
Abstract
The ICH initiated talks in June 2012 to revise regulatory guidance for carcinogenicity assessment of pharmaceutical products, stimulated in part by a proposal called Negative for Endocrine, Genotoxicity, and Chronic Study Associated Histopathologic Risk Factors for Carcinogenicity in the Rat (NEGCARC) from the Pharmaceutical Research and Manufacturing Association (PhRMA). The 2012 STP Town Hall Meeting focused on the need for change in carcinogenicity assessment strategies for pharmaceuticals. Dr. Todd Bourcier from the Division of Endocrine and Metabolic Products, U.S. FDA and a member of the FDA’s Alternative Carcinogenicity Assessment Committee, was the guest speaker and a panelist. Dr. Bourcier is also one of FDA’s representatives to the ICH S1 Expert Working Group that is discussing changes to regulatory guidelines for carcinogenicity assessment. Drs. Carl Alden and Dan Morton also participated in the panel discussion.
Introduction by Dr. Carl Alden
Two successive FDA commissioners have called on the toxicology sciences community to work to improve their practices based on the deluge of new understanding of the carcinogenicity process. Dr. Janet Woodcock called toxicology sciences (including those of us in industry) the least changing discipline in pharmaceutical R&D in response to new learning. Dr. Margaret Hamburg wrote:
Most of the toxicology tools used for regulatory assessment rely on high-dose animal studies and default extrapolation procedures and have remained relatively unchanged for decades, despite the scientific revolutions of the past half-century. We need better predictive models to identify concerns earlier in the product development process to reduce time and costs. (Science 2011)
This was embarrassing to me. I may sound critical of the past, but actually I credit regulatory agencies, industry, and the people in this room for decisions in introducing new chemicals (across industrial applications) in the workplace that have delivered continuous increases in life expectancy over the past century as well as a decrease in age-adjusted cancer rates in the past 20 years. However, we must critique the past to build foundations for the future and we need to know how our current testing paradigms are performing. For pharmaceutical chemicals, there is a 15% false negative rate and an 80% false positive rate using the lifetime rat and mouse bioassay models. This does not consider that Gottman et al. in 2001 reported on repeat testing of 121 chemicals and only 57% of those assays were repeatable. A regulatory authority in Europe reported similar false positive rates with the current test paradigms. So what have we learned over the past 50 years? We recognize now that pharmaceutical carcinogenesis occurs through one of four mechanisms: genotoxicity, hormonal dysregulation, immunosuppression, or chronic toxicity. Obviously, we do not need to do a cancer bioassay to assess for these biological attributes, so we probably could eliminate the false negative responses and drastically reduce the false positive response if we simply assess for these parameters and create an appropriate label based on the short term and chronic data. Recently, a PhRMA group of 13 pharmaceutical companies has worked together to create a new testing paradigm based on an intensive review of the toxicological attributes of 182 pharmaceuticals. They derived a paradigm that states if there is no evidence of hormonal disruption or genotoxicity and an absence of histopathologic risk factors in chronic toxicology studies, then a rat bioassay need not be conducted. If this proposal is adopted, it would result in a 40% reduction in the number of cancer bioassays without compromise to effectiveness. This proposal would also include a 6-month rasH2 mouse bioassay. The rasH2 model predicts as effectively for human cancer as the combined lifetime rat and mouse bioassays. My only critique of the PhRMA initiative is that it predicts carcinogenicity for rats and mice, not for humans. The ILSI initiative leading to transgenic alternatives asked if the models predicted for humans, not rodents. That is important to keep in mind when evaluating the PhRMA initiative.
Dr. Todd Bourcier
I am going to present two approaches (perhaps better described as visions) of how to test for carcinogenicity of pharmaceuticals, and how the current paradigm for carcinogenicity testing that is described in the current ICH S1 guidance might be changed. Carl introduced NEGCARC, and this diagram (Figure 1) represents NEGCARC in abstract form the way the FDA understands the proposal. With NEGCARC, one considers three triggering criteria to decide if one should do a carcinogenicity study. The first is called histologic “risk factors” in the 6-month rat study. The second is evidence of hormonal perturbation derived from the entire preclinical data profile available at the time. The third is the profile from the standard genotoxicity battery of assays (using the old ICH S2 guidance that has changed recently). If any one of these criteria is considered “positive,” then one would follow the existing paradigm of testing in which one would do a 2-year rat study plus a 2-year mouse study or a transgenic mouse study. If one follows this route, you will be right a little better than half the time. That is, after considering these criteria, you would be right about half the time in predicting that a compound is going to be positive in the 2-year rat study. If you flip a coin, you would do just as well. On the other hand, if all three criteria are “negative,” then one would conclude that the pharmaceutical under question poses little carcinogenic risk; therefore, one can reasonably waive the 2-year rat study. The sponsor then only needs to conduct a transgenic mouse or 2-year mouse study in this scenario. If we go along that route, we would be right about 82 to 88% of the time, but you would be wrong about 12 to 18% of the time. That is, 12 to 18% of the time you predict the 2-year rat study would be negative, but it would (if the study were actually conducted) turn out positive. These latter cases are considered “false negatives,” but what do they really mean? In the paper by Sistare et al. (2011) in Toxicologic Pathology, it was argued that the 18% error rate (the false negatives) can be disregarded because when one really looks at those false negative cases, none of them had any particular human relevance. Therefore, the authors concluded that one could safely ignore or dismiss all the false negatives that would occur in the future under a NEGCARC testing paradigm. The crux of NEGCARC comes down to how we define these three triggering criteria (histologic risk factors, genotoxicity, and hormonal perturbation). The histologic criteria are considered “positive” if there is any treatment-related incidence of hyperplasia, hypertrophy, foci, dysplasia, or tumors in a 6-month rat study. That means any hyperplasia or hypertrophy. Genotoxicity is considered a triggering factor if there is a “clear” positive finding in any one assay of the standard battery, including the in vitro chromosomal aberration assay. The hormonal criteria would be considered positive if there is a treatment-related microscopic change in endocrine cells or a macroscopic change in endocrine organs or a change in hormones or a hormonal mode of action. That is, different streams of data can determine that a compound causes disturbance of some hormonal system. As defined, these criteria put into practice can lead to some interesting situations if you ask the question “What are the findings in cases that would be triggered by just one of these criteria?” Here is such an example from the FDA data set. In addition to the PhRMA data set, the FDA constructed their own data set based on 51 compounds. The Japanese Pharmaceutical Manufacturers Association (JPMA) also created its own database. Remarkably, the predictive properties of all three data sets were very similar, and the organizations were not talking to each other when the data sets were being analyzed. That is quite amazing. As for cases where one positive criterion would trigger a 2-year rat study, in the FDA data set, histology was the sole triggering factor in 11 cases, and the predominant triggering signal in 9 of these 11 cases was simple microscopic liver hypertrophy. So if two of your high-dose animals in the chronic study have liver hypertrophy, you are doing a 2-year study under NEGCARC. Genetic toxicology was the only triggering signal in two cases, and in both cases the signal was positive only in a single in vitro assay. One was a chromosomal aberration assay and one was a mouse lymphoma assay, both at the limits of acceptable cytotoxicity. One of these cases proved to be a multi-organ tumorigen and the other was negative in the carcinogenicity study. Hormonal criteria alone triggered three cases. One was based on mode of action. The second was based on microscopic findings in reproductive organs. The third was based on changes in hormone levels for a hormone that is not routinely assayed in standard toxicology studies, but was measured because tumors were observed in the 2-year study. One observation about NEGCARC is that signals that are not commonly considered indicative of carcinogenic potential would nevertheless be triggering 2-year rat studies in at least one-third of cases.
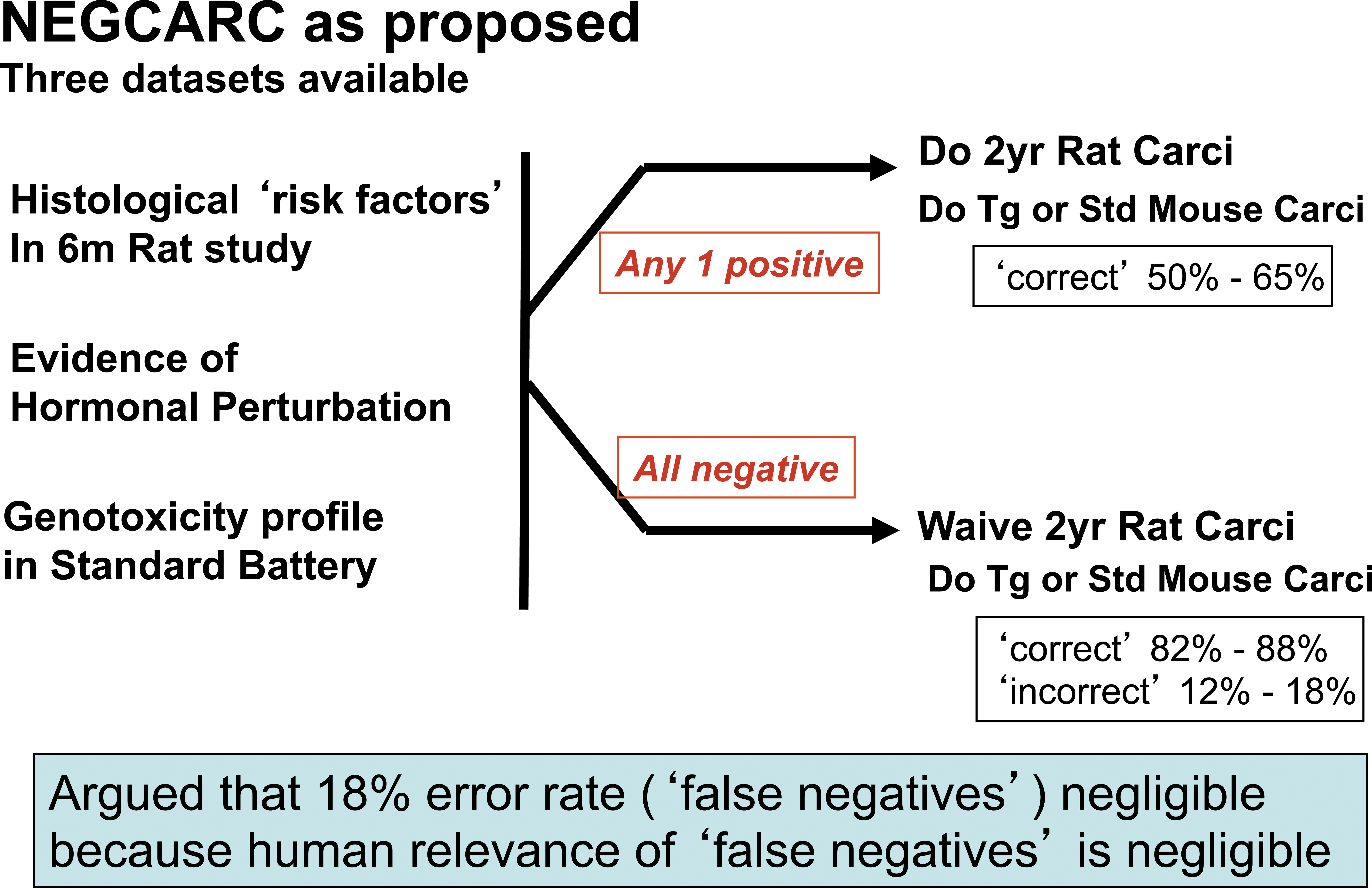
The “Negative for Endocrine, Genotoxicity, and Chronic Study Associated Histopathologic Risk Factors for Carcinogenicity in the Rat” (NEGCARC) proposal as described by Sistare et al. (2011) provides an empirical decision tree that would eliminate approximately 40% of rat carcinogenicity studies supporting registration of small molecule pharmaceuticals. The false negative rate is the percentage of compounds in a retrospective study that had no evidence of histopathologic risk factors for carcinogenicity (hyperplasia, hypertrophy, altered foci, or neoplasia) in 6-month or 12-month rat studies, evidence of hormonal perturbation, or positive genetic toxicology findings yet were positive in the rat carcinogenicity studies. This figure was provided by Dr. Todd Bourcier and presented at the STP Town Hall Meeting.
Another vision that has been offered is more of a weight-of-evidence approach similar to that found in the ICH S6 guidance. This guidance describes how one can approach carcinogenicity testing of biologics with their many unique characteristics. This approach can perhaps be modified for small molecules (Figure 2). If a carcinogenicity evaluation is needed according to the current ICH S1A guidance, the sponsor would submit a weight-of-evidence evaluation (essentially a white paper) arguing for why or why not a 2-year study would add value to the carcinogenic assessment for the pharmaceutical being developed. The content of the white paper will be a matter of great debate, and the audience is encouraged to offer ideas. Certainly, the white paper should consider data from all sources, including published literature and proprietary data. It should certainly include the drug’s primary and secondary pharmacology and a description of target expression and the biology of the target. The NEGCARC criteria are valid criteria conceptually: we need to look at preneoplastic lesions in the chronic rat study, genotoxicity, and hormonal effects, but perhaps a bit differently than how NEGCARC defines them. Maybe a transgenic mouse study would be useful to determine if a 2-year rat study should or should not be conducted. And then we probably could list other exploratory toxicology end points that may be useful as part of the weight-of-evidence argument. If there is sufficient product-specific and mechanistic evidence in the white paper, then maybe one could triage compounds across a spectrum of risk, with one extreme being that all parties agree that no carcinogenicity risk has been identified and then no further carcinogenicity testing would be required. On the other extreme, there may be a clear risk identified based on theoretical mode of action or real data. In this case, sponsors can always conduct a 2-year study if they think this would add value to human risk assessment. Or perhaps no further studies are needed, but meaningful risk communication must be prepared in the label and (possibly, but unlikely) in risk management plans post-market. That leaves the great middle where most compounds would probably fall. Some data may suggest that there may be a risk and other data suggest not. That presents a challenge. Perhaps, additional studies would be required and could be useful to specifically address a certain concern, but in most cases, conducting a 2-year study would be in the best interest of all concerned. The big caveat of this approach is obvious—small molecules are not biologics. Small molecules have issues that biologics do not: ADME-related issues, metabolites, metabolic pathways, and specificity on the target (a major challenge).
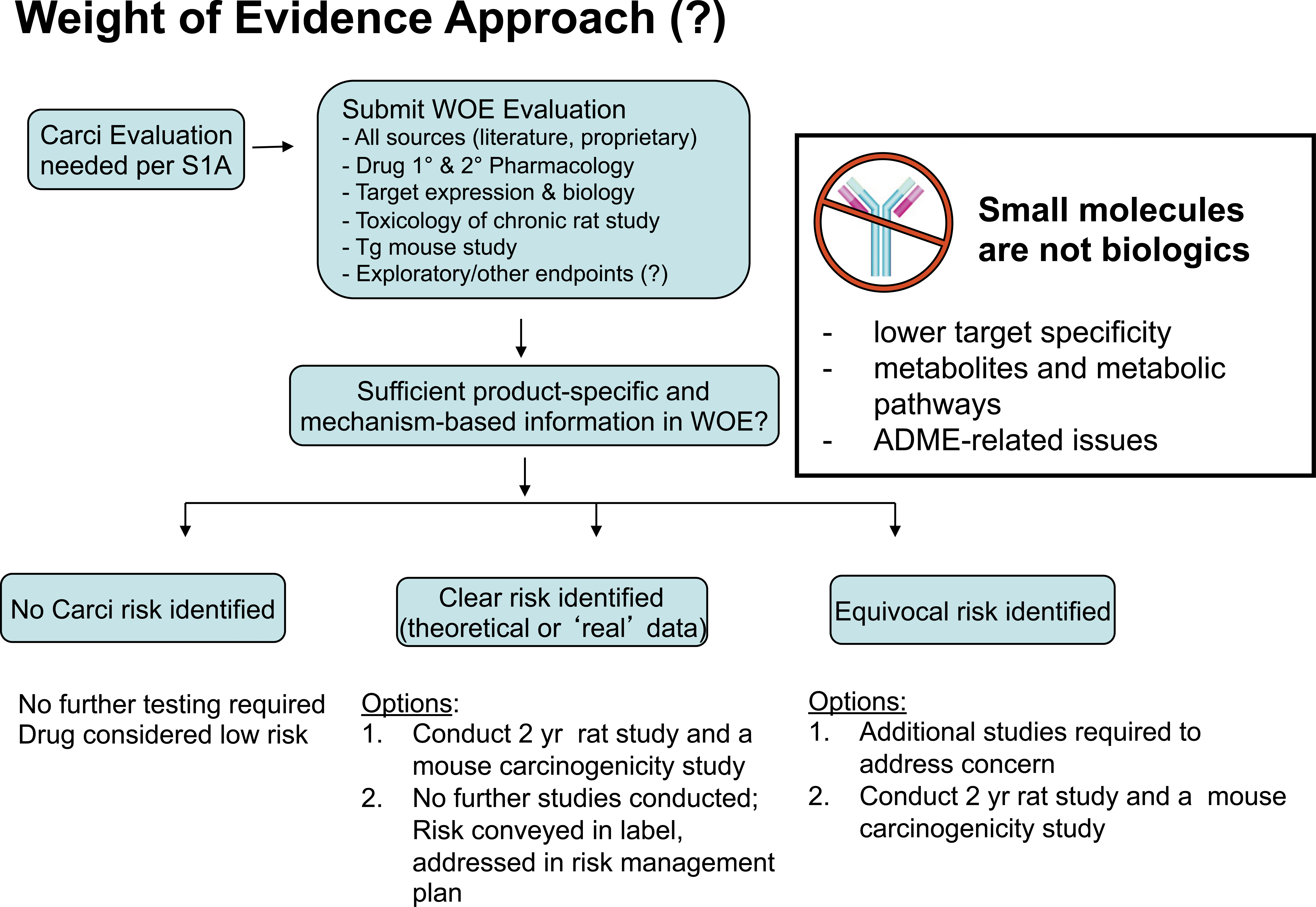
A weight of evidence (WOE) decision tree is offered as an alternative to the NEGCARC proposal. This figure was provided by Dr. Todd Bourcier and presented at the STP Town Hall Meeting.
We have discussed two visions, leaving us with choices and many questions that we need to ask before we change the current paradigm. There is an empirical approach proposed by NEGCARC. There is a weight-of-evidence approach that is more like ICH S6. Keep in mind factors of importance, particularly to us at the FDA: which approach improves assessment for potential human carcinogens? We want to predict and screen for human carcinogens, not rat carcinogens. NEGCARC was designed to predict what happens in the 2-year rat study. What level of uncertainty are we (regulatory authorities and industry) willing to assume if we do not have carcinogenicity studies to consider? What other pharm/tox end points can improve either of these approaches? Is it possible that we could approach this with a prospective period of testing and a safe-harbor period? Why would that be important? Because we really want to know that tomorrow’s approach will be better than what we do today. Second (from a regulatory view), which would work best in reaching harmonization across all regulatory regions? How often will we achieve concordance for specific compounds between industry and regulatory agencies? We do not want a situation in which we are arguing back and forth on what to do with every drug. This introduces uncertainty. Whatever we do, it must provide a reasonable rationale for regulatory decisions. Regulators must defend why they ask for specific studies, particularly in an environment in which we may be choosing or discriminating between sponsors when it comes to conducting 2-year rodent studies for small molecules.
Questions and Answers
The issue with the weight-of-evidence approach is “What is the cancer risk and to whom?” You indicated that we want to address the issue of cancer risk for humans, not for rats. Therefore, the question is: Is FDA going to change from considering carcinogens to be any chemicals that cause tumors in any species, or are we going to accept the definition that there are certain metabolic pathways that lead to carcinogenicity in rats and mice that are proven not be a risk for humans (e.g., phenobarbital)? That is, how will we address the issues of liver hypertrophy, metabolic activation, etc., that are not relevant for humans? Should all of those liver hypertrophies be eliminated from the NEGCARC false negatives because we all know that they do not apply to humans? To a great extent, we do that already. If we have seen a tumor finding before with a specific drug class and we know something about how those tumors come about, this is folded into the FDA risk assessment regarding whether this tumor is relevant or not. That is an important aspect of what we do. When a more unusual tumor is encountered, it is incumbent upon the sponsor to provide some information, if the tumor occurs at near clinical exposure, about why the tumor occurs and why should we be (or not be) concerned. Most times we do get sufficient mechanistic information to make a decision one way or another. When we see liver hypertrophy in a 6-month study, CYPs are induced, there is a lot of liver metabolism, and liver tumors occur at the end of 2 years, we at the FDA do not get excited about a human risk. One of the things that the National Toxicology Program did and now is available is to generate a lot of data about gene expression, proteomics, and so forth. I suggest that we move forward to collect specific tissues in 90-day or 6-month studies to generate gene expression data because it can be done easily with microarrays or next generation sequencing. After a few years, we may be able to tell that certain gene pathways are harbingers of potential risk. This does not prevent us from moving away from the 2-year studies now, but provides for collection of additional data that will improve future strategies. Gene expression has been suggested as a possible technology to predict carcinogenicity. Some comments from others are that it tends to be organ-specific, so it provides information for tissues that are analyzed, but not for others. I am open to the idea to collecting data prospectively to see how regulatory agencies can use the information. Fielden’s paper (Fielden et al. 2011) using 9 to 12 genes showed a correlation with tumor risk, but a standardized evaluation of 50 or more compounds is information that people would like to see before taking the concept into regulatory decisions. I am in favor of this. Of the 18% false negatives, how sure can we be that these are irrelevant to human risk? If they are ever relevant to human risk, who will be responsible? There is no paradigm that will eliminate all false negatives. Cigarette smoke is an example of a known human carcinogen that has been missed in 13 animal bioassays designed according to standard practice in experimental design. We are having repeated episodes of the discovery of carcinogens based on human data that were not detected in animal studies, and that is embarrassing. We have too many false negatives with the current paradigm. A new paradigm is needed. Labeling genotoxins, immunotoxins, hormonal disrupters, and chronic toxins (without sufficient margin) as carcinogenic risks would nearly eliminate false negatives and greatly reduce the false positive rate. No matter what testing strategy is used, there will be false negatives. We need to accept that. Using NEGCARC, we are wrong almost 20% of the time in predicting carcinogenicity for the rat. Pharmaceutical companies do not want to develop human tumorigens. We want to screen for a low-frequency event (the nongenotoxic compound that is a human carcinogen). Have we looked at enough compounds to say that we can rule out with X percentage of confidence that we would have picked up a low-frequency event? We have false negatives now, and we do not do a very good job of post-market surveillance of human cancers associated with drug treatment. As to who takes responsibility, the company must always take responsibility for their compound. The worst case scenario is if FDA waives the 2-year studies and a human cancer signal is discovered later on. Regulators are open to criticism (rightly or wrongly) that the compound was not evaluated well enough because the “gold standard” 2-year rodent study was not conducted. In the last 30 years, we created, based on chemical structure, a decision scheme on how to proceed if, for example, there are reactive moieties in small molecules. We can advance decision making based on very concrete end points, regarding all of the other liabilities, including DNA reactivity, adduct formation in various target tissues, exposure (area under the curve, etc.), other ADME data, and 3-month rat and 6-month rat studies (including recovery). All of these things can be built into a decision tree so that we can predict, using an extrapolatable database and pure science, if a compound presents a carcinogenic risk to human subjects. If we could construct such a decision tree, and the ICH would agree, there could be a purely scientific decision process. I think that you are describing the ideal situation. Filling in the branches on the tree will be extraordinarily difficult because we are trying to extrapolate to certain modes of action that are associated with human cancers, but when it gets into the realm of nongenotoxic carcinogens, I am less confident that science will allow us to create a decision tree that can be extrapolated with high confidence to the human condition. So a weight-of-evidence approach is at least scientific and justifiable taking into account all that is known about the compound including chemical structure, biology of the target, and secondary pharmacology. Capturing all of this in one decision tree would be challenging. It would be a multiple page decision tree.
Follow-on comment from audience:
I agree it is a challenge. We have been doing this relatively successfully for 15 years. There are so many models that are very helpful, such as accelerated bioassays that you can promote or tweak here or there, and there is a database already of DNA reactive or promoting compounds. At some point in the ICH process, whatever the ICH produces goes out for public comment. I encourage everyone here to submit comments, including the decision tree you propose. All comments and suggestions will be considered. An old version of Robbins and Cotran states emphatically that silica is not a carcinogen. Of course we know now that silica is a human carcinogen. Negative human data is absence of evidence which is never evidence of absence. In regards to silica, the mouse was always negative and the rat was always positive. We are looking at a new class of pharmaceuticals—the nanoparticulates—and the lesson from silica may be useful. When it comes to new nanopharmaceuticals, these compounds have significant distribution to the lung. I worry about discarding the rat when there may be significant distribution in the lung. The rat has been conservative in providing the early signal for human lung carcinogens. Thank you for those comments. One thing that I would like to see is a set of circumstances in which 2-year studies would always be done. So for example, new technology, a first-in-class pharmaceutical for a new drug target, and other cases in which one would always want to see a 2-year study. We can make progress in this area by starting at the extreme ends. When a clear risk is identified, there is ample room to say we don’t need a 2-year study because we would still consider the compound a potential tumorigen regardless of the rodent study outcome. On the other extreme, it will be trickier to agree on cases where this is no risk. The bar in this case needs to be higher. Progress can be made particularly if there is experience with the drug class. It is in the great middle (including nanopharmaceuticals) that we don’t know what will happen in a 2-year study, so perhaps we should find out rather than guess at the outcome. We need to guard against clinging to the past just because that is the most comfortable approach. Toxicological science disciplines need to leverage learnings from our past to create better paradigms in the future. After thousands of cancer hazard identification studies and identification of the biologic attributes of human carcinogens, I think we are now able to do that. In the specific circumstance cited, silicosis causes chronic inflammation in the lung of mice. Through review of all human carcinogenic influences, we are now able to recognize chronic inflammation as a risk factor for carcinogenesis in humans and animals. NEGCARC is based on well-established and reproducible findings. While not perfect, we know how to deal with it. In the ideal world, we would evaluate human carcinogenic risk, but we do not know how to get there. The NEGCARC paradigm is a pragmatic and robust way forward, but the weight-of-evidence paradigm does not seem to be quite so clear. With drug classes that are really novel, we all feel more comfortable conducting the 2-year studies to gain experience until we are confident in a new paradigm. One point I would like to see included in the NEGCARC proposal is an understanding of the target biology, mode of action, and potential mechanistic risk. When I saw the results from the three databases and they were within a few percentage points of each other regarding predictability, I nearly dropped. These efforts were done independently. There is a certain beauty in NEGCARC. It works reproducibly if you take the criteria and apply them to a set of compounds. But one difficulty is “How well will it work in a regulatory environment?” The genetic toxicology and hormonal criteria were added reduce false negatives in NEGCARC that resulted if one considered only histological end points in the 6 month rat study. These additional criteria are important because they flagged tumor responses for drugs that we would not want to miss. One of those was a drug that was positive in a genotoxicity assay, and this finding was the only trigger. The problem is we will get a set of data that show a positive result in an in vitro chromosomal aberration assay and not in any other genetic toxicology assay. According to NEGCARC, a 2-year rat study would be required even if we believe that the drug is not genotoxic. So my concern is if we were to adopt NEGCARC, prospectively one could argue that liver hypertrophy should not be a risk factor for human tumors, so why are we using liver hypertrophy or muscle hypertrophy as triggers for 2-year carcinogenicity studies? These are legitimate questions. I find it difficult to justify the conduct of a 2-year study based on some of these triggers. That is why I prefer the weight-of-evidence approach. The three criteria outlined in NEGCARC are certainly legitimate. They just need to be applied differently. If we decide to use genotoxicity as a criterion, let’s decide if a compound is genotoxic or not. If the trigger is histopathology, what kinds of hyperplasia or hypertrophy should concern us? I think that one of the problems with NEGCARC is that it does not always make sense. There are times when it is based on findings that we do not think should predict carcinogenicity. It does not take into account all that we know about the principles of carcinogenicity. In applying NEGCARC principles, there would be constant battles between regulators and sponsors regarding whether findings like minimal or mild hyperplasia in one tissue in a few animals should trigger a 2-year study. The FDA receives carcinogenicity data, and then sponsors often conduct mechanistic studies to explain the findings. It is very hard to provide convincing mechanistic data that a specific neoplasm is not relevant to humans. On the other hand, the weight-of-evidence approach may require so much more mechanistic information than the FDA now receives that it may be easier for the sponsor to simply run the 2-year rat study. Hormonal data and proliferative signals are often produced in response to carcinogenicity findings rather than prospectively. Should these now be required prospectively, and what tissues should be assessed? How do we use gene expression? And how would we label drugs without conventional carcinogenicity study data? You recall a presentation last fall about renal disease and a false positive renal tumor finding in a rat carcinogenicity study. Are you aware of the recent publication by Gordon Hard et al. describing data from F344 rats from the NTP database and the association of low levels of renal tumors and advanced spontaneous renal disease in rats? They identified (in my opinion) a series of false positives that were reported as renal carcinogens, but they were not carcinogens because there was the interaction with spontaneous disease. The other discussion addressed the SD rat, and when the background disease issue was addressed by managing the animals differently (using caloric restriction), the result was negative. What should we do about the false positives that are caused by a poor model system (the ad libitum fed rat)? That is our most common challenge. Sixty percent of the time these 2-year studies turn out positive in one way or another, and the challenge has always been “What do we do with that 60% of drugs that are rodent tumorigens?” You do the high-level cutting. At what dose does the tumor occur? What is the exposure margin to clinical dosing? What is the indication? And most of those tumor responses go away. Yet there is a sizable number of compounds that produce neoplasia in rodents at clinically relevant exposure, and if there is not much known about the mechanism, the sponsor should show some key events that occur in the tumorigenic response. In your opinion, what should sponsors be expected to show in the case of solid renal tumors? An association with chronic nephropathy?
Follow-on from audience
First of all, NTP has used a flawed inbred rat model for many years, but this issue of ad libitum food consumption seems widespread across rat models. If we are going to assess cancer risks in long term studies, we are relying on historical data that are flawed. That is a good point. We take historical control data into consideration. Occasionally, we see dose-dependent increases in some tumors that fall in the high dose group at the upper range of the historical control range. We are not quick to dismiss these just because they are within historical limits. When there is a concurrent control with a lower than normal (historical) incidence, we pay attention. Another observation is that some obesity drugs have remarkably lower incidences of tumors in certain organs.
Follow-on from audience
That observation is consistent with the pharmaceutical industry experience using highly toxic doses. Survival profiles for controls were much worse than for high dose animals because toxicity at the high dose required energy for repair. Until you leveled off that curve, there were big differences between high dose and controls, and the high dose animals lived much longer than controls. The “safest” group to be in was the high-dose group. With the obesity drugs, it is not a toxicologic effect, it is a pharmacologic effect. Treated animals with some but not all of these drugs live longer. Did we in fact get the same answer from the three different data sets reviewed by PhRMA, FDA, and PDMA, or did we get the same answer because of extensive overlap in the compounds examined? The FDA’s 51 compounds are unique and there is no overlap with the PhRMA database. PhRMA provided FDA with the key to the PhRMA data, so we know that the FDA compounds are unique. We do not know if some of the JPMA compounds overlapped.
Question
What do you do about metabolites? Does the industry group or FDA believe that metabolites will be a stumbling block going forward? It will be a stumbling block. NEGCARC is empirical and pragmatic. There will be a need for some pragmatic aspect that goes blindly on association because it is very difficult to determine in vivo exactly what metabolites are formed and what they will do. And then you have the difficulty of cross-species metabolite formation and reactivity. It will be difficult. We already attempt to evaluate metabolites. It may take years and lots of money to characterize metabolites extensively. Minor metabolites have never been demonstrated to present human cancer risk. What is worthwhile? What does the speaker propose?
Follow-up from audience
I can argue both sides of this issue. We already deal with metabolites. We analyze metabolites in genetic toxicology studies and animal studies. On the other side, how much do we know about these metabolites? We do not know how much of each metabolite is in the genetic toxicology assays. We do not know the pharmacology of many of these metabolites. This point requires additional consideration and discussion, but should not be a major stumbling block.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.