
Abstract
Cytokines are critical in allergic intercellular communication networks, and they contribute to disease pathology through the recruitment and activation of pro-inflammatory leukocytes and in chronic disease to pro-fibrotic/remodeling events. Th2 cytokines predominate primarily in mild to moderate allergic asthma, although clinical trials with inhibitors of IL-4 and IL-5 have not provided the robust efficacy observed in animal models of allergy. These results not only highlight the complexity of allergic disease, but they also point to the importance of other cytokine networks in driving pathology. The heterogeneous nature of the disease is emphasized by the fact that the Th2/Th1/Th17 cytokine balance can be influenced by the initiating allergic trigger. For example, the house dust mite allergen Der p 2 mimics the activity of MD-2 by presenting lipopolysaccharide to Toll-like receptor-4 for the activation of inflammatory genes including innate-type cytokines. Here we discuss the functions of the novel cytokine players, thymic stromal lymphopoetin (TSLP), IL-33, IL-25, and IL-9 and delineate nonredundant roles for IL-4 and IL-13 in allergic disease. Persistent efforts in the characterization of these and other cytokine networks will be essential for understanding the complex pathogenic mechanisms that underpin allergic disease and for guiding targeted therapeutic interventions.
Introduction
Over twenty years ago, Th1 and Th2 subtypes were first described on the basis of their restricted cytokine profile and different functions (Mosmann et al. 1986; Mosmann and Coffman 1989). Since Th2 cells produce IL-4, which is essential to IgE production; IL-5, which drives the terminal differentiation and survival of eosinophils; and IL-13, which promotes mucus secretion, airway hyperresponsiveness (AHR), and tissue remodeling, the hypothesis that allergy and asthma were primarily Th2-mediated diseases began to gain credence. Indeed, allergic asthma is characterized by high levels of serum IgE with increased reactivity to allergens (Hamid and Tulic 2009). Most asthmatics also present with eosinophilic disease, although some of the more severe patients exhibit high neutrophil counts within the airways (Hamid and Tulic 2009). Following an appropriate allergen challenge, an influx of Th2 cells and Th2-type cytokines is observed in the airways of allergic asthmatics and the numbers of infiltrating Th2 cells positively correlates with disease severity (reviewed in Larché et al. 2003). Thus it has been reasonable to suspect the Th2 cell as the key culprit in the pathophysiology of allergic disease.
Murine models of allergic airway inflammation have been useful in exploring the pathogenesis of asthma, although the majority have involved intraperitoneal injections of protein allergen, commonly ovalbumin (OVA), emulsified in aluminum hydroxide (alum) during the sensitization phase followed by direct allergen challenge to the airways in the challenge phase to induce a profoundly biased Th2-type response within the airways (Lloyd and Hessel 2010). The disease phenotype is characterized by features of AHR; high lung eosinophil counts; high levels of IL-4, IL-5, and IL-13 within the bronchoalveolar lavage fluid; and serum IgE production. Under these conditions, many researchers have shown a profound role for IL-4, IL-5, and IL-13 in allergic airway disease (Finkelman et al. 2010). However, with the realization that blockade of IL-5 with mepolizumab and SCH55700, which dramatically reduce blood eosinophils but show little clinical improvement (Hendeles et al. 2004; Leckie et al. 2000), and the recent cluster analysis of asthma clinical phenotypes showing the existence of eosinophilic and mixed/non-eosinophilic subtypes, there has been an emergence of interest in the role of other T cell phenotypes, cytokines, and innate cells in disease pathogenesis (Lloyd and Hessel 2010). Here we discuss important, nonredundant roles for the classical Th2 cytokines IL-4 and IL-13 in allergic atopy and asthma and introduce the relatively new players, IL-33 and TSLP, as well as IL-25 and IL-9, and review their potential contributions to disease pathogenesis.
Cellular Sources of IL-4 and IL-13
IL-4 and IL-13 are produced in abundance by activated CD4+ Th2 cells (de Waal Malefyt et al. 1995; Jung et al. 1996; Secrist et al. 1995), mast cells (Bradding et al. 1992; Brightling et al. 2003; Gessner et al. 2005; Seder et al. 1991), eosinophils (Gessner et al. 2005; Kay et al. 1997; Woerly et al. 2002), and basophils (Gessner et al. 2005; Li et al. 1996; MacGlashan 1994). Alternatively, activated alveolar macrophages also release substantial amounts of IL-4 (Gordon and Martinez 2010; Pouliot et al. 2005) and IL-13 (Kang et al. 2005). In addition, type-2 CD8+ T cells and invariant natural killer (NK) T cells have been reported to produce IL-4 in murine models of tumor rejection (Dobrzanski et al. 2001) and allergic airway disease (Lisbonne et al. 2003), whereas NK cells can produce IL-13 in response to IL-2 (Hoshino et al. 1999).
IL-4/IL-13 Receptor Engagement and Signal Transduction
IL-4 signals through the type I receptor that is made up of the IL-4Rα chain and the common γchain (γc), whereas both IL-4 and IL-13 signal through the type II IL-4 receptor, which consists of the IL-4Rα chain and the IL-13Rα1chain (Mitchell et al. 2010; Oh et al. 2010). IL-13 can also bind to the IL-13Rα2 chain, which has been well characterized as a decoy receptor in its secreted form. Until recently, the bound form of IL-13Rα2 was not thought to signal, since it has a short cytoplasmic tail that does not bind janus-associated kinases (JAKs) or the signal transducer and activator of transcription (STAT) family of transcription factors; however, others have demonstrated that cell-bound IL-13Rα2 can signal in a STAT6-independent, AP-1–dependent manner to induce activation of the TGFB1 promoter and promote tissue fibrosis in some situations (Fichtner-Feigl et al. 2006).
Following binding of IL-4 and IL-13 to their respective partners, a signal transduction cascade is induced. The initial signal is triggered by receptor dimerization and activation of the JAKs, which are associated with the cytoplasmic tails of the receptors. Type I receptors activate JAK1 and JAK3, whereas type II receptors activate JAK1, JAK2, and Tyk2. Tyrosine residues in the IL-4Rα chain become phosphorylated and act as docking sites for signaling molecules. For example, the Tyr497 tyrosine residue on the transmembrane domain of IL-4Rα is necessary for the docking of insulin receptor substrate (IRS)-2. JAK1 and JAK3 then phosphorylate IL-4Rα–bound IRS-2, which in turn binds to the p85 subunit of PI3 K and to the adaptor protein growth factor receptor–bound protein 2 (Gordon and Martinez 2010; Nelms et al. 1999). This pathway is linked to the proliferation of Th2 cells and the induction of genes associated with alternatively activated macrophages in response to IL-4 (Gordon and Martinez 2010).
Other IL-4Rα tyrosine residues interact with proteins containing Src homology 2 domains that are found in the STAT family of transcription factors (Nelms et al. 1999). For example, three closely clustered tyrosine residues serve as docking sites for STAT6, a transcription factor selectively coupled to the IL-4Rα chain. The binding of IL-13 to IL-13Rα1 also activates STAT6 because the IL-13:IL-13Rα1 complex binds with high affinity to IL-4Rα. Phosphorylated STAT6 dimerizes, migrates to the nucleus, and binds to the promoters of IL-4– and IL-13–responsive genes such as those associated with IgE class switching (Chatila 2004). STAT3 activation is also associated with IL-4 and IL-13 signals in human B cells (Umeshita-Suyama et al. 2000), and more recently, both IL-4 and IL-13 have been shown to induce a significant increase in the levels of phosphorylated STAT1 in human bronchial smooth muscle cells (Chiba et al. 2009).
Type I receptors are expressed on hematopoietic cells of both the myeloid (monocytes, macrophages, and fibroblasts) and lymphoid (T and B cells) lineage, whereas myeloid cells express both type I and type II IL-4Rs. Non-hematopoietic cells, such as smooth muscle cells and epithelial cells, predominantly express the type II receptor. The separate functions of IL-4 and IL-13 arise from (i) differential receptor expression on hematopoietic/non-hematopoietic cells; (ii) on local concentrations of cytokines at the site of inflammation; and (iii) on local levels of expression of type I and type II receptors.
IL-4 Potently Initiates Th2 Differentiation in Vitro
In vitro studies clearly demonstrate that Th2 cells are generated by activating naïve T cells through cross-linking the type 1 T cell antigen receptor (TCR) in the presence of exogenous IL-4 (Paul 2010). Type I receptor signals are transduced by STAT6, which, together with NFAT (nuclear factor of activated T cells), AP-1, NF-κB, and other TCR-induced signals, activate transcription of Il4 as well as the gene encoding the zinc-finger transcription factor GATA3, a major regulator of Th2 lineage commitment. The resulting autocrine feedback loop acts positively to favor Th2 differentiation. GATA3 also promotes the expression of MAF, a transcription factor that selectively transactivates IL-4, but not other Th2 cytokines. The IL-4R/STAT6 signaling pathway also leads to silencing of IFN-γ expression and thereby enhances Th2 immunity by inhibiting Th1 responses. During this process, the Il4, Il5, and Il13 genes, which reside together within the Il4 locus, are potentiated for extremely strong transcriptional activation upon restimulation. This secondary burst of cytokine gene transcription occurs independently of instructive cytokine signals.
IL-4/STAT6 Signaling in Vivo
Interestingly, in certain situations, neither IL-4 nor STAT6 signaling is required for the induction or differentiation of Th2 cells within the lymph nodes, as demonstrated using green fluorescent protein reporter transgenic techniques, which showed that IL-4–producing Th2 cells can develop in IL-4R- and STAT6-deficient animals undergoing certain Th2-mediated helminth infections and allergic immune responses with equivalent magnitude and timing to their IL-4/STAT6 sufficient counterparts (Paul 2010; van Panhuys et al. 2008). Nevertheless, these studies re-emphasize the importance of IL-4/STAT6 at mucosal sites and highlight the importance of IL-4/STAT6 in the induction of type 2 effector responses, including Th2 effector cell migration, eosinophil migration, and B cell IgE class switching (Harris et al. 2002; Kaplan et al. 1996; Kopf et al. 1993). For example, at the gastrointestinal (GI) mucosal surface, IL-4 and STAT6 have a critical role in the induction and effector phase of experimental oral antigen–induced intestinal anaphylaxis via the generation of optimal intestinal mastocytosis, antigen-specific IgE, oral antigen sensitization, and intestinal anaphylaxis (Forbes et al. 2008). Recent data have also shown that Th2 effector responses in the skin are highly IL-4/STAT6 dependent, whereas Th2 responses in the draining lymph nodes are not (Forbes et al. 2010).
IL-4/Type I Receptor Mediated Macrophage Activation
In analogy to the dichotomic characterization of Th cells (Th1, Th2), macrophages have also been characterized by their marked phenotypic heterogeneity depending on their microenvironmental stimulation. Classical activation by microbial agents and/or Th1 cytokines, in particular by IFN-γ, is associated with the production of TNF-α, IL-12, IL-8, IL-6, and IL-1; the expression of NOS2; and the down-regulation of CD14 expression. These classically activated macrophages (also called M1) typically take part in the initial immune response to invading microorganisms and promote Th1 immunity. On the other hand, alternatively activated macrophages (M2 macrophages) that are stimulated by the Th2 cytokines IL-4, IL-13, and IL-10 reveal a different gene signature that is characterized by a specific expression pattern of membrane receptors, including scavenger receptors A and B, mannose receptor (CD206), CD36, CD163, chemokine receptors (CXCR1, CXCR2, CCR2), and the expression of several cytokines and chemokines, including IL-1 receptor antagonist (IL-1RA), IL-10, CCL1, CCL17, CCL18, and CCL22 and arginase expression (Gordon and Martinez 2010). M2 cells are thought to play a critical role in allergy and parasitic infections and appear to be involved in the control of tissue repair. Several years ago, it was demonstrated that IL-4 induced a more robust expression of a panel of M2 genes (arginase I, chitinase-like-3 [also known as YM-1], and resistin-like molecule-α [RELM-α/FIZZ1]) than did IL-13. Of note, the response was type I receptor dependent, and IL-4–mediated induction of FIZZ1, but not YM-1 or arginase, was suppressed by wortmannin, a specific PI3 K inhibitor. Since IL-4–induced tyrosine phosphorylation of IRS-2 leads to the recruitment and activation of PI3 K, the results imply that IL-4 type I receptor, IRS-2–dependent signaling pathways, may lead to amplified expression of a subset of genes in alternatively activated macrophages (Heller et al. 2008).
In support of a dominant role for IL-4 type I receptor activation in the induction of an M2 phenotype, others have shown that allergen-mediated induction of these same genes is IL-13Rα1 independent in vivo (Munitz et al. 2008). Although the exact role of alternatively activated macrophages in allergic disease is not well understood, the disassociation of the induction of these genes from allergen-induced AHR and mucus cell changes in IL-13Rα -deficient mice suggests that alternatively activated macrophages do not drive these phenomena.
IL-13 and Localized Th2-Mediated Tissue Inflammation
IL-13Rα1 is not found on T cells, NK cells, basophils, mast cells, or most mouse B cells (most human B cells express both γc and IL-13Rα1). Thus, IL-13, unlike IL-4, does not promote the differentiation of naïve T cells into Th2 cells, and IL-4 is much more important than IL-13 for the induction of mouse IgE responses. In contrast, some bone marrow–derived cells, including macrophages and dendritic cells, express both γcand IL-13Rα1 and consequently respond to both IL-4 and IL-13, whereas IL-13Rα1, but little or no γc subunit, is found on most non-bone marrow–derived cells, including smooth muscle and epithelial cells. Thus IL-13-mediated pathologies are strongly implicated at sites of chronic tissue inflammation, in which a Th2 microenvironment is favored (Akdis et al. 2011; Mitchell et al. 2010).
Unique Roles for IL-4 and IL-13 in Allergic Lung and Skin Inflammation
Studies in our own laboratory and those of others have demonstrated unique roles for both IL-4 and IL-13 in allergic disease (Finkelman et al. 2010). In a set of studies in which we investigated the roles of IL-4 and IL-13 in OVA-induced allergic lung inflammation, we demonstrated that inhibition of IL-4 during the allergic sensitization phase strongly attenuated serum IgE levels, allergen challenge–induced pulmonary inflammation, and mucus production, whereas when IL-4 was blocked during the effector phase (i.e., at the time of allergen challenge) there was no impact on pulmonary inflammation, AHR, or serum IgE levels. In contrast to these findings, blockade of IL-13 at the time of allergen challenge significantly attenuated bronchoalveolar lavage (BAL) inflammation, AHR, and mucus production (oral presentation, 2011 Symposium, Society of Toxicologic Pathology). These results highlight the importance of the temporal relationship of IL-4 and IL-13 in driving tissue inflammation. At least under experimental conditions, in which mice are actively sensitized with OVA emulsified in alum, it is clear that IL-4 plays an important role early in the initiation of the Th2 inflammatory response. Interestingly, IL-13 blockade with IL-13Rα2 at the time of sensitization also attenuated tissue inflammation following allergen challenge, although not to the same extent as IL-4 blockade; nevertheless, IgE levels were not ablated with IL-13Rα2. We also sought to explore unique roles for IL-4 and IL-13 in allergic skin inflammation and demonstrate that blockade of IL-4 but not IL-13 is sufficient to attenuate a late-phase swelling response in the skin of OVA-sensitized mice that were challenged intradermally with OVA (oral presentation; 2011 Symposium, Society of Toxicologic Pathology). These results highlight the unique and overlapping roles of IL-4 and IL-13. Importantly, in allergic inflammation, there appears to be both a balanced temporal relationship between the two closely related cytokines as well as unique roles that are dependent on cytokine receptor expression in specific cell types involved in the inflammatory response and on the local tissue concentrations of available IL-4 and IL-13.
The New Kids on the Block
Recently, IL-1 family cytokines (IL-1, IL-18 and IL-33), epithelial-derived thymic stromal lymphopoietin (TSLP), and IL-25 have received attention with regard to their role in allergic tissue inflammation. These cytokines are upstream of both IL-4 and IL-13 and may have greater therapeutic potential, since they play an important “gate keeper” role in mucosal homeostasis. IL-9 has also received a resurgence of interest since an IL-9–producing CD4+ T cell subset designated Th9 has recently been implicated in asthma pathogenesis.
Thymic Stromal Lymphopoietin (TSLP)
TSLP is related to IL-7 and is produced by epithelial cells at barrier surfaces, fibroblasts, mast cells, and keratinocytes (Reche et al. 2001; Ziegler and Artis 2010). TSLP is expressed constitutively in intestinal epithelial cells with highest expression in colonic epithelium (Taylor et al. 2009). a Ongoing interactions with commensal bacteria may explain constitutive TSLP expression and account for the tolerogenic response to commensals in the gut. Thus, TSLP appears to play an important role in maintaining gut homeostasis. In addition, TSLP may contribute to allergic inflammation, since expression can be induced by proteases that interact with PAR-2. Notably, common airborne fungal allergens have been shown to act through PAR-2, which suggests that TSLP might have a function in early activation of the innate host defense response at the initial site of exposure in the epithelium, leading to Th2 polarization (Ziegler and Artis 2010).
The TSLP receptor complex is made up of an IL-7 receptor α chain and a distinctive TSLP receptor (TSLPR) chain, which is expressed by myeloid dendritic cells (mDCs), monocytes, pre-activated T cells, NK cells, mast cells, basophils, and eosinophils (Friend et al. 1994; Quentmeier et al. 2001; Reche et al. 2001). In humans, TSLP potently stimulates mDCs, resulting in upregulated expression of HLA-DR, CD40, CD80, CD86, OX40 L, and CD83 and production of chemokines, including CCL17 (TARC) and CCL22 (MDC; Reche et al. 2001; Ziegler and Artis 2010). Both of these chemokines are part of the Th2 axis of inflammation and potently attract Th2 cells via interaction with CCR4. TSLP-activated DCs in turn promote naive CD4+ T cells to differentiate into a Th2 phenotype and promote the expansion of Th2 memory cells. TSLP also suppresses the expression of the IL-12 p40 receptor in DCs, which further down-modulates Th1 responses (Rimoldi et al. 2005). Other TSLP-related functions include the enhancement of IL-13 production from mast cells, recruitment of eosinophils, amplification of basophil responses, and promotion of IL-4 transcription in Th2 cells (Ziegler and Artis 2010).
TSLP gene expression is significantly up-regulated in the epidermis of lesional skin, but not in the uninvolved skin in atopic dermatitis patients (Soumelis et al. 2002). Later, others demonstrated that skin-specific inducible TSLP gene expression can drive a Th2-biased spontaneous skin inflammation in mice with disease characteristics that resemble those of human atopic dermatitis (Yoo et al. 2005). Additionally, in a Fluorescein isothiocyanate-induced contact hypersensitivity Th2-mediated model of human allergic contact dermatitis, TSLP receptor–deficient mice are protected from disease progression (Larson et al. 2010).
Up-regulated expression of TSLP has also been implicated in the pathogenesis of asthma. For example, in a mouse model of antigen-induced airway inflammatory disease, Zhou et al. demonstrated that elevated levels of TSLP directly correlated with the extent of pulmonary infiltrates (Zhou et al. 2005). Not surprisingly, TSLP-deficient mice experienced considerably reduced airway disease, whereas both lung epithelial–specific overexpression of TSLP and direct intranasal delivery of TSLP with antigen resulted in the development of a Th2-biased CD4+ T cell infiltration, elevated serum IgE, and eosinophilia, as well as AHR and airway remodeling. Human studies also point fingers toward TSLP as a prime suspect in asthma pathogenesis. For example, in patients with asthma, TSLP expression correlates with Th2 chemoattractants and disease severity (Fang et al. 2010; He and Geha 2010; Ying et al. 2008). Additionally, several genome-wide studies have found polymorphisms in the TSLP gene that are associated with asthma pathophysiology (Harada et al. 2009; Harada et al. 2011; Hunninghake et al. 2010). Elevated levels of TSLP have also been linked to allergic rhinitis (Miyata et al. 2008; Mou et al. 2009), food and pollen allergy (Blázquez et al. 2010; Zhu et al. 2009), and allergic (kerato) conjunctivitis (Matsuda et al. 2010; Zheng et al. 2010). TSLP may therefore play a pivotal role in the initiation of allergic disease and contribute to the pathophysiology of asthma, rhinitis, and atopic dermatitis.
IL-33 and Allergic Airway Disease
IL-33 is another upstream cytokine and a member of the IL-1 family of cytokines that can function both as a secreted cytokine and within the nucleus by binding to chromatin via a homeodomain and nuclear localization signal in its amino-terminus (Moussion et al. 2008). The pathophysiological role of IL-33 as a nuclear factor is not fully understood, but in vitro studies have shown it can bind to the acidic pocket of a dimeric histone (H2A-H2B) on the surface of nucleosomes, resulting in suppression of gene transcription (Roussel et al. 2008). However, following cellular damage or appropriate pattern recognition receptor (PRR) activation, IL-33 may be released into the extracellular environment to act as an “alarmin” and induce a cascade of pro-inflammatory events. Extracellular IL-33 signals through a heterodimeric receptor that is made up of ST2 and the ubiquitously expressed IL-1R accessory protein IL-1RAcP (Chackerian et al. 2007) that activates NF-κB and MAPK pathways. Diverse receptor expression on innate immune cells including basophils, mast cells, eosinophils, innate lymphoid cells, and NK and NKT cells, as well as Th2 lymphocytes and B cells (Akdis et al. 2011; Bourgeois et al. 2009; Chackerian et al. 2007; Eiwegger and Akdis 2011; Liew et al. 2010), supports the notion that IL-33 may bridge innate and adaptive immune responses in allergic disease.
IL-33 may play a pivotal role in mast cell–mediated allergic responses, since it has been reported to directly induce cytokine and chemokine (IL-1β, IL-6, IL-13, TNF-α, and CCL2 [MCP1]) secretion from mouse bone marrow–derived cultured mast cells without affecting their degranulation (Moulin et al. 2007). Like its murine counterpart, human IL-33 can induce cytokine and chemokine production, prolong survival, and promote cell adhesion in human cord blood stem cell–derived cultured mast cells (Allakhverdi et al. 2007; Iikura et al. 2007). Mast cells also secrete IL-33 following activation through IgE receptors; thus, mast cell–derived IL-33 may provide a positive feedback loop that could maintain mast cell activation for an extended period of time (Hsu et al. 2010).
IL-33 directly activates human eosinophils, as demonstrated by superoxide anion production and degranulation, and increases eosinophil survival (Cherry et al. 2008). Similarly, IL-33 up-regulates ICAM-1 expression on the eosinophil cell surface, as well as the release of IL-6, CXCL8 (IL-8), and CCL2. IL-33–mediated eosinophil survival, ICAM-1 expression, and cytokine/chemokine release are regulated by NF-κB, p38 MAPK, and ERK pathways (Chow et al. 2010). IL-33 also appears to switch alveolar macrophages to the alternatively activated form (Kurowska-Stolarska et al. 2009) and may play a similar role to that of TSLP in that it can act as a maturation factor for bone marrow–derived dendritic cells via up-regulation of CD80, CD40, and OX40 L with concomitant release of pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and CCL17 (Besnard et al. 2011). Again, analogous to TSLP, IL-33-pretreated dendritic cells are significantly more potent in the skewing of naïve T cells to a Th2 phenotype following allergen challenge. In vivo studies have also demonstrated that intratracheal administration of OVA-pulsed dendritic cells with IL-33 significantly enhances eosinophil counts and mucus secretion in the lung as compared with OVA-pulsed DCs alone (Besnard et al. 2011). Other recent studies have implicated IL-33 in the activation of new innate type-2 immune effector leukocyte, the nuocyte, which is defined as lineageneg, ICOSpos, ST2pos, IL-17RBpos, and IL-17RApos (Neill et al. 2010). The nuocyte expands in vivo in response to IL-25 and IL-33, and it represents the predominant early source of IL-13 during helminth infection with Nippostrongylus brasiliensis (Barlow and McKenzie 2011). Thus, the nuocyte may be a key upstream innate immune cell required for the induction and amplification of Th2-mediated immunity in worm expulsion. Nevertheless, whether or not the nuocyte plays a prominent role in Th2-mediated allergic responses has yet to be determined.
In vivo evidence for a role for IL-33 in allergic asthma comes from several sources. For example, administration of IL-33 to wild-type mice induces AHR and goblet cell hyperplasia associated with induction of IL-4, IL-5, and IL-13 in the lungs. This pro-Th2 inflammatory effect appears to be independent of the adaptive immune response, since RAG-2 -/- mice develop a comparable response to IL-33 that is dependent on binding to ST2 and signaling via MyD88 (Kondo et al. 2008). Similarly, IL-33 transgenic mice develop spontaneous allergic airway inflammation characterized by pulmonary eosinophilic infiltration and goblet cell hyperplasia, whereas BAL samples show overall increased cellularity dominated by lymphocytes (Zhiguang et al. 2010). Furthermore, soluble ST2 blocks IL-33 signaling in a murine OVA model of allergic airway inflammation (Hayakawa et al. 2007), and in OVA models of allergic airway disease, monoclonal antibody treatment against ST2 (Kearley et al. 2009) or polyclonal antibody treatment against IL-33 (Liu et al. 2009) largely abrogate allergic airway inflammation and AHR. In our own laboratory, we also demonstrated similar results, using two different rat monoclonal antibodies against mouse IL-33 in murine allergic lung inflammation models induced via OVA or the house dust mite.
Human data point to a role for IL-33 in allergic disease. Increased systemic levels of the decoy receptor sST2 are found in asthmatic patients and correlate with asthma severity and/or exacerbations (Oshikawa et al. 2001). Furthermore, higher IL-33 mRNA levels are consistently found in lung biopsies from asthmatics and at even higher levels in severe asthmatics compared to healthy controls. Interestingly, cultured airway smooth muscle cells collected from these specimens show baseline IL-33 expression that can be synergistically enhanced by addition of TNF-α and/or IFN-γ. Of note, IL-33 up-regulation induced by TNF-α cannot be abolished by dexamethasone treatment, suggesting linkage to steroid insensitivity that is observed in certain forms of severe asthma (Préfontaine et al. 2009). IL-33 is also expressed by respiratory epithelial cells in the lungs of patients with moderate to severe asthma (Préfontaine et al. 2010). In addition to asthma, the IL-33/ST2 pathway has been implicated in atopic dermatitis. Nucleotide polymorphisms in the promoter of the ST2 receptor gene are correlated with atopic dermatitis (Shimizu et al. 2005) and the levels of IL-33 expression are substantially increased in the lesional skin tissue of patients with atopic dermatitis (Pushparaj et al. 2009).
IL-25
IL-25 (IL-17E) was initially reported to be derived from highly polarized Th2 cells (Fort et al. 2001; Reynolds et al. 2010); however, further studies have demonstrated expression in IgE-activated mast cells (Ikeda et al. 2003), alveolar macrophages (Kang et al. 2005), microglia (Kleinschek et al. 2007), eosinophils (Dolgachev et al. 2009; Wang et al. 2007), basophils (Wang et al. 2007), epithelial cells (Angkasekwinai et al. 2007; Zaph et al. 2008), and endothelial cells (Sonobe et al. 2009).
IL-25 uses a heterodimeric receptor complex consisting of IL-17RB and IL-17RA (Rickel et al. 2008) that is expressed on both innate and adaptive immune cells (Reynolds et al. 2010), including NKT cells (Terashima et al. 2008), eosinophils (Wong et al. 2005), monocytes (Dolgachev et al. 2009), and T cells (Iwakura et al. 2011). Furthermore, human Th2 central memory cells selectively up-regulate receptor expression when stimulated with TSLP-activated dendritic cells or when triggered by a specific antigen (Wang et al. 2007). Th9, a novel T helper lineage, can also respond to IL-25, which functions to augment IL-9 production (Neill and McKenzie 2010), and, as discussed above, IL-25 along with IL-33 can expand and activate the nuocyte population (Saenz et al. 2010).
Numerous studies in mice demonstrate a role for IL-25 in allergic lung inflammation. For example, IL-25 knockout animals exhibit fewer infiltrating inflammatory cells and Th2 cytokines in the BAL fluid after repeated allergen challenge (Angkasekwinai et al. 2010), whereas IL-25 overexpression in lung epithelium induces epithelial cell hyperplasia, mucus hypersecretion, airway infiltration of eosinophils and macrophages, and up-regulation of eotaxin-1 (Ccl11), eotaxin-2 (Ccl24), and macrophage-derived chemokine (mdc/Ccl22) which play crucial roles in the recruitment of eosinophils and Th2 cells, respectively (Angkasekwinai et al. 2007). Tslp was also up-regulated in IL-25 transgenic mice (Angkasekwinai et al. 2007).
IL-25 appears to be important in both the sensitization and the challenge phases of allergic airway disease (Ballantyne et al. 2007), and IL-25 blockade can result in attenuated levels of Th2 cytokines, along with a reduction in goblet cell hyperplasia and serum IgE levels (Ballantyne et al. 2007). Furthermore, neutralization of IL-25 at the time of antigen sensitization or during the challenge phase can prevent AHR in a mouse model of allergic lung inflammation (Ballantyne et al. 2007). It is also interesting to note that pneumonia virus infection may enhance IL-25 expression, which suggests that IL-25 induced by early life virus infection may promote further development of allergic airway responses (Siegle et al. 2010). In regard to human studies, increased expression of IL-25 and receptor has been detected in bronchial mucosa of asthmatic patients compared to healthy controls (Corrigan et al. 2011; Wang et al. 2007). The IL-25 signaling pathway may also contribute to pathologies associated with atopic dermatitis. For example, IL-25 gene expression is elevated in the lesional skin of atopic dermatitis patients (Wang et al. 2007), whereas IL-25 produced by DCs/mast cells can regulate skin barrier function via down-modulation of filaggrin expression by keratinocytes (Hvid et al. 2011).
IL-9
IL-9 was first identified in 1989 as a T cell growth factor in a mouse T helper cell line (Uyttenhove et al. 1988), and since then, it has been shown to have pleiotropic functions in the immune system. Mast cells (Hültner et al. 2000; Stassen et al. 2000), eosinophils (Shimbara et al. 2000), and neutrophils can produce IL-9 (McNamara et al. 2004; Shimbara et al. 2000). However, mounting evidence suggests that a large majority of the cytokine is secreted from specialized T lymphocytes. Quite recently, two independent groups demonstrated that naïve CD4+ T cells primed in the combination of TGF-β and IL-4 or Th2 cells additionally cultured in TGF-β can produce high levels of IL-9 with diminished expression of other lineage-specific cytokines and transcription factors (Dardalhon et al. 2008; Veldhoen et al. 2008). This IL-9–secreting phenotype was dependent on transcription factors shared with the Th2 cells, including GATA3, STAT6 (Dardalhon et al. 2008; Veldhoen et al. 2008), and IFN regulatory factor 4 (IRF4), as well as PU.1, an ETS family transcription factor known to have a repressive effect on the production of Th2 cytokines (Chang et al. 2005; Chang et al. 2009).
IL-9 signals through the IL-9 receptor, which is composed of the IL-9–specific α-chain (Il9r) and γc. Following ligand binding, a single tyrosine residue (Tyr407) in the IL-9Rα is phosphorylated, resulting in activation of JAK1, whereas engagement of the γc chain also results in activation of JAK3. Activation of STAT1, STAT3, and STAT5 further contributes to signal transduction (Druez et al. 1990; Goswami and Kaplan 2011).
The IL-9R is expressed on T cell lines and on effector T cells, but not naïve T cells (Cosmi et al. 2004; Druez et al. 1990). Among the Th cell subsets, IL-9R has greatest expression in Th2 and Th17 cells (Nowak et al. 2009). In asthma patients, IL-9R is found on mast cells and polymorphonuclear cells in the lung (Abdelilah et al. 2001; Kearley et al. 2011). The IL-9R also appears to be expressed on nonhematopoietic cells such as airway epithelium and airway smooth muscle cells, although the exact receptor complex on these cell types has not yet been clarified.
IL-9 is emerging as a significant player in allergic lung inflammation. For example, transgenic mice that overexpress IL-9 in the lung demonstrate airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness (Temann et al. 1998), whereas blockade of IL-9 in a chronic allergic lung inflammation model resulted in attenuation of mastocytosis and airway remodeling (Kearley et al. 2011). Several groups have established a link between IL-9 and IL-13 (Steenwinckel et al. 2009; Steenwinckel et al. 2007; Temann et al. 2007; Temann et al. 2002; Whittaker et al. 2002) with both direct and indirect effects on the airway epithelium, resulting in induction of mucus production genes in goblet cells (Longphre et al. 1999; Louahed et al. 2000; Reader et al. 2003; Steenwinckel et al. 2007; Vermeer et al. 2003). With regard to airway smooth muscle cells, coupling of IL-9 to the receptor was found to release CCL11 and IL-8 in an ERK-dependent manner. Human data also suggest a role for IL-9 in allergic lung inflammation. IL-9 expression is increased in lungs of asthmatic patients (Shimbara et al. 2000), and IL-9R expression is found in the lungs of asthmatic individuals, but not healthy controls (Bhathena et al. 2000). In addition, several SNPs in either IL-9 or the IL-9R are associated with an allergic phenotype, including lung function, allergen sensitization, and IgE levels.
Summary
Allergic asthma and atopic dermatitis are complex tissue inflammatory disorders that involve many cytokine pathways with both overlapping and unique roles. Thus, delineating a master cytokine switch in allergic disease, akin to TNF-α in rheumatoid arthritis, has remained elusive. Both IL-4 and IL-13 have received much attention as key players in the adaptive immune arm of the allergic response, although more recently identified and/or relatively understudied “older” members of the cytokine family may provide a critical link between early innate immune activation and the later-stage Th2-mediated allergic response (Figure 1). Furthermore, with the identification of new innate type 2 immune effector leukocytes such as nuocytes, which can secrete IL-4 and IL-13 following expansion and activation with IL-33 and IL-25, the interplay between early innate immune cellular sources of IL-4 and IL-13 and later-stage adaptive Th2-derived sources is highlighted. These studies also point to the significance of a set of early response cytokines such as IL-33 and IL-25, as well as TSLP and IL-9, which can be secreted from epithelial cells and/or resident tissue innate immune cells following allergen challenge or activation via toll-like receptors. Both TSLP and IL-33 can potentiate the effectiveness of DCs in skewing toward a Th2 phenotype, whereas early innate immune cellular sources of IL-9 may promote mast cell growth and differentiation at sites of tissue inflammation. Likewise, in addition to a role in nuocyte activation, early sources of IL-25 may contribute to a first wave of type 2 innate immune responses, including goblet cell mucus production and early eosinophil recruitment.
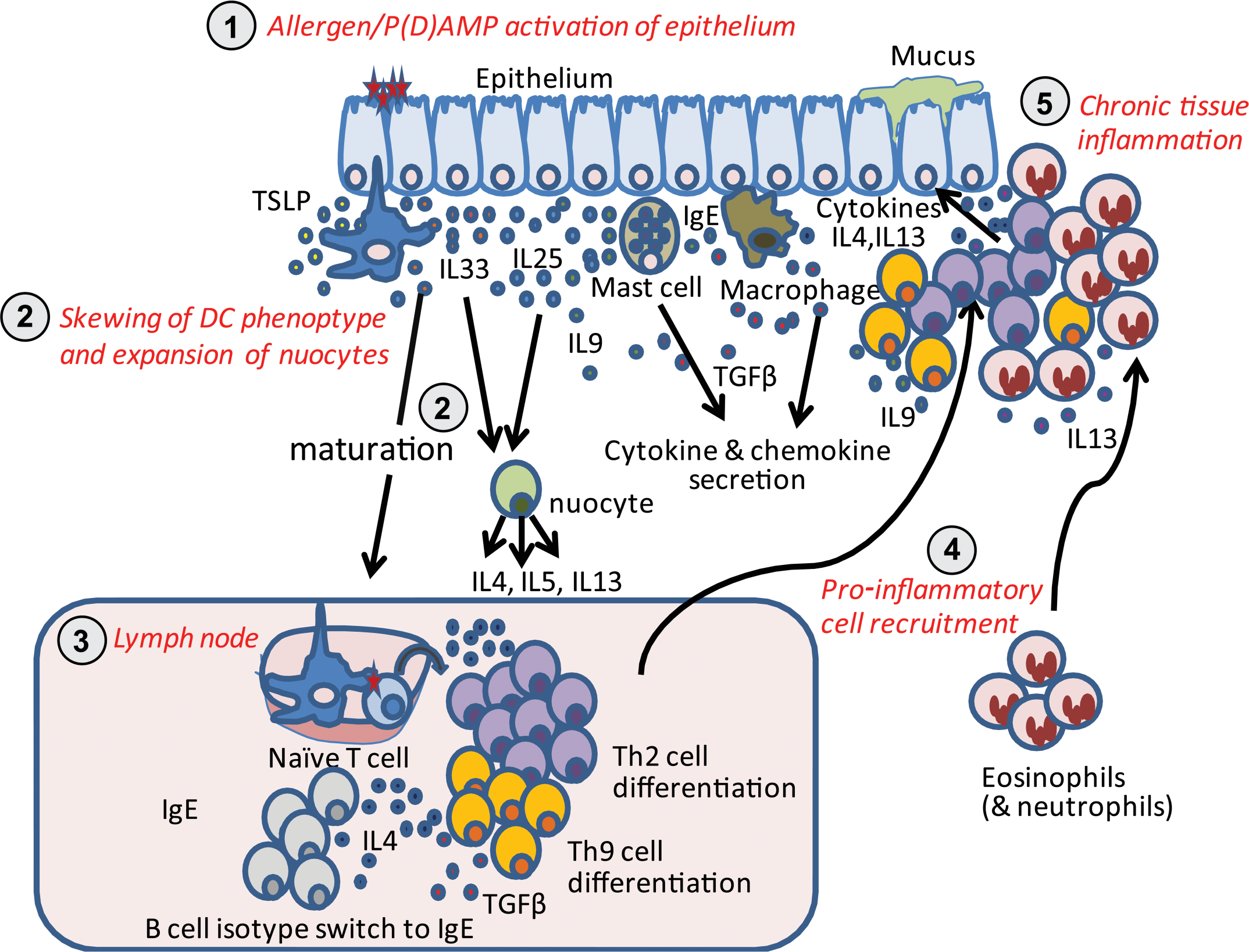
Emerging cytokine pathways in allergic disease. (1) Allergen/pathogen (damage)-associated molecular patterns activation of epithelium: following activation with allergen and/or pathogen (damage)-associated molecular patterns via interactions with toll-like receptors on the surface of epithelial cells, a set of early response cytokines including TSLP, IL-33, and IL-25 are released into the local tissue environment. Resident innate immune cells including mast cells, dendritic cells, and macrophages may also become activated via IgE crosslinkage (mast cells) and/or toll-like receptor engagement, inducing the release of cytokines. Early innate sources of IL-9 may promote mast cell growth and differentiation for further participation in the allergic response. (2) Skewing of DC phenotype and expansion of nuocytes: exposure of dendritic cells to thymic stromal lymphopoietin and IL-33 can promote the development of a dendritic cell phenotype that supports the differentiation of Th2 cells within the draining lymph nodes. IL-33 and IL-25 also promote the expansion and activation of tissue-resident nuocytes, which can then secrete IL-4, IL-5, and IL-13 to potentiate a Th2-type inflammatory response, whereas IL-4 along with TGF-β can promote the differentiation of naïve T cells to a Th9 phenotype. (3) Lymph node: differentiation of Th2/Th9 cells and B cell immunoglobulin isotype switch. (4) Proinflammatory cell recruitment: cytokine/chemokine gradients released by activated innate immune cells (mast cells, macrophages) and epithelium promote inflammatory cell recruitment. (5) Chronic tissue inflammation: recruited pro-inflammatory leukocytes further contribute to chronicity of allergic disease via release of Th2-type cytokines.
Footnotes
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. The authors received no financial support for the research, authorship, and/or publication of this article.