
Abstract
Food is not only vital for the health and well-being of any living being, but it is a potential source of harmful chemicals, both natural and man-made. Further complicating this is the fact that most nutrients themselves are potentially toxic when consumed in excess. Deficiencies in some of these same nutrients may cause effects that resemble toxicosis or enhance the toxic potential of other nutrients or exogenous chemicals and drugs. This review discusses some of the nutritional and metabolic mechanisms involved and the implications of excess and deficiency in macronutrients and micronutrients in toxicologic pathology. In addition, we review the adverse effects of ad libitum (AL) overfeeding on metabolic, endocrine, renal, and cardiac diseases, and many cancers and the healthful effects of moderate dietary restriction (DR) in modulating obesity and controlling spontaneous and induced diseases of laboratory animals used in toxicology and carcinogenicity studies for human safety assessment.
Keywords
Introduction
Food and water are not only vital for health; they are a potential source of harmful microbes and chemicals, both natural and man-made. Further complicating nutrients is that they are themselves potentially toxic and induce metabolic disease when consumed in excess. Foods are among the most complex mixtures of exogenous organic and inorganic chemicals to which animals and humans are exposed. The diet needs to be optimized for the species, sex, age, and physiological status (growth, maintenance, reproduction, lactation, exercise, heat loss) because the dietary composition and the feeding methods can affect the animal’s physiology, metabolism, and response to test substances in toxicological and carcinogenicity studies (Hart, Neumann, and Robertson 1995).
Laboratory animals are fed two types of diets, chemically defined or natural ingredient. Purified or chemically defined diets are formulated using a single nutrient or nutrient class (e.g., starch and sugars, casein and soybean protein, vegetable oils, lard, and cellulose) or using elemental ingredients (e.g., specific amino acids, sugars, fatty acids, vitamins, trace elements, and salts). These defined diets are usually used in short-term bioassays for comparative purposes with similar diets containing specific additives or having deficiencies for short-term, controlled experimental protocols but induce adverse effects with long-term feeding.
In contrast, natural ingredient diets are formulated with agricultural products and by-products such as whole grains, grain meals, high-protein meals, and natural mineral sources as well as other livestock ingredients and supplements. These are typically used in toxicology studies. Closed formula natural ingredient diets can have fixed percentages of ingredients, but these diets risk changes in nutritional content over time as specific nutrient levels change from batch to batch of ingredients. Open formulation, natural ingredient diets avoid these risks by altering the amounts of specific ingredients from batch to batch to maintain a more consistent concentration of protein, fat, carbohydrate, and other nutrients over time. All natural formulations will have low levels of natural and artificial contaminants present that can include pesticide residues, heavy metals, and natural plant and fungal toxins, carcinogens, and phytoestrogens. For toxicity studies in which natural ingredients are used, the nutrients and potential contaminants should be measured by the feed manufacturer on each batch. Furthermore, the chemical and nutrient assay results on each batch of certified diet should be obtained to know whether there are contaminants present and, if so, whether they are at acceptable low levels. Special diets should also be assayed for essential nutrients such as vitamin C content in diets of guinea pig and primate, and vitamin D3 levels needed for primates, along with the shelf life for those diets containing labile nutrients and additives (Keenan et al. 2000; Ziegler and Filer 1996; Jones, Hunt, and King 1996; Newberne and Sotnikov 1996; Carpenter, Harper, and Olson 1997; National Research Council [NRC] 1995, 2003).
The most authoritative sources of information on nutritional requirements for animals are the current editions of the NRC; Nutrient requirements of Laboratory Animals (see NRC references listed). The NRC also publishes nutritional requirements for other terrestrial and aquatic animals used in experimental studies. However, the values given in the NRC publications and most textbooks are estimates of minimal nutrient levels required, with no safety margins. In addition, most requirements are based on changes in the growth curves of rapidly growing weanlings fed ad libitum (AL) at varying concentrations of one nutrient while keeping all other nutrient levels constant. This is typically determined in a 1- or 2-month feeding study that defines optimal nutrient concentration as the level that does not statistically decrease the maximal growth curves of groups of rapidly growing weanling animals or birds. These studies do not consider physiological age, sex, reproductive status, lactation status, strain, or any variation in nutrient intake or bioavailability. Therefore, many diet formulations are at best educated estimates of nutrient requirements, with conservative safety margins added to ensure minimal requirements will be met and maintained for the shelf life of the diet (Carpenter, Harper, and Olson 1997; Keenan et al. 2000).
Neurobiology and Neurotransmitters in Nutrition
Food is necessary for life and energy balance and, like sleep and reproduction, is controlled by complex neuroendocrine networks between the brain’s hypothalamus, brain stem, higher centers, and in peripheral organs including the adipose tissue (Bagchi and Preuss 2013; Bidlack and Rodriguez 2012; Bray 2011), the adipose hormone leptin acts in the hypothalamus to reduce appetite and increase energy expenditure in response to a meal in healthy animals. Obese animals have elevated leptin levels due to their greatly increased adipose tissue mass but are resistant to leptin’s anorexigeneic effects. Mechanisms that have been suggested include impaired leptin transport to the brain, impaired receptors, or altered downstream signaling.
The act of eating also stimulates mechanisms that involve neurotransmitters such as norepinephrine (NA), dopamine (DA), serotonin (5-HT), orexins, melanocortins (MCs), neuropeptide Y (NPY), and others that produce neural and hormonal signals that adjust nutrient intake and metabolism. Other chemicals, such as endocannabinoids and their receptors, are also involved in the regulation of obesity. Many pharmacological compounds are being developed to modulate these pathways with the intent of medical management of obesity.
Serotonin or 5-hydroxytryptamine (5-HT) has several actions in the brain regarding food intake. 5-HT and its precursors, tryptophan and 5-HTP, decrease food intake in animals by several specific receptors in the brain. This helps explain the hypophagic and antiobesity actions of serotonin reuptake inhibitors such as fluoxetine and fenfluramine. It is thought that increased 5-HT is acting as a 5-HT receptor agonist that can decrease hypothalamic NYP-induced hyperphagia. This is thought to be an important part of mechanism of action of compounds like lorcaserin and similar specific 5-HT receptor agonists being developed for antiobesity therapy. In addition, 5-HT interacts with the MC system, with both systems being effective in decreasing food intake.
Of the catecholamines, DA and NA are involved in the regulation of food intake. DA has a reinforcing action on food intake, acting at the level of the brain nucleus accumbens. DA levels increase in the lateral hypothalamus during feeding and decrease in the ventromedial hypothalamus during fasting. It appears that DA is essential for feeding behavior. Moreover, neuropetides like orexin, NPY, and the MC system interact with DA in the regulation of appetite and feeding behavior. The neurotransmitter NA is released from sympathetic nerves and acts through receptors that induce a reduction in feeding activity and increased energy use by hyperactivity and thermogenesis. This is thought to be the mechanism involved in the antiobesity actions of many sympathomimetics, such as amphetamine, phentermine, and sibutamine.
Histamine receptor antagonists, such as thioperamide, can also suppress food intake in rodents by acting centrally on specific histamine receptors.
NPY mediates its actions in the hypothalamic paraventricular nucleus and arcuate nucleus through actions on at least three (Y1, Y2, and Y5) of its six receptor subtypes. While it appears that NPY has a central orexigenic effect, and NPY receptor agonists may have a role in preventing obesity, the body can adjust by other pathways to compensate for NPY receptor blockade. These and other neurotransmitters and neuropeptides are involved in complex ways in regulating food intake and appetite. Modulation of one pathway can be compensated by other pathways, making the development of antiobesity compounds a challenging problem (Bray 2011; Bagchi and Preuss 2013).
Caloric Excess and Obesity
Obesity is a leading cause of human morbidity and mortality worldwide. Dietary energy is derived from fats, carbohydrates, and to a lesser extent dietary protein. Metabolizable energy equals the gross energy of the diet minus the energy lost in fecal matter, urine, and combustible gases. It is calculated as 4 kcal/g for carbohydrates and proteins, 7 kcal/g for alcohols, and 9 kcal/g for fats. Deduction of energy loss as body heat equals the net energy used for all body processes and the nonnitrogenous, organic matter of tissues and secretions. Without adequate energy intake, other important organic nutrients cannot be used for normal maintenance and other functions. Dietary energy sources are needed in excess for maintenance, growth, reproduction, lactation, and “work,” and heat production are stored as glycogen in the liver and as body fat stores. When fat stores are not utilized, a dietary-induced obesity and other metabolic diseases, such as type 2 diabetes, can develop into the metabolic syndrome sometimes referred to as diabesity (Keenan et al. 2005).
The incidence of obesity, diabetes, and related metabolic disorders due to intake of caloric excess and their related comorbidities such as cardiovascular andrenal disease as well as cancer have become a major human health care issue worldwide and a major morbidity and mortality issue in developed countries. In humans, at least 17 genes are associated with obesity and 18 genes have been associated with type 2 diabetes (Bray 2011; Ramachandrappa and Farooqi 2011). In addition, epigenetic factors and intrauterine imprinting as well as some environmental agents are added risk factors (Seki et al. 2012; Wang et al. 2012). But a positive energy balance in excess of essential caloric requirements is necessary to induce excessive storage of lipids in body fat and to induce a person or animal to become overweight and obese (Bray 2011). Animal models have been discovered that have isolated some genetic factors responsible for hyperphagia and obesity as well as polygenetic models that appear to be just as complex as the typical polygenetic human diseases they try to model (Shafrir 2007; Toxicol Sci 1999; Table 1).
Animal models of obesity and type 2 diabetes.
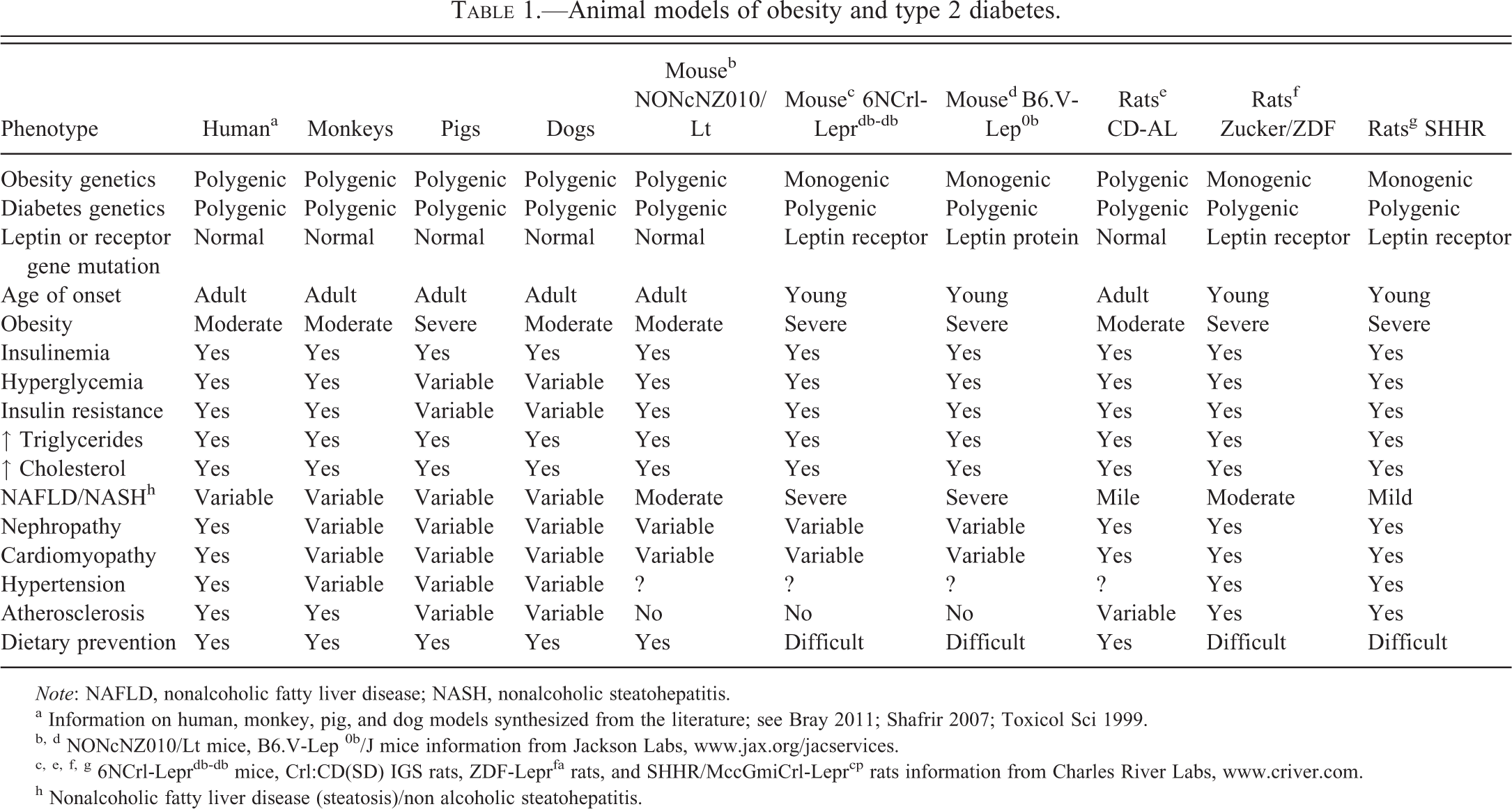
Note: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis.
a Information on human, monkey, pig, and dog models synthesized from the literature; see Bray 2011; Shafrir 2007; Toxicol Sci 1999.
b, d NONcNZ010/Lt mice, B6.V-Lep 0b/J mice information from Jackson Labs, www.jax.org/jacservices.
c, e, f, g 6NCrl-Leprdb-db mice, Crl:CD(SD) IGS rats, ZDF-Leprfa rats, and SHHR/MccGmiCrl-Leprcp rats information from Charles River Labs, www.criver.com.
h Nonalcoholic fatty liver disease (steatosis)/non alcoholic steatohepatitis.
It was the discovery of the hormone leptin in 1994, and the massive obesity produced in the leptin-protein-deficient Ob/Ob mouse, that moved obesity to the “molecular diseases” group rather than a vague “lack of willpower” behavioral condition. But the rodent models of obesity due to a single mutation in the leptin gene, such as the Ob/Ob mouse, or in the leptin receptor gene, such as the db/db mouse or the Zucker, ZDF, and SHHR rats, while helpful in understanding signaling pathways, do not model the more complex polygenetic syndromes of obesity and type 2 diabetes seen in heterozygous animal and human populations. For this purpose, outbred primates, pigs, dogs, and various rodents are better models because they develop the phenotype of obesity and type 2 diabetes and have the polygenetic and epigenetic disease trait loci that can interact with each other and thus be modulated by diet, environment, and potential treatments. Outbred animals better represent the clinically relevant human conditions of diabesity (Shafrir 2007; Keenan et al. 2005). Interestingly, only small laboratory rodents are managed by free-choice feeding or AL food consumption, while other laboratory animals, especially monogastric species (rabbits, pigs, dogs, primates, etc.), are usually fed controlled amounts of food to prevent the numerous comorbidities of diabetes and obesity, and other health problems induced by uncontrolled over nutrition.
Both obesity and type 2 diabetes (diabesity) are associated with a state of insulin resistance characterized by a reduced ability of insulin to exert its metabolic effects on key target tissues leading to multiple chronic degenerative diseases. It also is thought to increase the risk of multiple cancers. Insulin resistance could increase cancer risk through a chronic state of hyperinsulinemia, elevated levels of insulin-like growth factor (IGF-1), adipokines, and other growth factors. Insulin is both a growth factor and a metabolic hormone, and cancer cells often express insulin receptors, IGF-1 receptors, and other growth factor receptors. It is likely that hyperinsulinemia and insulin resistance, as seen early in diabesity, play an important role in the early stages of induced and spontaneous cancer development (Bray 2011; Molon-Noblot et al. 2003; Keenan et al. 2005). The basic cause of obesity or diabesity is an imbalance in ingested energy as food and expended energy for physiological functions. The excess energy is stored in fat cells that undergo hypertrophy and hyperplasia and become the initial pathological lesion in the obese individual. Enlarged fat cells produce clinical problems by their mass and increased secretion of free fatty acids (FFAs) to the liver that modulate the metabolism of insulin. This hepatic overload of FAA can induce nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH), diabetes mellitus, insulin resistance, and the metabolic syndrome (visceral obesity, hyperinsulinemia, glucose intolerance, hypertension, hypertriglyceridemia, high low-density lipoprotein [LDL] cholesterol, and low high-density lipoprotein [HDL] cholesterol) in humans. Similar disease patterns occur in rodent and larger animal models fed unlimited amounts of routine or specialized diets in excess of their nutritional and caloric needs (Table 1).
Hypertrophied and hyperplasic visceral adipocytes also secrete adipokines (adiponectin and visfatin) and cytokines (tumor necrosis factor-α [TNF-α] and interleukins (ILs) -1, -6 and -18) to the liver to increase circulation of C-reactive protein that adds to the inflammatory stimulation associated with obesity (Romeo, Lee, and Shoelson 2012). Adipocytes also secrete regulators of acute phase reactant and inflammatory responses (α-1 acid glycoprotein, serum amyloid A) and vascular endothelial growth factors (VEGFs, monobutyrin), matrix components (type IV collagen), chemokines, and steroid hormones. As endocrine cells, adipocytes secrete leptin that signals the brain as to the size of fat stores and whether or not to diminish food intake in normal animals, but this becomes defective and results in “leptin resistance” in obese animals. In obese animals and humans, the large adipose mass is dysfunctional as a metabolic, inflammatory, and endocrine tissue. This has complex consequences with many comorbidities that increase mortality in the obese. Obesity-related nephropathy and cardiovascular diseases develop in humans and in rodents and other animal models related to excessive growth, metabolic overload, and elevated inflammatory processes (Manabe 2011). The excessive body weight, increased inflammation (TNF-α secretion increases systemic C-reactive protein), dyslipidemia, and cardiac overload can lead to a cardiomyopathy and heart failure, with or without hypertension and atherosclerosis, depending on the species. The increased and enlarged fat cell mass produces increased activity of aromatase that converts adrenal androsteredione into estrone that can bring about reproductive senescence and lead to increased risk for pituitary and mammary gland tumors in both male and female rats. In rats and other rodents, the excessive growth from caloric excess induces hypersecretion of pituitary prolactin and growth hormone that increases IGF-1 production that in turn induces systemic growth effects (Molon-Noblot et al. 2003). Combined with pancreatic islet cell insulin hypersecretion and insulin resistance, these events induce a potentially tumorogenic state, with increased cell proliferation and decreased apoptosis in numerous tissues in both animals and humans (Arcidiacono et al. 2012).
In addition to increases in the visceral and subcutaneous adipose mass, dietary or caloric excess increases somatic body growth of many nonadipose tissues. This excessive growth appears to reflect the overall effect of total excess energy intake since all the dietary ingredients of animal diets are consumed at the same levels when expressed as food consumed as a percentage of body weight, even as the ratio of lean to fat tissue changes. The higher energy intake correlates with ever increasing levels of growth-promoting hormones to include growth hormone, IGF-1, prolactin, insulin, and many of the adrenal steroids, thyroid, and reproductive hormones (Keenan et al. 2005).
AL fed laboratory animals do not show the normal daily changes in metabolic rate and respiratory quotient (RQ) that occurs in animals fed single meals. Thus, AL fed animals have a constant ratio of protein, fat, and carbohydrate metabolism. This is one of the major metabolic reasons AL-fed animals store large amounts of energy as excessive visceral body fat (Hart, Neumann, and Robertson 1995; Keenan et al. 2005). The large visceral adipose mass in obese animals becomes dysfunctional as a metabolic and endocrine tissue. The secretory products of adipocytes in the obese include abnormal production of the regulators of energy homeostasis (leptin, adiponectin, resistin); regulators of inflammation, acute phase reactants; and the innate immune system (alpha-1 acidic glycoprotein, serum amyloid A, TNF-α, IL-6, etc.), vascular regulators (VEGFs, monobutyrin), and matrix components (type IV collagen), in addition to their overinduced metabolic enzyme systems. In obesity, increased leptin levels fail to prevent overconsumption, and this is considered leptin resistance. The overproduction of adipocyte TNF-α increases C-reactive protein secretion and increases systemic inflammation associated with obesity (Bray 2011; Bagchi and Preuss 2013).
In laboratory animals, the systemic effects of overeating and obesity must be recognized as complexly interactive and possibly confounding events that can alter or mask the effects of a given test article in a toxicology or carcinogenicity study. This determinant error in nutrition should be controlled by measuring food intake in toxicity studies to maintain the levels in laboratory rodents, as is done in well-controlled studies using rabbits, dogs, pigs, and other laboratory animals (Shafrir 2007; Chandra 1997; Keenan et al. 2000; Toxicol Sci 1999).
While a large number of fad diets, natural supplements, bariatric surgical interventions, pharmaceutical treatments, and exercise programs have been used to try to control obesity in humans and animals, the most effective intervention remains simple caloric restriction of total food intake to maintenance levels in all species. While dietary control by dietary restriction (DR) can be readily accomplished in confined laboratory animals and farm animals, its use in humans and companion animals has proven to be a very difficult effort in cognitive therapy. Current pharmacotherapies are largely ineffective and many have serious side effects. For example, in obese patients, the lipase inhibitor orlistat can only achieve weight losses of 12% of placebo treatment over 1 year in 30% of cases and has very unpleasant gastrointestinal (GI) side effects. Several other drugs have more significant safety issues, such as fenfluramine and dexfenfluramine, that have been associated with valvulopathy and aminorex that was withdrawn due to its effects on pulmonary artery hypertension. While the complex pathways of energy homeostasis have identified many potential peripheral and central drug targets, the development of safe compounds for chronic use remains a major safety and therapeutic challenge.
Caloric Restriction
The opposite of caloric excess and diabesity is severe caloric or food restriction leading to malnutrition and starvation. However, as it pertains to toxicology studies, caloric or DR without malnutrition refers to controlled feeding of balanced, healthy diets in maintenance amounts to support the adult health span. The classic study by McCay and colleagues at Cornell University in 1935 showed that controlling food intake in rats significantly ameliorates age-related diseases and increases life span. Since that time, numerous studies worldwide have confirmed those data and shown that a 25 to 50% reduction in maximal adult food consumption (without malnutrition) consistently increases both mean and maximum life span of laboratory rodents and other animals by delaying the development of obesity, diabetes, renal, cardiovascular disease, and cancer. These observations have been extended to various invertebrates, fish, birds, other small mammals, dogs, pigs, and are now being tested in primates (Hart, Neumann, and Robertson 1995; Mattison et al. 2012). This methodology has been called caloric restriction, food restriction, and DR, but its effects appear due to the decreased intake of total dietary calories, rather than the reduction of a specific macronutrient or micronutrient, such as a specific dietary protein, carbohydrate, or fat.
The healthful effects of DR can be contrasted with protein-energy malnutrition (PEM), which is a serious and often fatal disease seen in chronic starvation, severe organ toxicity, chronic infectious diseases, immunosuppression, and advanced cancers (Chandra 1997). In cancer, the process is often referred to as cachexia and results in extreme weight loss, muscle atrophy, anemia, anorexia, edema, and depression. Death usually results from atrophy of the diaphragm and other respiratory muscles. The precise causes of cachexia are not known in most cases, but in advanced cancers several cachectic agents and mechanisms are known. Proteolysis-inducing factor (PIF) is a glycosylated polypeptide detected in the urine of individuals with some pancreatic, mammary gland, colon, and other cancers. PIF and proinflammatory cytokines cause the breakdown and atrophy of skeletal muscle involving mechanisms that break down myosin heavy chains and alter myofibriles, with loss of dystrophin, similar to the mechanism seen in some muscular dystrophies. Lipid-mobilizing factor (LMF) is produced by some tumors and increases fatty acid oxidation, and proinflammatory cytokines such as TNF-α (formally called cachectin), IL-2, and IL-6 that increase the secretion of C-reactive protein and fibrinogen and decrease plasma albumen. In addition to PIF and LMF, other cachectic factors are thought to be operative in specific cancers and in other chronic disease processes that induce primary and secondary PEM syndromes.
In 2-year chronic rodent studies, at about 80 weeks of a 104-week study in rats, a gradual decline in maximal body weights and obesity peaks is observed, with the advanced development of age-related terminal weight loss, decreased food intake, poor body condition, and mortality. These changes are due to multiple comorbidities (renal, cardiovascular, endocrine, and musculoskeletal diseases, as well as multiple tumors) occurring in these rodents (Keenan et al. 2005). Determining the specific cause of death can be challenging for the pathologist when presented with aging animals that have multiple serious organ diseases. This is because a given specific disease may not, by itself, account for the death, but the numerous comorbidities all contribute to the animal’s death. The default usually taken is to select the most severe lesion as the cause of death, which in most rat studies are advanced pituitary tumors or severe renal disease (Molon-Noblot et al. 2003; Keenan et al. 2000).
In subacute and subchronic studies, body weight loss and food consumption decrements are the most common indicators of general systemic toxicity, and test material-related effects often show a dose–response pattern. Reduced body weight gain or loss is usually considered of toxicological significance if the reduction is at least a 10% reduction of the mean control value. The role of reduced food intake in reducing weight gain can be assessed by calculating the “food efficiency index” (food consumed/body weight gain) in young rapidly growing animals. A reduction in food intake is not considered the main cause of reduced weight gain if the index is similar between control and treated groups. However, most cases of decreased food intake and body weight gains in a toxicology study will have more complex mechanisms that cause the weight loss or lack of weight gain, rather than simple decreased food intake as seen in DR per se. This should not be confused with DR that controls body growth gain directly without malnutrition. Weight loss due to disease and toxicity should not be confused with controlled growth rates with DR feeding or the excessive and abnormal growth rates as seen in AL overfeeding.
In long-term DR feeding studies, the mechanisms involved in extending healthy life span and controlling body weight and obesity have been well studied but are very complex. The most studied signaling pathways indicate reduction in chronic oxidative damage/oxidative stress is important; other signaling pathways of importance include the insulin/IGF-1 signaling pathway, the redox-signaling pathway as indicated by gene expression studies, the target of rapamycin (TOR), and possibly the sirtuin-signaling pathways. This list is not all inclusive and, since almost all metabolic, inflammatory, and growth mechanisms change in AL versus DR feeding, the definitive proximal mechanisms remain to be determined and are likely to be multifactorial and interactive (Hart, Neumann, and Robertson 1995; Bidlack and Rodriguez 2012; Bray 2011; Bagchi and Preuss 2013; Seki et al. 2012).
In adult AL-fed rodents, it takes about 6 weeks to stabilize the animal’s body weight and metabolism following a sudden switch to 40% DR feeding. Initially, metabolic rates drop as the animals catabolize excess body fat and adjust somatic growth to the new nutritional levels provided. However, once stabilized, the DR animals have equal or slightly higher metabolic rates than animals fed AL and consume the same amount of food/unit of body weight as their AL counterparts. What appears to differ is the manner of utilization of the nutrition in DR animals versus animals fed AL. After feeding, the DR animals have a rapid rise in RQ as they shift to carbohydrate metabolism from other macronutrients. Later, when their glycogen reserves have been used, a lowering in RQ occurs, and the animals metabolize dietary proteins and fats. The drop in RQ indicates the DR animals have less dependence on carbohydrates for energy and have a more diverse utilization of other macronutrients throughout the day. This shift in feeding from carbohydrate metabolism to fat and protein use after feeding maintains better glycemic control, and this prevents the development of visceral obesity in DR animals. In contrast, animals fed AL maintain a constant RQ and a constant ratio of macronutrient metabolism that does not aid glycemic control but favors the storage of excess energy as visceral adipose tissue. This leads to subsequent development of chronic obesity and type 2 diabetes and their associated comorbidities.
DR animals, as compared to animals fed AL, have a lower body weight, smaller skeletal size, decreased adipose tissue mass, and decreased weight of most internal organs except for brain and testis. This indicates that malnutrition has not occurred. This reduction in body size, adipose mass, and organ growth rates correlates with the delay or prevention of degenerative disease and tumors and the extended life or health span of the animals. As would be expected in a method that prevents endocrine disruption, moderate DR does not have adverse effects on estrous cyclicity or the onset of reproductive senescence in rodents as long as severe PEM that will impair animal reproduction is avoided. Since pregnancy and lactation typically increase protein and energy needs by over 30% of maintenance requirements, breeding females of most species should not be fed DR. However, DR feeding in rats to control body weight to as much as 70% of that fed AL had no adverse effects on fertility, number of fetal implants/dam, and sperm parameters. These data suggest that changes in reproductive indices associated with lower body weight in toxicity studies should not be discounted as being simply secondary to body weight loss due to reduced food consumption. In contrast, breeding mice with a similar sudden reduction in food consumption and body weight can experience major adverse metabolic, reproductive, and endocrine effects and should be evaluated as having experienced an adverse event (Chapin et al. 1993; Keenan et al. 2005).
Nutrients
Dietary components traditionally are divided into macronutrients and micronutrients. Macronutrients, which make up the bulk of the diet and provide energy, include lipids, carbohydrates, and protein. Fiber, while originally identified as indigestible in monogastric species, is now known to be digested partly by colonic microbiota, resulting in short-chain fatty acids that can provide an important energy source. This is just one example of how the intestinal microbiota of a given species is critical in utilization and production of most of the nutrients required for homeostasis and survival. If a microbial imbalance occurs, changes in the species microbiome can contribute to the development of obesity, metabolic disease, and inflammatory abnormalities (Bidlack and Rodriguez 2012).
Micronutrients include both vitamins and minerals, the latter being divided into the major minerals such as calcium (Ca) and potassium (K) and trace elements such as copper (Cu). The list of elements still fluctuates as new evidence emerges, supporting or opposing their essentiality; the element fluorine (F) is a good example. Although F does not appear essential for full growth, the American Dental Association supports a role for F in maintaining dental health, and it has proven to be a significant aid in preventing inflammatory oral disease and its systemic consequences. While a study from the National Toxicology Program suggests that even less than 1 ppm F, the dose provided in many municipal water supplies, has adverse effect on experimental rodent bone health and perhaps a small segment of the population, the significant public health benefits of supplemental F in drinking water far exceed any theoretical risk to the overall human and animal population.
Nutrients not only play an essential role in optimal growth of the organism but prevent the biochemical and pathological lesions of deficiency diseases, the characteristic nature of which can be used to diagnose specific nutritional imbalances. The dose required for optimal growth in humans is used to calculate the recommended daily allowance (RDA). The Food and Nutrition Board of the NRC of the National Academy of Sciences has set RDAs for 15 different age and gender groups, including pregnant and lactating women and domestic and laboratory animals. While the end point for production of optimal growth appears quite clear, determining the dose needed to overcome a deficiency disease becomes a matter of interpretation. The NCR Food and Nutrition Board do not extend its area of concern to the potentially adverse effects of doses of nutrients that greatly exceed the RDA. However, the availability of vitamin and mineral supplements on the market today permits ready opportunity for excess intake, resulting in overdose. Some nutrients have specific biological effects at doses in excess of the RDA, and most have nonspecific toxic effects if the dose is sufficiently large. A good example of a nutrient with a specific, pharmacologically useful effect at a higher dose is niacin, used to lower elevated plasma cholesterol levels but now only used when other drugs are contraindicated. This effect is unrelated to the effect of niacin as a vitamin and requires gram quantities rather than the milligram quantities needed for vitamin action. Even at the optimal pharmacological dose of niacin, one starts to see toxicity. Toxicity includes flushing and pruritus in response to unregulated prostaglandin production, which can be allayed by the use of aspirin prior to ingestion of these massive doses of niacin. If the dose is raised still further, hepatic toxicity can ensue.
For many minerals, excess ingestion competitively inhibits the uptake of other minerals, precipitating a secondary nutritional deficiency. For example, excess molybdenum (Mo) produces Cu deficiency, and excess manganese (Mn) produces iron (Fe)-deficiency anemia. In chronic zinc (Zn) overdose, first Cu deficiency is seen, and if the excessive intake persists, then Fe deficiency develops, both deficiencies due to competitive inhibition of intestinal uptake by Zn.
The concept of nutrient overdose, so apparent to the pharmacologist and toxicologist, rarely has been addressed through nutritional research or food regulatory channels. For food additives, such as the antioxidants butylated hydroxytoluene (BHT) and butylated hydroxyanisole (BHA), the concern for potential toxicity is recognized and regulated carefully. A few dietary components that are nutrients at low dose are recognized as particularly toxic or even carcinogenic at higher doses. For “noncarcinogenic” compounds, results from dose–response studies are used to calculate a no observable adverse effect level (NOAEL) or lowest observable adverse effect level (LOAEL). Determination of safe dietary levels (acceptable daily intake [ADI]) is carried out by dividing the experimental value by 100 in most cases. This calculation is based upon the theoretical idea that there is a threshold below which toxicity does not occur and takes into account possible variation in sensitivity among the species tested as well as among different genders and/or ages. An example of a nutrient that has been found to be toxic at higher doses is sodium (Na). The LOAEL leading to hypertension is considered to be 60 g/day. The ADI, if set two orders of magnitude below this, would be 0.6 g/day. Yet many apparently healthy individuals take in 10 g/day or more, suggesting that the DI/100 safety margin used to calculate the ADI is larger than necessary. For contaminants and other foreign chemicals not used intentionally, the term tolerable daily intake, TDI, is often used.
This exaggerated safety calculation may go far in explaining the dichotomy that has arisen for a number of trace elements, where the calculated ADI is of similar magnitude as the RDA. Selenium is an extreme example of this dichotomy, where inclusion in the diet (at no less than 0.04 ppm) is recommended to avoid deficiency diseases, even though higher doses have proven carcinogenic in rodents. The RDA for selenium (Se) is 55 µg/day for adult women and 70 µg/day for adult men; yet the Environmental Protection Agency (EPA) has set the maximum contaminant level in drinking water at 10 µg/L, even though the average daily consumption of water is only 2 L. Thus, the safety limit sits below the RDA. For nonnutrient carcinogens, the Delaney clause of 1954 required that no amount of a carcinogen is acceptable as a food additive, based on the now incorrect assumption that there can be no threshold for carcinogens. As the sensitivity of our methods for measurement of contaminants, such as pesticides, has increased, the Delaney clause has become more and more difficult to uphold. Today a tolerable increment in the risk for cancer has been set at the dose of 10−6 per lifetime.
Macronutrients
Proteins
Protein deficiency usually is combined with carbohydrate and fat (i.e., energy) deficiency or PEM as discussed earlier. In malnourished humans and animals PEM presents a range of clinical syndromes. Functionally, two differentially regulated protein compartments exist in the body, the somatic portion, represented by proteins in skeletal muscle, and the visceral portion, represented by protein stores in the visceral organs, primarily the liver. When the somatic component is mainly affected and energy (calories from carbohydrates and fats) is the predominant limiting factor, the condition is termed marasmus. When the visceral component is mainly affected and PEM is the predominant limiting dietary factor, then kwashiorkor is the resulting disease syndrome, a condition with more ominous consequences since both visceral and somatic protein reserves are depleted. This leads not only to wasting of muscle but also to atrophy of internal organs. Plasma protein concentrations decrease in kwashiorkor, leading to generalized edema in addition to generalized wasting, serous atrophy of visceral and peripheral fat stores, anemia, abnormal bone growth, endocrine atrophy, cerebral atrophy, bone marrow hypoplasia, fatty liver, thymic and lymphoid depletion, intercurrent infections, and skin lesions. In domestic and laboratory animals, generalized edema may not be evident, but serous atrophy of fat, visceral organ atrophy, anemia, poor hair coat, and predisposition to infectious disease are prominent features of protein and/or protein/carbohydrate malnutrition (PEM). Adverse changes attributable to PEM can be subtle and difficult to assess in a qualitative fashion histologically. Changes such as atrophy of fat can be discerned as an increase in myxomatous tissue with high ground substance in places where mature adipose tissue normally would be found, but other changes, such as atrophy of adipose cells within organs, can be more difficult, and sometimes impossible without ancillary techniques such as organ to body weight ratios and morphometry.
However, moderate protein restriction without malnutrition is associated with the reduction in or moderation of chronic diseases such as chronic renal disease. The reduction in protein intake can be difficult to differentiate from overall caloric restriction (DR) since both usually occur together. In relation to cancer, DR (usually involving caloric restriction as well) can reduce the incidence and/or susceptibility to spontaneous or induced cancer in most species tested. Decreased protein-energy intakes generally inhibit or delay the onset of spontaneous and chemically induced tumor growth as well as the growth of transplantable tumors. The evidence for protein restriction as a major means of preventing cancer is far from unequivocal, and the complicated interactions among protein, carbohydrate, and fat intake, and therefore overall caloric or energy restriction leading to prevention of cancer have yet to be resolved.
Protein excess seldom is of pathological relevance in most domestic species except in the context of individual amino acids since, in most cases, excessive protein is degraded, individual amino acids deaminated, and the carbon backbones enter the Kreb’s cycle or gluconeogenic pathways. However, excess protein is important in dietary management of animals with chronic renal failure since excessive protein intake, particularly of milk proteins such as casein, appears to increase the incidence and severity of chronic renal disease leading to the development of uremia.
The general rule that excess dietary protein is not harmful to an animal with adequate renal function may not be the case in rodents, where excess dietary protein has been thought to be a major factor in chronic progressive nephropathy (CPN), a common problem in most laboratory rats. The mechanism of excessive dietary protein in CPN was thought to be an enhancement of the glomerular filtration rate and glomerular hypertrophy and overall nephron growth. However, the hemodynamic basis of CPN has been disproven in most normotensive strains and stocks of rats (Baylis 1994; Hard, Betz, and Seely 2012). While the precise pathogenesis of CPN remains unknown, the causes appear to be under the control of physiological factors that depend on both dietary factors (increased protein and caloric intake) and hormonal growth factors such as excessive androgens, steroids, and growth hormone, and especially IGF-1 (Keenan et al. 2000; Molon-Noblot et al. 2003). The hypertrophied glomerulus leaks excessive proteins that accumulate as mesangial deposits inducing glomerular sclerosis, overwhelms the tubular protein reabsorption system, and eventually compromises overall nephron function, leading to glomerular and tubular dysfunction and loss and finally to renal failure. While the role of excessive dietary protein (especially milk proteins like casein) with excessive calorie intake cannot be discounted in the pathogenesis of CPN, its role appears to be excessive kidney growth stimulation through hormonal effects rather than a hemodynamic effect. But there is still a strong indication that dietary protein content should be controlled to just that required for maintenance in adult rodent diets (Keenan et al. 2000; Adams 2011).
Amino Acids
Deficiencies of specific amino acids are rare, although many diets can be relatively deficient in certain groups of amino acids. Removal of a single indispensable amino acid results in an immediate reduction in food consumption. Many corn-based diets are limited in lysine and tryptophan content, while peanut- and soy-based diets are often low in methionine content. A lack of tryptophan can result in cataract formation, corneal vascularization, and alopecia. Diets deficient in lysine lead to dental caries, impaired bone formation, and ataxia. A deficiency of methionine will induce NAFLD. General signs of amino acid deficiency are similar to those of kwashiorkor or PEU (Keenan et al. 2000; Adams 2011; NRC 1995).
Arginine is one amino acid in which a deficiency can result in disease, particularly in dogs and rodents, which as adults seem to have a requirement for this amino acid, normally not essential in adults of other mammalian species. A lack of arginine causes increased urinary excretion of urea, citrate, and orotate and decreased plasma and liver glutamate and glutamine. In dogs, the major manifestation is hyperammonemia, probably due to an inability to deaminate amino acids in the urea cycle. This ultimately leads to neurologic dysfunction and death (NRC 2006).
Taurine, 2-aminoethanesulfonic acid, derived from cysteine, is a free intracellular amino acid that generally is synthesized in sufficient quantities in all species except the cat, which must obtain additional taurine from the diet. This additional requirement may be due to the fact that the cat must rely exclusively on taurine for conjugation of bile acids. The cat will deplete its taurine stores to perform this conjugation function at the expense of other pathways. In cats with taurine deficiency, the principal lesion is progressive central retinal degeneration, characterized by degeneration of the photoreceptor portions of first cones and then rods. Eventually the entire retina is involved. Dilative cardiomyopathy also has been linked with subclinical taurine deficiency in cats, although the mechanism is unclear. Taurine mechanistically has a major role in maintenance of mitochondrial function in cardiomyocytes, maintaining respiratory chain function via regulation of mitochondrial protein synthesis as well as abrogating oxidant stress and maintaining Ca homeostasis (NRC 2006).
The addition of excessive amounts of individual amino acids at concentrations that are disproportionate to appropriate ratios in which they are required for normal growth causes a depression in food intake and growth in addition to any toxicity specific to the particular amino acid in question. Depression of food intake occurs soon after animals are fed the imbalanced diet. DA metabolites may be involved in this adverse feeding response as they are altered with amino acid–imbalanced diets. Inclusion of excess methionine or tryptophan in rat diets produces a severe depression of food intake and growth. Feeding excess amounts of methionine or N-acetyl-
An excess of dietary leucine retards rat growth rates. Dietary excess of leucine may be a precipitating factor in pellagra (niacin deficiency) when tryptophan is limiting and unable to meet a part or all of the requirements for the synthesis of nicotinamide nucleotides.
Phenylalanine is an amino acid with high toxic potential in the brain. The likely mechanism of phenylalanine toxicity involves inhibition of entry into the brain of large neutral amino acids. Phenylalanine toxicity is not a concern for most species except for humans with phenylketonuria, an inborn error of metabolism resulting from a lack of phenylalanine hydroxylase with a relatively high incidence in certain Caucasian populations. Since only about one-half of phenylalanine in children is utilized for protein synthesis, any inborn error in the alternative pathways of metabolism (usually the tyrosine pathway) can result in hyperphenylalaninemia and accumulation of toxic intermediates. Mental retardation that results from phenylalanine excess may be due to a combination of impaired lipid synthesis and altered neurotransmitter synthesis (Bidlack and Rodriquez 2012).
Lysine is a major constituent of hyperalimentation solutions and is known to inhibit tubular reabsorption of protein. Both lysine and aminoglycoside antibiotics are nephrotoxic, and when the two are combined, toxicity is additive. Lysine competes with aminoglycosides for a common receptor site on the proximal tubular brush border. Lysine is known to inhibit proximal tubular reabsorption of protein, an effect that promotes the formation of obstructive tubular casts. Also, excessive lysine is toxic to pancreatic acinar cells in the rat, with mitochondrial accumulation of Ca identified as an early abnormality. This results in the release of cytochrome c from the electron transport chain and activation of the mitochondrial permeability transition. Subsequently, focal areas of necrosis develop and lead to pancreatitis (NRC 1995).
Carbohydrates
Generalized deficiency of carbohydrates is of little consequence in most species, provided adequate lipids, proteins, vitamins, minerals, and micronutrients are present in the diet. Many carnivores do not require a carbohydrate source in the diet. However, most noncarnivorous domestic animals and most rodents require carbohydrates for successful reproduction and lactation. Absolute or relative carbohydrate deficiency in ruminants, rabbits, and guinea pigs, on the other hand, usually associated with starvation or inanition, additionally can lead to ketosis and fatty liver. This is particularly true during late pregnancy or lactation, when demand for glucose to nourish the rapidly developing late-term fetus (or fetuses) and to synthesize lactose for milk production is high. This results in excessive mobilization of fat and the formation of ketone bodies to meet the demands of other tissues in the body for energy. The deficiency of readily available glucose due to the peculiarities of ruminant carbohydrate metabolism sets in motion a complicated series of events that ultimately leads to severe fatty liver, even to the point of hepatic dysfunction and death (Jones, Hunt, and King 1996; NRC 1995).
Fatty liver in cats probably has a somewhat similar pathogenesis but this is not yet fully understood, although a relative carbohydrate deficiency that results when an obese cat abruptly stops eating probably has a role in triggering the excessive mobilization of peripheral fat stores that overwhelms the liver's capacity to process the fat, leading to its storage (NRC 2006).
Fatty liver in diabetes mellitus has a similar pathogenesis to fatty liver in ruminants, only in this case the glucose deficiency is due to inability of peripheral tissues to take up glucose, leading to mobilization of peripheral fat stores and dependence on the liver to process vast amounts of fat for production of ketone bodies (Bray 2011; Bagchi and Preuss 2013).
Severe caloric deficiency, whether due to insufficient carbohydrates or to combined carbohydrate–protein (PEM) deficiency, leads to fairly typical changes across nonprimate species. As discussed above, these changes include wastage (atrophy) of musculature and serous atrophy of fat in the diaphyseal bone marrow, perirenal and renal pelvic fat depots, in the coronary grooves of the heart and the abdominal mesentery. Thymic atrophy is a frequent finding in neonatal animals as well. In very small animals, serous atrophy may not be readily apparent but will instead manifest itself merely as a complete absence of visceral fat, including the epididymal fat pads in male rodents. Affected animals, especially the young, are prone to viral and bacterial infections and often will have manifestations of infectious disease, in particular pneumonia (Jones, Hunt, and King 1996).
It is difficult, if not impossible, to produce carbohydrate toxicity in the classical sense (excluding obesity). However, toxic responses to specific carbohydrates under certain situations, often when there is an inherited deficiency in a carbohydrate metabolizing enzyme, have been reported. An example of a toxicosis from feeding a particular carbohydrate is that of sucrose and fructose, both of which are toxic to neonatal piglets. Sucrose toxicity is a result of the low activity of intestinal sucrase (a disaccharidase) in the intestine of young pigs. Therefore, neither glucose nor the fructose is available as an energy source. Although neonatal pigs absorb fructose intact during the first week of life, they are not able to phosphorylate the fructose. Consequently, this hexose is not catabolized to trioses for energy utilization. As a result, animals experience osmotic diarrhea and limited growth (Jones, Hunt, and King 1996).
Another example of a toxic response to a specific carbohydrate is the inability of the laboratory rat to metabolize efficiently high concentrations (50% or greater) of sucrose (and fructose derived from sucrose) in the diet, leading to fatty liver disease and hypertriglyceridemia. The lesions are typical of hepatic lipidosis or NAFLD seen in other species and may be the result of enhanced FFA synthesis with a concomitant reduction in apolipoprotein synthesis, triggered by the exaggerated conversion of sucrose into acetate. In addition, high sucrose diets induce marked mineralization in the renal tubules of the outer and inner stripe of rodent kidneys, leading to renal failure. These findings have led to substantial changes in “purified” dietary formulations that utilize sucrose or fructose as a sole carbohydrate source for laboratory rodents (NRC 1995; Keenan et al. 2000).
Many neonatal mammals are unable to digest lactose to galactose and glucose due to a lack of intestinal lactase, leading to malabsorption and osmotic diarrhea. Some human infants do not have the enzymes needed for galactose catabolism (e.g., galactose-1-phosphate uridyl transferase and UDP-glucose-4-epimerase); therefore, they experience a toxicity syndrome characterized by vomiting, diarrhea, failure to thrive, icterus, brain damage, and cataracts. The specific mechanism of toxicity is unclear. The end result is fatty liver disease, cataracts, cerebral edema and gliosis, and gonadal dysfunction. Cats develop cataracts as a result of a buildup of galactitol, from an alternative pathway of galactose catabolism. The enzyme UDP-glucose-4-epimerase has an absolute requirement for nicotinamide adenine dinucleotide (NAD). Therefore, a dietary excess of leucine, which increases the requirement for tryptophan, necessary for in vivo synthesis of nicotinamide nucleotides, in conjunction with aberrant galactose metabolism, may result in toxicity. Excess dietary galactose results in food refusal by adult rats. Unlike lactose intolerance, due to intestinal malabsorption, galactose-induced flavor avoidance may be due to incomplete postabsorptive metabolism (NRC 1995, 2006; Bidlack and Rodriguez 2012).
Dietary fiber is the sum of polysaccharides and lignin which are not digested by the endogenous secretions of the GI tract. Dietary fiber includes substances not broken down by digestive enzymes and includes not only cellulose and lignin but also hemicellulose, pectins, gums, and mucilages. Fiber may be divided into soluble fiber and insoluble fiber. Soluble fiber can be metabolized by the microflora of the lower gut in nonruminants; whereas, insoluble fiber, or roughage, generally is nondigestible in monogastric species. This nondigestible fiber serves to retain water in the lumen of the lower gut and enhances the passage of ingesta, increases the size of the cecum and colon in rodents, and increases fecal bulk. The chemical constitution of fiber is not homogenous. Cellulose, hemicelluloses, pectin, and lignin are present in varying percentages in various types of diets. Fermentation of different fiber constituents by gut microbiota differs from substance to substance and depends on relative amounts of individual components. Short-chain fatty acids, such as butyric acid, released by microbiota fermentation of fiber in the cecum and colon, provide not only energy to colonic epithelium but also serve as important mediators of cellular differentiation in this portion of the gut. Increased fiber intake increases excretion of fecal nitrogen and decreases urinary nitrogen due to microbiota fermentation and utilization of dietary protein. This effect can lower the bioavailability of dietary protein but also may be beneficial to animals prone to chronic renal disease (NRC 1995; Keenan et al. 2000, 2005; Ziegler and Filer 1996).
Dietary fiber possesses underappreciated antitoxic effects. Diets high in fiber reduce gut transit time and have binding effect on secreted bile and secreted bile-bound metabolites, thus reducing exposure time of harmful substances to the intestinal mucosa. Also, fiber has the ability to retain water resulting in reduced concentrations of carcinogens so the direct effect on intestinal mucosa is reduced. Insoluble fiber undergoes minimal change in the digestive tract and primarily affects digestion by increasing stool bulk and promoting a shorter transit time.
Increased intake of insoluble fiber may result in binding of essential dietary minerals; therefore, ingestion of large amounts of fiber may interfere with intestinal absorption (Keenan et al. 2000; Newberne and Sotnikov 1996). For example, animals that have a higher intake of total dietary fibers may have lower magnesium levels. Theoretically, a loss of Ca and the development of osteoporosis might be enhanced under these conditions. The reduction in absorption of minerals and vitamins could have adverse nutritional consequences, particularly in populations fed high-fiber diets inherently deficient in these nutrients. Fiber may reduce and delay monosaccharide absorption, thus reducing the postprandial hyperglycemic phase, which can affect animals with insulin-dependent diabetes mellitus. The ingestion of dietary fiber may affect drug absorption directly, by reducing gastric emptying, by inhibiting mixing in the small intestine or by bile acid binding and therefore reducing enterohepatic recycling of excreted drugs and drug metabolites.
Lipids
Dietary lipids (fats) are important because certain fatty acids are essential for many species and lipids are needed as carriers for the absorption of certain essential nutrients, such as lipid-soluble vitamins. Metabolizable fat, in the form of triglycerides, is required to provide fatty acids from the n-6 family for optimal membrane-bound enzyme function and as a precursor for prostaglandin and leukotriene formation. The requirement for n-6 fatty acids can be met by dietary linoleic acid. The need for n-3 fatty acids can be provided by dietary oils that contain linolenic acid. Most of these requirements can be met for rodents in diets with 4 to 6% dietary fats. In reality, in developed countries, excess dietary fat, in the range of 25 to 40% total dietary calories, has contributed to the “epidemic” of obesity in humans, with all its unwanted consequences (NRC 1995, 2003; Bray 2011; Bagchi and Preuss 2013).
Whereas deficiencies of certain essential fatty acids, in particular linoleic (omega-6), linolenic (omega-3), and arachidonic acids, can be induced under experimental conditions, naturally occurring fatty acid deficiency is rare and requires a long time to develop. For most mammalian species, linoleic acid is essential, while for linolenic and arachidonic acids, there is considerable species variability in essentiality. Fatty acid deficiency may occur if biliary or pancreatic dysfunction leads to impairment of fat digestion and absorption, which also may lead to deficiencies in fat-soluble vitamins. Fatty acid deficiencies typically manifest themselves as skin abnormalities, including scaling and dermatitis with seborrhea, poor hair quality, and secondary bacterial infections, leading to dehydration and failure of wounds to heal. Dogs and cats, in particular, are sensitive to dietary deficiencies of linoleic acid, since they lack the enzyme necessary to synthesize this fatty acid. Dietary fatty acid deficiencies are typically accompanied by vitamin deficiencies, such as vitamin E, and commonly linked with excessively low-fat, dry, poor quality diets (NRC 1995, 2006).
There are numerous references to the enhancement of carcinogenesis resulting from ingestion of high-fat diets (Ames and Gold 1998; Bray 2011; Hart, Neumann, and Robertson 1995; Keenan et al. 2005). However, there is considerable confounding evidence when high fat intakes and low carbohydrate intakes are taken into account; therefore, the issue of separating high fat intake from high caloric intake is yet to be resolved. In studies with experimental animals, feeding diets high in total fat as a rule enhances carcinogenesis, especially colon carcinogenesis. In general, increased tumor incidence is observed in skin, lung, colon, liver, and/or pancreas when diets contain high levels of specific polyunsaturated fatty acids, including n-3 and n-6 omega fatty acids. However, mono- and disaturated fatty acids (e.g., oleic and linoleic acids) have been associated with decreased carcinogenesis. Monounsaturated fatty acids, in most studies, have had the reverse effect, enhancing carcinogenesis. Increasing dietary fat concentrations per se increases the incidence and multiplicity of mammary cancers in a wide variety of chemical carcinogen induced, radiation induced, and spontaneous, transplantable, and metastatic rodent cancer models. The effect of dietary fat on mammary tumor promotion may depend on the specific characteristics and peculiarities of the model under investigation and on the particular fatty acid composition and ratios. Experimental design often is predicated on the addition of a particular fat or fatty acid to a purified diet which is then fed to the experimental animals of choice; however, this does not account for the variety of fats ingested by humans.
Micronutrients
Major Vitamins
Vitamin A
Severe vitamin A deficiency, virtually unknown in Western countries, is still a disease of high morbidity in many underdeveloped countries. Severe vitamin A deficiency is the second most prevalent nutritional disease in humans behind protein-calorie deficiency. Marginal deficiency may be more common than generally thought, mainly because standard methods for assessing vitamin A status often do not take into account the varying activities and potencies of the various provitamin forms of vitamin A (e.g., carotenoids).
Vitamin A deficiency also can occur in cases of severe protein deficiency, where synthesis of retinol-binding protein essential for transport of vitamin A to peripheral tissues is inadequate to meet transport needs. Poor dietary intake associated with prolonged illness is a common cause of vitamin A deficiency. Since vitamin A uptake from the diet is dependent on bile, pancreatic enzymes, dietary lipids, and chylomicron formation, longstanding GI disease with malabsorption or maldigestion can result in deficiency. Liver damage (e.g., secondary to alcoholism or severe NAFLD) can also lead to vitamin A deficiency because >90% of the body's stores of this vitamin are present in the liver within perisinusoidal or stellate cells (Ito cells). In addition, the liver is responsible for producing the binding protein necessary for transport of vitamin A to peripheral tissues. The active form of the vitamin at the cellular level is retinoic acid. The main morphologic effect of vitamin A deficiency that underlies most of the lesions seen grossly is squamous metaplasia of pseudostratified columnar epithelium. This reflects vitamin A's essential role in the differentiation of mucus-secreting, ciliated, and noncornified epithelium. The metaplastic epithelium eventually cornifies and loses any ciliary structures as well as any mucus secreting capacity. The most common and perhaps the earliest outward manifestation of deficiency is night blindness (nyctalopia) followed by keratinization of the conjunctival epithelium and lacrimal glands, leading to “dry eye” or xerophthalmia. Both conditions are reversible in the early stages if the deficiency is reversed. If deficiency persists, however, perforation of the cornea and permanent loss of vision occur. Likewise, retinal lesions, although less obvious, are manifested as degeneration of rods, ultimately leading to atrophy of the entire retina. Unlike the corneal lesions, the retinal lesions and associated nyctalopia are not due to impaired differentiation but rather due to the role of 11-cis-retinal, a biologically active form of vitamin A, in the formation of rhodopsin, the light-sensitive pigment essential for night vision.
Keratinization is not confined to the eye in vitamin A deficiency, however, and squamous metaplasia with secondary loss of function is also present in the genitourinary, upper respiratory, and GI tracts. Impaired mucociliary clearance in the respiratory tract, due to loss of cilia and decreased mucus secretion, and malabsorption/maldigestion in the digestive tract due to metaplastic changes in absorptive epithelium may be responsible in large part for the increased susceptibility to respiratory infections and diarrhea noted in severely deficient individuals. Squamous metaplasia of glandular ducts, for example in the pancreas and salivary glands as well as cholangioles (experimentally in rats), is also common. Skin also can be affected, especially in domestic animals, with hyperkeratosis and hyperplasia leading to raised plaques on the skin. This change apparently is due to an overproduction of basal cells in the epidermis, leading to death of overlying epithelial cells and replacement with keratinizing squamous cells. Immune function is also affected, with impaired function of lymphocytes and macrophages. A classic example of the cutaneous effects of vitamin A deficiency is the historical “X disease” disease in cattle. This occurred after exposure to chlorinated naphthalenes once commonly used as an additive to petroleum-based lubricants and wood preservatives, which deplete vitamin A. Bone development and maintenance is frequently abnormal, for vitamin A is required for the normal functioning of osteoclasts. The subsequent imbalance between osteoclast and osteoblast function leads to the formation of excessive cancellous bone, often leading to narrowing of nerve foramina and resultant nerve dysfunction. Odontoblast and ameloblast function also may be affected, leading to defective tooth formation. Perhaps the most definitive manifestations of deficiency are impaired growth and death, which can occur fairly rapidly after overt signs of deficiency develop, especially in young animals. The link between vitamin A deficiency and cancer is not clear at this point although experimental evidence in rodents would suggest that there is an enhanced susceptibility to cancer in deficient individuals. There is accumulating epidemiological evidence for a link between smokers with low intakes of vitamin A and increased chances for carcinoma of the lung. This has become controversial in recent years when several intervention studies were terminated early because vitamin A supplementation was associated with an increased risk of cancer. Many large-scale epidemiological studies have failed to find a correlation between vitamin A status and cancer (Ames and Gold 1998; Ziegler and Filer 1996; Abelson 1995).
The toxicity of vitamin A (hypervitaminosis A) is manifested in acute and chronic disease. As an example of acute toxicity, adult humans can become intoxicated after eating an excess of marine fish livers or livers of animals in the food chain where marine fish are heavily consumed (e.g., seal and polar bear livers). Proforms of the vitamin, such as the carotenoids, generally are not toxic, even at very high doses, although individuals consuming excessively high doses may develop a yellow or orange skin. Retinoids used for control of skin conditions can be vitamin A agonists as well, binding to vitamin A receptors and activating them, producing a hypervitaminosis A type condition (Jones, Hunt, and King 1996).
Chronic hypervitaminosis A is much more common and insidious. It is generally associated with self-prescribed oversupplementation by humans or improperly mixed diets in domestic animals. Aqueous preparations of vitamin A are most prone to overdose, since absorption is better and there is less loss in the feces than with traditional oil-based preparations. Symptoms are variable and may not necessarily correlate with blood levels or even dosage above a certain point.
Toxicity at the cellular level is manifested by metaplasia of many types of epithelium into more complex forms, especially mucous respiratory epithelium. Accompanying this is decreased cohesion between epithelial cells in the skin. Accordingly, most affected humans report skin changes such as pruritus, erythema, eczema, and dermatitis with bleeding and cracking of the skin, especially around lips and gums, as well as hair loss and nosebleeds. Double vision and headache are frequent, probably due to increased cerebrospinal fluid pressure due to blockage of fluid outflow. In domestic animals there can be narrowing of the spinal canal and brain case due to bony changes. These changes are the result of increased proliferation of periosteal bone and bone near the growth plate. The bone initially becomes thicker and then less dense, weaker, and more prone to fracture as the osteoblasts become dysfunctional and die.
Enlargement of the liver has been reported due to an accumulation of vast numbers of vitamin A–laden perisinusoidal Ito cells with activation of perisinusoidal cells leading to collagen production and sinusoidal fibrosis and ultimate loss of hepatocytes and eventual liver failure. There is evidence that vitamin A also can be teratogenic, apparently because of its biological role in inducing apoptosis at the appropriate time in mesenchymal and epithelial development in the fetus, in particular the face, ears, eyes, digits, and brain (Ziegler and Filer 1996; NRC 1995, 2003, 2006).
Vitamin D
Vitamin D, the “sunshine” vitamin, is also a hormone. Vitamin D deficiency can result from three factors: insufficient exposure to sunlight (biosynthesis occurs in the epidermis by the action of ultraviolet light on 7-dehydrocholesterol to form cholecalciferol [nonhydroxylated vitamin D3], the first step in active synthesis), inadequate dietary intake of vitamin D3 in lieu of epidermal synthesis, or chronic renal disease due to impaired conversion by the renal convoluted tubules of the intermediate but inactive monohydroxy form of D3 (formed in the liver) to the active dihydroxy form. Occasionally, GI disease can cause deficiency. The main source of vitamin D (also known as calcitrol) is the skin where it is produced via activation by sunlight. Apart from the skin, vitamin D must be absorbed from the diet, and dietary sources are usually inadequate in regard to optimal vitamin D content (NRC 1995, 2003, 2006).
Vitamin D exerts effects at the cellular level via the vitamin D receptor (VDR), a receptor that forms a heterodimer with the vitamin A receptor (RXR) in similar fashion to retinoic acid. The complex then binds to and activates the vitamin D response element in the promoter regions of several genes, including the gene for calbindin D9k, the enterocyte Ca-binding protein that allows for increased uptake of Ca from the gut, and transport into the blood without altering free intracellular Ca concentrations. The link of vitamin D with cell differentiation (and apoptosis) has been most definitively defined in colon. The VDR also has epigenetic effects, many of which have been linked with maintenance and enhancement of muscular function and immunity.
In recent years, vitamin D deficiency (or rather insufficiency or marginal deficiency) has been implicated in osteomalacia (mineralization defect) in patients receiving long-term total parenteral nutrition. Deficiencies have also been reported in pigs and monkeys. Occult vitamin D deficiency has been identified in postmenopausal women with hip fractures, with increased healing and decreased recurrence in those supplemented with the vitamin (NRC 2003).
Major manifestations of deficiency are rickets in the young and osteomalacia in the adult. Although these conditions traditionally are associated with vitamin D deficiency, similar changes occur in dietary Ca or phosphorus (P) deficiency, although the biochemical mechanisms may differ. Characteristic features of rickets are enlargement of the metaphyseal regions of long bones and the nodular expansion of ribs at the costochondral junctions (i.e., rachitic rosary), with bending or bowing of the diaphyses and softening of the osseous matrix. Microscopically there is thickening and disorganization of the cartilaginous growth plate, with delayed ossification, retention of cartilage, and widening of the growth plate transversely. Osteoid matrix that does get deposited is unmineralized, and the marrow often is fibrous rather than hematopoietic. Osteomalacia in adults occurs by similar mechanisms but results in different lesions since the growth plate is no longer present; the primary change is an increase in deposition of unmineralized osteoid, resulting in a progressive thickening (and softening) of the diaphyseal portions of long bones and thickening of flat bones. The unmineralized bone is resistant to the action of osteoclasts; therefore, the osteoid progressively accumulates (Jones, Hunt, and King 1996).
Toxicity (hypervitaminosis D) is rare in both humans and domestic animals. In humans and domestic animals, clinical signs are similar to those of renal failure. In animals, often the animal is found dead either without any clinical signs or in terminal stages of kidney failure. The manifestations of vitamin D toxicity are due to vitamin D's primary effect on the small intestine, leading to increased synthesis of calbindin D9k. This results in excess absorption of Ca (and P) from the gut. Also, there is reabsorption of Ca (as well as P) by the kidney (via calbindin D28k) and from resorption of bone, especially if the animal survives the initial onset of toxic signs. This massive uptake of Ca and phosphorus leads initially to a profound hypercalcemia and hyperphosphatemia which may return to normal over time as the excess Ca and P (as phosphate) precipitate out as hydroxyapatite in peripheral tissues. Ca deposition is most significant in the kidney; affected individuals usually die of renal failure due to massive mineralization of tubular epithelium. Arterial tunica media also is prone to calcification, as are myocardium and skeletal muscle, all tissues with a high Ca turnover and which also have high phosphorus content. Deposition of Ca salts in the joint capsules may occur in long-standing cases; pulmonary mineralization in alveolar walls is a common lesion in young dogs that have ingested rodenticides that are cholecalciferol analogs (Jones, Hunt, and King 1996).
Unique bone lesions, considered by some to be pathognomonic for vitamin D toxicosis, are characterized by initial rarefaction of spongy bone, due to increased osteoclastic activity, and followed by deposition of intensely basophilic, disorganized fibrillar matrix. This defective matrix is produced by osteoblasts and is deposited on both endosteal and periosteal surfaces as well as on trabecula. The matrix becomes mineralized at some point well after its deposition by osteoblasts.
Under most circumstances, vitamin D toxicity is not a concern, but certain disease states can lead to a predisposition to vitamin D toxicity even when the ingested amount is only slightly above the recommended levels. In humans with chronic mycobacterial infections or certain chronic inflammatory conditions involving large numbers of activated macrophages, cytokines can be produced that result in increased bone resorption and hypercalcemia. Vitamin D exacerbates the situation by promoting absorption of Ca from the intestine. Renal failure patients being treated with vitamin D itself or vitamin D analogues, such as 1α-hydroxy-vitamin D, can become intoxicated. In domestic animals, there are three major causes of hypervitaminosis D: overzealous supplementation of diet with vitamin D, ingestion of rodenticides containing cholecalciferol (vitamin D3), and ingestion of plants containing cholecalciferols that can be metabolized into vitamin D or active vitamin D analogs (Cestrum and Solanum species). Vitamin D toxicity has been reported in dogs given excessive amounts of vitamin D–containing supplements (Jones, Hunt, and King 1996; Ziegler and Filer 1996).
The interactions of vitamin D with Ca, P, and parathyroid hormone (PTH) are complex and often seemingly paradoxical. Secondary hyperparathyroidism is a common feature of vitamin D deficiency, further exacerbating the osteomalacia associated with the decreased intestinal and renal absorption although PTH also stimulates the increased renal conversion of monohydroxy into the active dihydroxy form of the vitamin. Low dietary Ca as well as low dietary P will also enhance the renal conversion of the monohydroxy form to the dihydroxy form.
Vitamin E
Vitamin E (D-α-tocopherol) has attracted a large amount of attention in the popular press in past years as the “antiaging” vitamin and has been linked with protection against cancer, as well as a whole host of other positive effects. Biochemically, vitamin E has an antioxidant role in cell membranes that vitamin C and the glutathione/glutathione peroxidase/glutathione reductase system perform in the cytosol by lodging in cell membranes and protecting unsaturated lipids from free radical peroxidation. Although there are numerous studies reporting effects of vitamin E on regulation of antioxidant-signaling pathways, there is continued disagreement about whether these are direct effects or manifestations of vitamin E’s direct suppression of biological lipid free radicals, also regulators of gene pathways, as they are generated. Two major signaling pathways putatively influenced or regulated by vitamin E are the protein kinase C (PCK) and phosphoinositide 3-kinase (PI3K) pathways. Genes whose regulation are influenced by vitamin E independent of PKC and PI3K include genes that code for antioxidant or biotransformation proteins like CYP3A isoforms, the heavy subunit of glutamate cysteine ligase, and certain glutathione S-transferases; genes that code for proteins associated with lipid uptake (e.g., CD36, SR-BI, and SR-AI/II); genes that code for proteins involved in inflammation such as E-selectins, intergrins, TGF-β, and certain ILs (e.g., IL-4); genes that code for cell cycle proteins like peroxisome proliferator-activated receptor (PPAR-γ), cyclin D, cyclin E, and p27; and finally genes that code for matrix protein (e.g., matrix metalloproteinase [MMP]-1 and MMP-19). There is also some evidence that vitamin E can bind to and directly regulate the activity of some enzymes, notably protein kinase C (Bidlack and Rodriguez 2012).
Vitamin E deficiency diseases often respond to selenium supplementation as well as vitamin E; the precise etiology of these diseases often is complex and only partially understood. In some cases, the deficiency may be relative in the sense that increased dietary polyunsaturated fatty acids, which are prone to lipid peroxidation, may increase the requirement for vitamin E. In these cases, a deficiency state occurs even if the individual is consuming otherwise adequate doses of the vitamin. Many of the well-known diseases associated with vitamin E deficiency in humans and domestic animals, such as Keshan disease (necrotizing cardiomyopathy followed by fibrosis) in humans and white muscle disease (nutritional myopathy), hepatosis dietetica (hepatic necrosis), and mulberry heart disease (dietetic microangiopathy) in domestic animals can also occur due to deficiency in dietary Se (see Se discussion below). There are, however, several deficiency syndromes that respond to vitamin E supplementation alone and/or which can be produced in situations of “pure” vitamin E deficiency. The primary cellular effects of such a deficiency are in skeletal muscle and myocardium with swelling of mitochondria, dissolution of myofibrils, fragmentation of sarcoplasmic reticulum, and degeneration of the sarcolemma, leading to cell swelling, hyaline change, and oncotic necrosis of the affected cell. Precipitation of Ca salts is a frequent occurrence as the cell dies.
In animals, vitamin E deficiency results in a steatitis consisting of yellow, firm, subcutaneous, and abdominal deposits of ceroid-laden necrotic adipocytes, surrounded by neutrophils and macrophages. This syndrome has been reported in dogs, cats, mink, horses, pigs, and rats due to high dietary polyunsaturated lipids in the face of low dietary vitamin E consumption. Encephalomalacia (gray matter necrosis) in the cerebellum, triggered by necrosis of vascular endothelium in the brain, and axonal degeneration in the brain stem and spinal cord have been reported in chicks, rats, dogs, and monkeys with vitamin E deficiency. Testicular atrophy and aspermatogenesis are additional conditions connected with vitamin E deficiency in rats and guinea pigs, as is fetal resorption in rats and mice (NRC 1995, 2003, 2006).
Unlike its mineral counterpart, Se, vitamin E is relatively nontoxic; what toxic effects do occur typically are reversible. Toxic effects reported in humans include headache, nausea, fatigue, and double vision, with severe muscle weakness, GI disturbances and elevated serum creatine kinase, but these symptoms are primarily anecdotal in nature. Vitamin E toxicosis is rarely reported in animals; but in humans, at daily doses >1,000 mg, serious physiological effects have also been reported. The most critical is depression of prothrombin levels and decreased clotting time in patients undergoing anticoagulant therapy or deficient in vitamin K, apparently because excessive vitamin E impairs vitamin K utilization, either through competitive absorption or by prevention of oxidation. Another possibility is quenching of the endoperoxide intermediates during thromboxane production. Hemorrhagic tendencies have been shown experimentally in rats and mice, in both cases associated with a suppression of vitamin K–dependent reactions. Impaired wound healing has been reported as a consequence of hypervitaminosis E, probably because excess vitamin E may impair membrane turnover essential for macrophage activation and generation of prostaglandins and leukotrienes.
Vitamin K
Vitamin K has been found to serve as a cofactor in the posttranslational modification of two sets of proteins: (1) factors II, VII, IX, and X of the clotting cascade (as well as proteins C and S) and (2) the bone proteins, osteocalcin (bone Gla protein), matrix Gla protein (MGP), and periostin. These posttranslational modifications involve the selected carboxylation of glutamic acid residues necessary for these proteins to function properly, usually binding Ca. Vitamin K1, also known as phylloquinone, is derived mainly from leafy green vegetables and is primarily responsible for activation of coagulation factors. Vitamin K2, on the other hand, termed menaquinone, is derived mainly from bacterial action on K1, forming the MK-7 subtype, or by tissue cells themselves, forming the MK-4 subtype from K2. Both MK-4 and MK-7 appear to have key roles in promoting osteoblast deposition in bone and inhibiting osteoclast-mediated resorption of bone, in particular in the elderly. It is difficult to achieve a deficiency of vitamin K unless an individual's intestinal microbiota’s production of menaquinones (source of vitamin K2) is eliminated or disrupted (e.g., prolonged oral antibiotic therapy) and ingestion of fruits or vegetables (source of vitamin K1 or phylloquinone) is inadequate. Impaired lipid absorption due to chronic intestinal disease or cholestasis may contribute to deficiency, as with the other lipid-soluble vitamins.
Ingestion by cattle of sweet clover hay (Meliotus spp.), containing coumarin, a substance that frequently undergoes further chemical rearrangement to form dicoumarol, results in a potent vitamin K antagonism that over the course of a month or so results in severe hemorrhagic tendencies, including uncontrolled bleeding from nose and mouth, extensive subcutaneous and intramuscular bleeding from the slightest trauma, and bleeding into joints and abdominal organs as well as in the endocardium. Dicoumarol is the active component of warfarin, used not only as a rat poison but therapeutically in cardiac patients with thrombotic tendencies or postoperatively to prevent hypercoagulation of blood. Such use in actuality produces a relative but regulated vitamin K deficiency that must be monitored carefully and assessed in relation to the patient's dietary intake of vitamin K containing fruits and vegetables.
Perhaps the greatest cautionary note for humans and animals regarding vitamin K is for those being treated with vitamin K antagonists to prevent thrombosis. Ingestion of vitamin K rich vegetables (especially cruciferous vegetables) can lead to reversal of the vitamin K antagonism (with subsequent thrombosis) if the dose of antagonist is not adjusted accordingly.
Vitamin C
Water-soluble vitamins present a somewhat different picture toxicologically from fat-soluble ones, since their toxicokinetics are not the same. Water-soluble vitamins tend not to accumulate in tissues, partly because they are not lipid soluble, allowing them to be filtered out of plasma and excreted in the urine. For this reason, it is hard to “saturate the system” with these vitamins. They are not taken up or stored by any organ and any excess is excreted into the urine. Therefore, toxicities of water-soluble vitamins, when they occur, tend to be acute and regress rapidly if the animal survives the acute episode.
Vitamin C has a variety of functions; the most important, from a deficiency disease standpoint, is its role in hydroxylation reactions, in particular the hydroxylation of proline in collagen to stabilize its triple helical structure. Vitamin C also serves as a water-soluble antioxidant in a variety of vitamin- and mineral-dependent metabolic pathways and as a quencher of cytosolic free radical reactions. Vitamin C is an essential vitamin only in guinea pigs, fruit bats, and primates; all other mammals, birds, reptiles, and amphibians can synthesize their own vitamin C. Deficiency is seen in primates and guinea pigs in association with diets poor in fruits and/or leafy green vegetables. “Scurvy” (or scorbutus) is the hallmark feature of vitamin C deficiency; it takes approximately 1 month to develop in guinea pigs and up to several months in primates. Lesions of deficiency consist of gingivitis with hemorrhage and loosening of teeth, subperiosteal hemorrhages, periosteal proliferation without deposition of osteoid or mineral, and abnormal ossification characterized by calcification without ossification at growth plates (with resultant pathologic fractures). Poor wound healing is another hallmark feature of vitamin C deficiency (Jones, Hunt, and King 1996; NRC 1995, 2003).
Acute toxicity does not appear to occur after megadoses of vitamin C, although long-term high doses (generally above 1 g/day in humans) have been associated with GI disturbances such as nausea, diarrhea, and abdominal cramps, probably due to the osmotic effect of having a large bolus of slowly absorbed carbohydrate present in the gut. Perhaps of more import is that megadoses of vitamin C can cause an increase in the excretion of oxalic acid into the urine, which can precipitate as oxalate uroliths. This does not occur in most individuals, where this pathway of vitamin C metabolism is a minor one; however, there are those unfortunate few who have extremely efficient enzymes in their oxalate conversion pathway and urolithiasis can occur with doses of 1 to 2 g/day over the course of weeks to months.
Although humans and animals can tolerate enormous doses of vitamin C, adverse effects can occur in individuals with predisposing conditions. Excessive vitamin C actually can raise oxygen requirements for normal cell function, resulting in a loss of high altitude resistance in individuals taking megadoses. Excessive vitamin C increases the requirement for vitamin E, probably because the extra vitamin E is required to counteract the pro-oxidant behavior of vitamin C at high concentrations. Excessive vitamin C is dangerous to individuals with hemochromatosis, for vitamin C increases the uptake of Fe and therefore enhances the likelihood of liver or heart damage in affected individuals. Vitamin C can enhance turnover of bone and cause extensive remodeling of bone, which may not be desirable in elderly individuals. Individuals with chronic renal disease may not be able to excrete the increased acid load associated with megadoses of vitamin C (after all, it is an acid) and may suffer from acidosis.
The B Vitamins
Thiamine (Vitamin B1)
Thiamine was the first of the B vitamins to be discovered, and the results of its deficiency are well known, although its role in several diseases in domestic animals has been at times controversial and unclear. Thiamine is essential for several key metabolic reactions, among them are reactions linking the glycolytic pathway and the Kreb's cycle, reactions within the Kreb's cycle itself, and as a cofactor in the hexose monophosphate shunt, essential for the maintenance of axonal membranes. Overt deficiency disease in humans can manifest itself as “beriberi.” In “wet” beriberi, the initial abnormality is peripheral vasodilatation. This eventually leads to overload of the heart and bilateral failure with resultant edema. The “dry” form, on the other hand, leads to nonspecific peripheral neuropathy, with demyelination and loss of function. A unique neurological form of thiamine deficiency, Wernicke–Korsakoff syndrome, is observed most often in prolonged deficiency, especially in alcoholics, and is the result of hemorrhage and concomitant necrosis of neurons in the paraventricular regions of the thalamus, midbrain, medulla, and cerebellum. Thiamine deficiency is an ever present concern in elderly individuals or the chronically ill, where food intake may be insufficient to meet the daily thiamine requirements. A relative thiamine deficiency (thiamine-responsive encephalopathies) has been reported after pancreatitis, pancreatectomy, and prolonged parenteral nutrition.
The counterpart of Wernicke–Korsakoff in domestic animals has been termed Chastek paralysis, first described in cats, foxes, and mink consuming raw fish rich in thiaminase (NRC 2006). The lesions are similar in morphology and distribution as those in humans, although cerebral cortical gray matter may be involved as well. Myocardial lesions also have been described. Recently the Food and Drug Administration (FDA) or pet food companies themselves have recalled several types of canned meat- or fish-based cat food preparations due to the occurrence of clinical thiamine deficiency in pet cats fed diets exclusively composed of the particular preparations. Some studies have linked the occurrence of thiamine deficiency in some of these preparations to use sulfur dioxide as a preservation agent, resulting in the unintentional inactivation of thiamine in the food. Ruminants, whose microbiota normally produces ample amounts of thiamine, develop a thiamine-responsive disorder characterized by polioencephalomalacia, a laminar necrosis of gray matter of the cerebral cortex. Horses may suffer deficiency upon consumption of thiaminase-rich plants such as bracken fern (Pteridium aquilinum) or horsetail (Equisetum arvense; Jones, Hunt, and King 1996).
Choline
Choline is a component of lecithin, sphingomyelin, and the neurotransmitter acetylcholine. It is also important in one-carbon metabolism. Choline is required by most species and can be synthesized from methionine. Its metabolism is influenced by folic acid and vitamin B12 so a minimum requirement is hard to determine (NRC 1995).
Dietary choline deficiency causes the rapid development of a form of nonalcholic fatty liver characterized histologically by small (microvesicular) fat droplets within hepatocytes present initially in a centrilobular distribution that becomes panlobular with large (macrovesicular) fat droplets and ballooning degeneration. Initially it appears to be due to partial loss of phospholipid function in lipoprotein micelles with massive accumulation of triglycerides and abnormal membrane formation in hepatocytes. Chronic choline deficiency progresses from NAFLD to NASH and then to severe inflammation, bridging fibrosis and cirrhosis, vascular shunting, and hepatic failure. In addition, choline deficiency has severe, necrotizing effects on the heart and kidney tubules of young rodents and can be fatal in just a few weeks. Partial supplementation of choline can protect rodents from fatal kidney, heart, and liver disease but will not prevent NAFLD. This form of NAFLA/NASH has been used in rodents as a model of the human disease, but dietary choline deficiency causes a fatty liver by a different mechanism than that seen in diabetes and the metabolic syndrome. A chronic deficiency of major dietary methyl group donors—methionine, choline, folic acid, and vitamin B12—can induce development of liver cancer in rodents. Feeding methyl-deficient diets causes several molecular alterations, including altered lipid metabolism, and a number of epigenetic abnormalities that result in progressive liver injury culminating in the development of primary liver tumors. Importantly, this methyl-deficient model of endogenous hepatocarcinogenesis is one of the most relevant models of human liver carcinogenesis (Pogribny, James, and Beland 2012; Newberne and Sotnikov 1996).
Choline toxicity has been reported to depress growth rate and cause acute and chronic damage to the heart, lungs, liver, and lymph nodes. It can cause alterations in sensorimotor and brain development in rodents. The margin of safety between required and toxic doses is small for choline, so caution is needed in dietary choline supplementation.
Major Minerals
Calcium
Ca is the most abundant divalent cation in the body, constituting 1 to 2% of body weight. While 99% of total body Ca is found in the bone, maintenance of extracellular (10−3 M) and intracellular (10−7 M) Ca is so vital that, if intake of Ca is compromised, bone will demineralize to prevent hypocalcemia. Typically only about 30% of ingested Ca is absorbed. However, a small decrease in serum Ca triggers PTH- and calcitriol-dependent upregulation of absorption from the gut and decreased urinary excretion. If the diet remains low in Ca, the chronic hyperparathyroidism that results leads to loss of bone mineral density causing osteomalacia in adults and rickets in growing individuals. The lesions are very similar to those of vitamin D deficiency (see above). Osteomalacia is most pronounced in vertebrae, mandible and the proximal and distal ends of the long bones, and areas rich in trabecular bone, which turns over far more rapidly than cortical bone of long bone shafts. Diets high in protein or fiber can cause chronic Ca loss, the former through increased renal clearance of Ca, the latter through lumenal chelation of Ca and subsequent decreased absorption from the gut. Unless severe, these conditions are compensated for by increased uptake and decreased clearance, respectively. Acute Ca loss can occur in the presence of a chelator such as EDTA, precipitating convulsions, tremor, and tetany. For this reason, EDTA chelators typically are administered as the Ca salt. Ca, P, and Mg metabolism are related intimately, in that high dietary phosphate and low dietary Mg can lead to hypocalcemia. Thus, all three conditions result in the same pathological condition: renal failure, with loss of bone Ca and osteomalacia.
If dietary Ca is increased above 1%, daily food intake decreases, fecal weight increases, and growth is retarded. In one experiment, rats fed a 2% Ca lactate diet exhibited a sharp decrease in food intake, and growing rats (60 g at starting) died within the 15-day experimental period. Both protein and fat absorption are decreased by elevated dietary Ca. The mechanism is unknown, and, while it has been linked to the decrease in intestinal pH following ingestion of inorganic Ca salts, it is not clear that organic Ca salts cause any pH change, even though they also disrupt nutrient absorption. Experimental diets high in Ca alone, or in both Ca and P, cause renal tubular mineralization, commencing at the corticomedullary junction and progressing throughout the inner cortex. Initial deposits of Ca salts are present in the lumen of the descending portion of the proximal tubule, rather than intracellularly. This progresses to loss of the brush border with intracellular deposits appearing after extensive lumenal calcification. In the absence of concomitant increases in dietary P, a high Ca diet leads to phosphate deficiency, due to chelation and precipitation in the lumen of the GI tract. Phosphate deficiency then leads to vitamin D production. Therefore, in many species, rather than renal calcification, excess Ca produces osteosclerosis due to inhibition of osteoclastic activity by vitamin D. High dietary Ca also aggravates Mg deficiency.
Phosphorus
Under normal circumstances, most (70–80%) dietary P (as phosphate) is absorbed regardless of the dose, and regulation is by excretion in urine or feces, which in most cases is extremely efficient, especially by the kidney. Dietary phosphorus tends to be relatively abundant, so pure dietary deficiencies without concomitant deficiencies (e.g., K, Mn or Zn) or complicating factors (e.g., starvation) are rare. Much of the P in natural product diets is from plant products in the form of phytate P, which is largely unavailable to the animal. The NRC-recommended P requirement is based on highly available inorganic P, so when evaluating the P levels in the diet, one should consider the nonphytate P rather than the total dietary P concentrations (Keenan et al. 2000; NRC 1995). Dietary P deficiency in domestic animals is most commonly seen in ruminants, grazing on substandard pastures with acidic soil, soil leached by excessive rain or soils with a high clay content, leading to low phosphorus content in the forage. Pica or abnormal appetite is a common feature of phosphorus deficiency in both humans and domestic animals. Prolonged deficiency can result in osteomalacia with pathologic fractures. In less severe cases, there are less specific signs such as poor skin and hair quality, lethargy, poor growth, and poor reproductive performance. In phosphorus deficiency, vitamin D production is increased and is associated with development of a mild hypercalcemia. While this is thought to trigger a compensatory decrease in renal P excretion, the same change in renal P excretion is seen in parathyroidectomized animals, casting doubt on the mechanism of regulation of P excretion. If uncompensated, hypophosphatemia results in osteomalacia, cardiomyopathy, and a decrease in erythrocyte adenosine triphosphate (ATP) levels, precipitating an oxidative state and hemolytic anemia (Jones, Hunt, and King 1996).
The RDA for P (as phosphate) is 800 mg/day, but no adverse effects are observed even when P intake is raised to 2,000 mg/day, although this high P intake decreases Ca intake, renal clearance of Ca is decreased to compensate. However, in experimental animals when the P (as phosphate) to Ca ratio is raised above 1:2 for an extended period of time, secondary hyperparathyroidism results in osteomalacia and fibrous osteodystrophy, most obvious in the mandible. The use of orthophosphates and polyphosphates, both as animal feed additives and as food additives to perform numerous functions in the food industry, including acidifiers, emulsifiers, leavening agents and water binders, has led to a high dietary intake of phosphate in the Western world. This is of particular concern in relation to the high intake of phosphate in carbonated beverages that are so popular in America today. Similarly phosphate concentrations are quite high in such popular foods as hamburger and pizza. Although humans are relatively tolerant of a dietary imbalance in Ca and P, it is unknown whether the persistent Ca:P imbalance in most Western diets is causative in the osteoporosis of old age common in Western society. Furthermore, in chronic renal disease, in which serum phosphorus is already abnormally high, even normal dietary phosphate levels have the potential to induce toxicity, mainly manifested as vascular calcification.
Sudden, acute hyperphosphatemia from excessive pharmacologic administration of phosphates can cause such a rapid decrease in plasma Ca that tetany results. The common response to chronic hyperphosphatemia, on the other hand, is a mild hypocalcemia with hyperphosphatemia, which occasionally produces renal mineralization and, ultimately, renal failure. While this hypocalcemia causes fibrous osteodystrophy, intracellular Ca actually rises, producing pathological calcification, particularly in blood vessels, heart, and kidney. In rats fed a normocalcemic diet of 0.6% Ca, raising dietary P to 1.2% precipitated renal and aortic calcification. Therefore, pharmacologic use of P salts to lower blood Ca should only be undertaken with great caution, especially when renal insufficiency is present, where hyperphosphatemia is likely to be present. The problem is even more severe when vitamin D supplementation is used. In experimental studies as well as in clinical situations, excessive dietary intake of phosphorus, whether relative, as in chronic renal disease patients, or absolute (experimentally), plasma phosphorus levels are directly correlated with vascular mineralization and mortality.
Na/Cl/K
Diets composed solely of plants generally do not provide sufficient Na or chloride (Cl) for optimal health. Consequently from early times, humans as well as many mammals have sought environments where not only food and water are in abundance, but where there is a ready source of NaCl (salt). Since salts often must be added to animal diets, especially for animals in confinement or in restricted environmental locations (i.e., pasture), deficiencies and toxicoses have been associated with incorrect mixing of confinement diets or dietary supplements. Extracellular fluid has Na as its major cation, while potassium (K) is the major cation in intracellular fluid. Na, Cl, and K are absorbed fully from the GI tract and regulated primarily by urinary excretion, although sweat and diarrhea can contribute to excessive losses under certain circumstances. Homeostasis is lost in renal disease, when body Na, Cl, and water content increase, and in Addison’s disease, when decreased aldosterone production permits excretion of Na to increase over intake, slowly leading to volume contraction, hypotension, shock, collapse, and death. Salt deficiency is accompanied by a salt craving (licking of rocks in animals) followed by loss of appetite and signs of dehydration. Signs of K deficiency are weakness, lethargy, and cardiac arrhythmia, leading to death. Either increasing or decreasing the plasma K level disrupts depolarization across the cell membrane. The signs and symptoms of hyperkalemia and hypokalemia are therefore similar, both being due to disruption of myocardial function.
Salt restriction is found to lower blood pressure in many hypertensive humans, suggesting, but not proving, that excess salt will increase blood pressure. In contrast to Na, dietary K is related inversely to blood pressure. The mechanism is unknown, although it may be secondary to changes in blood Na, since urinary Na and K excretion are inversely related. In the Western world, blood pressure increases with age. It is not clear whether there is a potential health benefit from lowering salt intake (or raising K) as one ages. Animals can withstand very large doses of salt, provided adequate water is made available. Cattle given water containing 12 g NaCl or more per liter exhibit anorexia, have decreased water intake, lose weight, and finally collapse. In sheep, 13 g NaCl/L causes decreased reproductive rates, poor weight gain in lambs, some diarrhea, and an increase in mortality. Swine have been given as much as 30 g/L in water, with no obvious adverse effects. However, 6 or 8% NaCl in the diet will precipitate staggering, weakness, paralysis, and histopathological changes in the liver. Four or 5% NaCl in the diet causes reduced weight gain, diarrhea, and increased mortality in poultry. The greatest risk for toxicity is in ruminants, pigs, and poultry where excess salt in feed or water, or deprivation of water can lead to neurotoxicity. Lesions develop as a result of the inability of the cerebrospinal fluid to maintain ionic equilibrium with extracellular fluid, leading to cerebral edema and inflammation characterized by the accumulation of eosinophils (pigs) or neutrophils (ruminants) in Virchow-Robins spaces and necrosis of neurons in the cerebral cortex (polioencephalomalacia), especially in pigs (Jones, Hunt, and King 1996).
Dietary Contaminants
The issue of pesticides and other contaminants in the diet is a complex and controversial one for both animal and human diets and may not be a biological issue for a given contaminant when one considers the dose consumed and the increased sensitivity of current detection methods (Abelson 1995; Ames and Gold 1998; Newberne and McConnell 1980; World Health Organization 2011). Much attention has been focused on estimating an ADI or TDI for humans, a judgment based on the NOAEL for a particular compound. The calculation of an NOAEL is based on determining the dose threshold in the most sensitive test or assay in the species most sensitive to the compound in question, with an additional “safety factor” multiplied into the equation. The calculation of an ADI is complicated by a variety of things, one of which from the nutrition aspect is the fact that children consume more food on a body weight basis than adults and that neonates and females of reproductive age have other considerations that must be taken into account when making the calculation. In the context of estimating ADIs for “contaminants” in the diet, it must be remembered that fruits and vegetables contain “naturally” occurring carcinogens, yet the overall consumption of such foods consistently has been identified in epidemiologic and experimental studies as “beneficial.”
Pesticides
With regard to pesticides much of the current research has been focused on the carbamates and organophosphates, with much debate centered on which rodent assay should take precedence when determining NOAEL and which uncertainty factor should be used for estimating the ADI—the rodent uncertainty factor of 100× or the human-based uncertainty factor of 10×. Complicating the debate is the observation that not all carbamates or organophosphates have the same mechanism of action, requiring modification of old or development of new assays. Some scientists have concluded that covalently bound pesticide residues in foods (e.g., edible animal tissues) are either not bioavailable or nonreactive in their covalently bound form and hence of little concern to humans. Much of the evidence to date has been nebulous or inconclusive regarding low levels of pesticides in the diet and the risk of toxicity or cancer in animals or humans. Many of the pesticides (e.g., chlorinated hydrocarbons) that have been shown to be carcinogenic in rodent assay systems have yet to be shown to be associated with cancer in humans, and studies using mixtures of common pesticides at levels which might be encountered realistically in the diet have been negative for genotoxic or other DNA damage in in vitro systems or rodents.
Mycotoxins, Heavy Metals, Phytoestrogens, and Other Contaminants
Of more relevance to the toxicological pathologist is the contamination of diets used to feed animals in experimental situations. Harmful naturally occurring substances as well as man-made contaminants are routinely found, generally at very low concentrations, in commercial animal diets. This is a potentially important problem when “natural” ingredients are used, the composition of which may vary considerably from batch to batch (Newberne and McConnell 1980). This is particularly true when using plant material that may have widely varying concentrations of phytochemicals, complex organic compounds produced for various reasons such as protection against predators, sequestration of harmful substances absorbed from soil, and storage of unusable metabolic waste. Phytochemicals have no nutritional value for animals consuming them but are often “biologically active,” meaning at the very least they can and will trigger phase I or phase II (or both) biotransformation systems. In some cases, phytochemicals are highly toxic; in others, much less so but nevertheless affecting biotransformation systems, hence altering baseline metabolism. Phytochemicals vary widely among plant species and even among plants within a plant species, depending on the growth conditions, variety of plant grown, and processing for dietary inclusion. Thus, when plants known to contain large quantities of phytochemicals are used in diets, it is often critical to know what the phytochemical composition and concentrations are, especially when using different batches of dietary material from different sources or harvest times (Keenan et al. 2000).
The problem of pesticide contamination is sometimes not considered when purchasing materials for inclusion in the dietary formulations, if the diets are not tested by the manufacturer for contaminants. For laboratory animals used in toxicological studies only certified diets should be used so that each batch is assayed for suspected environmental contaminants, and the chemical composition of the diet, including the maximum concentrations of any known or suspected contaminants present are documented. Since blanket analysis for all types of pesticides is impossible and would be prohibitively expensive, a careful and thorough knowledge of the source of the material and any potential pesticides used is essential (Keenan et al. 2000).
In addition, certain nutrients can assume exaggerated relevance when some experimental compounds are studied. For example, dietary folate becomes an important covariate when studying contraceptives or anticonvulsants. In rodent cancer studies, low dietary folate increases the risk of colonic carcinogens and depresses the safety and efficacy of chemotherapeutic agents like cyclophosphamide. In similar fashion, high dietary Fe can exacerbate aminoglycoside ototoxicity due to the generation of free radicals and depletion of glutathione in the ear, while high dietary Ca can affect Zn absorption and metabolism, altering appetite and a variety of other metabolic processes. Phytochemicals become potentially important when studies involving induction or suppression of phase I or phase II drug metabolizing enzymes are being performed or when the toxicity (as well as safety and/or efficacy) of a drug is dependent upon induction or suppression of a particular detoxification enzyme or endocrine disruption studies are being performed.
Phytoestrogens in laboratory animal diets have attracted attention regarding the potential deleterious effects of high doses of these substances on a number of biological systems. Phytoestrogens are nonsteroidal compounds (e.g., isoflavones like genistein, daidzein, glycitein, coumestans, and lignans) produced by many plants, including soybeans, wheat, barley, corn, alfalfa, and oats. Although not steroids, phytoestrogens mimic or antagonize some of the actions of endogenous estrogens, but their potency is much lower than steroidal estrogens. At concentrations found in plants such as in soybeans, phytoestrogens have been shown to have potential health benefits against heart disease, osteoporosis, and certain cancers. However, experimental studies of high doses have shown certain adverse effects and endocrine disruption that cause some to advocate phytoestrogen-free diets. Removal of these substances from natural foodstuffs would not be advisable; however, knowledge of the types of phytoestrogens and their concentrations might be critical for appropriate interpretations of results. Besides the health benefits of normal levels of phytoestrogens in plants, very low levels may be deleterious to the animal’s health and have the potential to retard normal growth and maturation. Manufacturers can provide phytoestrogen profiles and concentrations present in each batch of certified diets and this approach is much more sensible from a scientific point of view for toxicity studies. Similar concerns are present for mycoestrogens consisting of zearalenone and its metabolites. These are also nonsteroidal compounds that interact with estrogen receptors. They are produced primarily by the fungus Fusarium graminearu that infects corn, wheat, and other grains. Pigs are the most sensitive species.
More attention has been paid to sources of components for dietary formulations in recent years, and the use of certified diets has minimized potential contamination problems. Furthermore, by paying closer attention to component sources, major variations in dietary concentrations of various bioactive compounds that have complicated past bioassays are now less likely to be major problems in the Good Laboratory Practices (GLP) environment. Past variation has resulted in appreciable variations in response in certain species to specific drugs or chemicals both within studies and between studies. This is especially relevant in the context of mycotoxin contamination of dietary components, where minuscule concentrations can cause significant alterations in response to test compounds or even trigger toxicity in a sensitive species (e.g., aflatoxins, fumonisin B1, and trichothecenes). Common contaminants of experimental and commercial animal diets that have been identified in the past include mycotoxins such as aflatoxin, heavy metals (lead, mercury, cadmium, and arsenic), nitrates, nitrosamines, chlorinated hydrocarbons, and polychlorinated biphenyls. In addition, microbial contamination remains a potential danger in all foodstuffs, including bacterial contamination from pathogenic Escherichia coli, salmonellosis, listeriosis, and other microbial pathogens. Even sterilized animal protein sources have been associated with contamination from heavy metals like organic mercury in fish meal and prion proteins (scrapie, bovine spongiform encephalopathy, and chronic wasting disease) in animal protein products. Therefore, it is of prime importance that all dietary components, either natural or formulated, be evaluated, tested, analyzed, and certified before inclusion in any experimental diet intended for a GLP toxicological studies for human safety assessment.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.
Authors’ Note
Portions of this review are presented in more detail in M. A. Wallig and K. P. Keenan (in press) Nutritional Toxicologic Pathology (chapter 36). In: Handbook of Toxicologic Pathology, 3rd edition, Haschek, Rousseaux, and Wallig (Eds), Academic Press.