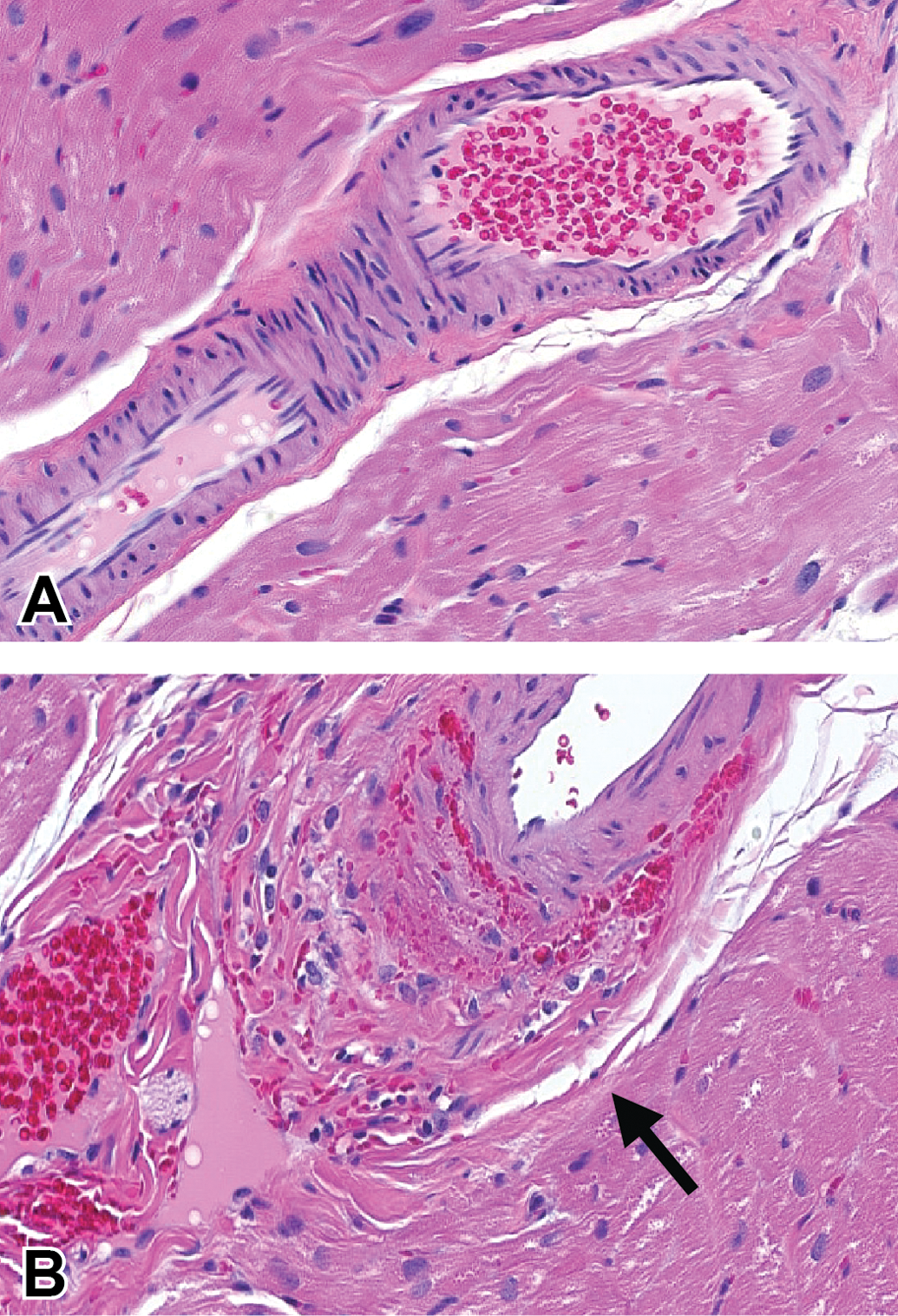
Representative photomicrograph (hematoxylin and eosin) of the left ventricular coronary artery of the heart from monkeys administered vehicle or 8/32/48 mg/kg/day piragliatin. (A) Vehicle: Normal coronary artery. (B) Piragliatin: Arteriopathy (arrow) consisting of fibrinoid necrosis, hemorrhage, and degeneration/loss of smooth muscle cells within tunica media.
PF-04937319
Male rats received 50, 250, and 1,000 mg/kg/day and females received 10, 50, and 1,000 mg/kg/day for 1 month. Different low and mid doses were selected for males and females because systemic exposure was shown to be higher in female as compared to male rats in an earlier exploratory toxicity study. Systemic exposure increased with increasing dose and exposure was higher in female as compared to male rats for a given dose level (Table 2). There were no treatment-related effects on clinical signs, body weight, and food consumption. In addition to standard clinical chemistry tests (end of study at 24 hr after last dose), serial whole blood sampling of toxicokinetics rats was performed on days 1 and 28 at 1-, 3-, 7-, and 24-hr post dose. Results from these glucose time course studies revealed consistently prolonged lowered glucose values postdose, with the lowest individual values occurring 1- to 3-hr postdose (Figure 8a and b). The lowest reported glucose value in an individual rat was 26 mg/dl 1-hr postdose in a female at 1,000 mg/kg/day on day 28. Biologically significant lower glucose levels were noted in all dose groups at 1- and/or 3-hr postdose on days 1 and 28, in males at 250 and 1,000 mg/kg/day at 7- and 24-hr postdose on day 1, in females at 50 and 1,000 mg/kg/day at 7-hr postdose on day 1 and 28, and in females at 1,000 mg/kg/day 24-hr postdose on day 1. In the peripheral (sciatic) nerve, minimal to mild axonal degeneration, characterized by axonal digestion chambers visible on longitudinal sections of hind limb peripheral nerves, was present in 10/10 females at 1,000 mg/kg/day and in 2/10 females at 50 mg/kg/day.
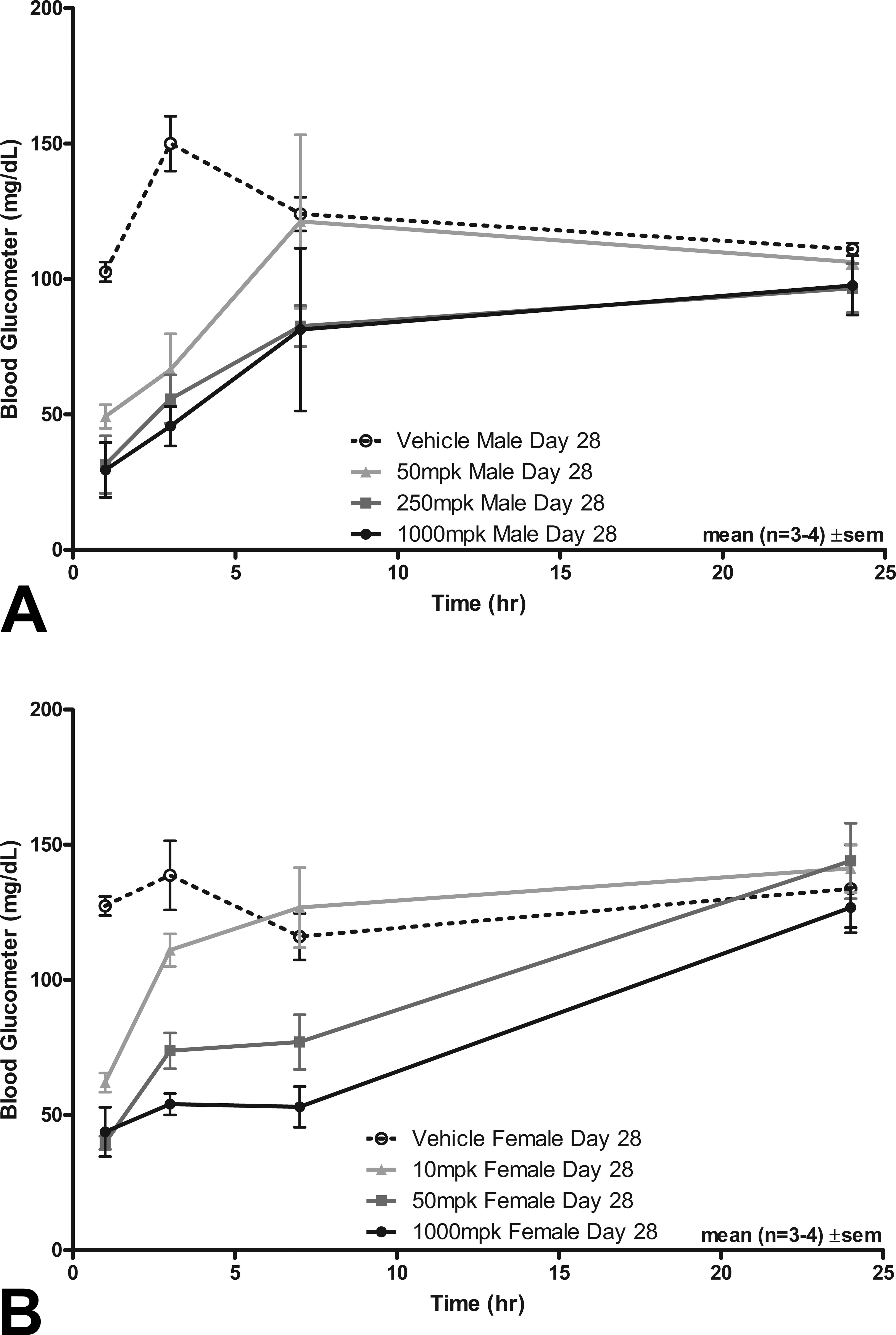
The time course for plasma glucose in (A) male and (B) female rats administered PF-04937319 for 1 month. Each point represents the mean ± standard deviation (N = 3 males + 3 females/group).
Male and female dogs received doses of PF-04937319 at 5, 50, or 500 mg/kg/day for 1 month. Two unscheduled deaths occurred at 500 mg/kg/day. The first unscheduled death was a male dog that was euthanized on day 12 after exhibiting multiple episodes of convulsions and emesis. The second unscheduled death was a female dog that was euthanized on day 22 after developing ataxia and convulsions. In surviving dogs, there were no treatment-related effects on body weight, food consumption, electrocardiograms, or urinalysis parameters. Hematological changes in surviving dogs included slight decreases in red cell count, hemoglobin concentration, and hematocrit at 500 mg/kg/day; this was considered most likely secondary to prolonged reduced serum glucose. Treatment-related clinical chemistry changes included a decrease in serum glucose and a slight increase in insulin at ≥5 mg/kg/day. Time course data (Figure 9) showed a dose-responsive decrease in plasma glucose concentration with the 50 and 500 mg/kg/day dose groups falling into the hypoglycemic range. Microscopic changes in the 2 unscheduled deaths included neuronal necrosis in the brain in one dog and peripheral nerve degeneration in the other dog. Other findings were consistent with severe stress and/or moribundity, with the possible exception of an arteriopathy (characterized by fibrinoid necrosis and intramural hemorrhage) affecting a few myocardial arteries in the dog euthanized on day 12. Since this finding occurred in a single moribund dog, these changes are difficult to interpret, but a direct effect of treatment cannot be ruled out. Treatment-related microscopic changes in the dogs surviving the treatment period were limited to minimal or mild degeneration in the peripheral nerve of 1/2 male and 2/3 females at 500 mg/kg/day, and 1/3 females at 50 mg/kg/day.
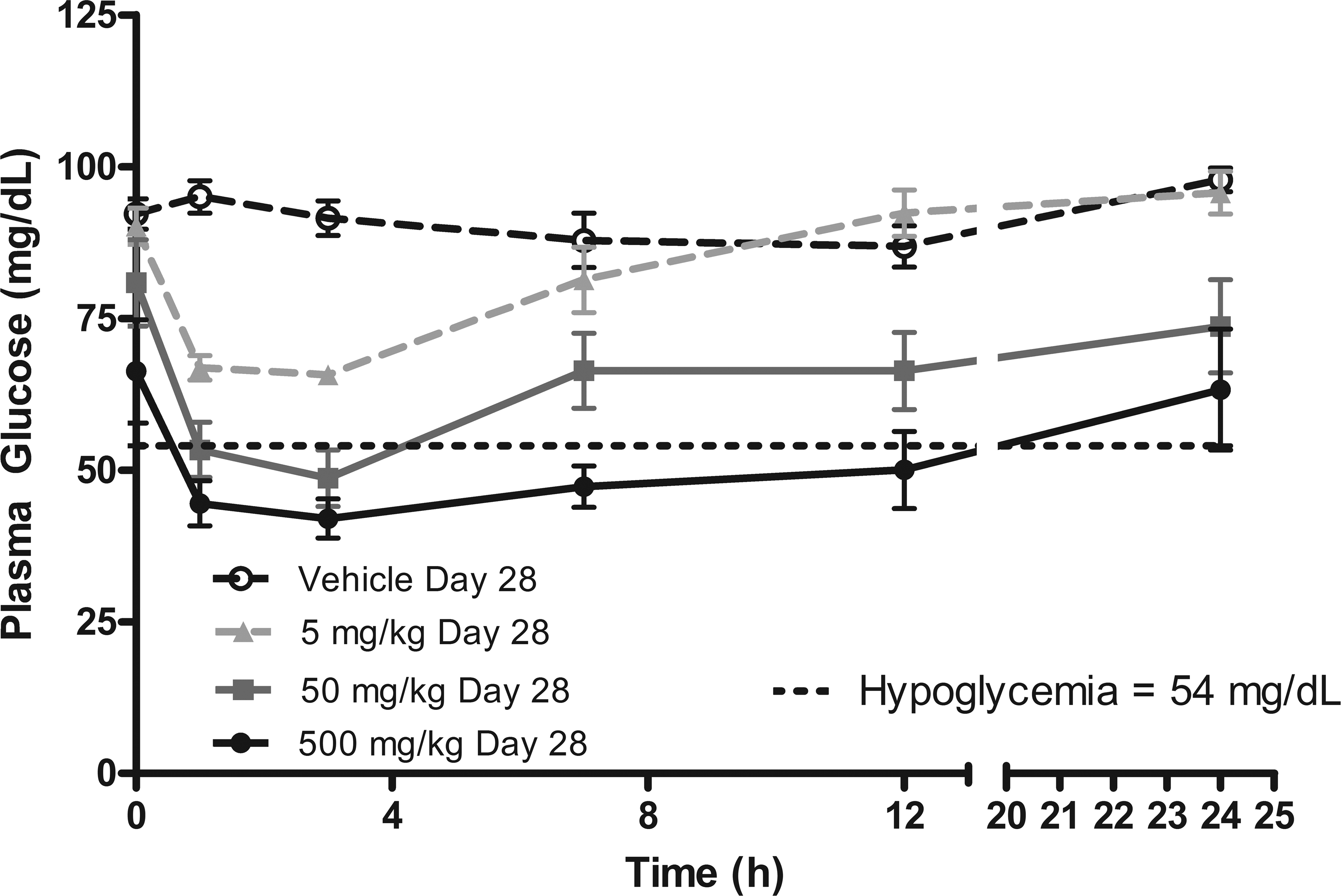
The time course for plasma glucose in dogs administered PF-04937319 for 1 month. Each point represents the mean ± standard deviation (N = 3 males + 3 females/group).
Discussion
To understand whether the vascular lesion observed with PF-04279405 in monkeys resulted from a chemotype-specific toxicity or was instead pharmacologically mediated, we conducted toxicological evaluations of 2 additional, structurally distinct GKAs in female cynomolgus monkeys (PF-04651887 and piragliatin). Like PF-04279405, these activators also induced disproportionately severe and prolonged hypoglycemia in monkey relative to either rat or dog. Both compounds were found to cause coronary arteriopathy in the monkey similar to that observed for PF-04279405. Peripheral neuropathy and brain neuronal necrosis, findings that are well documented in association with hypoglycemia, were also observed in these monkey studies of PF-04651887 and piragliatin. Low blood glucose levels observed in this study have been shown to cause peripheral nerve pathology in several species (Mohseni 2001; Tabata 2000) and are the likely pathogenesis of the peripheral nerve axonal degeneration in this study. There is some evidence that female rodents may be more sensitive to hypoglycemic neuropathy than male rodents (Tabata 2000), which is consistent with our findings in rodents treated with PF-04279405 or PF-04937319. Myocardial inflammation and necrosis were additional findings in 2 animals treated with PF-04651887, and subendocardial hemorrhage in 2 animals treated with piragliatin. Toxicological evaluation of PF-04651887 in dog or rat at equivalent or greater free drug exposures achieved in monkey studies did not result in the same levels of prolonged hypoglycemia and there were no vascular or myocardial lesions observed.
In the previously mentioned studies, lesions consistent with severe hypoglycemia (brain neuronal necrosis, axonal changes) were consistently observed. Cardiovascular changes were also consistently observed, independently of the compounds tested. Hence, just like the neuronal and axonal lesions, these cardiovascular lesions might also be related to hypoglycemia rather than a direct effect of GKAs. These lesions bear some morphological similarities with catecholamine stimulation/administration (fibrinoid necrosis of coronary arteries, myocardial inflammation/necrosis, subendocardial hemorrhage; Jellinek et al. 1966; Khullar et al. 1989; Szakacs and Mehlman 1960) and strongly suggest that the vascular/heart lesions seen with our GKAs may be related to a counterregulatory catecholamine response to hypoglycemia in these species (Havel and Valverde 1996; Molina and Abumrad 2001; Yamaguchi, Briand, and Gaspo 1990), rather than a direct effect of GK activators. Therefore, these cardiovascular changes (myocardial necrosis and hemorrhage and arteriopathy) could be the direct manifestation of marked severe hypoglycemia and/or indirectly the consequence of a transient catecholamine surge or a combination of both. Due to this suspected pathogenesis, norepinephrine and epinephrine were measured from blood samples taken at the time of peak hypoglycemia in some of our studies. In general, the results were consistent with a counterregulatory response but were highly variable due to the small number of animals and the variable nature of these measurements (data not shown). Therefore, this main hypothesis could not be definitely confirmed in this set of studies but could be further characterized and studied by additional biomarker measurements and functional cardiovascular investigative studies.
Endothelial dysfunction resulting from hypoglycemia is another potential mechanism for the arteriopathy. This hypothesis is substantiated by reports in patients with type 2 diabetes of acute hypoglycemia-related impairment of flow-mediated dilatation by nitric oxide (NO)-dependent and NO-independent mechanisms as well as changes in inflammatory and pro-atherothrombotic biomarkers (Chow and Heller 2012; Gogitidze et al. 2010). Endothelial dysfunction and alterations in NO signaling may be an important feature in the vascular injury observed in this study since recent reports indicate a link between endothelial nitric oxide synthase and vascular injury in mesenteric arteries (Sheth et al. 2011; Brott, Richardson, and Louden 2012). Acute hypoglycemia can also induce the release of potent vasoconstrictors such as endothelin which may have a role in vascular injury (Chow and Heller 2012; Wright et al. 2007).
This connection between hypoglycemia and arteriopathy has not been formally documented to date, presumably due to the 2 phenomena being investigated independently, that is, detailed histopathological evaluation of the heart is not typically conducted in animal hypoglycemia physiology studies, and hypoglycemia is not used as a mechanism for inducing catecholamine-related cardiac lesions in animals. This link is also very difficult to confirm experimentally, because the doses needed to produce heart lesions via systemic administration of catecholamines will not necessarily reflect the physiological condition of elevated catecholamines in hypoglycemic animals with heart lesions since the latter effect is mainly mediated by direct release of catecholamines into the cardiac vascular synapses during episodes of hypoglycemia as opposed to systemically released catecholamines (Bevan, Bevan, and Duckles 1980). However, there are accumulating reports suggesting that cardiac infarction and other adverse cardiac and vascular events may infrequently follow insulin-induced hypoglycemia in humans (Hsueh et al. 2012; reviewed by Desouza, Bolli, and Fonseca 2010; Chow and Heller 2012).
Even in toxicology studies involving agents that induce hypoglycemia and where the heart is also examined histopathologically, arteriopathy is unlikely to be seen. This is because the doses are typically set (for survival and animal welfare considerations) below the level where significant hypoglycemia occurs, and in the case of dogs (the most common nonrodent species), convulsions further limit dosing and generally keep glucose levels above those necessary to produce these cardiovascular lesions. In primates in general, profound hypoglycemia is more often associated with loss of consciousness and coma. The brain neuronal necrosis that was observed in several animals from these monkey and dog studies is supportive that the levels of hypoglycemia were below those typically reached in most toxicology studies of anti-diabetic agents, including insulin. Moreover, the arteriopathy occurs inconsistently within the affected dose groups (and hence may not be attributed to treatment if seen in a single hypoglycemic animal) and is also extremely subtle and easily overlooked if not anticipated by the study pathologist.
Toxicological evaluation of a fourth GK activator PF-04937319 was subsequently conducted in the dog and rat, as this compound was thought to have the best qualities for study in human clinical trials. In the dog study (doses of 5, 50, and 500 mg/kg), animals in the high-dose group were found to experience prolonged episodes of significant hypoglycemia and 2 were euthanized early after experiencing hypoglycemia-induced seizures. Coronary arteriopathy and neuronal necrosis in the brain were noted in 1 of these early termination dogs. Moreover, peripheral neuropathy was observed in some animals at the 50- and 500-mg/kg doses. A parallel evaluation of PF-04937319 in rats revealed similar peripheral neuropathy but no coronary arteriopathy.
Toxicological evaluation of glucose lowering drugs for the treatment of type 2 diabetes can present unique challenges due to hypoglycemia (e.g., dose level limitations, use of normal euglycemic animals, species-related differences in sensitivity to hypoglycemia). The use of normal euglycemic animals and dose levels that approach the maximum tolerated dose in our GKAs toxicology studies were the major reason for the observed pharmacologically related reductions in plasma glucose concentrations and increases in serum insulin concentrations. Unlike the toxicology animals studies, there is also less risk of producing marked and prolonged hypoglycemia in human clinical studies that use patients with type 2 diabetes since they typically have hyperglycemia and insulin resistance. Thus, a reduction in blood glucose levels in hyperglycemic patients would pose less risk for producing hypoglycemia relative to a similar dose in normal euglycemic laboratory animals. This increased sensitivity of normal euglycemic animals to hypoglycemic complications is supported by a recently published abstract (Horvath et al. 2013) in which a GK activator was reported to cause degenerative changes in several tissues (stomach, sciatic nerve, heart, and skeletal muscle) in normal euglycemic rats but not in a Zucker Diabetic Fatty rat model. Thus, a good understanding of the pathological responses of normal euglycemic animals to marked and prolonged hypoglycemia is crucial when assessing the risk of these findings to human patients.
Additional toxicological testing challenges include the relative sensitivity of different species to drugs that induce hypoglycemia and species-related differences in drug metabolism. In our studies, profound decreases in glucose were clinically well tolerated by monkeys (i.e., no clinical signs of hypoglycemia) relative to dogs. Since PF-04279405 exhibited metabolic instability in both rat and dog, toxicological evaluation of this GKA was conducted in mouse and monkey.
In conclusion, we have observed low-frequency occurrences of arteriopathy in toxicological studies of GKAs in monkey and dog. These lesions occurred in multiple studies utilizing structurally different activators suggesting that this is not a chemotype-specific toxicity, and based on the characteristic arterial histopathology, but likely reflects the catecholamine-affected animal responses to exaggerated hypoglycemia in a similar way to peripheral neuropathy and brain neuronal necrosis in the affected animals. This is supported by showing a close correlation between coronary arteriopathy and the magnitude and duration of hypoglycemia during the course of the study. Accordingly, this lesion is not anticipated to occur in human clinical studies with GKAs where such levels of prolonged hypoglycemia should not be approached and should be avoided by appropriate actions and monitoring.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.