
Abstract
Human exposure to capsaicin, the most abundant pungent chili pepper component, is ubiquitous. Evaluation of capsaicin’s carcinogenic potential has produced variable results in in vitro and in vivo genotoxicity and carcinogenicity assays. The capsaicin tested in older studies was often from pepper plant extracts and included other capsaicinoids and diverse impurities. Recent studies utilizing high-purity capsaicin and standardized protocols provide evidence that the genotoxic and carcinogenic potential of capsaicin is quite low and that the purity of capsaicin is important. Several small epidemiological studies suggest a link between capsaicin consumption and stomach or gall bladder cancer, but contamination of capsaicin-containing foods with known carcinogens renders their interpretation problematic. The postulated ability of capsaicin metabolites to damage DNA and promote carcinogenesis remains unsupported. Anticancer activities of capsaicin have been widely reported, as it inhibits the activity of carcinogens and induces apoptosis in numerous cancer cell lines in vitro and explanted into rodents. Diverse mechanisms have been postulated for capsaicin’s anticancer properties. One hypothesis is that inhibition of cytochrome P450 enzymes—particularly CYP2E1—retards carcinogen activation but is contradicted by the low potency of capsaicin for CYP inhibition. The potential for dietary capsaicin to act as a chemopreventative is now widely postulated.
Introduction
Capsaicin, the most abundant pungent molecule produced by pepper plants, represents an important ingredient in spicy foods consumed throughout the world (European Commission Scientific Committee on Food, 2002). The capsaicin content of chili peppers ranges from 0.1 to 1% w/w (Govindarajan and Sathyanarayana 1991). Substantial human exposure to capsaicin in the form of prescription or nonprescription topical analgesics, self-defense products (e.g., pepper spray), cosmetics, and oral herbal supplements also occurs.
Capsaicin is a highly selective agonist for the transient receptor potential cation channel, subfamily V, member 1 (TRPV1), a ligand-gated, nonselective cation channel, preferentially expressed on small-diameter sensory neurons (C-fibers and to a lesser extent Aδ-fibers; Alawi and Keeble 2010). Initially, activation of TRPV1-expressing nociceptors by capsaicin results in burning and itching sensations, hyperalgesia, and allodynia. The transient erythema produced by capsaicin is due to a neurovascular response stemming from the release of vasoactive neuropeptides from nociceptors (Geppetti et al. 2008). These acute effects are followed by a reversible defunctionalization of nociceptive sensory axons. Defunctionalization of hyperactive nociceptors is thought to underlie the pain relief that follows topical application or intra-articular injections of capsaicin (Anand and Bley 2011). For instance, Qutenza®, a cutaneous patch containing a high concentration (8%) of synthetic capsaicin, has been approved recently in the European Union and United States for the management of peripheral neuropathic pain syndromes (McCormack 2010).
Much of the published toxicological literature on capsaicin relates to extracts of capsaicin derived from peppers; these extracts are typically a mixture of capsaicin, norhydrocapsaicin, dihydrocapsaicin, homocapsaicin, homodihydrocapsaicin, and nonivamide. The actual percentage of capsaicin and other capsaicinoids varies depending on the pepper source and method of extraction (Johnson 2007). The capsaicin concentration can range (on a by weight basis) from about 65% (USP grade natural capsaicin) to >99% (in the case of synthetically pure capsaicin) in medical and cosmetic products. Pepper extracts may also contain chemical entities other than capsaicinoid compounds. Toxicology results with pure capsaicin may differ from results obtained with extracts because of variable contents and potentially toxic impurities in the extracts.
The relationship of capsaicin to genotoxicity, carcinogenicity, and cancer prevention and treatment has been widely explored. A PubMed search using the terms “capsaicin” and “cancer” or “carcinogenicity” or “neoplasm” returns nearly 400 references. We have attempted to include all directly relevant references in this review, as well as some publications which clearly pertain to the subject matter but were not captured by the above PubMed search. Publications reviewed range from in vitro and in vivo studies, chemoprevention and metabolic inhibition, to apoptosis of cancer cell lines and epidemiological studies. The carcinogenic potential of capsaicin, other capsaicinoids, and capsaicin extracts has been reviewed previously (e.g., Surh and Lee 1995; Johnson 2007). Although comprehensive and very thorough, the most recent reference cited in Johnson (2007) is from 2003, and roughly half of the publications on capsaicin and carcinogenicity have appeared since 2003.
This review focuses on carcinogenic and anticarcinogenic potential of capsaicin and not on pepper extracts or other capsaicinoid molecules. When interpreting toxicological evidence, weight was given to data from nonclinical studies for which protocols were consistent with drug regulatory agency guidelines and/or utilized high-purity capsaicin. Interpretation of metabolism studies was based upon available human pharmacokinetic information, with particular attention paid to the physiological relevance of capsaicin concentrations and exposures. Data regarding the numerous mechanisms postulated to underlie capsaicin’s anticancer activities were summarized. Finally, human epidemiological studies were examined for completeness with respect to accounting for known dietary carcinogens.
In Vitro Genotoxicity
Published information on the potential genotoxicity of capsaicin has been inconsistent, as both positive and negative effects have been found in classical genetic toxicology assays. Brief summaries of eighteen in vitro genotoxicity studies are provided in Table 1.
In vitro genotoxicity assays with capsaicin.
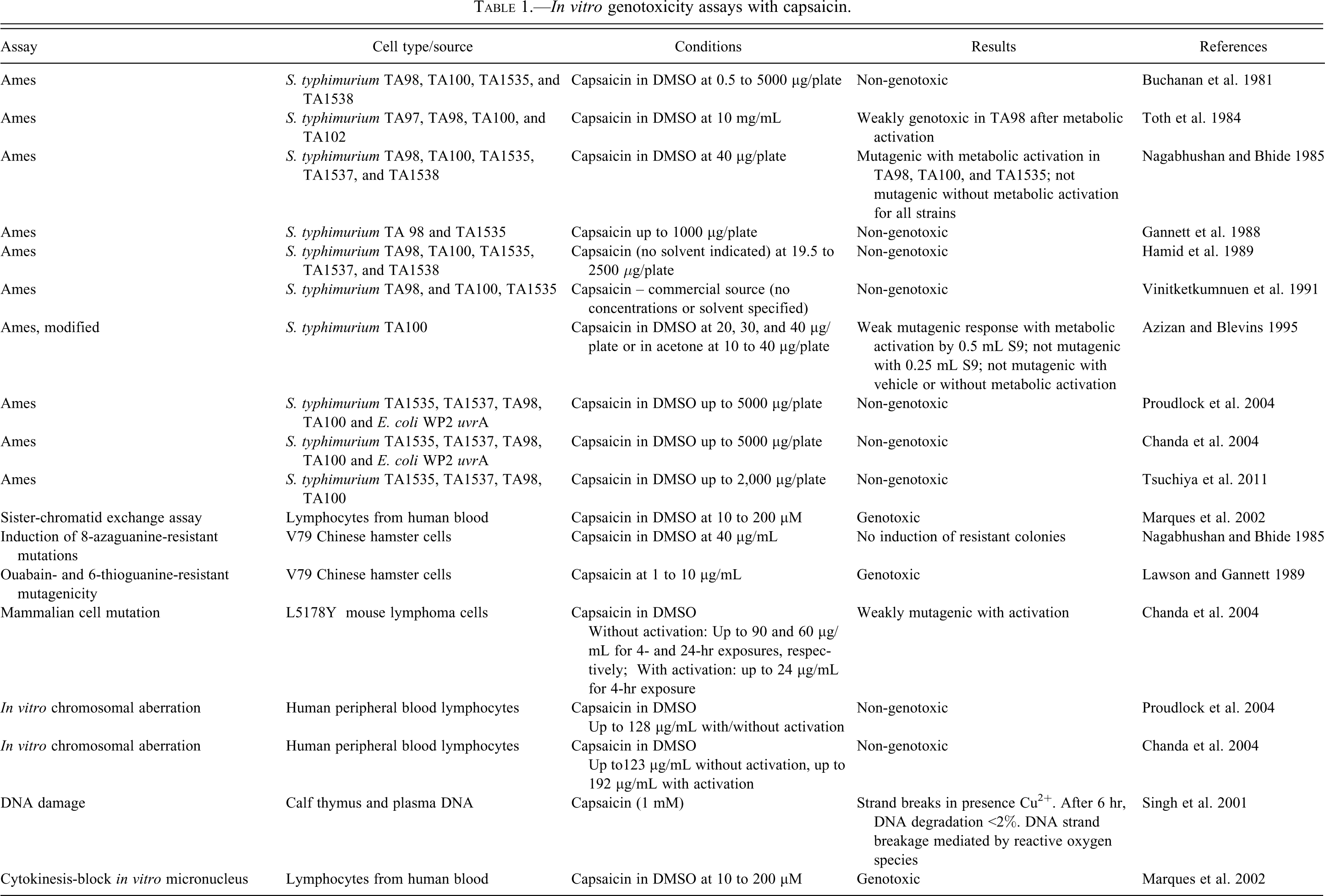
Bacterial point mutation tests (Ames assays; Ames et al. 1975) were performed with and without metabolic activation using various strains of Salmonella typhimurium; seven of these tests resulted in negative responses and three in positive or weakly positive responses. Importantly, two most recently completed studies were conducted in compliance with Good Laboratory Practices and International Committee on Harmonization Guidelines (Mueller et al. 1999) and used either highly pure synthetic capsaicin (Chanda et al. 2004) or highly purified capsaicin extracted from chili peppers (Proudlock et al. 2004); data from these two studies appear to be of high quality and quite consistent. Chanda and colleagues (2004) performed the Ames test with S. typhimurium TA1535, TA1537, TA98, and TA100, and Escherichia coli WP2uvrA, both in the absence and presence of metabolic activation, by both preincubation and plate incorporation methods. No mutagenic activity was observed in any of the five bacterial strains, in any of the activation conditions. Essentially identical results were reported by Proudlock et al. (2004). Tsuchiya et al. (2011) demonstrated mutagenicity of chili pepper extract in strain TA98, while pure capsaicin was inactive in the same assay; subsequent analysis of the chili pepper samples identified the presence of carcinogenic aflatoxins B1 and G1.
In vitro chromosomal aberration assays using human peripheral blood lymphocytes (HBPLcs) were conducted twice using pure capsaicin. No significant increases in chromosomal aberrations, polyploidy, or endoreduplication were observed, with or without metabolic activation (Chanda et al. 2004; Proudlock et al. 2004). Previously, capsaicin had been studied using HBPLcs in the cytokinesis-block micronucleus (CBMN) assay and the sister chromatid exchange (SCE) assay in the presence or absence of external metabolic activation (Marques et al. 2002). Capsaicin induced the formation of micronuclei in a concentration-dependent manner in the CBMN assay; this increase was more evident in the absence of metabolic activation, with a maximum of 3.4-fold increase above the background. These results were interpreted by Marques et al. (2002) to indicate that capsaicin is genotoxic.
Chanda and colleagues (2004) found a positive result in the mouse lymphoma assay conducted with L5178Y tk+/tk− mouse lymphoma cells, in the absence and presence of metabolic activation. Capsaicin was found to be weakly mutagenic in the presence of S9, when tested at concentrations extending into the toxic range. Limited evidence of very weak activity was also noted in the absence of S9. However, the clinical significance of the weak positive result in the mouse lymphoma assay is unclear: the capsaicin molecule contains a catechol moiety, and other naturally occurring catechol compounds such as
Capsaicin was also reported to induce DNA strand breaks in human neuroblastoma cells SHSY-5Y (Richeux et al. 1999). However, because of the established ability of capsaicin to selectively induce apoptosis in cancerous cells (discussed below), this study should not be considered as evidence for mutagenic potential and is not included in Table 1. Point mutation tests in Chinese hamster V79 cells were conducted, resulting in positive (Gannett et al. 1988; Lawson and Gannett 1989) and negative (Nagabhushan and Bhide 1985) responses. While a positive finding was reported for V79 mammalian cells by Gannett et al. (1988), it was noted that a greater response was observed for red pepper extract as compared to pure capsaicin. Although V79 cells have been used in studies of DNA damage and repair and are not classified as cancerous cells, they share several properties with cancer cells (Chaung et al. 1997). Therefore, data derived from V79 cells should be interpreted with caution for molecules such as capsaicin which can induce apoptosis in cancerous cells.
In Vivo Genotoxicity
Non-mutagenicity of capsaicin in albino mice was first shown by Muralidhara (1988) upon intraperitoneal administration of pure capsaicin at doses up to 1.6 mg/kg (one-fifth of the LD50 ). At least five in vivo micronucleus tests have been conducted with capsaicin in bone marrow cells from different strains of mice, yielding three negative and two positive results. Nagabhushan and Bhide (1985) showed a positive result at a dose near the LD50 but not at a lower dose (one quarter of the LD50 ), but the capsaicin used was only 80% pure. Arceo et al. (1995) also showed a significant increase in number of peripheral blood cells with micronuclei. The micronucleus assay was repeated by groups with >98% pure capsaicin, both times yielding negative results: Chanda et al. (2004) administered capsaicin orally to male mice at dose levels up to 800 mg/kg and females received 200 mg/kg; Proudlock et al. (2004) administered capsaicin subcutaneously using an escalating dosing regime up to 500 mg/kg. Earlier, Villaseñor et al. (1993) had also shown that capsaicin at a maximum tolerable dose of 1.22 mg/kg (intraperitoneally) was not mutagenic in the same assay. In another study, De et al. (1995) showed that capsaicin significantly inhibited cyclophosphamide-induced chromosomal aberrations and DNA strand breakages in bone marrow cells of Swiss albino mice. These groups concluded that capsaicin was non-clastogenic. The results for nine in vivo genotoxicity studies are briefly summarized in Table 2.
In vivo genotoxicity assays with capsaicin.
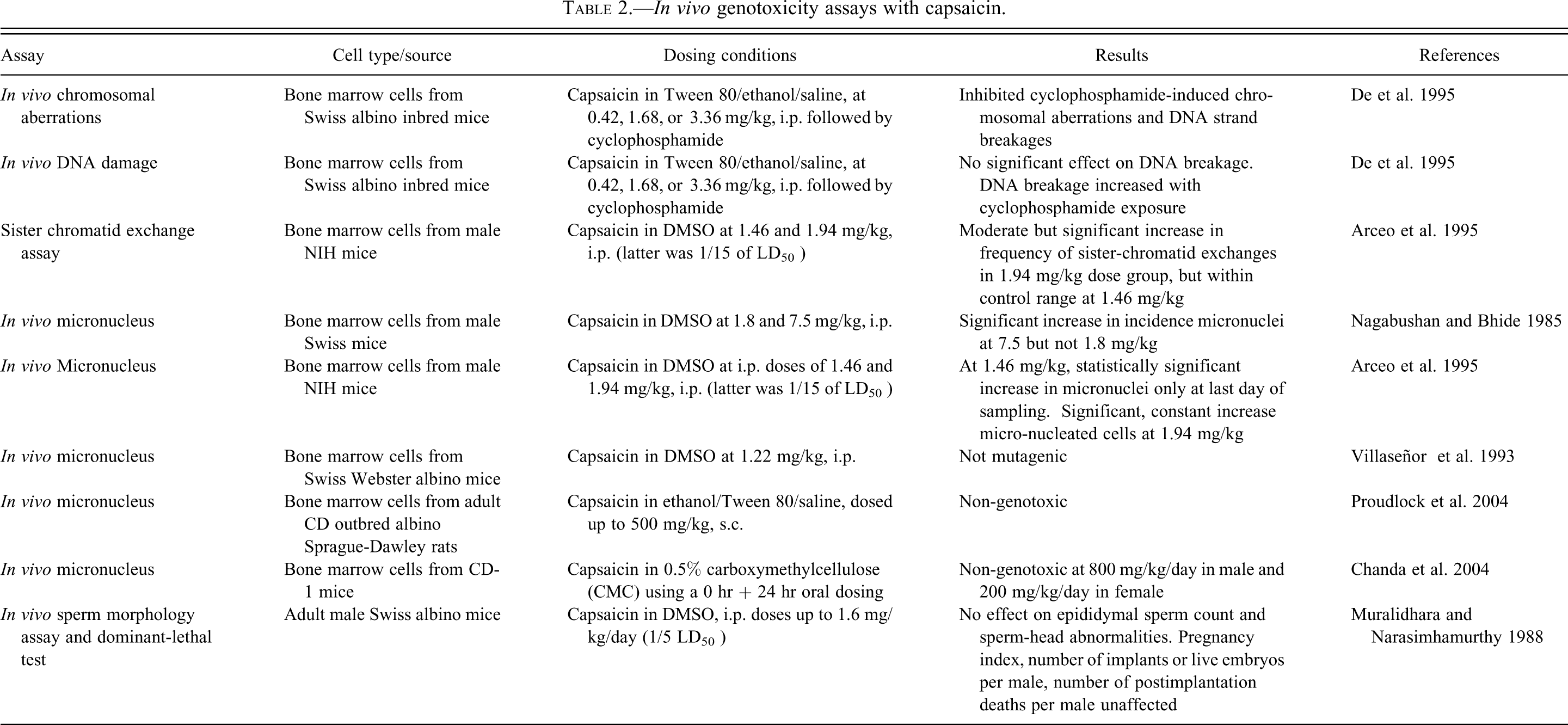
These in vitro and in vivo results point to the absence of genotoxic activity of high-purity capsaicin, even with metabolic activation. It is possible that the genotoxic activity observed in some of the studies was associated with other compounds or mutagenic impurities in the test articles, as is suggested by the results of Tsuchiya et al. (2011). Moreover, at the high concentrations used in some of these assays, capsaicin itself displays antimicrobial activities and induces cytotoxicity—properties which are shared with many other naturally occurring phenolic and flavonoid compounds (Ozçelik et al. 2011).
In Vivo Carcinogenicity
Fewer investigations of the carcinogenic potential of capsaicin in in vivo animal models (Table 3) are reported, in contrast to the numerous genotoxicity studies. Capsaicin-containing materials tested in these studies were always natural extracts and never highly pure capsaicin, with exception of the Tg.AC mouse study (Chanda et al. 2007).
Studies evaluating capsaicin in in vivo carcinogenicity models.

Capsaicin was administered at concentrations of 1, 0.5, 0.25, 0.125, and 0.0625% in powdered diet for 35 days to five groups (four/sex/group) of Swiss albino mice (Toth et al. 1984). Capsaicin had no effect on survival. Three males and one female developed adenocarcinomas of the duodenum (one tumor in each of the four lowest capsaicin dose level groups), while no such tumors occurred in the highest dose level group. No direct comparison with capsaicin-treated controls was possible, as data for control groups were presented in another report (Toth et al. 1980). Pathological descriptions of the tumors were not provided. In a subsequent study using fifty animals/sex/group, capsaicin was administered in a semisynthetic powdered diet at 0.03125% level (the capsaicin extract contained 65% capsaicin) for the lifespan of Swiss albino mice, starting from 6 weeks of age (Toth and Gannett 1992). Again, capsaicin had no effect on survival. Adenocarcinomas of the duodenum were not seen in females and the one tumor in the males was not considered related to capsaicin. The authors also noted that duodenal tumors were not significantly increased in their previous (Toth et al. 1984) study. Instead, benign polypoid adenomas of the cecum were considered to be increased, with 7/50 in treated males versus 4/50 in the control males and 11/50 in treated females versus 4/40 in the control females.
Muralidhara (1988) administered intraperitoneally capsaicin to adult male mice at 0.4, 0.8, or 1.6 mg/kg body weight/day (1/20, 1/10, or 1/5 of the LD50 ) on five consecutive days; these doses did not induce any clinical signs of toxicity. No significant alterations were observed in epididymal weights, caudal sperm counts, testicular weights, or testicular histology. In the sperm morphology assay, sperms at 1, 3, 5, and 7 weeks did not reveal any treatment-related increase in the incidence of sperm head abnormalities. Capsaicin also failed to induce dominant lethal mutations during an 8-week sequential mating schedule of males treated at the highest dose level.
Akagi et al. (1998) evaluated the carcinogenicity of a mixture of 64.5% capsaicin and 32.6% dihydrocapsaicin using groups of fifty male and female B6C3F1 mice fed 0.025%, 0.083%, and 0.25% capsaicinoids for 79 weeks, and then a standard diet for 4 weeks. No effect on survival was seen and body weights were lower in females in a dose-related fashion. The numbers of tumor-bearing females in the high-dose groups were significantly lower than the controls, and the incidences of hepatocellular neoplasms in both sexes were negatively correlated with the dose of capsaicinoids. The authors concluded that a mixture of capsaicinoids was not carcinogenic in B6C3F1 mice.
The dermal carcinogenic potential of pure synthetic capsaicin was evaluated in the Tg.AC transgenic mouse model (Chanda et al. 2007). Capsaicin was administered weekly via topical application to the dorsal skin of 25/group of male and female Tg.AC mice for 26 weeks. The positive control tetradecanoylphorbol-13-acetate (TPA), was dosed twice per week. Analysis of the macroscopic observations after the final sacrifice revealed no noteworthy treatment-related findings. The frequency of dermal masses in the capsaicin-treated groups (at dose levels of up to 102.4 mg/kg and an application rate of 25.6 mg/cm2/kg/week) was not elevated in comparison to either concurrent vehicle or untreated controls. Increased incidence of dermal masses was observed in the TPA-treated mice compared to both control groups. Topical application of capsaicin resulted in no increased incidence of pre-neoplastic or neoplastic skin lesions. Spontaneously occurring neoplasms were also not increased in capsaicin-treated animals. Capsaicin-related non-neoplastic microscopic findings were seen sporadically in males and females and included acanthosis, hyperkeratosis/parakeratosis, epidermal crusts, subepidermal fibrosis, epidermal ulcerations/erosions, and chronic-active inflammation. No evidence of a dose response in either the incidence or severity of these findings was found and the authors concluded that capsaicin was non-oncogenic in the Tg.AC dermal mouse model.
Co-Carcinogenicity, Multiple-Stage, and Anticarcinogenicity Models
Studies on the activity of capsaicin in in vivo co-carcinogenicity, multiple-stage, and anticarcinogenicity models are summarized briefly in Table 4, and selected publications are discussed below. Studies with chili pepper extracts have generally been excluded. For instance, Agrawal conducted a large study in BALB/c mice (Agrawal et al. 1986). However, the source of the capsaicin extract was dried chilies purchased from a local market in India. As discussed below in context of human epidemiological studies, known carcinogens have been found in chilies from India (Siddiqi et al. 1988) and Chile (Tsuchiya et al. 2011).
Studies on capsaicin in in vivo co-carcinogenicity, multiple-stage, and anticarcinogenicity models.

Abbreviations: ACF, aberrant crypt foci; AOM, azoxymethane; BP, benzo(a)pyrene; DBN, N,N-dibutylnitrosamine; DEN, diethylnitrosamine; DMBA, 9,10-demethyl-1,2-benzanthracene; HCAs, heterocyclic amines; KO, knock-out; MNNG, N-Methyl-N'-nitro-N-nitrosoguanidine; MNU, N-methylnitrosourea, NDMA, N-nitrosodimethylamine; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; 4-NQO, 4-nitroquinoline 1-oxide; TPA, 12-O-tetradecanoylphorbol-13-acetate; VC, vinyl carbamates; WT, wild-type.
LaHann (1986) evaluated the effect of capsaicin on TPA-induced tumorigenesis in the skin of CD-1 mice in which 25 nmol 7,12-dimethylbez(a)-arthracene (DMBA) was applied for 7 days during the initiation phase. Mice were then treated twice weekly with 10 nmol TPA or vehicle, and one group was also treated with capsaicin (13.5 µmol) 0.5, 1.5, and 24 hr prior to each TPA dose. The rate and incidence of papillomas were recorded weekly for 20 weeks. Capsaicin did not reduce the incidence or rate of TPA-induced tumor formation but may have reduced the time required for tumor development.
Newborn Swiss Webster mice were injected subcutaneously with 1 mg of benzo(a)pyrene BP or 40 µg of DMBA to study lung carcinogenesis (Jang et al. 1989). Capsaicin was dissolved in dimethyl sulfoxide (DMSO) and included in food pellets at 0.01% for 6 weeks after weaning, with an average daily intake of about 13–18 mg/kg. Capsaicin had reduced significantly the mean number of lung tumors in both BP- and DMBA-treated animals at 9 weeks after birth. The same author (Jang et al. 1991) evaluated the effect of capsaicin on F344 rats pretreated with three carcinogens (diethylnitrosamine [DEN], N,N-dibutylnitrosamine [DBN] and N-methylnitrosourea [MNU]). The rats were initially administered a single dose of DEN (100 mg/kg, i.p.), then MNU (20 mg/kg, i.p.) on days 2, 5, 8, and 11, and then DBN was at 0.05% in drinking water during weeks 3 and 4. A diet containing capsaicin (0.01%) until week 20 significantly inhibited the induction of glutathione S-transferase π hepatic foci, significantly decreased the incidence of lung adenoma (from 56% to 13%), but enhanced the incidence of papillary or nodular hyperplasia of the urinary bladder. Incidences of carcinogen-induced lesions of the thyroid and kidney were not significantly altered by capsaicin.
Surh, Lee, and colleagues have been key contributors to the examination of the pro- and anticarcinogenic potential of capsaicin. Surh, Lee, et al. (1995) studied the antitumorigenicity of capsaicin using Imprinting control region (ICR) mice. Capsaicin (2.5 µmol) in 0.2 ml acetone was applied 10 min prior to topical application of vinyl carbamate (VC; 5.8 µmol), which was followed by promotion with TPA. Pretreatment with capsaicin lowered tumor multiplicity by 62%. One µmol topical capsaicin 24 and 1 hr before initiation by topical BP (0.3 µmol) and then promoted with TPA also significantly reduced the number of tumors. Park and Surh (1997) evaluated the tumor initiating and promotional effects of capsaicin using female ICR mice. Topical capsaicin (10 µmol) followed by treatment with TPA for 22 weeks did not produce an increase in the incidence and multiplicity of skin papillomas. In the same paper, after initiation with DMBA, capsaicin was applied topically twice weekly for 22 weeks, again with no significant enhancement of skin tumor formation. In another experiment, treatment with capsaicin 30 min prior to each topical application of TPA lowered the incidence of DMBA-initiated mouse skin tumors by 22% at week 15 (the multiplicity of skin tumors was only slightly affected). In a study by the same group under similar conditions in female ICR mice (Park et al. 1998), a single topical application of DMBA was used for initiation. At 1 week after initiation, controls or capsaicin (10 µmol) were applied twice weekly for 22 weeks. One group received simultaneously TPA and capsaicin during the promotion stage. In addition to tumor counts, induction of epidermal ornithine decarboxylase (ODC) activity was evaluated as a marker of tumor promotion. Neither capsaicin alone nor combined with DMBA resulted in increased papillomas. Compared to the DMBA-TPA group, simultaneous application of capsaicin and TPA after DMBA initiation resulted in a slightly higher tumor incidence and multiplicity during the first 12 weeks of tumor promotion (no statistics provided), but then the tumor count was clearly reduced after the first 12 weeks. Microscopic examination indicated that capsaicin did not alter the morphological features of untreated skin, while TPA resulted in striking changes, including epithelial hyperplasia and chronic dermal inflammation. Application of capsaicin did not cause any increase in ODC activity, while TPA caused a dramatic increase which was not reversed with capsaicin.
Female A/J mice were dosed intragastrically with capsaicin (5 mg/kg, in 0.2 mL corn oil or corn oil alone) for 3 consecutive days (Teel and Huynh 1999). In both groups, the last dose was followed by a single i.p. injection of 10 µmol NNK. A small but not statistically significant decrease in lung tumors in the capsaicin-dosed animals was observed after 21 weeks.
Kim et al. (1999) evaluated the effects of capsaicin on N-methyl-N’-nitro-N-nitrosoguanidine (MNNG)-induced c-jun proto-oncogene expression in male Fisher-344 rats. Rats were fed a capsaicin-containing (20 ppm) diet for 13-weeks. After 6 weeks, some rats were dosed orally with MNNG (200 mg/kg) via gavage. In another 6-week study presented in the same paper, rats were given a single oral dose of MNNG (200 mg/kg) via gavage, and then at 1 week post-dosing, capsaicin (20 ppm) was given to half the animals. MNNG enhanced c-jun expression 1.5- to 3.0-fold in tissues of the spleen, heart, and intestine; however, c-jun transcripts were reduced in the liver, kidney, stomach, and lung. Capsaicin decreased c-jun mRNA levels in most organs, except for the spleen and intestine, with the most significant transcription reductions in the stomach and liver. Kim et al. (1999) suggested that capsaicin uptake in the diet could play a role in inhibition of tumorigenesis induced by MNNG.
Bode et al. (2009) reported a striking increase in skin carcinogenesis in TRPV1 knock-out (TRPV1-/-) mice in a two-stage carcinogenesis experiment. When TRPV1-/- mice were initiated with DMBA and then promoted with TPA over 21 weeks, a significant enhancement of tumor development relative to wild-type controls was found. Subsequently, the same group (Hwang et al. 2010) showed that capsaicin did not induce tumors when applied topically to DMBA-initiated mice twice a week for 21 weeks, as treatment with only capsaicin or vehicle did not induce any skin tumors in either wild-type or TRPV1-knockout mice. Tumors appeared only following treatment with TPA, with the combination of TPA and capsaicin producing even more tumors than TPA alone. Unfortunately for the interpretation of these data, the potential effects of variable skin pharmacokinetics of TPA were not evaluated. Capsaicin is known to enhance delivery of compounds into skin, with efficacy similar to the widely used skin penetration enhancer Azone® (Degim et al. 1999; Parhi et al. 2011). Hence it is possible that the putative tumor promotion by capsaicin was due to increased delivery of TPA and not a direct effect of capsaicin. Regarding the ability of TRPV1 to make mouse skin more susceptible to the effect of either TPA or TPA plus capsaicin, it must be remembered that activation of TRPV1 produces a neurovascular response associated with increased skin blood flow (Geppetti et al. 2008). TPRV1 knock-out mice do not respond to capsaicin (Caterina et al. 2000) and in consequence may be missing enhancement of drug clearance from skin due to capsaicin-mediated blood flow increases. It is possible that TPA remains longer in the skin of TRPV1 knockout mice, resulting in increased tumor formation. None of these pharmacokinetic complexities were considered in a recent review of these data (Bode and Dong 2011).
The effects of dietary capsaicin on azoxymethane (AOM)-induced colon tumorigenesis were investigated in male F344 rats (Yoshitani et al. 2001). Gavage with capsaicin significantly elevated phase II enzymes (glutathione S-transferase and quinone reductase) in the liver and colon. In an aberrant crypt foci (ACF) bioassay, feeding of capsaicin at 500 ppm for 4 weeks significantly inhibited ACF formation induced by AOM (20 mg/kg body weight, once a week for 2 weeks). In a subsequent long-term study designed to examine possible protective effects on ACF development, groups were treated with AOM alone or AOM plus 500 ppm capsaicin for 4 weeks (initiation phase) and for 34 weeks (post-initiation phase). Capsaicin during the initiation phase was found to significantly reduce (by 60%, relative to controls) the incidence of colonic adenocarcinoma. The same group next studied the effects of dietary capsaicin on 4-nitroquinoline 1-oxide (4-NQO)-induced tongue tumorigenesis in male F344 rats (Tanaka et al. 2002). Ten weeks of feeding 500 ppm capsaicin together with 4-NQO inhibited the occurrence of tongue dysplasia. In a subsequent long-term study, groups were treated with 4-NQO alone (20 ppm in drinking water for 8 weeks) or 4-NQO plus 500 ppm capsaicin in diet for 10 weeks (initiation phase) or for 28 weeks (post-initiation phase). At the termination of the study (38 weeks), capsaicin feeding during either the initiation or promotion phase reduced the frequency of tongue squamous cell papillomas and squamous cell carcinomas (NQO alone, 9/15 rats with tongue tumors; NQO plus 10 weeks capsaicin, 5/17 rats with tongue tumors [not significant]; NQO plus 28 weeks of capsaicin, 3/14 rats with tongue tumors [p<0.05]).
In a series of closely related experiments using Swiss albino mice, capsaicin was shown to exert its chemoprotective effects via several different mechanisms against benzo(a)pyrene (BP)-induced lung cancer (Anandakumar et al. 2008; Anandakumar et al. 2008a, 2008b; Anandakumar et al.2009a, 2009b; Anandakumar, Jagan, Kamaraj, et al. 2009; Anandakumar, Kamaraj, Ramakrishnan, et al. 2009; Anandakumar, P., Kamaraj, S., Jagan, S., Ramakrishnan, G., Naveenkumar, et al. 2009). Due to the closely related nature of methods and somewhat overlapping results, key results from all 8 publications are grouped into one entry in Table 4. In this model, BP was administered orally (50 mg/kg) twice a week for 4 weeks. Capsaicin treatment (10 mg/kg, i.p.) was initiated one week prior to the first dose of BP and continued once a week for 14 weeks, with outcomes appearing in separate publications. Generally, capsaicin reversed all BP-induced changes. For instance, capsaicin prevented pronounced oxidative damage caused by BP to biomolecules, preserved lysosomal stability, restored the activities of phase I and II biotransformation enzymes and the levels of tumor markers to near normalcy. BP-induced lung cancer animals showed abnormal changes in body weight, tumor incidence, low blood glucose and alterations in lipid levels, all of which were significantly reversed by capsaicin (Anandakumar et al. 2008; Anandakumar et al. 2008a, 2008b; Anandakumar et al.2009a, 2009b; Anandakumar, Jagan, Kamaraj, et al. 2009; Anandakumar, Kamaraj, Ramakrishnan, et al. 2009; Anandakumar, P., Kamaraj, S., Jagan, S., Ramakrishnan, G., Naveenkumar, et al. 2009).
These studies in which rodents were exposed to multiple compounds support the conclusion that capsaicin alone produces little to no increase in tumor incidence or multiplicity. When administered in combination with known carcinogenic substances, in almost all cases, capsaicin either reduced tumors or did not potentiate them. The only exception to this conclusion is the study of Hwang et al. (2010), which unfortunately fails to take into account indirect effects on dermal delivery and retention of the administered tumor promoter TPA. In order to substantiate a direct effect of capsaicin on tumor formation in this or in any other model, levels of carcinogens in the skin should be quantified and be comparable in the various treatment groups.
Metabolism, cyp Inhibition and Antimutagenicity
The in vitro metabolism of capsaicin has been investigated extensively and a number of metabolites have been characterized. A compilation of potential capsaicin metabolites are shown in Figure 1, based on figures which appeared in Surh and Lee (1995), Reilly and Yost (2006), and Chanda et al. (2008). It is important to note that while the metabolites depicted in Figure 1 have been reported under in vitro or ex vivo conditions, the in vivo metabolism of capsaicin in humans or animals following experimental, dietary, environmental or medical exposure has not been thoroughly investigated.
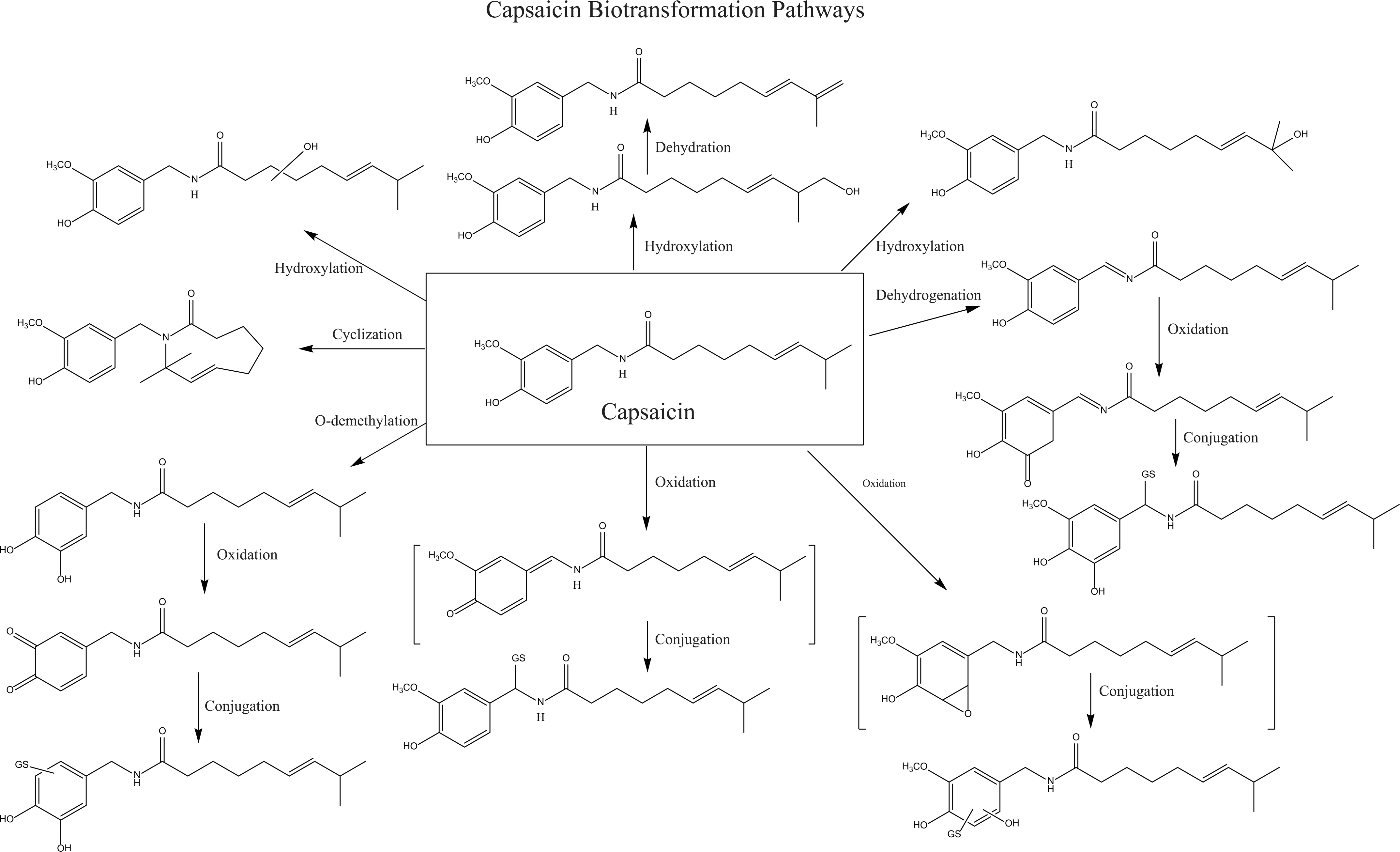
Compilation of all reported potential capsaicin metabolites, based on Surh and Lee (1995), Reilly and Yost (2006), and Chanda et al. (2008). No quantitative determination of relative abundance is available, although Chanda et al. found that 16-hydroxycapsaicin, 17-hydroxycapsaicin, and 16,17-dehydrocapsaicin were the major metabolites.
Capsaicin is metabolized by a number of cytochrome (CYP) P450 enzymes leading to aromatic and alkyl hydroxylation, O-demethylation and alkyl dehydrogenation (Surh, Ahn, et al. 1995; Reilly et al. 2003; Reilly and Yost 2006). It has been further suggested that capsaicin is bioactivated through the formation of several electrophilic metabolites that have the potential to lead to hepatotoxic and/or mutagenic responses (Surh and Lee 1995; Reilly et al 2003; Reilly and Yost 2006). However, the formation of reactive metabolites as identified through in vitro experiments such as glutathione trapping and covalent binding has not been directly linked to adverse drug reactions in the clinic (Park et al. 2011). For example, a number of hepatotoxic and non-hepatotoxic compounds have been tested for covalent binding to liver microsomal protein, and the results indicated that prediction of toxicity through the use of in vitro covalent binding experiments was unreliable due to the number of false positive findings (Obach et al. 2008). In fact, BEAS-2B and HepG2 cells displayed a greater sensitivity to cytotoxicity in the presence of capsaicin and 1-aminobenzotriazole (a nonspecific CYP450 inhibitor), indicating that capsaicin metabolism represented a detoxification mechanism, as opposed to bioactivation (Reilly et al. 2003).
Compounds can either directly inhibit CYP enzymes or be metabolized to compounds that can reversibly or irreversibly inhibit CYP enzymes (Madan et al. 2002). Inhibition of CYP enzymes, particularly CPY2E1, has been evoked by several contributors to explain the antimutagenic and anticancer properties of capsaicin, which occur at the same time as the putative pro-carcinogenic activities of other metabolites (see Figure 2). For instance, one recent review states that, “Capsaicin has been documented to modulate microsomal cytochrome P450-dependent monooxygenase activities with resultant alteration of metabolism of carcinogens and other xenobiotics, to [the] ultimate carcinogen” (Oyagbemi et al. 2010). The general acceptance of the hypothesis, which focuses on the inhibition of CYP2E1 by capsaicin, appears to be due to the influence of reviews by Surh and Lee (1995) and Reilly and Yost (2006).
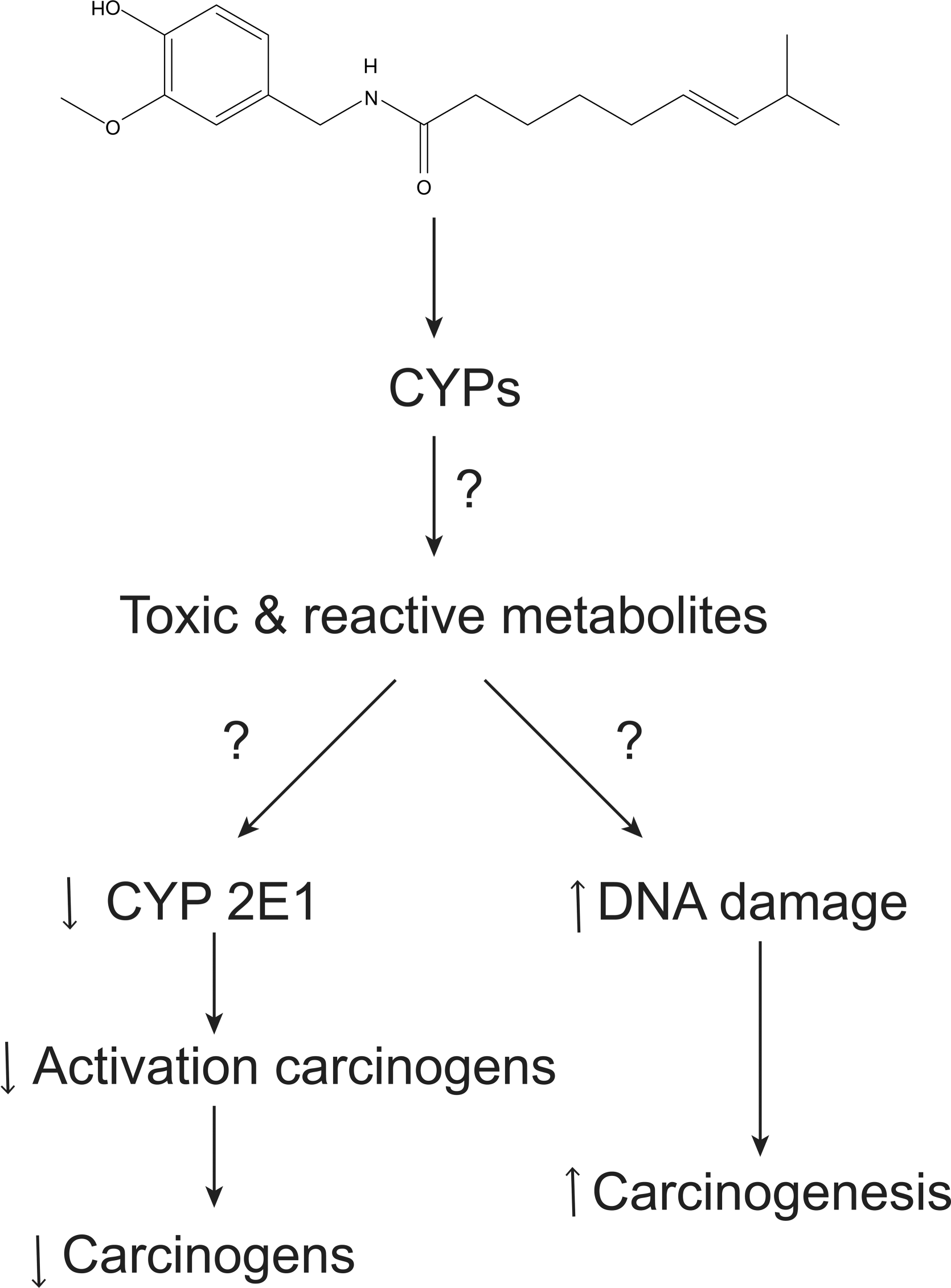
Conjectured roles of capsaicin metabolites in CYP-mediated carcinogenesis and anticarcinogenesis. Although often repeated in literature, little evidence suggests than any CYP-produced metabolite has either physiological or clinical relevance.
The origins of the hypothesis that capsaicin prevents cancer via the inhibition of CYP2E1 arose from studies with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Metabolic activation is required for the mutagenicity and carcinogenicity of NNK and its metabolism occurs via three major pathways: (1) carbonyl reduction, (2) N-oxidation of the pyridine ring, and (3) α-methylene hydroxylation (Richter et al. 2009). Zhang et al. (1993) evaluated the effect of capsaicin on the in vitro metabolism of NNK by hamster and rat liver microsomes. At each of the concentrations of capsaicin tested (250, 500, and 1000 µM), significant inhibition of all three major pathways for NNK metabolism was observed in hamster liver microsomes. Concentration-dependent inhibition of NNK reduction occurred in rat microsomes and 500 and 1,000 µM capsaicin also significantly inhibited α-hydroxylation of NNK. Zhang et al. (1993) also evaluated the effect of capsaicin (500 µM) on the formation of metabolites of 14C-testosterone by hamster and rat microsomes. As testosterone is hydroxylated by several CYP P450s, results indicated that capsaicin inhibited the activity of several enzymes. In a parallel in vitro study by the same group, capsaicin significantly inhibited the mutagenicity of NNK in S. typhimurium strain TA1535 at doses of 0.1, 0.2, and 0.4 µmol/plate (with metabolic activation; Miller et al. 1994). Therefore, it was suggested that the antimutagenic and anticarcinogenic properties of capsaicin are due to its inhibition of xenobiotic-metabolizing enzymes.
Dihydrocapsaicin, the saturated analog of capsaicin, was observed to inhibit CYP2E1 (Gannett et al. 1990); the tested concentration of dihydrocapsaicin was not disclosed. Subsequently, Surh, Ahn, et al. (1995) reported that capsaicin (420 µM) attenuated the mutagenicity of vinyl carbamate and N-nitrosodimethylamine (NDMA) in S. typhimurium TA100. The authors also suggested that capsaicin suppressed vinyl carbamate and NDMA-induced mutagenesis, in part, through inhibition of CYP2E1, which is responsible for the activation of these two carcinogens. Because inhibition of CYP2E1 is thought to prevent the metabolic activation of carcinogens such as NNK, it was inferred that capsaicin was a CYP2E1 inhibitor (Surh and Lee 1995).
A summarized listing of these antimutagenicity and anticancer (next section) activities of capsaicin—excluding the compound’s direct effects on cancerous cells in vitro and in xenograph mouse models—is provided in Table 5. This listing includes in vivo studies in which capsaicin’s ability to protect from known carcinogens was evaluated.
Chronological listing of studies evaluating the direct effects of capsaicin on cancerous cells

Abbreviations: AR, androgen receptor; BAPTA-AM, 1,2-bis(2-aminophenoxy)ethane-N, N,N',N'-tetraacetic acid tetrakis(acetoxymethyl ester); [Ca2+]i, intracellular calcium ions; C/EBPβ, CCAAT/enhancer binding protein beta; hEGFR, human epidermal growth factor receptor; ER, estrogen receptor; iNOS, inducible nitric oxide synthase; NAC, N-acetyl-L-cysteine; NADPH, nicotinamide adenine dinucleotide phosphate; NAG-1, nonsteroidal anti-inflammatory drug-activated gene-1, NF-κB, nuclear factor kappa B; PARP-1, polyADP-ribose polymerase 1; PPARγ, peroxisome proliferator-activated receptor gamma; PSA, prostate-specific antigen; ROS, reactive oxygen species; SODs, superoxide dismutases; STAT3, Signal transducer and activator of transcription 3; tNOX, tumor-associated NADPH oxidase; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRPV1, transient receptor potential vanilloid 1 receptor; ΔΨm, mitochondrial membrane potential.
Protocols for the prediction of potential drug-drug interaction via in vitro experiments with CYPs have evolved and are now governed by guidance from drug regulatory agencies (Singh 2006). Data derived from such protocols has recently appeared for capsaicin: capsaicin inhibits CYP1A2, CYP2C9, and CYP2C19, with IC50 values of 2.1, 2.0, and 3.2 µM, respectively, and inhibits CYP2B6, CYP2D6, CYP3A4, and CYP3A5 with extrapolated IC5 0 values of 24, 18, 38, and 12 µM, respectively (Babbar et al. 2010). Due to a lack of inhibition at the highest tested concentration (10 µM), an IC50 value for CYP2E1 could not even be extrapolated. Drugs are predicted to lack drug–drug interactions when the [I]/Ki ratio is less than 0.1, where [I] is the plasma Cmax value and Ki is inhibition constant that defines the affinity of the inhibitor for the enzyme is estimated from the IC50 values (US FDA 2006). In the case of capsaicin, assuming a competitive mode of inhibition where Ki ≈ IC50 /2 for each of the enzymes evaluated and 58 nM for the highest plasma capsaicin concentration reported in any publication (Babbar et al. 2009; Chaiyasit et al. 2009), all values are calculated to be less than 0.1. With the exception of CYP2B6 and CYP2D6, the IC50 values for metabolism-dependent CYP inhibition increased upon preincubation of capsaicin with each CYP (Babbar et al. 2010). Even for CYP2B6 and CYP2D6, they were still in the micromolar range and outside of concentrations considered relevant based on potential ingestion or medical use.
In summary, standard safety calculations indicate that capsaicin is unlikely to inhibit directly or even indirectly the metabolism of compounds or drugs by CYPs based on the pharmacokinetic values for the dietary and medical product exposure discussed above. Recent data suggest that concentrations of capsaicin outside of those readily generated in human tissues may be required to produce either substantial inhibition of CYP2E1 or inhibit metabolic activation of carcinogens such as NNK. In the study on capsaicin-mediated CYP inhibition by Reilly and Yost (2006), the lowest capsaicin concentration tested was 10 µM, which is already about 10,000-fold higher than required to activate TRPV1 (Alawi and Keeble 2010). We question the physiological relevance of millimolar capsaicin concentrations. Alternative possible mechanisms for protection by capsaicin against carcinogens, which require metabolic activation are discussed below.
Direct Effects on Cancerous Cells
Capsaicin has been recognized to inhibit the growth of or induce apoptosis in a wide variety of tumor and cancer cell lines (Surh 2002; Oyagbemi et al. 2010). More recent studies have confirmed and extended these observations to additional cell lines and to rodent in vivo xenograph tumor models. Table 5 briefly captures some key observations, in these studies, in approximate order of their publication. Note that this table excludes data generated from heterologous systems, such as cell lines stably transfected with TRPV1. Overall, capsaicin appears to inhibit the growth of or induce apoptosis in over forty distinct cancer cell lines, the vast majority of which are human in origin. Due to the preponderance of data from human cell lines, it is not possible to conclude whether cancers from rodents or other species display differential sensitivity to capsaicin.
The anticancer effects of capsaicin in vitro are both concentration- and time-dependent, as illustrated in Figure 3. In most publications, the effects of capsaicin appear in the low micromolar range and become maximal at approximately 200–300 µM. Duration of exposure enhances the potency of capsaicin, and the stability of capsaicin under specific experimental conditions may vary, so rigorous comparisons of IC50 values across studies is not possible. Most studies are limited to measurements of cell viability, growth rates or apoptosis within 48–72 hr of treatment. The only information about long-term or continuous exposure is provided by Macho et al. (1999), who found that incubation with 300 µM capsaicin for 5 days did not induce apoptosis in resting T cells.
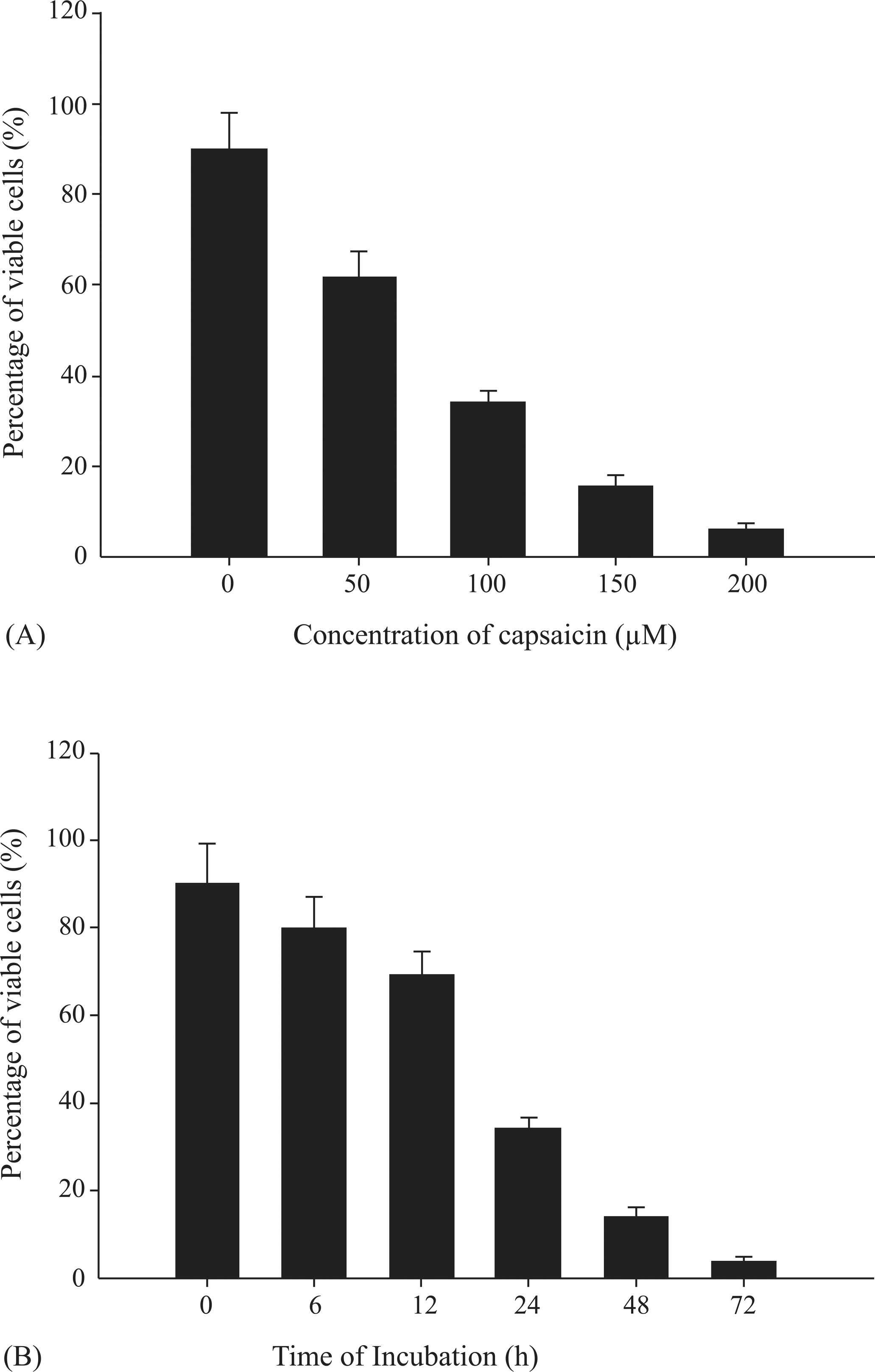
Representative concentration- and time-dependency (at 100 µM) of capsaicin for induction of apoptosis in cancerous cells. Study on human esophageal epidermoid carcinonama CE 81T/VGH cells. Taken from Wu et al. (2006).
Corresponding antitumor activity has been observed in in vivo mouse xenograph models in eleven publications in which capsaicin was administered orally, subcutaneously, and intraperitoneally, as well as directly into tumors. Source cells included mouse melanoma (Morré et al. 1996), human leukemia (Ito et al. 2004), human prostate (Mori et al. 2006; Sánchez et al. 2006; Díaz-Laviada 2010), human multiple myeloma (Bhutani et al. 2007), human pancreatic (Zhang et al. 2008; Pramanik et al. 2011), and human bladder (Yang et al. 2010) cancers. With the exception of skin ulceration and irritability noted by Sánchez et al. (2006), no appreciable toxic effects on mice by capsaicin administration were reported.
Normal or noncancerous cells appear to be significantly less sensitive to the apoptotic or growth inhibitory effects of capsaicin than cancerous cells (as described in fourteen studies listed in Table 5); the potential bases for this selective action will be discussed below. One important implication of this general observation is that mutagenicity or genotoxicity studies with capsaicin using cancer cells may have limited relevance for normal cells. For instance, Richeux et al. (1999) reported that cytotoxicty and DNA fragmentation was induced in human neuroblastoma SHSY-5Y cells by 25–100 µM capsaicin, with an IC50 of 60 µM for the inhibition of protein synthesis. The authors concluded that this finding could have, “important implications for the possible health threats of capsaicin, especially in the case of misuse of capsaicin preparations in pathological situations.” Similarly, Lawson and Gannett (1989) reported that oxidative metabolism of capsaicin by hepatic microsomes produced a metabolite that was mutagenic for Chinese hamster lung fibroblast V79 cells, even though capsaicin itself was not mutagenic. This study by Lawson and Gannett seems to have provided additional impetus for research into whether metabolic activation of capsaicin could yield mutagens or carcinogens (discussed above). Although V79 cells are widely used in studies of DNA damage and repair and are not classified as cancerous cells, they share several properties with cancer cells, including being immortal, not expressing functional p53 protein, failing to show induction of the mdm2 gene product in response to DNA damage, and being readily mutagenized to make stable mutant lines deficient in DNA repair enzymes (Chaung et al. 1997). Therefore, it seems prudent to interpret with caution DNA damage results derived from capsaicin exposure to cells, which display properties typical of cancerous cells. We have excluded the study of Richeux et al. (1999) in the listing of mutagenicity or genotoxicity studies in Table 1, but to be conservative, have included the two studies based on V79 cells.
A comprehensive review of the potential pro-apoptotic cascades initiated by capsaicin in cancerous cells is beyond the scope of this review, so the following constitutes a survey of possible explanations for the selective action of capsaicin. Reviews with more details regarding apoptotic pathways in cancerous cells have been assembled by others (Surh 2002; Hail and Lotan 2009; Ziglioli et al. 2009).
The first report on the selective action of capsaicin against cancer cells was published by Morré and colleagues (1995), and this group has continued to investigate the potential to use capsaicin and similarly acting molecules as cancer chemotherapeutic agents. Initially, they observed that capsaicin inhibited the growth of human HeLa, ovarian carcinoma, mammary adenocarcinoma, and HL-60 leukemia cells (Morré et al. 1995). Without added epidermal growth factor (EGF), 50% growth inhibition of HeLa cells was noted at 1 µM capsaicin, and with the addition of EGF, complete growth inhibition occurred at 1 µM. Capsaicin was largely without effect in mammary epithelial cells, rat kidney cells, or HL-60 cells induced to differentiate with DMSO. In a subsequent study, Morré et al. (1996) reported that capsaicin inhibited the growth of human A-375 melanoma cells with an EC50 value of ∼6 µM (but human melanoma cell line SK-MEL-28 was resistant to capsaicin). However, growth of primary melanocytes was not inhibited with 10 µM capsaicin and only ∼20% by 1 mM capsaicin.
One important early observation was that the NADPH oxidase activity in plasma membranes was inhibited by capsaicin in HeLa, ovarian carcinoma, mammary adenocarcinoma, and HL-60 cells (with an ED50 value of 5 nM in HeLa cells). In contrast, the NADPH oxidase activity in plasma membranes of normal human mammary epithelial, rat liver, normal rat kidney cells, or HL-60 cells induced to differentiate with DMSO was not inhibited by capsaicin (Morré et al. 1995). Subsequently, this group claimed that a cancer-specific form of NADPH oxidase modulated by capsaicin is present in sera from cancer patients, but absent from the sera of individuals free of cancer (Morré et al. 1997). Additional studies of plasma membrane NADPH oxidase by Morré and colleagues led to the designation of the cancer-specific and growth-related cell surface protein with both oxidative (hydroquinone) and protein disulfide-thiol interchange activity as tNOX (Morré et al. 1998; the official full name of tNOX is ecto-NOX disulfide-thiol exchanger). Chueh et al. (2004) found that vector-forced overexpression of tNOX in MCF-10A mammary epithelia or COS cells that lack tNOX or in COS cells that underexpress tNOX enhanced the susceptibility of apoptosis induction by capsaicin. Moreover, antisense tNOX DNA abrogated growth inhibition by capsaicin and reduced anchorage-dependent growth of HeLa cells. The authors concluded that cell surface expression of tNOX is both necessary and sufficient for the anticancer activities attributed to capsaicin. Subsequently, tNOX has been cloned (Jiang et al. 2008), but the existence of more than twenty different isoforms dependent on parental tissue and cancer type led to the conclusion that a single, highly effective treatment for all types of cancer is unlikely (Morré et al. 2009).
There are NADPH oxidases that are distinct from cell-surface tNOX enzymes and the plasma membrane electron transport system. NADPH oxidase also constitutes part of complex I of the mitochondrial electron transport chain, which is involved in oxidative metabolism (Kamata 2009). Capsaicin directly inhibits mitochondrial NADPH oxidase activity by binding competitively to the ubiquinone/coenzyme Q site on this enzyme and has been termed an “antagonist” to ubiquinone (Shimomura et al. 1989; Satoh et al. 1996). Capsaicin even more strongly inhibited activity of mitochondrial complex III in pancreatic cancer cells (Pramanik et al. 2011). Therefore, if capsaicin blocks electron transport in mitochondria, dissipation of transmembrane mitochondrial potential (ΔΨm) should follow. Indeed, capsaicin is widely reported to cause the dissipation of ΔΨm in cancerous cells, as is summarized in Table 5. Loss of ΔΨm is widely understood to initiate apoptosis through causing mitochondrial permeability, which leads to the release of cytochrome c and subsequent activation of pro-apoptotic pathways. ΔΨm dissipation is associated with hyperproduction of reactive oxygen species (ROS), although it is unclear whether this hyperproduction initiates or follows ΔΨm dissipation (see discussion below).
Several lines of evidence suggest that the selective action of capsaicin against some cancerous cells may be via convergence on mitochondrial pathways. Not only does capsaicin cause the dissipation of ΔΨm , but at least in some cancerous cells, it also induces large increases in intracellular calcium, mediated by activation of TRPV1, which is expressed both in plasma and in intracellular membranes (Han et al. 2007). Together, these multiple sources of calcium provide a substantial load for intracellular buffering capacity. Mitochondria are important buffers for high intracellular calcium, and it is widely appreciated that they can be overwhelmed by excessively high calcium loads (Chinopoulos and Adam-Vizi 2010). At this time, in cancerous cells that express TRPV1, there are scant data regarding the relative contributions of the inhibition of mitochondrial electron transport and excessive TRPV1-mediated calcium load to mitochondrial permeability, which leads to release of cytochrome c and subsequent activation of pro-apoptotic pathways. Figure 4 depicts the key elements of these pathways. The existence of multiple sites of action (inhibition of NADPH oxidases, mitochondrial electron chain transport, etc.) may make it difficult to determine whether one mechanism is generally more important for capsaicin’s anticancer actions. Given the heterogeneity between types of cancerous cells and the possible differential sensitivity to capsaicin, such a generalization may not even be possible.
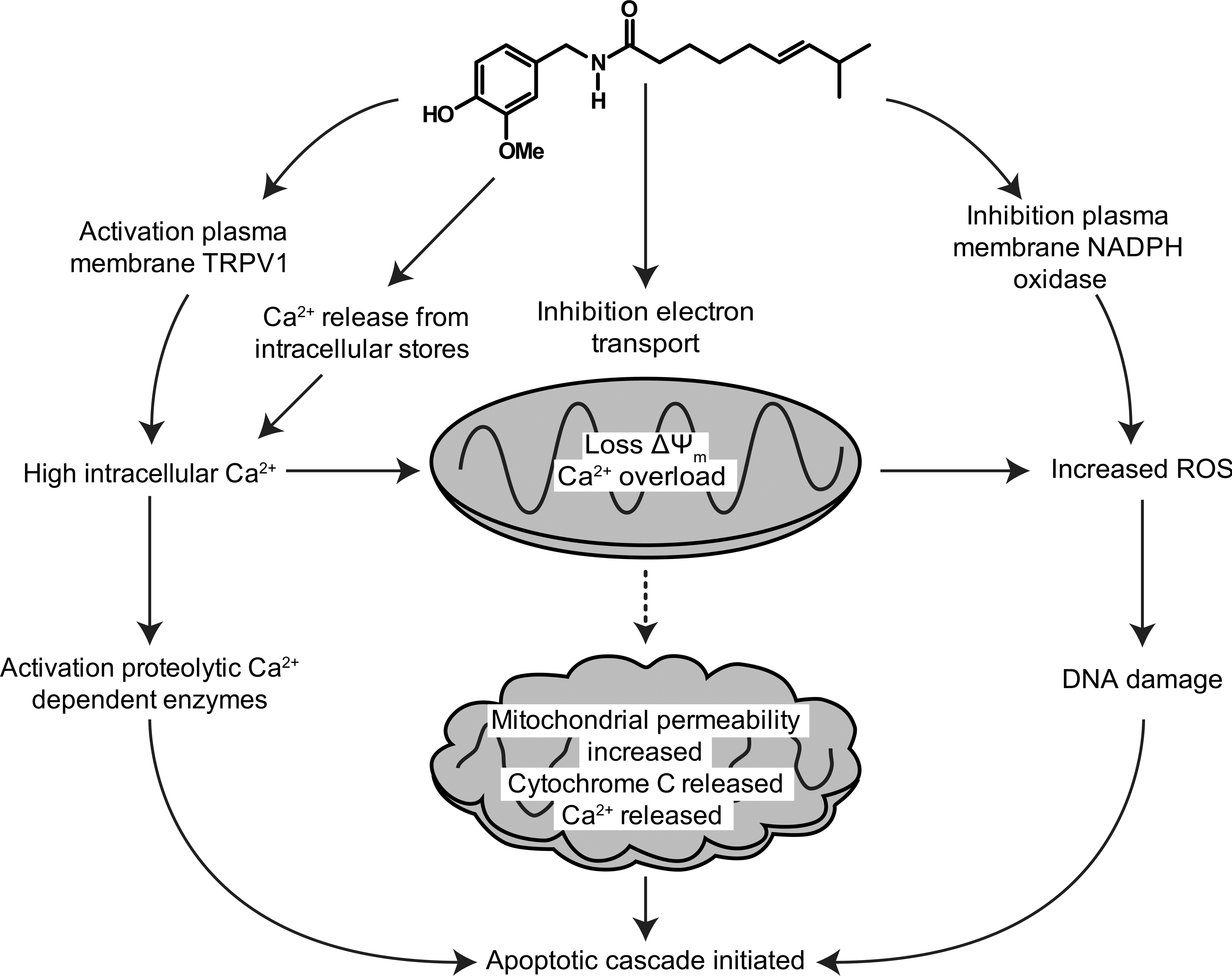
Schema showing pro-apoptotic pathways activated potentially by capsaicin in some cancerous cells.
The relationship between capsaicin exposure and generation of ROS is also quite complex. In normal cells, ROS are conventionally thought as cytotoxic and mutagenic, and at high levels they can induce cell death, apoptosis, and senescence (Hail and Lotan 2009). In contrast, ROS at low levels are understood to function as signaling molecules involved in cell growth, migration, differentiation, and gene expression. Relative to normal cells, tumors contain higher levels of ROS (Hail and Lotan 2009). Many groups have suggested that capsaicin induces apoptosis in cancerous cells via the generation of even higher levels of intracellular ROS (Table 5). For instance, Macho and coworkers addressed the relationship between capsaicin-induced apoptosis and ROS, generation by studying human T cell lymphoblast Jurkat cells. In these rigorous studies, they found that DNA disruption induced by 100–300 µM is preceded by ΔΨm dissipation, with ROS increases being independent of mitochondrial respiration, as pretreatment with cyclosporin A or N-acetylcysteine blocked ΔΨm disruption and apoptosis, but not the generation of ROS (Macho et al. 1998). In the second study, 250 µM capsaicin induced rapid increases of ROS, which were followed by a disruption of ΔΨm (Macho et al. 1999); blocking mitochondrial respiration did not reduce ROS induction by capsaicin. Consistent with the relative insensitivity of normal cells, which do not express TRPV1 to capsaicin, capsaicin (even at 300 µM for 5 days) failed to initiate ROS production, apoptosis or Ca2+ mobilization in resting T cells. In contrast, other laboratories have reported that the anticancer activities of capsaicin are due to a reduction of ROS levels (Patel, Varney, et al. 2002; Y. S. Lee et al. 2004; Qiao et al. 2005). For instance, Brar et al. (2001) found that capsaicin inhibited the growth of melanoma and other malignant cell lines. In these cells, O2 − generation was reduced by capsaicin, suggesting that electron transfer from NADPH oxidase may be an important source of endogenous ROS in tumor cells, which could constitutively activate NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) in an autocrine fashion. Therefore, the relationship between intracellular ROS levels and apoptosis of cancer cells may be dependent upon the differentiated phenotype of cancer cell, and despite the numerous studies in this area, the roles which the regulation of ROS play in the initiation and progression of cancer and the induction of apoptosis are not well understood.
NF-κB is a ubiquitous and evolutionarily conserved transcription factor that regulates the expression of genes involved in the transformation, survival, proliferation, invasion, angiogenesis, metastasis, and drug resistance of tumor cells (Gupta et al. 2010; Luqman and Pezzuto 2010). Constitutively, active NF-κB has now been identified in tissues from most cancer patients. A consistent observation is that capsaicin inhibits expression of the pro-inflammatory NF-κB in both normal cells exposed to stressors (Lee et al. 2007; Kang et al. 2007; Luqman et al. 2011) and cancerous cells (e.g., Mori et al. 2006). Capsaicin concentration dependently blocked TNFα-induced NFκB activation in stably transfected 293/NFκB-Luc human embryonic kidney cells with an IC50 value of 0.68 µM, while no cytotoxicity was observed at 65.5 µM (Luqman et al. 2011).
Another unresolved issue is to what degree the anticancer activity of capsaicin is mediated by TRPV1 receptors. In the studies listed in Table 5, some cancer cell lines were shown to express TRPV1, while many others show either no receptor mRNA and/or protein. Block of effects by receptor antagonists is an obvious way to suggest TRPV1 involvement, and several reports show that capsazepine or other TRPV1 antagonists block the pro-apoptotic effects of capsaicin. However, the interpretation of these data is clouded by the limited selectivity of the early antagonists (e.g., Ruthenium Red) and substantial species differences reported for the commonly used capsazepine (Valenzano and Sun 2004). Notably, unlike the TRP channels TRPM8, TRPV6 and TRPM1, there is little data to suggest either a causative role for TRPV1 in cancer pathogenesis or even increased expression with cancer (Lehen’kyi and Prevarskaya 2011).
Adding more complexity to the issue of TRPV1 involvement in capsaicin’s anticancer activities, multiple laboratories have reported that capsazepine actually mimics the effects of capsaicin on cancer cell viability (e.g., Kim et al. 2004). In a revealing set of experiments, Athanasiou et al. (2007) showed that capsazepine and one of the new highly selective antagonists (SB366791) were both able to induce apoptosis in the human non-small cell lung cancer line H460 and disrupt ΔΨm. Strikingly, capsaicin caused a decrease in mitochondrial OH- production, while capsazepine increased it. The authors concluded that, for apopotosis induction, whether vanilloid ligands increase or decrease ROS production is less important than the disruption of ΔΨm and the consequent increase in mitochondrial permeability and release of cytochrome c. Therefore, inhibition of any step in the mitochondrial electron transport chain — whether at complex I and/or III — is sufficient to make cancerous cells prone to apoptosis. They suggest that cancerous cells that express TRPV1 channels are even more susceptible to capsaicin-induced apoptosis, as the increased cytosolic calcium load further compromises the integrity of mitochondria.
In summary, the mechanisms of capsaicin’s anticancer activities are complex and require additional elucidation. Figure 4 briefly summarizes some of the TRPV1-dependent and -independent pro-apoptotic mechanisms described in this section. Because of the phenotypic and genotypic diversity intrinsic to cancer, it is quite unlikely that these potential pathways are generalizable to all cancerous cells. Moreover, as a pleitrophic molecule with potentially multiple mechanisms of action at high concentrations, it is quite likely that capsaicin works through multiple pathways, and several of these do not involve activation of the TRPV1 receptor. Other than inhibition of metabolic activation of carcinogens and induction of apoptosis in a wide variety of cancer cells, mechanisms suggested to account for capsaicin’s anticancer activities include antioxidant activity (Dairam et al. 2008), activation of peroxisome proliferator-activated receptor gamma (C. S. Kim et al. 2004), inhibition of angiogenesis (Min et al. 2004), modulation of lipid metabolism due to cancer (Anandakumar, Jagan, Kamaraj, et al. 2009) and inhibition of aromatase activity (Luqman et al. 2011).
Human Exposure and Epidemiological Studies
The relevancy of the dose levels administered to rodents in in vivo toxicology studies is provided context by reports on levels of actual human consumption and exposure. For instance, the maximum daily intake of capsaicin in the United States and Europe from chilies and paprika was calculated to be 1.5 mg/person/day (Govindarajan and Sathyanarayana 1991). Alternately, the mean and maximum intake of capsaicin from industrially prepared food products containing the recommended general limit of 5 ppm would be 0.77 and 2.64 mg/day, respectively (CREDOC/OCA 1998); this would translate into a maximum of about 0.05 mg/kg for a 60 kg person. Finally, in countries such as Thailand and Mexico, which consume relatively large quantities of capsaicin-containing peppers, the daily intake of total capsaicinoids for a 50 kg body person was estimated to be 0.5 to 4 mg/kg per day (Council of Europe 2001). Assuming capsaicin constitutes 60% of the total capsaicinoids, and using 60 kg as average body weight, the maximum intake would be about 2.9 mg/kg per day.
There is only one publication on plasma concentrations following the ingestion of peppers. In a study conducted in Thailand, the maximum average plasma concentration of capsaicin observed following oral administration of 5 g of standardized chili peppers was 8.1 nM (Chaiyasit et al. 2009). Similarly, there is very limited data on systemic exposure following topical administration of capsaicin-containing medical products. In the only published study, following a 60-min administration of cutaneous high-concentration (8% w/w) capsaicin patch for pain management, the mean population Cmax was 1.38 ng/mL and the highest plasma concentration measured in any patient at any time point was 17.8 ng/mL, or 58 nM (Babbar et al. 2009). In addition to low maximal levels, the plasma half life of capsaicin was quite short: about 25 min following oral administration (Chaiyasit et al. 2009) and about 98 min when delivered through the skin (Babbar et al. 2009). The rapid clearance of capsaicin is likely related to significant first-pass hepatic metabolism (Donnerer et al. 1990; Chanda et al. 2008). Adding to the expectation of very modest systemic exposure, the water solubility of capsaicin is very low, in the range of ∼60 ng/mL under saturated conditions (Turgut et al. 2004). As is generally the case with water-insoluble drugs, an assessment of protein binding of capsaicin in human plasma indicates that capsaicin is highly protein bound (∼93%; US FDA 2009). Therefore, it is likely that circulating levels of free capsaicin are quite low.
Several epidemiological studies pertain to the potential carcinogenic effects of capsaicin. A significantly increased incidence of gastric cancer amongst high chili pepper consumers in Mexico who were estimated to consume 90–250 mg of capsaicin per day (the equivalent maximum of about 4.2 mg/kg per day) has been reported (López-Carrillo et al. 1994; López-Carrillo et al. 2003). Interestingly, López-Carrillo and colleagues suggested that a previous report from Korea (Lee et al. 1995) corroborated the finding from Mexico. However, Lee et al. (1995) described only a positive relationship between stomach cancer and consumption of hot pepper-soybean paste stew based on a questionnaire administered to hospitalized patients in Seoul and the capsaicin content of this stew was not estimated. In a hospital-based study conducted in India, cases of stomach cancer were matched to control; food group analysis suggested that consumption of chilies was a significant risk factor (Gajalakshmi and Shanta 1996). More recently, two groups (Serra et al. 2002; Báez et al. 2010) have reported that gall bladder cancer patients in Chile consume significantly higher amounts of red chili pepper relative to controls. In an attempt to determine a causal relationship, Tsuchiya et al. (2011) not only failed to detect capsaicin-induced genotoxicity (discussed above) but also confirmed the presence of the carcinogenic aflatoxins B1 and G1 in chili pepper samples from the region. Finally, in an analysis of U.S. population and mortality data, significantly higher rates for stomach and liver cancer were found in counties inhabited by four ethnic-cultural groups that were conjectured to consume more black pepper and chili peppers (Archer and Jones 2002); this conjecture was unsupported by data, as there was no attempt to quantify capsaicin consumption.
Chili pepper plants and their fruits may be contaminated with pesticides, insecticides, fertilizers, herbicides, microbiocides, or heavy metals. For instance, an assessment of pesticide content of chili peppers in the United States found that 55% of samples contained pesticide residues, with an average amount of 0.290 ppm; fifty-one different pesticides were identified (Environmental Working Group, 2010). In California alone, 37,642 pounds of at least ten different classes of pesticides were applied to chili pepper crops in 2009 (Pesticides Action Network Pesticides Database, 2009). Many countries outside the United States appear to have much less stringent standards regarding the types of pesticides in use. There can be little doubt that some pesticides are human carcinogens (e.g., there is a clear relationship between cumulative exposure and lymphoma in farmworkers), although the extent of the public health issue remains controversial (Bassil et al. 2007).
Other non-pesticide carcinogens have also been found in chilies. When twenty chili samples from different suppliers obtained in Germany were analyzed for volatile N-nitroso compounds, over 75% of both dried, whole chilies and chili powder samples analyzed contained N-nitrosodimethylamine and N-nitrosopyrrolidine, at levels up to 6 µg/kg (Tricker et al. 1988). The same group also found in a small survey of foodstuffs from an area in India with high esophageal cancer rates that chilies purchased in that area contained N-nitrosopyrrolidine (Siddiqi et al. 1988). Subsequent analyses by the same research group found that consumption of chilies led to exposure to the amines dimethylamine, pyrrolide, and methylbenzylamine; nitrosamine may be formed from these compounds, which is genotoxic (Siddiqi et al. 1992). Directly relevant to the increased incidence of gallbladder cancer, chili peppers in Chile were contaminated with the genotoxic aflatoxins B1 and G1 at 4.4 ng/g and 0.5 ng/g, respectively (Tsuchiya et al. 2011).
Therefore, the modest epidemiological evidence suggesting an increased incidence of gastric or gallbladder cancer among high chili pepper consumers should allow for the possibility that contaminants that are carcinogens could have given rise to the putative signal. These studies should be considered inconclusive until additional larger studies exclude potential contributions from carcinogenic contaminants sometimes present in chili peppers.
Discussion
In their frequently cited 1995 review in that capsaicin was referred to as a “double-edged sword,” Surh and Lee (1995) proposed mechanistic bases for the putative carcinogenicity, co-carcinogenicity, and anticarcinogenicity effects of capsaicin. They suggested that although minute amounts of capsaicin display few or no deleterious effects, heavy ingestion of the compound had been associated with carcinogenesis. The presumed underlying basis was that capsaicin is metabolized by CYP P450s to electrophilic compounds, that may subsequently interact with DNA in an irreversible manner, thereby triggering mutagenicity and malignant transformation. For the anticancer mechanism, it was suggested that the ability of capsaicin to alter carcinogen metabolism via CYP inhibition (especially CYP2E1) provided the primary rationale for application of this compound to cancer chemoprevention and inhibitory effects on the metabolism, DNA binding, mutagenicity and tumorigenicity of various carcinogens.
Our literature review suggests that there is very limited evidence for the pro- and anticancer mechanisms described by Surh and Lee (1995) and subsequently Reilly and Yost (2006). We conclude that the in vitro data supporting the putative reactive metabolite genotoxic mechanism are weak: assumptions that formation of reactive metabolites inevitably lead to carcinogenicity are questionable, as the propensity for covalent binding of metabolites does not even predict hepatotoxicity, and the liver should be the organ most susceptible to damage by reactive metabolites (Park et al. 2011). Moreover, the presence of these putative highly reactive capsaicin metabolites has never been demonstrated in vivo, only in experimental situations that did not provide for quantification of the relative amounts of metabolites.
Regarding the putative anticancer mechanisms involving inhibition of carcinogen activation, based on recent human pharmacokinetic and in vitro metabolism data, no significant inhibition of any CYP—especially CYP2E1—is likely to occur following either ingestion of chili peppers or topical administration of a capsaicin-containing pain medicine. Given the very low levels of systemic exposure that are reported in humans, coupled with the low potency of capsaicin to inhibit CYPs, systemic inhibition of metabolic activation of carcinogens is unlikely to contribute significantly to the anticancer properties of capsaicin.
Very few of the studies summarized here were intended to define the activity of concentrations of capsaicin available to humans through diet, environment or medical products. Rather, the intention seems to have been to employ either a capsaicin concentration or a dose that would generate measurable and reproducible effects. For a number or reasons—not least of that is the low aqueous solubility of capsaicin—it seems quite unlikely that the high concentrations and dose levels administered to rodents or in vitro systems could be readily achieved in humans.
Ultimately, the ability of capsaicin or its putative reactive metabolites to induce pro-cancer effects would most likely depend upon a substantial local accumulation of those compounds in organs or tissues. At this time, there is no published data pointing to the accumulation of capsaicin or any of its metabolites in the liver, stomach, skin, or other organs. Clearly, much more information about capsaicin’s pharmacokinetic profile would be needed to render credible either the previously postulated pro- or anticancer mechanisms, for any potential target organ. Therefore, other explanations for capsaicin’s anticancer properties may be more important, such as the ability to inhibit selectively mitochondrial function in cancerous cells, modulate changes in lipid metabolism due to carcinogens, or serve as an antioxidant. Still, the absence of evidence for high levels of systemic exposure has not prevented speculation regarding usefulness as a cancer treatment or in chemoprevention and the subsequent inclusion of capsaicin in anticancer nutritional supplements. Recently, the US FDA warned a nutritional supplement manufacturer that a product that contains 55 mg of purified capsaicin could not be promoted to prevent or slow the development of cancer cells (US FDA 2011). At this time, the only data-driven recommendation is that a diet rich in vegetables, fruits, seeds, and spices, that will contain capsaicin and a variety of similar phenolic compounds, may be effective for the prevention of some types of cancer (Gupta et al. 2010).
Much remains to be learned about the relationship of capsaicin to carcinogenesis, despite the appearance of new information at an increasingly greater rate. Mechanistic or safety conclusions based on older studies that used capsaicin concentrations of questionable physiological relevance or capsaicin of unknown purity should be modified to give weight to more recent data that are based on capsaicin of known purity and concentrations more reflective of potential real-world human exposure. Small epidemiological studies in which capsaicin-containing foods were associated with increased cancer provide little basis for human health concerns because of potential contamination by known carcinogens. Of great interest would be carefully controlled studies comprehensively monitoring other dietary and environmental carcinogens sufficiently powered to detect capsaicin’s potential carcinogenic or cancer-protective effects.
Footnotes
The authors declared the following potential conflict of interest with respect to the research, authorship, and/or publication of this article: Keith Bley and Sunita Bubbar were formerly employees of NeurogesX, Inc., which markets Qutenza®, a prescription capsaicin pain medicine.
The authors received the following financial support with respect to the research, authorship, and/or publication of this article: Keith Bley and Sunita Bubbar were employees of NeurogesX during the time that this article was prepared.