
Abstract
Glucagon-like peptide-1 is an incretin hormone from the gastrointestinal tract, which enhances insulin secretion, slows gastric emptying, and reduces food intake. GLP-1 receptor agonists are being developed for Type 2 diabetes mellitus. GLP-1 is rapidly degraded by serum dipeptidyl peptidase IV, so analogues with a prolonged serum half-life are used clinically. Exenatide was the first GLP-1 agonist approved and is a synthetic version of exendin-4 derived from the Gila monster. Liraglutide was approved for clinical use in 2010. GLP-1 receptor agonists have been shown to increase calcitonin secretion and stimulate C-cell hyperplasia and neoplasia in rats and mice of both sexes. Rat C-cells are more sensitive to the effects of GLP-1 agonists than mice. The effects of GLP-1 agonists on C-cell proliferation or neoplasia have not been documented in nonhuman primates or humans. The proliferative C-cell effects may be rodent-specific. GLP-1 receptors have been demonstrated on normal rodent C-cells, but are either not present or occur in low numbers on C-cells of nonhuman primates and humans. Hyperplasia and neoplasia of C-cells in rodents treated with GLP-1 agonists represent a unique example of an on-target species-specific effect that may not have relevance to humans.
Introduction
Blood glucose concentrations are maintained in a narrow range for normal homeostasis. During fasting, insulin and glucagon are the most important hormones that work together to regulate blood glucose concentrations. Insulin and glucagon are secreted from the β-cells and α-cells, respectively, of the Islets of Langerhans of the pancreas. Insulin reduces blood glucose by binding to cell membrane insulin receptors and stimulating glucose uptake by multiple cell types. Glucagon increases blood glucose by increasing gluconeogenesis and glycogenolysis, especially in the liver.
In the postprandial state, additional gastrointestinal (incretin) hormones play an important role in the regulation of blood glucose. The gastrointestinal incretins include glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP or glucose-dependent insulinotropic polypeptide) and are secreted by intestinal mucosal endocrine cells (L cells; Baggio and Drucker 2007). Both GLP-1 and GIP increase insulin secretion after a meal, especially a high glucose meal (Yabe and Seino 2011). The actions of GLP-1 and GIP are codependent on increased blood glucose to stimulate insulin secretion from the β-cells. However, intravenous glucose challenges do not stimulate the secretion of the incretins. In contrast, oral glucose challenge will stimulate secretion of the incretins, and blood insulin levels are greater after an oral compared to an intravenous glucose challenge. The gastrointestinal incretins are responsible for 50 to 70% of the total postprandial insulin secretion.
Administration of GLP-1 or receptor agonists has been shown to increase insulin secretion in animal and humans with Type 2 diabetes mellitus (T2DM). In contrast, GIP does not increase insulin secretion in patients with T2DM. Therefore, GLP-1 receptor (GLP-1R) agonists have been developed as adjunctive therapy for T2DM (Garber 2012; Lovshin and Drucker 2009). GLP-1 is normally produced by the L cells of the intestines by alternative proteolytic processing of proglucagon (Figure 1). Proglucagon is processed to glucagon in the pancreatic α-cells by prohormone convertase 2, but is processed to GLP-1, GLP-2, glicentin, and oxyntomodulin by the intestinal L cells and cells in the brain by prohormone convertase 1 (Whalley et al. 2011).
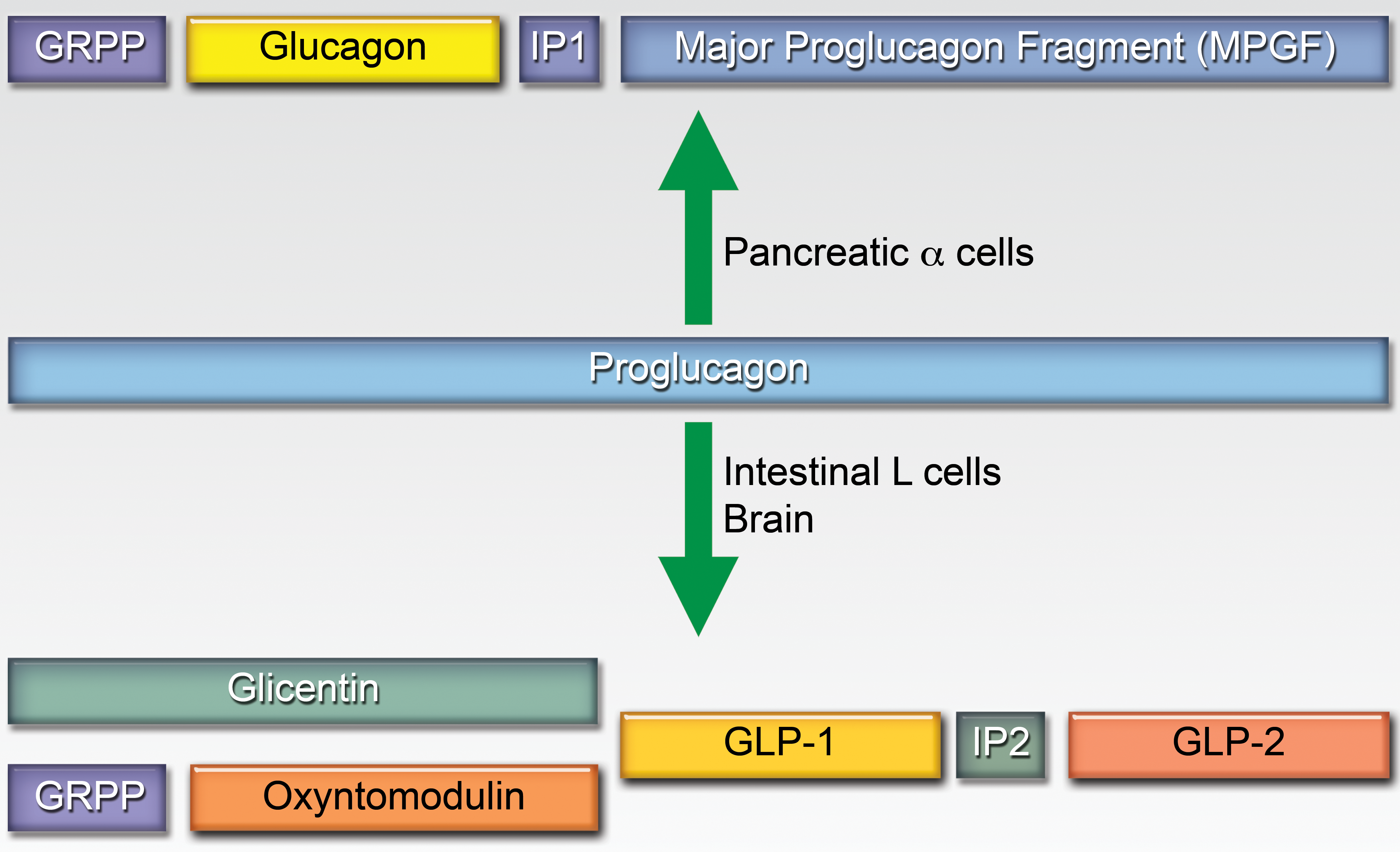
Alternative proteolytic processing of proglucagon in the α-cells of the pancreatic islets and intestinal L cells or brain.
GLP-1 has a very short half-life (1–5 min) in the blood and circulates as GLP-1 (7–36) amide and GLP-1 (7–37). GLP-1 is rapidly degraded by the ubiquitous enzyme, dipeptidyl peptidase-IV (DPP-IV). DPP-IV is present in the plasma and on the surface of many cell types. Inhibitors of DPP-IV have been developed for the treatment of T2DM and include sitagliptin, saxagliptin, vildagliptin, and linagliptin (Gupta et al. 2009). GLP-1R agonists that have been or are under development for treatment of T2DM have prolonged half-lives in vivo based on modifications or sequences that have reduced degradation by DPP-IV.
GLP-1 has multiple effects in vivo, in addition to stimulation of insulin secretion, which supports the beneficial effects of GLP-1R agonists as therapeutic agents for T2DM (Figure 2). In the pancreatic islets, GLP-1 increases insulin synthesis and secretion (in a glucose-dependent manner), increases β-cell proliferation, decreases β-cell apoptosis, and decreases insulin secretion. GLP-1 increases the peripheral sensitivity to insulin in organs such as skeletal muscle, decreases glucose production in the liver, is cardioprotective, and increases cardiac output (Axelsen et al. 2012; Mells et al. 2012). GLP-1R agonists also improve weight balance and reduce weight gain by directly reducing appetite stimulation in the brain and decreased gastric emptying (Velasquez et al. 2010). The effects on the stomach can lead to nausea as an undesirable side effect (Kanoski et al. 2012).
Actions of GLP-1.
GLP-1 Analogues for T2DM
The first GLP-1R agonist that was developed for clinical use in T2DM was exenatide (Byetta®, Amylin Pharmaceuticals, LLC). Exenatide is a synthetic form of exendin-4, which is a GLP-1 homologue isolated from Gila monster salivary glands and venom (Eng et al. 1992). Exendin-4 has 50% homology to human GLP-1 and a longer half-life in humans (Furman 2012; Thorens et al. 1993). It is used as an adjunctive therapy for patients with T2DM and requires twice daily injections. More recently a once-weekly formulation of exenetide (Bydureon®, Amylin Pharmaceuticals) has been approved from human use that utilizes a microsphere formulation (Barnett 2012).
Liraglutide (Victoza®, Novo Nordisk) is an approved GLP-1R agonist that consists of recombinant GLP-7-37 with a palmitic acid moiety attached to a mid-region amino acid. The palmitic acid increases the plasma half-life of liraglutide, which enables it to be administered once daily by injection as adjunctive therapy to patients with T2DM (Flint et al. 2011). Additional long-acting formulations in development include dulaglutide (GLP-7-37 linked to an Fc IgG fragment for once weekly injection; Eli Lilly), albiglutide (GLP-1 dimer fused to albumin for once weekly or biweekly administration; GlaxoSmithKline), lixisenatide (once daily injection; Sanofi), and glymera (PB1023; recombinant 636-amino acid polypeptide of GLP-1 fused to an inert repeating polymeric elastin-like peptide [ELP] formulated for SQ administration once weekly, PhaseBio Pharmaceuticals).
C-cell Proliferation Associated with GLP-1 Agonists
Administration of exenatide to rats in 2-year carcinogenicity studies resulted in development of an increased incidence of C-cell adenomas in female rats at the low (18 mg/kg/day), mid (70 mg/kg/day), and high (250 mg/kg/day) doses (Joffe et al. 2009; Madsen et al. 2012b). The low dose of exenatide had a human exposure multiple of approximately 5×. There was no increase in C-cell tumors in mice after 2 years with the same dosages. Subsequently, it was shown that liraglutide increased C-cell adenomas in male and female rats, C-cell carcinomas in male rats, C-cell adenomas in male mice, and C-cell adenomas and combined adenomas and carcinomas in female mice in 2-year carcinogenicity studies (Bjerre Knudsen et al. 2010). The human exposure multiples were 1 to 2×. These findings indicated that GLP-1R agonists were tumor-inducing in multiple rodent species, but rats were more susceptible than mice. Studies have not shown C-cell hyperplasia or tumors in Cynomolgus monkeys treated with liraglutide.
GLP-1 Receptors
The GLP-1 receptor is a classic G protein-coupled receptor (GPCR) and is expressed in cells of the pancreatic islets, GI tract, brain, lung, heart, and kidney (Bullock, Heller, and Habener 1996; Dunphy, Taylor, and Fuller 1998; Willard and Sloop 2012). It has been challenging to identify the GLP-1R on C-cells in the thyroid gland. Molecular analysis of thyroid C-cells is difficult because they represent a small portion of thyroid gland. The GLP-1R has been identified on C-cells of rats and mice by immunohistochemistry and in situ hybridization (Bjerre Knudsen et al. 2010). There may be a lower receptor number on C-cells compared to other cell types based on relatively weak signals using immunohistochemistry or in situ hybridization. In humans, it has recently been reported that the GLP-1R was consistently expressed in C-cells in neoplastic and hyperplastic lesions and in about one-third of normal C-cells in 5/15 thyroids using immunofluorescence and immunohistochemistry (Gier et al. 2012). This suggests that proliferative lesions of human C-cells may have greater expression of GLP-1 receptors. The GLP-1R was also identified in neoplastic follicular epithelial cells in 3/17 cases of papillary thyroid carcinoma in humans (Gier et al. 2012).
Function of GLP-1 in C-cells
Calcitonin Secretogogue
Calcitonin is stored in numerous cytoplasmic secretory granules and is available for rapid secretion. Calcitonin secretion is regulated both by cations and GI hormones (Rosol and Capen 1996). Ionized calcium (Ca2+) is an important regulator of calcitonin secretion. Increased blood concentrations of Ca2+ rapidly and dramatically increase calcitonin secretion and blood concentrations of calcitonin (Rourke et al. 2009). Calcimimetics and Mg2+ also stimulate calcitonin, but Mg2+ is less potent than Ca2+. Gastrointestinal hormones including gastrin, cholecystokinin, secretin, glucagon, enteroglucagon (glicentin or oxyntomodulin) are calcitonin secretogogues. Norepinephrine and calcitonin gene–related peptide can also stimulate calcitonin secretion. After consumption of a high calcium meal, the GI hormones (particularly gastrin and cholecystokinin) are responsible for stimulating calcitonin secretion to prevent a postprandial rise in serum Ca2+. Provocative testing of calcitonin secretion can be conducted with exogenous administration of pentagastrin, Ca2+, or omeprazole (Vitale et al. 2002). Long-acting GLP-1R agonists also stimulate calcitonin secretion in rats and mice (Bjerre Knudsen et al. 2010; Willard and Sloop 2012). GLP-1R agonists have been reported to have minimal or no ability to stimulate calcitonin secretion in normal C-cells of primates and humans (Bjerre Knudsen et al. 2010; Hegedus et al. 2011; Joffe et al. 2009).
C-cell Carcinogen
C-cell and ultimobranchial body tumors occur in a variety of species including rats, mice, bulls, dogs, horses, ferrets, Mouflon sheep, zebrafish, and humans (Capen 2007; Feitsma and Cuppen 2008). Ionizing radiation is a direct carcinogen of C-cells (Triggs and Williams 1977). Chronic hypercalcemia has an indirect carcinogenic action by stimulating calcitonin secretion and C-cell proliferation leading to diffuse and focal hyperplasia with progression to C-cell adenoma and carcinoma. Chronic hypercalcemia may be associated with certain forms of cancer, excess vitamin D, excess dietary calcium, and increased dietary absorption of calcium (Okada et al. 1994; Rosol and Capen 1992, 1997). Drugs or chemicals have been reported to cause C-cell hyperplasia and tumors in rats, but in most cases the mode of action is unknown (Joffe et al. 2009). Tumorigenic drugs in rats include a bisphosphonate (alendronate), β2 adrenergic receptor agonists (arformoterol and atenonol), a bile acid sequestrant (colesevelam), 5-hydroxytryptamine receptor antagonists (naratriptan and palonosetron), and GLP-1R agonists (exenatide and liraglutide).
Induction of focal C-cell hyperplasia, adenoma, and carcinoma in rats and mice is likely a class effect for the long-acting GLP-1R agonists. The proposed mode of action for the C-cell tumorigenic effect of the long-acting GLP-1R agonists is as follows:
Drug binding to GLP-1R on C-cells
Increased cytoplasmic cAMP and calcitonin secretion
Increased C-cell proliferation
Increased diffuse C-cell proliferation
Increased focal C-cell proliferation
Increased C-cell adenomas
Increased C-cell carcinomas
The mode of action, for the most part, has been supported by experimental data (Bjerre Knudsen et al. 2010; Chiu, Shih, and Tseng 2012; Madsen et al. 2012a). C-cell (medullary) carcinoma is an uncommon tumor of the thyroid glands in humans and is often associated with germ line or somatic mutations in the RET proto-oncogene that results in gene rearrangement and increased RET activation (La Perle, Jhiang, and Capen 2000). RET is a receptor tyrosine kinase and proto-oncogene that regulates cell proliferation and development. RET activation has not been associated with C-cell tumors induced in mice by liraglutide or exenetide (Madsen et al. 2012a) or in spontaneous C-cell tumors in WAG/Rij rats that have a high incidence of C-cell tumors (De Miguel et al. 2003).
GLP-1R knockout mice were reported by the Drucker laboratory in 1996 (Scrocchi et al. 1996). The mice are viable and reproduce and have mild glucose intolerance and fasting hyperglycemia. There is no change in serum Ca2+ and parathyroid hormone concentrations. There is cortical bone osteopenia, bone fragility, increased bone resorption, and reduced calcitonin mRNA in the thyroid gland (Yamada et al. 2008). These data suggest that the C-cell GLP-1 receptor and its downstream signaling through calcitonin are important for the regulation of bone resorption and bone mass in mice. Importantly, GLP-1R knockout mice were used to confirm the role of the GLP-1R in the actions of exenetide and liraglutide in mice (Madsen et al. 2012a). The mice did not have increased calcitonin secretion and did not develop C-cell hyperplasia in response to GLP-1R agonists.
The mode of action for C-cell tumorigenesis in rats and mice by long-acting GLP-1R agonists indicates that diffuse C-cell hyperplasia should precede focal C-cell hyperplasia as described for the actions of liraglutide in mice (Madsen et al. 2012a). Diffuse C-cell hyperplasia is a physiological response due to chronic stimulation of calcitonin secretion. Focal C-cell hyperplasia is considered a preneoplastic finding (Capen and Martin 1989). It may be particularly challenging to confirm the presence of diffuse C-cell hyperplasia in rat studies due to the prevalence of C-cells in rat thyroid glands, their regional distribution, and the normal increase in C-cell numbers with age. In contrast, mice have fewer C-cells and diffuse C-cell hyperplasia is more easily documented in mice. However, C-cells in mice cannot be readily identified in hematoxylin & eosin–stained sections and immunohistochemistry for calcitonin is required to identify the C-cells. C-cells in mice are usually represented as isolated cells in or adjacent to thyroid follicles in the central region of the thyroid gland. Diffuse C-cell hyperplasia in mice is characterized by increased numbers of C-cells in and between thyroid follicles that form small clusters of C-cells between the follicles or continuous rows of C-cells lining the follicle walls (Figure 3). In contrast, focal C-cell hyperplasia in rats or mice is characterized by a well-demarcated, solid nodule of C-cells that is less than 5 average thyroid follicles in diameter. Normal C-cells or C-cells with diffuse hyperplasia typically stain intensely positive for calcitonin by immunohistochemistry. Focal C-cell hyperplasia is not encapsulated, does not compress adjacent thyroid follicles, and may be graded as mild, moderate, or marked. C-cells in regions of focal hyperplasia may stain less densely for calcitonin by immunohistochemistry compared to normal cells.
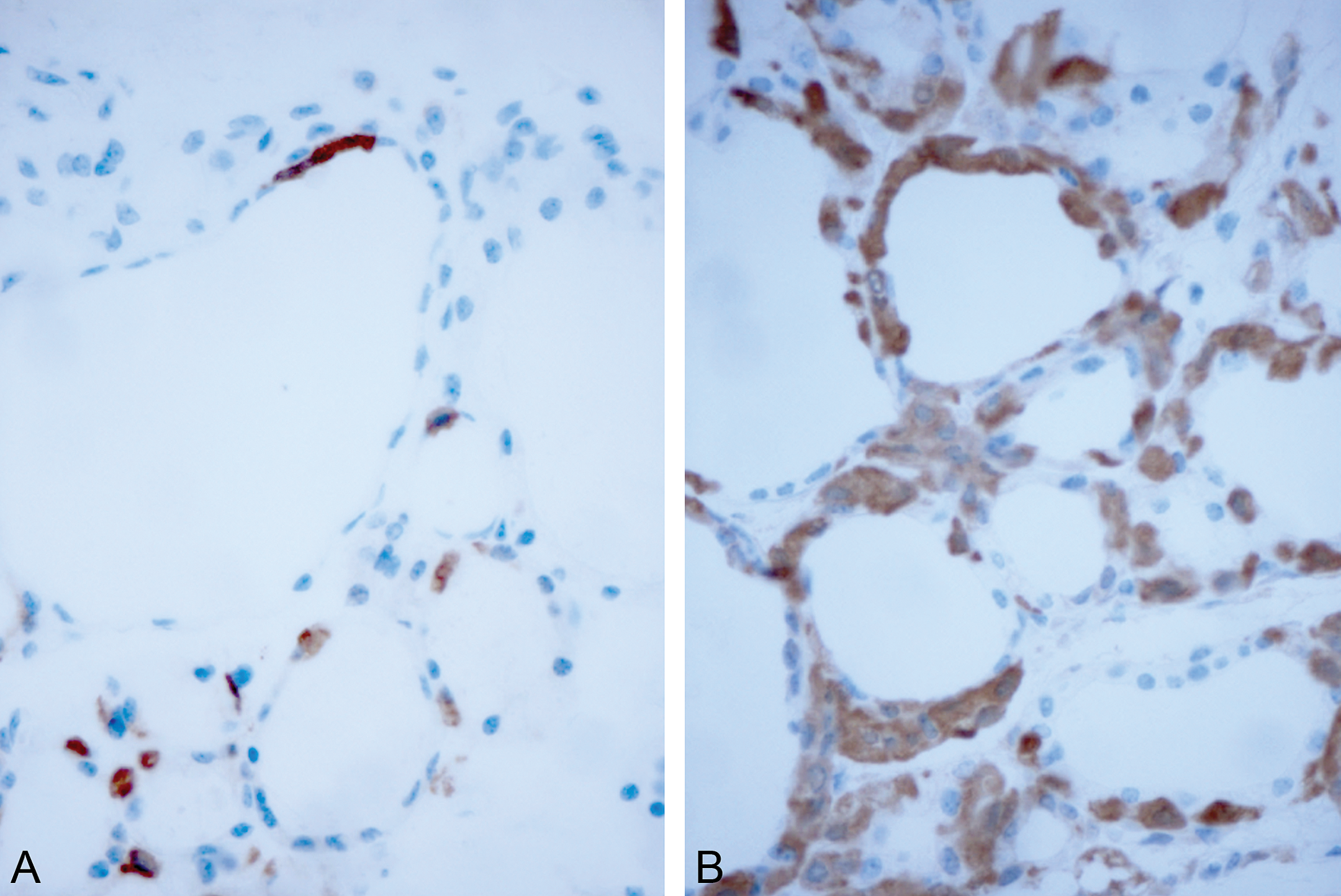
Thyroid glands (central regions) from 2-year-old mice immunohistochemically stained for calcitonin using diaminobenzidine (brown reaction product). A: Normal mouse. Note the scattered calcitonin-positive cells in and between follicles. B: Mouse with diffuse C-cell hyperplasia. Note the increased numbers of C-cells between and in the walls of the follicles. The C-cells form clusters between the follicles or contiguous rows of C-cells in the follicle walls.
Histomorphometry or stereology of C-cell numbers can be used to improve the sensitivity of discovering diffuse C-cell hyperplasia in rats, mice, dogs, or nonhuman primates (Filipovic et al. 2005). C-cells represent a greater proportion of the thyroid gland in rats (4–10%) compared to monkeys and humans (<1%) and C-cell numbers increase in rats as they age (Bjerre Knudsen et al. 2010; Kameda 1983; Martin-Lacave et al. 1992). The C-cells should be stained immunohistochemically for calcitonin. The methods for tissue collection, embedding, and sectioning need to be consistent and appropriate for the type of quantitation (Boyce, Boyce, and Gundersen 2010; Boyce et al. 2010). The total volume of C-cells in the thyroid gland can be measured using the Cavalieri stereological method, which involves nonbiased sampling, sectioning through the entire thyroid gland, collection of 6 to 12 sections at defined intervals, and measuring the area of C-cells in each section (using a manual grid or automated procedure).
Calcitonin as a Biomarker for C-cell Hyperplasia and Tumors
There is no recognized clinical condition that is associated with calcitonin excess or deficiency, although calcitonin is used as an effective acute therapy for hypercalcemia (Schenck et al. 2010). Even though there are no clinical conditions associated with hyper- or hypocalcitonemia, calcitonin is an excellent biomarker for C-cell hyperplasia and medullary (C-cell) carcinoma in humans. Calcitonin immunoassays are species-specific. Assays are commercially available for humans, primates (can use human assays, but should be validated), dogs, ruminants, and rodents. GLP-1 agonists will increase calcitonin secretion in rats and mice (Bjerre Knudsen et al. 2010; Madsen et al. 2012a). Calcitonin has not proven to be a robust marker for C-cell hyperplasia and tumors in rodents. This may be due to fact that immunohistochemical staining for calcitonin in C-cell hyperplasia and tumors in rodents is often less intense than normal C-cells. This suggests that focal C-cell hyperplasia and tumors in rodents produce less calcitonin or store less calcitonin in the cytoplasm of C-cells. Since there are few C-cells in nonhuman primates and humans, calcitonin is expected to be an effective biomarker for C-cell hyperplasia and neoplasia in nonhuman primates and is known to be effective for this purpose in humans. In large clinical trials of GLP-1R agonists in humans, a low incidence of increased serum calcitonin concentrations due to medullary thyroid cancer may be expected reflecting spontaneous cases of medullary cancer in the general adult population.
Summary
The on-target effects of long-acting GLP-1R agonists in rodents include the stimulation of calcitonin secretion and C-cell proliferation, induction of diffuse and focal C-cell hyperplasia, and development of C-cell adenomas and carcinomas. GLP-1R expression in C-cells of the thyroid gland is greatest in rodents and less in nonhuman primates and humans. GLP-1R agonists do not induce C-cell proliferation or calcitonin secretion in nonhuman primates. Mice with knockout of the GLP-1R do not secrete calcitonin from the C-cells and do not develop C-cell hyperplasia in response to GLP-1R agonists. Serum calcitonin concentration is an effective biomarker for C-cell hyperplasia and medullary carcinoma in humans. There is an equivocal or no increase in calcitonin secretion in humans in response to liraglutide, which suggests that GLP-1R agonists have minimal effects on human C-cells likely due to low receptor expression. In contrast, increased GLP-1 receptors have been reported in C-cell hyperplasia and medullary thyroid carcinoma in humans. Therefore, serum calcitonin concentration and the incidence of C-cell hyperplasia and medullary carcinoma should be monitored in humans during clinical trials and post marketing use of GLP-1R agonists until the effects of long-acting GLP-1R agonists on C-cells in humans are established.
Footnotes
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author received no financial support for the research, authorship, and/or publication of this article.