
Abstract
In this study of chemoprevention in the rat modified resistant hepatocyte model, preneoplastic cells were diminished by >60% with quercetin pretreatment compared with those rats treated with N-Diethylnitrosamine (DEN) to induce liver cancer. This decrease occurred associated with an abolished DEN-induced lipid peroxidation as well as activation of caspase 9 and increased caspase 3, as determined by increased expression of cleaved caspase 3 and 9, but not cleaved caspase 8 and increased fragmentation of Poly (ADP-ribose) polymerase (PARP) inducing apoptosis of presumed genetically injured cells, when quercetin was administered before the initiation agent.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common type of cancer and the third most common cause of cancer deaths worldwide (Ferlay et al. 2010). HCC is a major health problem due to its poor prognosis and lack of effective treatment options. The prognosis of HCC following surgery remains dismal due to high rates of recurrence and metastases. Based on several models and epidemiological data, the carcinogenic process involves the general aberrant actions of signaling pathways that are related to survival, differentiation, cell proliferation, the loss of antigrowth signals, evasion of apoptotic mechanisms (escape from senescence), and the capacity of cells to induce angiogenesis and to metastasize (Anisimov 2007; Hanahan and Weinberg 2011). There are multiple risk factors associated with malignant cell transformation, including aging, genetic predisposition, and physical inactivity. It is currently believed that 90 to 95% of all cancers are attributable to lifestyle and diet, with the remaining 5 to 10% linked to genetic defects (Benetou et al. 2008; Freedman et al. 2008; Aggarwal et al. 2009). It is generally accepted that aflatoxin, hepatitis B and C virus infections, and excessive alcohol consumption are the predominant risk factors worldwide for hepatocarcinogenesis (Perera 2000). Chemoprevention strategies are an important medical consideration relative to HCC, as well as for other cancers.
Chemopreventive agents are chemicals that prevent or reverse the onset of disease (Weinstein 1991). Numerous in vitro and in vivo studies have demonstrated the protective effects of natural and synthetic substances, fatty acids, and vitamins through a variety of mechanisms of action (Mikstacka and Ignatowicz 2010). Epidemiological studies have shown that a high intake of fruits and vegetables may reduce the incidence of cancer (Benetou et al. 2008; Freedman et al. 2008). Flavonoids are a family of polyphenols with low molecular weights that are present in vegetables and fruits. Quercetin is a prominent flavonoid in the human diet; it can act as an antioxidant and anti-inflammatory agent; it can reduce edema formation; it can induce cell death by apoptosis in tumor cells; and it efficiently chelates iron affecting redox cycling (Williamson and Manach 2005; Kim et al. 2010; Senthilkumar et al. 2010; Sun et al. 2010).There are considerable in vitro and in vivo data indicating that quercetin is an anticarcinogen; therefore, its role in early carcinogenesis is of significant interest (Gibellini et al. 2011; Li et al. 2012). The molecular mechanisms involved in the preventive effects of quercetin in cancer appear to be complex. To validate and further characterize the utility of quercetin as a chemopreventive treatment, its effects have been examined in animal models of carcinogenesis.
Using hepatocarcinogenesis models to evaluate the effects of quercetin allows observations of lesions at multiple stages and enables exploration of the mechanisms of action. Quercetin administration before the initiation stage of carcinogenesis has been shown to reduce tumor formation (Volate et al. 2005; Devipriya, Ganapathy, and Shyamaladevi 2006; Kamaraj et al. 2007). A modified resistant hepatocyte model (MRHM) has been utilized to further elucidate quercetin activity and its relevance in chemoprevention. In MRHM, three interventions are capable of reproducing the stages of liver cancer in rats in a relatively short time period (Marche-Cova et al. 1995). Administration of a necrogenic dose of N-Diethylnitrosamine (DEN) induces genetically altered hepatocytes during initiation. The initiated liver cells, resistant to the mitoinhibitory effect of 2-acetylaminofluorene (2-AAF) administration, are promoted and with the proliferative stimulus of hepatectomy develop into groups of altered hepatocytes and larger nodules, considered as preneoplastic lesions (Farber 1984; Rotstein et al. 1984), which are demonstrated using a gamma-glutamyl transpeptidase (GGT) histochemical assay. Induced liver preneoplastic lesions in rats are similar to human dysplastic lesions in terms of morphological, histochemical, and molecular characteristics (Pascale et al. 2002). Previous studies of HCC in rats and humans have identified similarities between the molecular changes that occur in both species and have described the overexpression of similar proteins (Lee et al. 2004). This model thereby allows for the study of the carcinogenic process and the capacities of various agents to prevent or reverse disease progression (Perez-Carreon et al. 2006). This study investigates the capacity of quercetin to diminish the formation of preneoplastic lesions when administered before carcinogenesis treatment and characterized some important molecular alterations involved in the anticarcinogenic activity of quercetin.
Materials and Methods
Animals
All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee. Male Fischer 344 rats (180–200 g) were obtained from the Production Unit of Experimental Laboratory Animals at UPEAL-Cinvestav, Mexico City, Mexico. The rats had free access to water and standard laboratory diet. Carboxymethylcellulose (CMC) at 0.5% in an aqueous solution was used as the vehicle for quercetin, and H2O was used for DEN. A group of rats were subjected to the full procedure of the resistant hepatocyte model (FT group) for the first protocol (Figure 1A; Marche-Cova et al. 1995). In this protocol, the HCC process was initiated in rats by intraperitoneal administration of 200 mg/kg DEN. At 7, 8, and 9 days after initiation, 20 mg/kg 2-AAF was orally administered to the rats. On day 10 after initiation, they underwent partial hepatectomies. The group of rats that received the full resistant hepatocyte model treatment and pretreatment with 10 mg/kg of quercetin for 2 hr before initiation was named Q + FT (Figure 1A). All animals in this first protocol were euthanized 24 days after initiation, livers preserved in formalin, and preneoplastic lesions were quantified. For the second protocol, Group D was treated with DEN only (Figure 1C). The quercetin plus D group (Q + D) was exposed to DEN after treatment with 10 mg/kg of quercetin for 2 hr before initiation. Two more groups were included as controls of CMC and quercetin administration and were named the CN and Q groups, respectively (Figure 1C). All rats in this second protocol were euthanized 24 hr after treatment with the initiating agent, DEN. The livers were removed under ether anesthesia 24 hr after DEN administration, washed in a physiological saline solution, flash-frozen in 2-methyl butane with liquid nitrogen, and stored at −80°C until they were analyzed. Additional portions of liver were fixed in formalin. There were five animals in each treatment group in both protocols.
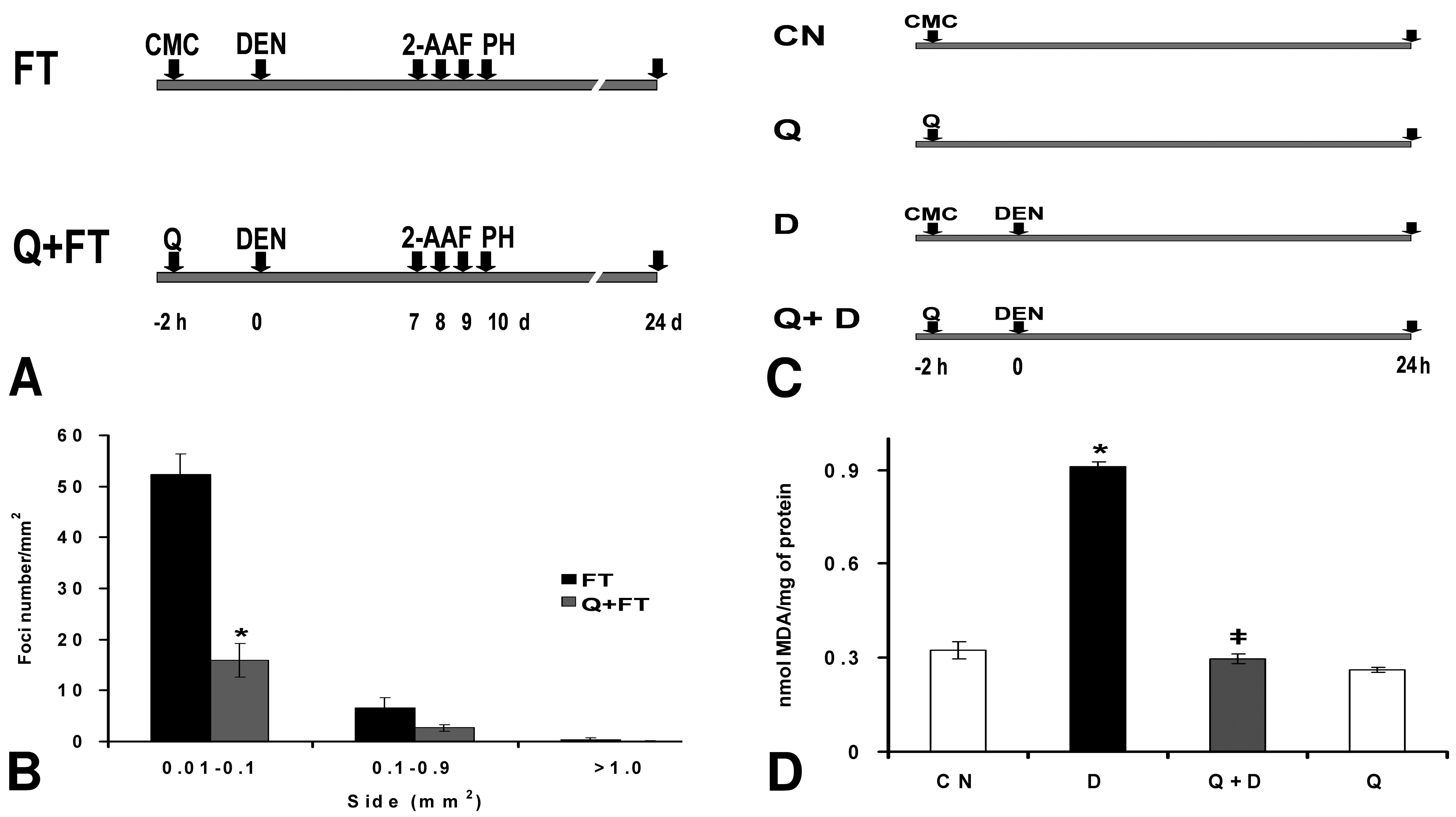
Effect of quercetin on chemoprevention of preneoplastic lesions. A. The groups were subjected to the full procedure of the resistant hepatocyte model (FT group) and sacrificed 24 days after exposure to DEN. B- Foci number/mm2 at intervals of 0.01 to 0.1 mm2, 0.1 to 0.9 mm2, and greater than 1 mm2. CMC+FT denotes the group with the quercetin vehicle (CMC) and the complete carcinogenic treatment; Q+FT indicates the group that was treated with quercetin at a dose of 10 mg/kg and the complete carcinogenic treatment. C. The experimental design used in this project to evaluate the effects of quercetin on the initiation of hepatocarcinogenesis. The rats were sacrificed at 24 hr after exposure to DEN. D. The effect of quercetin on the inhibition of lipid peroxidation as caused by DEN. Lipid peroxidation was determined in terms of TBARS at 24 hr after exposure to DEN. *p < .0001 versus the CMC+FT or CN group. ‡p < .0001 versus the D group.
Chemicals and Reagents
DEN, 2-AAF, quercetin, GGT, tetramethoxypropane (TMP), and phenylmethanesulfonyl fluoride (PMSF) were acquired from Sigma Chemical Co. (St. Louis, MO). The Lowry Assay Kit for protein concentration determination was purchased from BioRad (Richmond, CA). COMPLETE™ protease inhibitor cocktail tablets were provided by Roche Molecular Biochemicals (Mannheim, Germany). The DAB-Plus Substrate kit and Cas-Block (00-8120) were obtained from Zymed (San Francisco, CA). A DeadEnd™ Colorimetric TUNEL System Kit was purchased from Promega (Madison, WI). Anti-caspase 3 and anti-PARP reagents were obtained from Cell Signaling (Danvers, MA). Antibodies against caspases 8 and 9 were purchased from Santa Cruz (Santa Cruz, CA).
Histology
Frozen sections of 20-µ thickness were prepared from the liver slices and stained to detect GGT (Rutenburg et al. 1969). The stained liver images were captured, and GGT-positive labels were quantified using analysis software (AnalySIS Soft Imaging System GmbH, Germany). In addition, 3-µm-thick paraffin-fixed sections were processed for routine histological examinations by staining with hematoxylin and eosin.
Determination of Thiobarbituric Acid Reactive Substances (TBARS)
Lipid peroxidation was measured using the TBARS method (Buege and Aust 1978). The frozen liver samples were homogenized, and 650 µg of liver homogenate protein and 300 µl of 0.4% thiobarbituric acid in 20% acetic acid (pH 3.0) were mixed and heated at 100°C for 45 min. The samples were then cooled, and 200 µl of 1.2% KCl and 0.5 ml of 1:15 pyridine/butanol were added. The samples were centrifuged at 7,500 rpm for 10 min. The absorbances of the supernatants were measured at 532 nm. The data were interpolated from a TMP standard curve, which was used as a reference control. The TBARS levels are expressed as nmol of malondialdehyde (MDA) per mg protein.
Liver TUNEL Assay
Paraffin-embedded liver tissue sections of 4-µ thickness were processed using the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay to assess DNA fragmentation (Promega, WI). Based on the manufacturer's instructions, tissue that had been treated with DNase I was used as a positive control. The sections were lightly counterstained with hematoxylin, dehydrated, and mounted. The tissue images were captured by optical microscopy (Olympus 1X70, Olympus Europa GmbH, Hamburg, Germany). Cells that were positive for TUNEL were then quantified in ten randomly selected fields (20× magnification) per individual sample and were recorded using image analysis software (AnalySIS Soft Imaging System GmbH).
Western Blot (Caspase 8 and 9, Procaspase 3, and Cleaved PARP)
The liver tissue samples (∼100 mg) were homogenized in a 10 mM HEPES buffer containing 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, and 0.1 mM EDTA at pH 7.4 and supplemented with 0.1 mM PMSF and a protease inhibitor cocktail that had been prepared according to the manufacturer’s directions. The proteins were separated by SDS-PAGE and then transferred to a Polyvinylidene fluoride (PVDF) membrane. The protein of interest was visualized using the indicated antibody in a chemiluminescence system. Actin was used as a protein loading control.
Results
Quantification of Preneoplastic Lesions
To assess the preneoplastic lesions, each of the carcinogenesis stages can be followed using different molecular markers known as tumor markers. One well-described tumor marker is the gamma-glutamyltranspeptidase (GGT). GGT is not expressed in adult at hepatocytes but is highly expressed in most of the chemical carcinogenesis models; its induction can be seen to alter the administration of carcinogens and is present until tumor formation. Due to its pattern of expression from the first until the last stages of liver tumor development, GGT is actually considered as an early tumor marker (Pitot et al. 1996).
A single dose of 10 mg/kg quercetin (Q + FT Group) introduced 2 hr before the administration of the initiation agent, DEN, reduced the development of preneoplastic lesions at day 24 in the MRHM. The GGT-positive total area and number of GGT-positive foci were significantly reduced. We examined the effectiveness of quercetin in reducing the formation of preneoplastic lesions in three size ranges (0.01 to 0.1 mm2, 0.1 to 0.9 mm2, and >1 mm2) and found that it reduced the formation of lesions measuring 0.01 to 0.1 mm2 by 70.1% (p < .0001) and those measuring 0.1 to 0.9 mm2 by 60% (Figure 1B) when compared with the MRHM only (FT) treatment group. This effect was analyzed at day 24, when lesions >1 mm2 were rare in the FT group precluding determination of an effect in this size range.
Assessment of Oxidative Stress
DEN administration (Group D) caused oxidative stress with significantly increased (2.8-fold) lipid peroxidation compared with the CMC controls (CN Group) at 24 hr (Figure 1D). Pretreatment with quercetin (Q + D Group) was able to prevent the increased oxidative damage that was present in the DEN-treated group, resulting in a 68% reduction in lipid peroxidation. These results indicate that quercetin inhibits the lipid peroxidation levels at 24 hr caused by the metabolism of DEN. Furthermore, the Q + D Group had similar levels of lipid peroxidation to those of the CMC controls (CN Group). The single administration of quercetin (Q Group) did not induce changes in the lipid peroxidation levels compared to the CN Group. Thus, quercetin clearly protected cells from DEN-induced lipid peroxidation.
The carboxymethylcellulose control group (CN) presented with normal hepatic lobules, each having a normal hepatic vein and containing blood vessels and bile ducts comparable to the quercetin control group (Q).
Analysis of Apoptosis
Induction of apoptosis in initiated cells was evaluated using the TUNEL assay. Administration of quercetin 2 hr before administering DEN clearly enhanced apoptosis in hepatocytes (Groups Q + D vs. D). Quercetin did not increase apoptosis in the livers of nontreated rats (Groups Q vs. CN). Representative tissue sections from each treatment are depicted in Figure 2. Significantly increased apoptotic nuclei and apoptotic bodies were observed in the D group, and an even greater increase was observed in the Q + D group. At 24 hr post-initiation, the Q + D group had 3.7 times the number of apoptotic nuclei compared with the D group. A 1.9-fold increase occurred in the number of apoptotic bodies in the Q + D group when compared with the D group at 24 hr post-initiation (Figure 3A and B). These results indicate that the chemopreventive effects of quercetin occurs, at least in part, by induction of apoptosis in the liver.
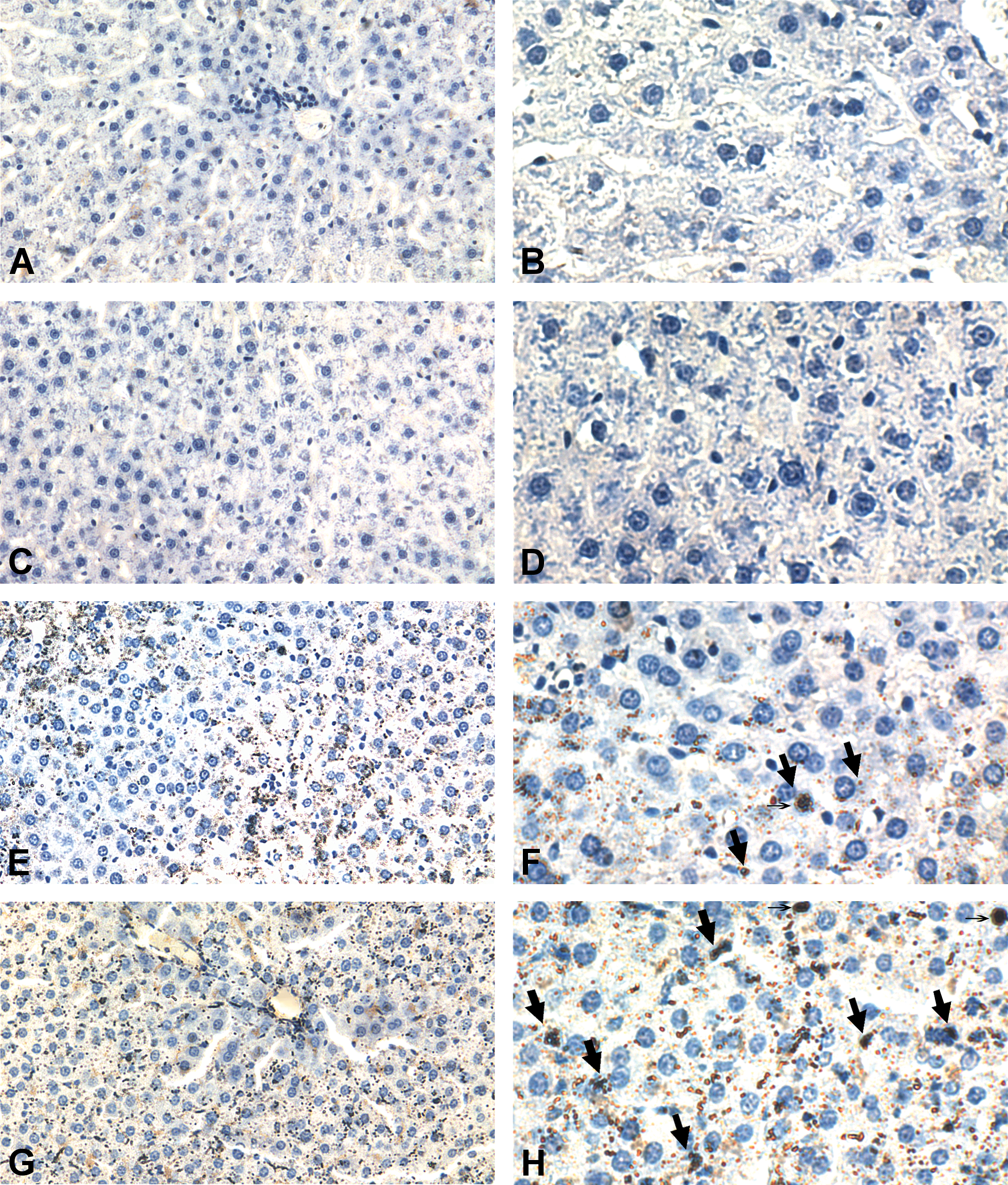
Chemopreventive treatment of quercetin induces post-initiation apoptosis. Representative liver sections from each treatment. A and B. CN group. C and D. Q group. E and F. D group. G and H. Q + D group. Right, 20×; left, 40×. The apoptotic bodies are defined only by size. Areas below 15 µm2 were counted as apoptotic bodies. The arrows indicated positive brown staining in the TUNEL assay. Also with thin arrows we indicate apoptotic nucleus.
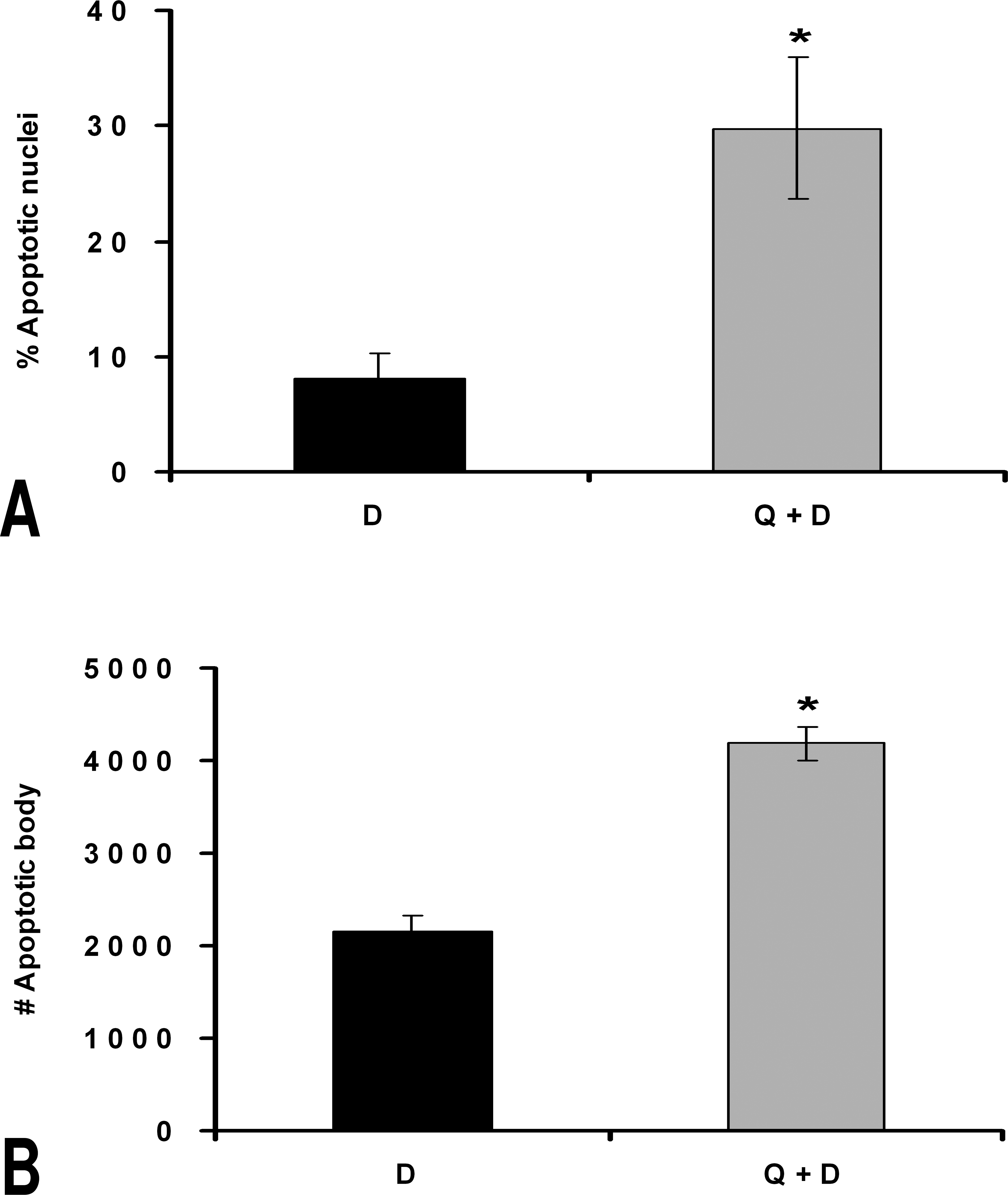
Percentage of apoptotic nuclei and number of apoptotic bodies in the D and Q + D groups. A. Percentage of apoptotic nuclei in the D and Q + D groups. B. Number of apoptotic bodies in the D and Q + D groups. We did not find nuclei or minute bodies in the tissue sections of quercetin treated rats. CN groups presented a minimum of brown staining in the TUNEL assay, with a 0.3% of apoptotic bodies and 1.4 minute bodies by selected fields. *p < .00001 versus the D group.
Evaluation of Mechanisms of Apoptosis
Apoptosis is an important event in cell development and the homeostasis of organisms. However, for cancer to progress, cells must resist the programmed cell death of senescence as well as proliferate. The results revealed a difference in the cleavage of pro-caspase 3 to caspase 3 at 24 hr in the Q + D group compared with the D group. Pretreatment with quercetin increased the cleavage of pro-caspase 3 in the Q + D group by 3.8-fold. In addition, levels of cleaved PARP were increased 1.6-fold in the Q + D group compared with those in the D group. Caspase 9, which is activated by the intrinsic pathway, exhibited a 2.5-fold increase in the Q + D group compared with the D group (Figure 4). Caspase 8, which is activated exclusively by the extrinsic signaling pathway that begins at the cell surface with membrane receptor activation, was not activated (Figure 4). Therefore, these results indicate that mitochondrial apoptotic signaling in the DEN-treated rats is increased by quercetin pretreatment.
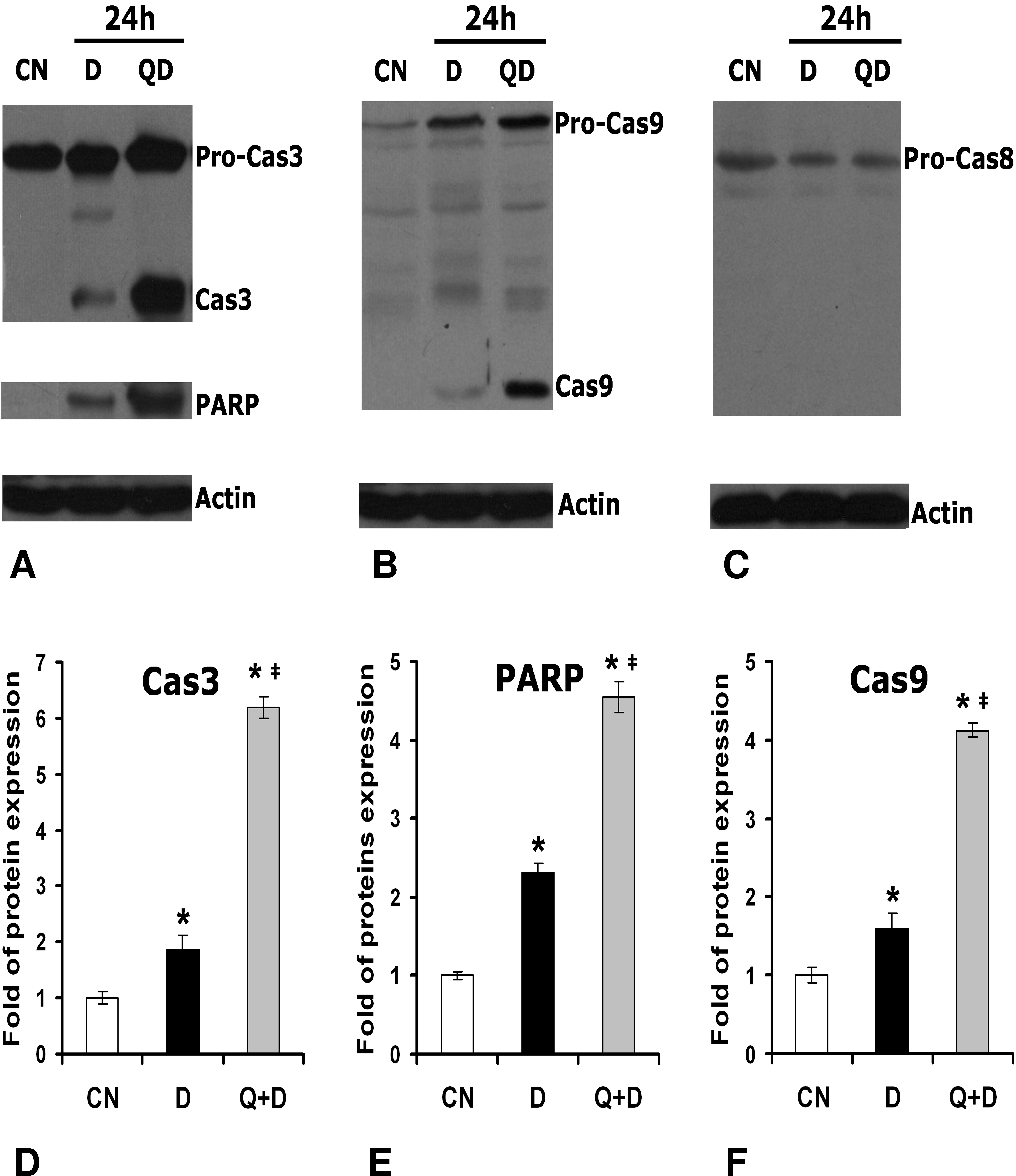
Effect of quercetin on caspase 3, PARP, caspase 9, and caspase 8 protein expression after 24 hr of DEN administration. A, B, and C. Western blot analysis of caspase 3, PARP, caspase 9, and caspase 8 expression. D, E, and F. Densitometry analysis of caspase 3, PARP, and caspase 9. Actin was used as the loading control. *p < .001 versus the D group. ‡p < .001 versus the D group.
Discussion
In recent years, studies have indicated that quercetin, the main flavonoid in the human diet (Kaldas et al. 2005), is a promising chemopreventive agent. This study assessed the chemopreventive properties of a single dose of 10 mg/kg quercetin administered to rats 2 hr before an initiating agent, DEN, in the MRHM. Results were analyzed according to the presence and size of liver preneoplastic lesions after 24 days or molecular and biochemical parameters 24 hr after administration of the initiator, DEN. Each preneoplastic lesion arises from the clonal expansion of an initiated cell (Campbell et al. 1986); thus, we can suggest that pretreatment with quercetin 2 hr before treatment with DEN diminished the number of initiated cells. Quercetin pretreatment resulted in a decrease in the size and number of preneoplastic lesions by >60% in the MRHM reflecting a significant decrease in the numbers of initiated cells.
A single administration of quercetin did not alter histologic morphology in normal rats. The inability of quercetin to induce toxicological damage, particularly at low doses, has been reported previously (Jeong et al. 2009; Vasquez-Garzon et al. 2009). Escape from senescence, avoiding apoptosis, is a basic biologic attribute of cancer cells and was thus evaluated early (24 hr) after DEN intiation. Pretreatment with quercetin in DEN-treated rats induced significant increase in apoptotic cells in liver tissue compared to DEN treated rats. Pretreatment with quercetin activated caspase 9 and increased caspase 3 levels and PARP fragmentation but did not affect caspase 8 in DEN treated rats. These data from the TUNEL assay and Western blot analyses clearly show that quercetin has the capacity to induce programmed cell death in DEN-injured hepatocytes via the intrinsic mitochondrial pathway.
To determine whether the chemopreventive effects of quercetin were, in part, due to its antioxidant properties, the oxidation levels of unsaturated fatty acids were assessed via measurement of the TBARS. When DEN is metabolized, there is an increase in reactive oxygen species (ROS; Yamada, Yamamiya, and Utsumi 2006; Sivaramakrishnan et al. 2008). Results clearly demonstrate that a single administration of DEN induces oxidative damage. Administration of a single dose of quercetin before DEN prevented DEN associated lipid peroxidation. The lipid peroxidation products are generally considered mutagenic and carcinogenic and in some cases cause macromolecular damage.
Previously, in 2009, Gupta et al. treated Sprague-Dawley rats with DEN and quercetin for 5 days in serial doses of 10, 30, and 100 mg/kg (Gupta et al. 2009). The AST, ALT, GSH, and MDA levels increased by DEN were prevented at doses of 10 and 30 mg/kg and were accompanied by decreased DNA fragmentation and apoptosis, but not at the 100 mg/kg dose of quercetin. In the MRHM, celecoxib (Marquez-Rosado et al. 2005), an anti-inflammatory agent, and CAPE (Carrasco-Legleu et al. 2004), an antioxidant and anti-inflammatory agent, prevent the upregulation of NFkB. There are several rat studies that have shown that antioxidant substances such as caffeic acid, phenethyl ester, and anti-inflammatory agents such as Celecoxib prevent or reverse cancer development (Beltran-Ramirez et al. 2008; Arellanes-Robledo et al. 2009). This current study demonstrates increased apoptosis in DEN injured cells at 24 hr after initiation and further correlates these early changes with the effect on numbers of initiated cells at 24 days.
Studies of cellular growth of adenoid cystic carcinoma (ACC) have shown that quercetin decreases viability and metastasis in ACC cell line–induced cancer in a concentration- and time-dependent manner and significantly increases apoptosis (Sun et al. 2010). JC-1, a potentiometric dye, revealed that ACC cell death is dependent on cell depolarization in cells treated with quercetin, indicating that quercetin could induce apoptosis via effects in the mitochondria. The release of cytochrome C from the mitochondria and its binding to Apaf-1 in the cytosol, which enables the recruitment of caspase 9 to form the apoptosome, results in the activation of caspase 3 and subsequent cell death. In ACC cells that were treated with quercetin, cytosolic cytochrome C increased in a dose-dependent manner. In other studies using PC-3 cells, quercetin increased the levels of cytochrome C and apoptosis through the activation of both intrinsic and extrinsic pathways (Senthilkumar et al. 2010). In HT-29 cells that have been treated with quercetin, apoptosis is mediated by the mitochondrial pathway. In addition, p53 and p21 are activated at 48 hr posttreatment (Kim et al. 2010). Studies using HepG2 cells have illustrated the effect of quercetin on the activation of the apoptotic pathway (Granado-Serrano et al. 2006). In contrast to what occurs in normal hepatocytes, the exposure of HepG2 cells, an immortalized cancer cell line, to quercetin for 18 hr induced dose-dependent cell death. The activation of apoptosis was mediated by activated caspase 9, decreased Bcl-2 levels, increased Bax levels, and inhibition of the major survival signals, Akt and ERK. Additionally, in cells that have been treated with quercetin, apoptosis has been shown to increase signaling in the AMPK and p53 pathways and to inhibit PI-3K, JNK, and NF-kB (Kim et al. 2010; Sun et al. 2010). The current study confirms that the quercetin apoptotic effect in cancer cells occurs via the intrinsic mitochondrial pathway. The results in the HepG2 cell line in conjunction with data from this current study indicate a quercetin effect in genetically injured cells. Finally, generation of oxidative stress in this model upregulates the transcription factor NFkB (Carrasco-Legleu et al. 2004; Marquez-Rosado et al. 2005). The upregulation of NFkB is associated with inflammatory, anti-apoptotic, and proliferative processes (Sun et al. 2010; Carrasco-Legleu et al. 2004).
In conclusion, this study demonstrates the chemopreventive effects of quercetin, evidenced by the inhibition of the development of preneoplastic lesions in the MRHM, reflecting a significantly decreased number of initiated cells. These effects are achieved by quercetin induction of apoptosis in presumably genetically injured cells by activation of caspase 9, the intrinsic mitochondrial pathway. Additionally, quercetin markedly decreases the amount of lipid peroxidation that was induced during the metabolism of the initiator DEN 24 hr after treatment. A third potential factor can be identified, specifically that the antioxidant capacity of quercetin prevents the upregulation of NFkB thereby creating a permissive microenvironment for apoptosis in those injured cells that would have become initiated cells. The evasion of apoptosis or escape from senescence contributes to tumor formation and treatment resistance; thus, the identification of a mechanism for quercetin chemoprevention (i.e., induced programmed cell death in genetically injured cells) introduces the possibility that combining this drug with anticancer drugs might enhance efficacy in clinical oncology.
Footnotes
Acknowledgments
We would like to acknowledge the animal technical support at UPEAL-Cinvestav, including Rafael Leyva-Muñoz, Ricardo Gaxiola-Centeno, and Dr. Jorge Fernandez.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a scholarship (VGVR173007), grant contribution (39525-M) from CONACYT, and postdoctoral scholarship of multidisciplinary project 3 from the grant SVT of CINVESTAV.