
Abstract
Peroxisome proliferator-activated receptors (PPARs) represent therapeutic targets for the management of type 2 diabetes mellitus and dyslipidemia. Rodent carcinogenicity studies have revealed a link between γ and dual γ/α PPAR agonist treatment and the increased incidence of subcutaneous (SC) liposarcomas/fibrosarcomas or hemangiosarcomas, but very little has been reported for potent and selective PPARα agonists. We present a mode of action framework for the development of SC mesenchymal tumors in rodents given PPAR agonists. (1) Tumor promotion results from pharmacologically mediated recruitment (proliferation and differentiation), thermogenesis and adipogenesis of stromovascular cells, and subsequent generation of oxidative free radicals. (2) Tumor initiation consists of chemotype-driven mitochondrial dysfunction causing uncontrolled oxidative stress and permanent DNA damage. Promotion is characterized by enhanced adipogenesis in the SC adipose tissue, where the baseline PPARγ expression and responsiveness to PPARγ ligands is the highest, and by thermogenesis through expression of the uncoupling protein 1 (UCP-1) and the PPARγ co-activator 1 α (PGC-1α), two factors more highly expressed in brown versus white adipose tissue. Initiation is supported by the demonstration of mitochondrial uncoupling and OXPHOS Complexes dysfunction (Complexes III, IV and V) by compounds associated with increased incidences of sarcomas (muraglitazar and troglitazone), but not others lacking malignant tumor effects (pioglitazone, rosiglitazone).
Keywords
Introduction
Peroxisome proliferator-activated receptors (PPARs) are involved in the pathogenesis of insulin resistance and in the regulation of lipid metabolism. Consequently, their activation is seen as a potential therapeutic target for the management of type 2 diabetes mellitus and dyslipidemia. PPARs are ligand-activated transcription factors that bind, as heterodimers, with members of the retinoid X receptor (RXR) subfamily, to PPAR-responsive elements in the promoter region of responsive genes. Among the three receptor subtypes, PPARδ expression is ubiquitous, whereas PPARα is expressed only in tissues that exhibit a high rate of fatty acid metabolism such as the BAT, liver, kidney, and the heart. Peroxisome proliferator-activated receptor-γ1 and PPARγ2, derived by alternative splicing of PPARγ, are differentially expressed; PPARγ1 exhibits widespread but low levels of expression in adipose tissue, pancreatic β islets, macrophages, vascular endothelium, skeletal muscle, and heart, whereas PPARγ2 is almost exclusively found in the adipose tissue in both rodents and humans. Both PPARγ isoforms are more highly expressed in SC than OM preadipocytes (Sewter et al. 2002). PPARγ2 plays a key role in driving the early events in adipogenesis (Saladin et al. 1999).
Available data from rodent carcinogenicity studies have demonstrated that PPAR agonists can be tumorigenic in one or more species of rodents at multiple sites. For twelve of those PPAR agonists (six γ and six dual γ/α agonists) reviewed by the United States Food and Drug Administration, the tumor types are consistent with the wide PPAR tissue distribution, in particular, the increased incidence of liposarcomas/fibrosarcomas in rats, mice, and hamsters in the subcutis and the increased incidence of hemangiosarcomas in mice and hamsters at sites of spontaneous tumor formation such as the liver, spleen, kidneys, bone marrow, adipose tissue, and skin (El-Hage 2005). The carcinogenic effects of PPARδ agonists have not been extensively investigated and have produced varying and conflicting results. Therefore, as a result of these toxicopathological findings, the FDA requested the completion of 2-year rodent carcinogenicity studies prior to the initiation of clinical studies longer than 6 months in duration. In response, the Health and Environmental Sciences Institute PPAR Agonist Project Committee was established by pharmaceutical companies to investigate modes of action and human relevance of PPAR-mediated tumors (Cohen et al. 2009). Since the most commonly observed tumors in rodents were hemangiosarcomas, fibrosarcomas, and liposarcomas, the PPAR Agonist Project Committee also approved a Pathology Working Group (PWG) to develop consensus on the morphologic criteria to be used for diagnoses and consistency of tumor diagnoses across the pharmaceutical industry (Hardisty et al. 2007).
In humans, soft tissue sarcomas are a heterogeneous group of rare tumors, representing less than 1% of all malignant tumors and showing cellular differentiation toward the smooth muscle (leiomyosarcoma), adipocyte (liposarcoma), striated muscle (rhabdomyosarcoma), endothelium (angiosarcoma), and fibroblast (dermatofibrosarcoma) lineages (Toro et al. 2006). Half (47.9%) of these sarcomas arise in the soft tissues, only 14.0% in the skin, and 7.0% in the uterus. Little is known about their etiology, but both environmental (radiation, herbicides) and genetic factors (familial cancer syndromes) seem important. According to a recent epidemiologic review of 26,758 cases (1978 to 2001), liposarcoma (11.5% of the cases) was one of the top 6 histologic types (the more frequent leiomyosarcoma represents 23.9% of the cases). The most frequent subtypes were the myxoid/round cell liposarcomas (32.9%), with 90% of the cases characterized by pivotal translocations of the FUS and DDIT regions. Liposarcomas were rarely diagnosed in childhood and adolescence, but the rate rises exponentially in adulthood and peaks in the elderly population. In control rats used in two-year cacinogenicity studies, the reported incidence of cutaneous tumors is 6.5%, and males are 3 times more likely to be affected than females (Zwicker et al. 1992). In those tumors, 61.3% were epithelial and 22.6% mesenchymal in origin. The most frequently diagnosed mesenchymal tumor was the fibroma (71.4%) followed by the lipoma (52.4%) and fibrosarcoma (33.3%). Liposarcoma and hemangiosarcoma represented only 4.8% and 9.5% of the cases, respectively. In PPAR agonist–treated rats (ninety-one cases submitted to the PPAR Pathology Working Group) at the end of the two-year carcinogenicity studies, the diagnoses ranged from non-neoplastic proliferation of adipose tissue (hyperplasia) to mesenchymal cell tumors (benign or malignant) in various sites/organs, but more particularly in the white adipose tissue (WAT) of the skin and subcutis. The majority of the benign tumors were classified as lipomas, whereas the malignant neoplasms were primarily fibrosarcomas and liposarcomas. There were only a few cases of malignant fibrous histiocytomas, malignant schwannomas, and nonspecified sarcomas. The rare cases of malignant liposarcomas and fibrosarcomas from the vehicle-treated control groups were composed of a more uniform and differentiated population of spindle cells, whereas neoplasms in PPAR agonist–treated rats were more pleomorphic (fibroblastic spindle cell proliferation in the liposarcomas and vacuolation/myxomatous appearance of the fibrosarcomas).
Adipocytes are organized in a multidepot organ called adipose tissue (Avram et al. 2005; Cinti 2002). Only one-third of the adipose tissue contains mature adipocytes. The stromovascular islet—a combination of blood vessels, nerve tissue, fibroblasts, and preadipocytes (adipocyte precursors)—accounts for the remaining two-thirds. Adipose tissue is further distinguished as WAT or brown (BAT) based on its color, function, and histology. White adipocytes are spherical, up to 70 µm in diameter, and contain few elongated mitochondria with randomly oriented cristae and one large, unilocular lipid droplet that compresses the cytoplasmic content into a visible peripheral thin rim. Immunohistochemically, white adipocytes express the calcium-binding protein S-100 at the pre-adipocyte stage (Atanassova 2001). The primary function of the WAT is to store excess energy (fatty acids) in the form of triacylglycerol, which can then be redeployed to other tissues in response to metabolic needs (Avram et al. 2005). Fatty acid uptake is facilitated by the activities of lipoprotein lipase, cholesterol, and fatty acid transporter, fatty acid binding protein, and other factors. White adipocytes also contribute to glucose metabolism and perform important endocrine functions. In most species, the development of the WAT begins in the embryonic stage, but the majority of differentiation occurs after birth in two anatomically distinct areas: the visceral depot (subdivided into the intraperitoneal and retroperitoneal regions) and the subcutaneous (SC) depot (superficial and deep). Brown adipocytes are smaller, up to 40 µm in diameter, and polygonal in shape with centrally located nuclei, and they contain numerous small (multilocular) lipid droplets and cristae-packed mitochondria. Brown adipocytes are characterized by intense expression of the uncoupling protein 1 (UCP-1) and its mRNA (Kozak and Anunciado-Koza 2008). The principal role of BAT is nonshivering thermogenesis, the utilization of food lipids as a source for chemical energy that can be released from the cell in the form of heat (Brees et al. 2008). Thermogenesis is mediated by UCP-1, an enzyme located in the inner mitochondrial membrane, where it allows protons to reenter the mitochondrial matrix instead of being pumped out, thereby uncoupling the oxidation of fatty acids from ATP synthesis, which results in heat generation. This adipose organ is considered “dynamic” because of its lifelong potential for expansion through pre-adipocyte replication and transdifferentiation, a reversible phenomenon by which differentiated white adipocytes can convert phenotypically and functionally into differentiated brown adipocytes or the reverse, without undergoing de-differentiation. In particular, rodent brown adipocytes can be induced by low temperatures through the sympathetic nervous system, retinoic acid, and the thyroid hormone triiodothyronine (Cinti 2006). Norepinephrine, via the β-adrenergic pathway, is considered the most important factor in the regulation of BAT proliferation and thermogenesis (Brees et al. 2008; Lean et al. 1986). Despite this dynamicity and unlike rodents in which the BAT continues to develop rapidly after birth and remains important in thermogenesis throughout the lifespan, the development of human BAT seems to terminate shortly after birth (at which time it accounts for only 1% of the body weight and is only found in the cervical, axillary, perirenal, and periadrenal areas) and the brown adipocytes become devoid of mitochondria and thermogenic capacity.
Experimental models of chemical carcinogenesis have led to the distinction of two concomitant or sequential processes during tumorigenesis, namely, initiation and promotion. In contrast to initiators, the cellular changes resulting from application of the promoters do not affect DNA directly through mutations and are reversible. Initiation alone, which is usually mutagenic and therefore irreversible, is not sufficient for tumor formation. Promotion is indeed required to enhance the development of neoplastic cells by further enhancing proliferation, disrupting cellular differentiation, and causing additional mutations. With PPAR agonists, it is well established that drug candidates are nongenotoxic in vitro and in vivo. The most accepted mode of action only partially explains the promotion phase and does not expand on human relevance: PPAR agonists stimulate adipogenesis with subsequent release of cytokines and growth factors leading to mesenchymal or endothelial cell proliferation in the skin and associated adipose tissue (Cohen et al. 2004; Cohen et al. 2009). In this mode of action, one of the gaps is that PPARγ binding appears necessary but not sufficient; that is, thiazolidinediones such as the PPARγ agonist troglitazone, but not rosiglitazone, produce hemangiosarcomas in mice. We therefore propose to complete this published information by adding to the above promotion phase (adipogenesis) the concept of PPAR-mediated recruitment (replication and differentiation) of stromovascular islets in the subcutis, PGC-1α-dependent thermogenesis, and associated oxidative stress. We also wish to hypothesize that initiation consists of chemotype-driven mitochondrial dysfunction leading to permanent oxidative DNA damage of the recruited stromovascular cells in the SC adipose tissue rich in oxidation-prone residues (unsaturated fat). With regard to the human risk, we propose to eliminate the initiation event by screening the chemotypes in vitro using two high-throughput mitochondrial assays. We also highlight the steps of the promotion phase that are tissue/species specific and therefore not relevant in humans.
Mode of Action Framework
The proposed mode of action framework for γ and dual α/γ PPAR agonists is presented below and illustrated in Figure 1. During tumorigenesis, we hypothesize that promotion and initiation occur concomitantly as a result of the combined pharmacological and structural property of some PPAR agonists.
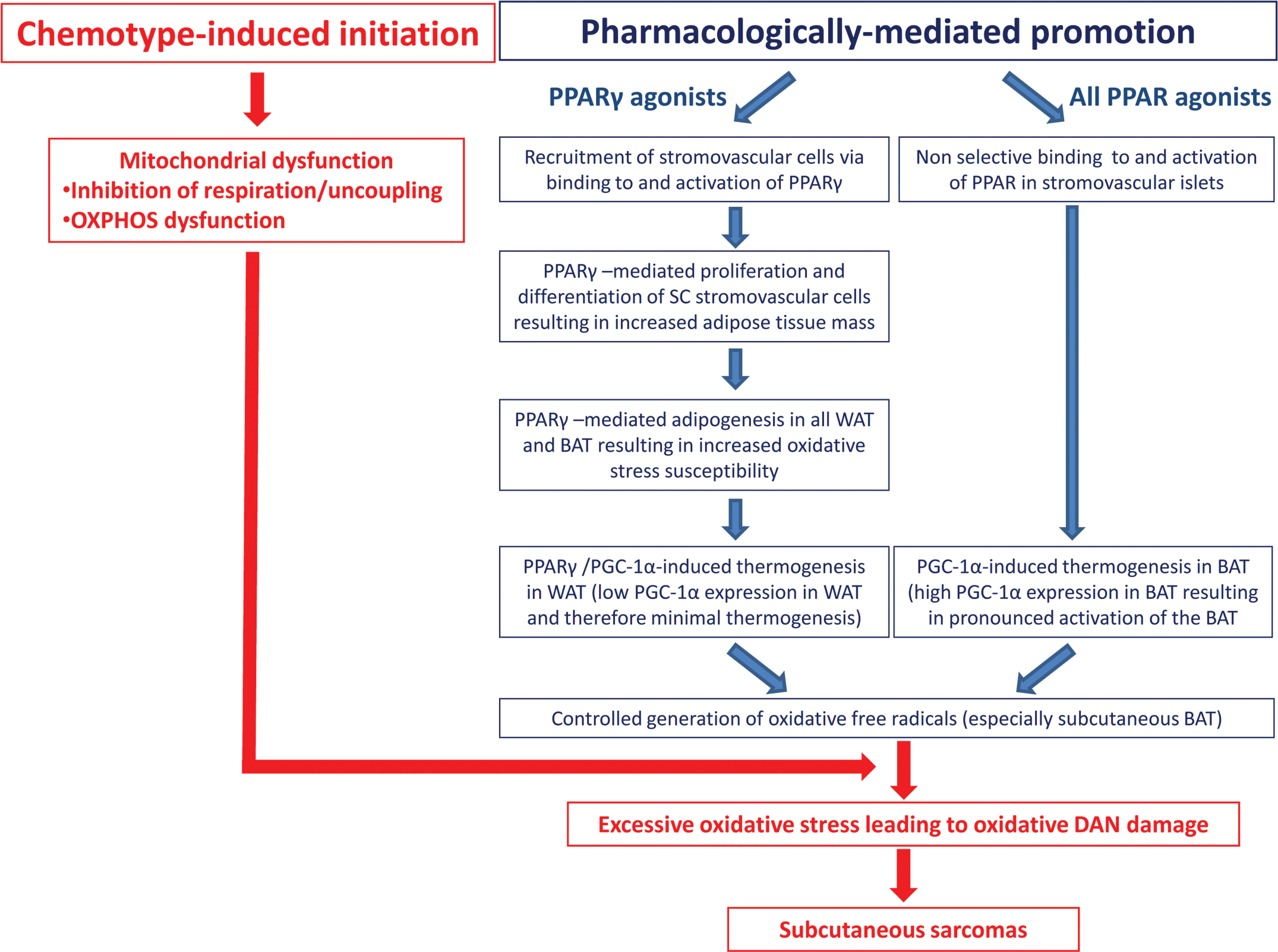
Proposed mechanism of action framework for PPARγ and dual α/γ agonist–induced subcutaneous sarcomas.
Promotion
PPAR-mediated recruitment (proliferation, differentiation, adipogenesis, and thermogenesis) of SC stromovascular cells (adipocytes, pre-adipocytes, neural, and vascular tissue) leads to controlled generation of free radicals.
PPARγ is required for adipocyte differentiation
The overall process of adipocyte differentiation is driven by the expression and activation of three transcription factor families: the CAAT/enhancer binding proteins (C/EBPs), which are mainly expressed in the early phases of mitotic clonal expansion occuring just before the differentiation process begins; the helix-loop-helix adipocyte differentiation and determination factor-1, which is expressed in the early stages of differentiation; and the PPARγ pathway. The fact that PPARγ-deficient, undifferentiated stromovascular cells do not differentiate in vitro in the presence of potent PPARγ agonists also confirms the pivotal role of this receptor in the process of differentiation (Vernochet et al. 2002).
In response to PPARγ agonist treatment, the expression of PPARγ-regulated genes (and in particular the PPARγ2 adipocytic-specific isoform) is upregulated in adipose tissue to halt clonal expansion, and to induce differentiation and adipogenesis of recruited stromovascular cells (Saladin et al. 1999). In vitro (3T3-L1 adipocytes) and in vivo (db/db hyperglycemic mice), the gene expression profile associated with these prodifferentiating effects of PPARγ agonists can be organized into thirty-two clusters (Gerhold et al. 2002). We propose that two of these cell cycle/growth markers could play a role during carcinogenesis in PPAR agonist–treated rats: vascular endothelial growth factor (VEGF, upregulated) and β-catenin (downregulated). Upregulation in VEGF is consistent with the previously published “dysregulated angiogenesis” hypothesis (Cohen et al. 2009). These authors postulate that VEGF upregulation could result from local tissue hypoxia from the growing fat pad (hypoxia activates HIF-1α, which in turn activates VEGF secretion by adipocytes and/or macrophage activation, leading to the release of cytokines and the generation of reactive oxygen species [ROS] and inflammation). With regard to β-catenin, we know that its function, in close association with Wnt, is to maintain preadipocytes in an undifferentiated state through PPARγ inhibition (Luo et al. 2009). When downregulated, however, β-catenin would drive preadipocyte differentiation and could also promote neoplastic transformation via its key role in cell-to-cell interactions (inhibition of contact theory; Kumar et al. 2005).
Subcutaneous preadipocytes are more susceptible to the prodifferentiation effects of PPARγ than their omental counterpart
In humans, the prodifferentiating effects of potent PPARγ agonists such as the thiazolidinediones are much more pronounced in SC preadipocytes (Adams et al. 1997; Sewter et al. 2002). This finding is consistent with the in vivo alterations in fat distribution that are seen in humans treated with thiazolidinediones. The relative insensitivity of omental preadipocytes is supported by the observation that in adipose tissue biopsies, PPARγ1 and PPARγ2 baseline expression is higher in the SC compared to OM preadipocytes. Second, the resulting increases in PPARγ receptor expression are also much greater for PPARγ2, the adipose tissue–specific isoform than for PPARγ1, and this increase is also greater in the SC than in the omental cells. Finally, the reduced responsiveness of omental cells to the prodifferentiating actions of RXR ligands and lower RXRα expression levels in the undifferentiated preadipocytes further point toward a more generalized resistance of omental preadipocytes toward differentiation.
PGC-1α is required for thermogenesis
In differentiating stromovascular cells, PGC-1α is a transcriptional coactivator for the type II class of nuclear hormone receptors (such as PPARγ) that is responsible for the concerted activation of genes determining mitochondrial biogenesis and oxidative metabolism in the liver, muscle, and adipose tissue (Hondares et al. 2006; Uldry et al. 2006). In particular, PGC-1α baseline expression can be induced by cold temperatures in the rodent BAT but is undetectable in the WAT with or without cold stimulation (Puigserver et al. 1998). However, when overexpressed in human and rodent white adipocytes, PGC-1α drives differentiation toward the brown phenotype with a resulting enhancement of the thermogenic genes required for uncoupling (UCP-1), electron transport (cytochrome c, cytochrome oxidase 4, mitofusin 2), fatty acid intake and fatty acid β oxidation (carnitine palimitoyltransferase 1, medium-chain acylcoenzyme A dehydrogenase), and mitochondrial biogenesis (e.g., mitofusin 2, mitochondrial transcription factor A). Glycerol kinase, the enzyme catalyzing the phosphorylation of glycerol into glycerol-3-phosphate, which constitutes the backbone of triglyceride, is also induced and generates futile oxidative cycles by reincorporating into triacylglycerols, fatty acids, and glycerols generated from the triacylglycerol hydrolysis. Altogether, these genes limit the use of fatty acids to the stromovascular bed, resulting in the generation of free radicals locally instead of releasing them into the blood stream. Finally, within PGC-1α-transduced white adipocytes, UCP-1 is selectively upregulated by troglitazone and other PPARγ agonists that directly act on the PPARγ-responsive element of the distal PGC-1α promoter. However, this activation of white adipocytes is not achieved with PPARα, PPARβ agonists, and 9-cis-retinoic acid (Tiraby et al. 2003). The selective PPARγ/PGC-1α association in the WAT contrasts with the nonselective activity of all PPAR agonists on PGC-1α in the BAT. This difference is also consistent with the expression levels of PPARγ1/γ2 in all adipose tissue components including endothelial cells, PPARα in BAT only, and the lower level of PGC-1α in the rodent and human WAT compared to the rodent BAT (Puigserver et al. 1998). Others have also demonstrated that this potent mechanism of BAT recruitment and thermogenesis was independent of sympathetic stimulation, namely, norepinephrine stimulation of the β3-adrenergic receptor on brown adipocytes (Petrovic et al. 2008).
PPARγ but not PGC-1α is required for differentiation and adipogenesis
Adipogenesis results from PPAR-mediated expression of genes involved in lipid transport and storage, such as fatty acid binding protein, adipocyte protein aP2, cholesterol and fatty acid transporters, lipoprotein lipase, leptin, and adipocyte lipid-binding protein. PPAR-driven adipogenesis is subtype-specific (PPARγ and especially PPAR γ2), is not PGC-1α-dependent, and is observed in all adipose tissues and all species. Accordingly, hypertrophy of BAT in the subcapsular, perirenal, and periaortic tissues, and WAT in the epididymal fat pad was reported in a three-month toxicology study in rats with the potent PPARγ agonist darglitazone (Aleo et al. 2003). Interestingly, the BAT appeared morphologically abnormal, with abundant accumulation of large lipid droplets around the numerous cristae-rich mitochondria (Figure 2), which is suggestive of adipogenesis in brown (numerous cristae-rich mitochondria) adipocytes. In mice, rats, and Beagle dogs, thiazolidinedione PPAR γ agonists such as troglitazone and rosiglitazone have also shown macrovesiculation of the BAT (increase in the lipid-filled cytoplasmic vacuoles indicative of adipogenesis) and microvesiculation of the WAT (reduction in the size of the lipid-filled vacuoles and increased number of cristae-rich mitochondria, which is indicative of thermogenesis) indicative of a phenotypic change so that the two types of fat resemble one another (Brieder et al. 1999; Toseland et al. 2001).
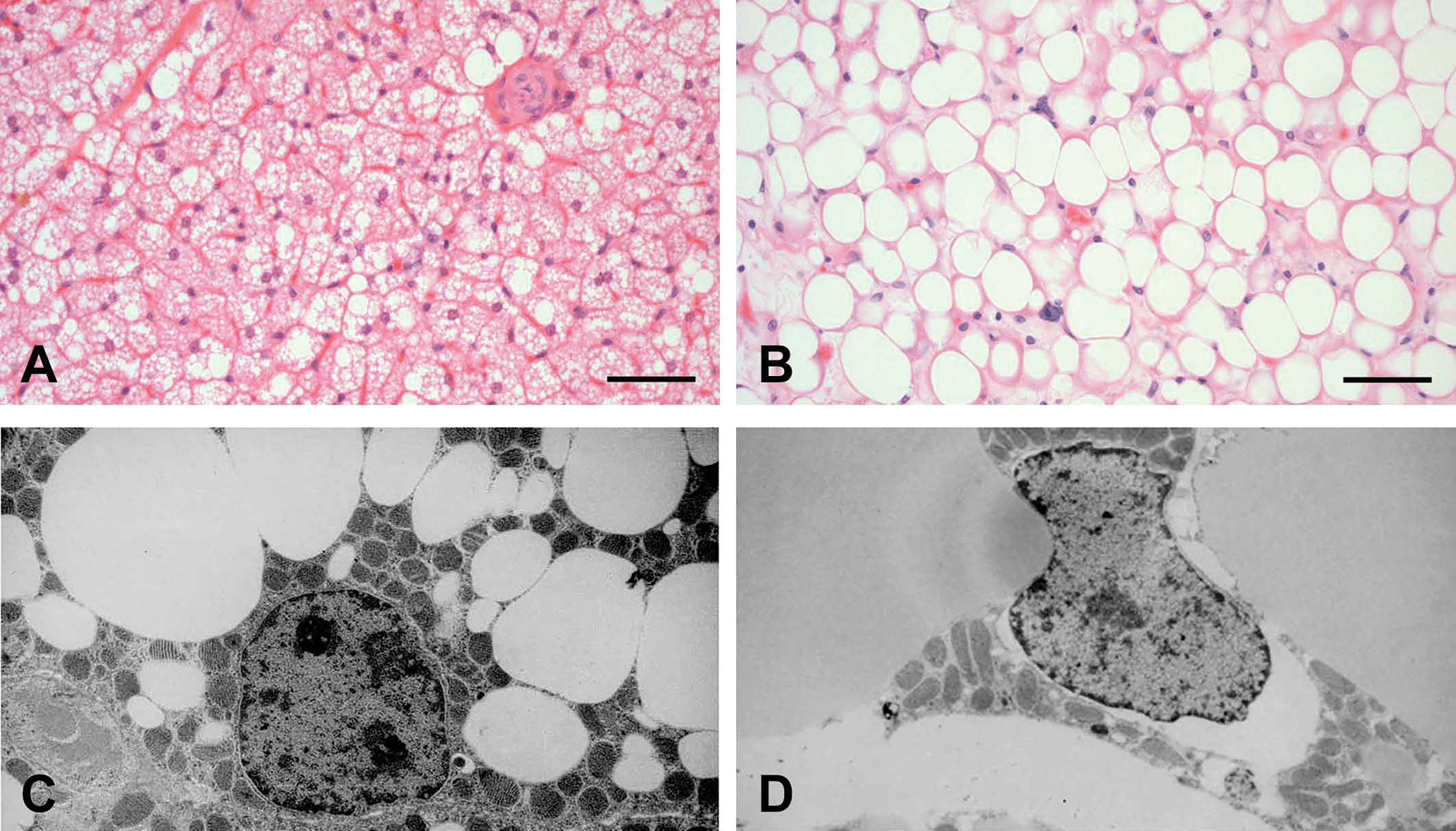
Interscapsular brown adipose tissue. (A and B) Hematoxylin and eosin staining, bar = 50 μm. In a control rat (A), brown adipocytes are multilocular. In darglitazone-treated rats (B), there is hyperplasia and hypertrophy of brown adipocytes, which are also becoming unilocular. (C and D) Transmission electron microscopy, 3,000× magnification. In a control rat (C), the brown adipocyte contains many small lipid vacuoles and cristae-rich mitochondria. In a darglitazone-treated rat (D), the cell is enlarged by a few large lipid vacuoles (reminiscent of white adipose tissue) and clusters of cristae-rich mitochondria.
Oxidative stress
Together, the above two metabolic changes (adipogenesis and thermogenesis) are likely to generate significant levels of free radicals in the differentiating and proliferating stromovascular cells. Although physiological thermogenesis is accompanied by the upregulation of antioxidant machinery, the rate of lipid peroxidation and oxidative stress should be proportionally higher during PPAR agonist treatment because adipogenesis is simultaneously ongoing and polyunsaturated fatty acids, which are more sensitive to peroxisomal β oxidation, accumulate in the triglyceride pool. The latter effect results from the fact that mitochondria, as opposed to peroxisomes, cannot oxidize the polyunsaturated fatty acids that result from enhanced triglyceride/fatty acid cycling.
De-risking the Promotion Phase
Brown adipose tissue, although present in most mammals, varies across species in its time of development, quantity, and function. In rodents, BAT develops rapidly after birth and remains important for nonshivering thermogenesis throughout the lifespan. Unlike rodents, the development of human BAT terminates shortly after birth and also loses its thermogenic capacity.
In humans and rodents, the expression of PGC-1α, the key regulator of adaptative thermogenesis through increased mitochondrial biogenesis and uncoupled oxidative metabolism, is strongly expressed in BAT (baseline expression and cold response) compared to WAT.
The rate of mitochondrial free radical generation by the mitochondrial respiratory chain is a better correlate of maximum longevity and aging rate than the basal rate of oxygen consumption (Barja 1999). This is the case across species, comparing animals according to the “rate of living theory” (inverse relationship between longevity and metabolic rate) or between species showing differences in longevities not explained by the rate of living theory (mammals and birds). Accordingly, the rate of mitochondrial free radical generation should be smaller in humans compared to rats.
Therefore, we propose that in rodents treated with PPAR agonists, differentiation is driven by PPARγ in the SC adipose tissue, adipogenesis is driven by PPARγ in all adipose tissue beds, and PGC-1α-dependent thermogenesis is upregulated in BAT in a nonselective manner by all PPAR agonists and to a lesser magnitude in WAT in a PPARγ-specific manner. Because (1) BAT does not persist after birth in humans and loses its thermogenecic capacity, (2) PGC-1α expression is lower in WAT in humans and rodents compared to BAT, and (3) oxygen radical generation by the mitochondrial respiration chain is lower in long-lived than in short-lived animals (humans vs. rodents), we suggest that thermogenesis and oxidative stress should not be significantly induced during PPAR agonist treatment in the human subcutis and associated adipose tissue.
Initiation
Chemotype-mediated mitochondrial dysfunction exacerbates PPAR-mediated oxidative stress (promotion), leading to oxidative DNA damage. Mitochondrial impairment is now accepted as the etiology of hepatotoxicity, myopathy, and cardiomyopathy associated with many therapeutics, including some lipid-lowering drugs. Nadanaciva et al. (2007) investigated the effects of multiple γ and dual α/γ PPAR agonists, including some thiazolidinediones, on the respiration of isolated mitochondria using a phosphorescent oxygen-sensitive probe with the goal of identifying uncouplers and/or inhibitors of respiration, and on the activity of individual OXPHOS complexes immunocaptured from isolated mitochondria on ninety-six–well plates coated with monoclonal antibodies raised against Complexes I, II, IV, and V. A Complex II+III activity assay, which does not rely on immunocaptured material, was also included to complete the profile of the drugs on oxidative phosphorylations.
Mitochondrial respiration
In the first experiment, oxygen consumption was measured in mitochondria isolated from rat liver over an eight-point, two-fold dilution dose response starting at a drug concentration of 100 nmol/mg mitochondrial protein. Respiration was measured in the presence of the oxidizable substrates glutamate and malate, but in the absence of exogenous ADP (state 2 respiration), whereas in state 3 respiration, both substrates and ADP were provided. At the maximum dose (100 nmol/mg protein), all drugs increased basal oxygen consumption. However, at a drug concentration of 25 nmol/mg mitochondrial protein, differences appeared, with ciglitazone, darglitazone, and troglitazone being undoubtedly more potent (with a depletion of oxygen by eight minutes) on states 2 and 3 respiration, whereas rosiglitazone, muraglitazar, and pioglitazone showed very little (approximately 75% less of an effect with rosiglitazone) to no effect (pioglitazone; Figure 3, Table 1).
UC50 and IC50 values of PPARγ and dual α/γ agonists during states 2 (Glutamate/Malate) and 3 (ADP-stimulated) respiration in isolated mitochondria.

Note: Nonlinear regression yields dose responses and a UC50 value (concentration at 50% uncoupling) for State 2 respiration and an IC50 (concentration for 50% inhibition) for State 3 respiration.
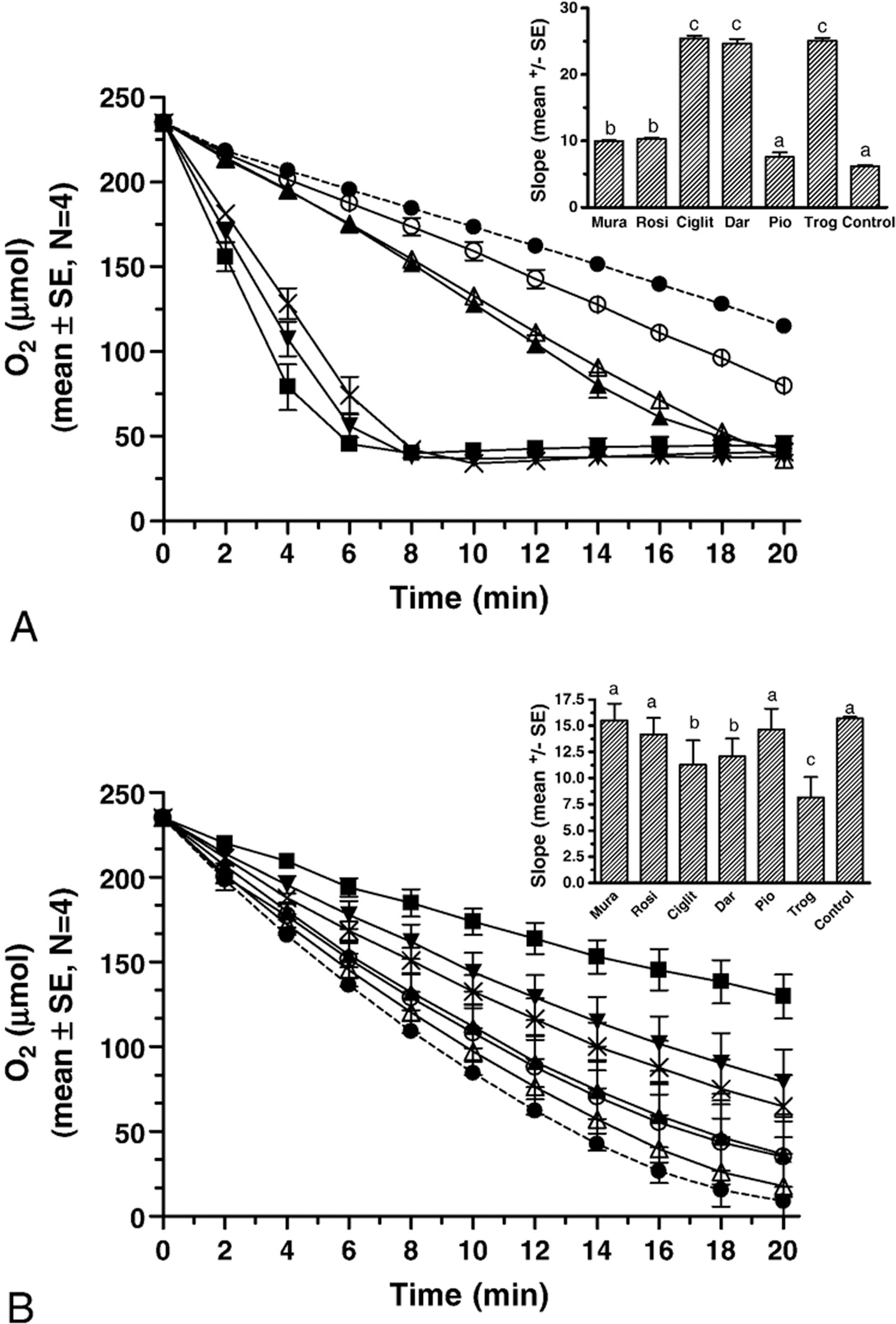
Effects of thiazolidinediones (25 nmol/mg mitochondrial protein) on basal glutamate/malate–driven state 2 mitochondrial respiration (A), and on ADP-driven state 3 respiration (B). Data are mean ± SE, n = 4 for drugs, and n = 32 for control. Symbols: ▾, ciglitazone; ▪, troglitazone; ×, darglitazone; ▴, rosiglitazone; ^, pioglitazone; ▵, muraglitazar; and •, control. Insert bar graph shows data expressed as slope during initial eight minutes. Columns not significantly different via ANOVA at p < .05 share superscripts.
Mitochondrial OXPHOS
In addition, the effects of thiazolidonediones on the individual OXPHOS complexes I to V were assayed at 150 µM in the immunocapture technique. When inhibition was seen, the compounds were reevaluated over a nine-point dose response using two-fold dilutions. Generated IC50 values are reported in Table 2. None of the compound affected complex II activity. The most potent inhibitor was troglitazone, which inhibited complexes II + III, IV, and V with IC50 values of 30 ± 1, 10.4 ± 4.7, and 13.2 ± 2.4 µM, respectively, and also caused 25% inhibition of complex I at 150 µM. Ciglitazone inhibited complexes II + III, IV, and V with IC50 values of 60 ± 1, 38.5 ± 1.5, and 74.9 ± 5 µM, respectively, and caused 15% inhibition of complex I at 150 µM. Darglitazone inhibited complex IV with an IC50 of 26.6 ± 8.6 µM and complex V with an IC50 of 105 ± 10. Muraglitazar caused 70% inhibition of complex V and 15% inhibition of complex II + III at 150 µM. Rosiglitazone caused 25% inhibition of complex I at 150 µM, whereas pioglitazone had no significant effect on any of the OXPHOS complexes at this concentration. Thus, the overall rank order of potency of the thiazolidinediones with regard to the inhibition of all mitochondrial oxidative phosphorylation complexes, and in particular complex V, was troglitazone > ciglitazone > darglitazone = muraglitazar > rosiglitazone = pioglitazone.
Effects of PPARγ and dual α/γ agonists on OXPHOS complex activity in the immunocapture assay and reported binding Potencies (Henke 2004).
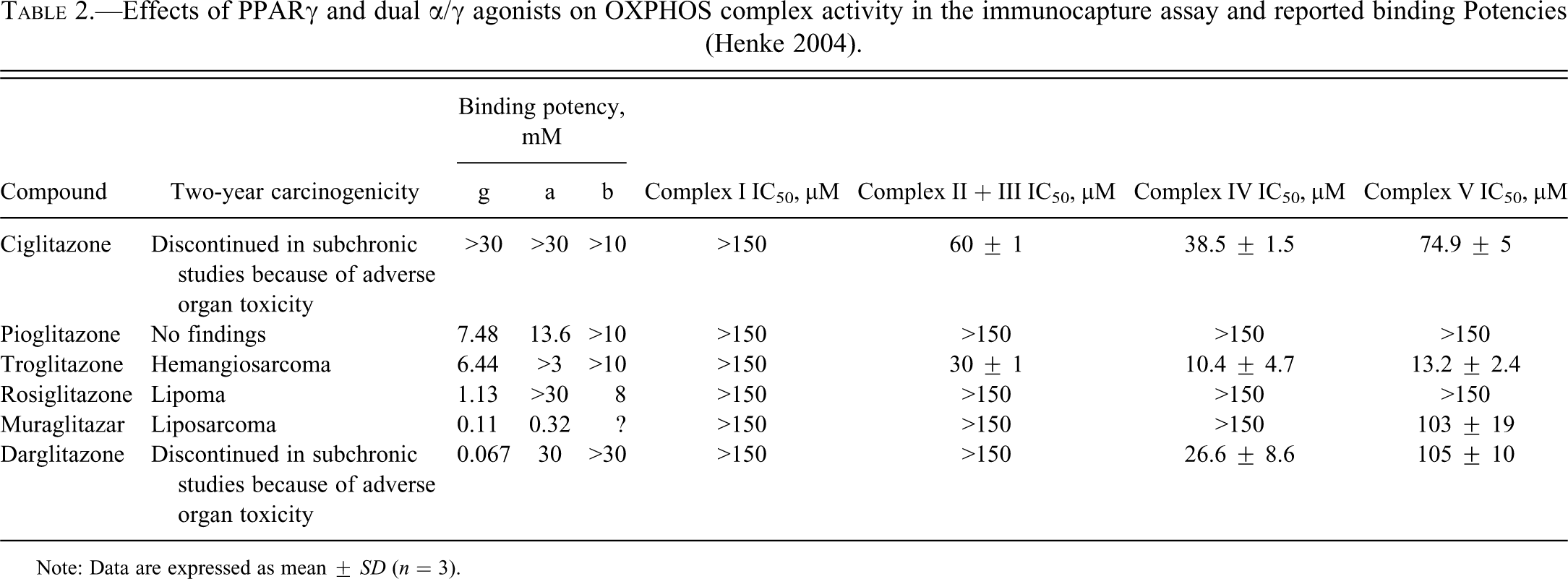
Note: Data are expressed as mean ± SD (n = 3).
Oxidative stress
Oxidative stress can be defined as the sum of all deleterious processes resulting from an imbalance between the excessive production of ROS and reactive nitrogen species (RNS) and the limited antioxidant defenses. In most tissues, the mitochondrial respiratory chain constitutes the main intracellular source of these reactive species. In health or under physiological stimulation, steady-state concentrations of oxidants are maintained at nontoxic levels by a variety of antioxidant defenses and repair enzymes. However, this delicate balance between antioxidant defenses and ROS/RNS production may be disrupted by deficient antioxidant defenses, inhibition of electron flow, or exposure to xenobiotics, leading to free radical–mediated chain reactions targeting proteins, lipids, polysaccharides, and DNA (Turrens 2003). In their sequential in vitro screening approach, Nadaciva et al. demonstrated that many of the γ and dual γ/α PPAR agonists with well-documented preclinical and clinical safety issues were indeed mitochondrial toxicants. The authors also postulated that this toxicity occurred via conformational alteration of mitochondrial transmembrane proteins or intercalation into the hydrophobic domain of the mitochondrial inner membrane, thereby compromising the impermeability of the mitochondrial membrane to protons. This statement is also consistent with the fact that the only two non-active thiazolidinediones (rosiglitazone and pioglitazone) have the lowest octonal-to-water partition coefficients.
De-risking the Initiation Phase
PPARγ and dual α/γ agonists such as troglitazone and muraglitazar induce sarcomas in rodents. Pioglitazone and rosiglitazone also bind and activate the same receptors, but they do not produce sarcomas in two-year carcinogenicity studies. Therefore, PPAR binding (potency) appears to be necessary but does not correlate/is not sufficient to produce sarcomas, which is consistent with the definition of a promoter (Table 2; Henke 2004).
In contrast, the rank order of inhibition of PPARγ and dual α/γ agonists with regard to mitochondrial respiration (oxygen consumption and uncoupling) and OXPHOS complex activity (particularly complexes III, IV, and V) complements very well the preclinical and clinical safety profiles of those drugs. For example, the cataractogenic potential of ciglitazone was demonstrated to result from chemotype-mediated alterations in lens bioenergics secondary to the inhibition of the mitochondrial calcium uniport inhibitor ruthenium red channel (Aleo et al. 2005). Troglitazone and muraglitazar were associated with an increased incidence of sarcomas (hemangiosarcomas and liposarcomas) at the end of the two-year rodent carcinogenicity studies (Cohen et al. 2009; Tannehill-Gregg et al. 2007). Darglitazone development was discontinued early because of severe adipose tissue hypertrophy and lipomas at the end of the three-month toxicology studies and because of lymphothorax caused by the compression of many lymphatics by this enlarged adipose tissue mass (Aleo et al. 2003). This chemotype-driven mitochondrial toxicity would triumph over the antioxidant defenses by worsening the generation of ROS and RNS already enhanced by the PPAR-mediated metabolic changes (adipogenesis and thermogenesis). This process would ultimately lead to irreversible mitochondrial and nuclear oxidative DNA damage, DNA mutation in recruited (i.e., differentiated and proliferating) stromovascular cells (including pre-adipocytes), and neoplastic transformation, which is the exact definition of initiation.
The sequential measurement of oxygen consumption (a process reflecting the performance of the respiratory chain and integrity of the inner mitochondrial membrane) and OXPHOS complex activity (to further characterize the mitochondrial impairment) highlighted that not all PPARγ and dual α/γ agonists equally undermine mitochondrial function. Therefore, we propose that in order to eliminate mitochondrial toxicity and therefore the initiation process, PPAR agonist chemotypes should be screened in two high-throughput in vitro mitochondrial assays, namely, mitochondrial respiration (using a phosphorescent oxygen sensitive probe) and mitochondrial OXPHOS complex activity.
Conclusions
Because PPARs are involved in the pathogenesis of insulin resistance and the regulation of lipid metabolism, their activation has been pursued as a potential therapeutic target for the management of type 2 diabetes mellitus and dyslipidemia. However, in two-year rodent carcinogenicity studies, many PPARγ and dual γ/α receptor agonists were found to increase the incidence of liposarcomas/fibrosarcomas in rats, mice, and hamsters in the subcutis and of hemangiosarcomas in mice and hamsters. Experimental models of carcinogenesis have indicated that, in general, the tumorigenesis process progresses through two ovelapping or consecutive stages, namely, initiation, where irreversible DNA damage occurs, and promotion, during which aberrant cell proliferation of the initiated cells is seen. After a careful examination of previously reported in vivo and in vitro preclinical and clinical studies with the γ and dual α/γ PPAR agonists ciglitazone, darglitazone, troglitazone, pioglitazone, rosiglitazone, and muraglitazar, we present a detailed mode of action that could explain the varied tumogenic properties of these drugs in rodent skin, subcutis, and adipose tissues. We also highlighted the sensitivity of the rodent for many of the promoting events and recommended a high-throughput in vitro screening strategy to prevent the initiation process.
Our mode of action framework can be summarized as follows. The tumor promotion response follows PPARα- and PPARγ-mediated recruitment of stromovascular cells, that is, the promotion event is pharmacologically mediated. During the promotion stage in rats, the recruitment, proliferation, and prodifferentiation effects of PPARγ agonists on stromovascular cells would be expected in the subcutis based on receptor expression and omental resistance to differentiation. Subsequently and in all adipose tissues, PPARγ and PGC-1α would drive adipogenesis and thermogenesis, respectively. PGC-1α-mediated thermogenesis would primarily be subtype specific (PPARγ) in WAT and nonselective (all PPAR) in BAT, whereas PPAR-driven adipogenesis would be noted in the WAT and BAT in a subtype-specific (PPARγ) but PGC-1α-independent manner. With a “safe chemotype,” thermogensis and adipogenesis would only cause controlled increases in the steady-state concentrations of free radicals. We hypothesize that initiation consists of chemotype-mediated mitochondrial dysfunction in proliferating SC stromovascular cells already undergoing profound oxidative stress secondary to the PPAR-driven adipogenesis and thermogenesis. This process would lead to uncontrolled oxidative stress and permanent oxidative DNA damage. In particular, the rank order for the inhibition of the mitochondrial OXPHOS complexes for the tested PPAR agonists correlated well with the safety data. The most potent inhibitor was troglitazone, which inhibited complexes III, IV, and V, whereas in contrast, pioglitazone had no significant effects on any of the OXPHOS complexes at the concentrations tested. The rank order for inhibiting mitochondrial respiration in isolated mitochondria also demonstrated that many of the discontinued thiazolidinediones behave as uncouplers of respiration.
Footnotes
Acknowledgments
The authors wish to thank Pfizer Inc. for their support, as well as David Goodwin and Majid Syed for outstanding photographic assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.