
Abstract
The complement system and toll-like receptors (TLRs) are two components of innate immunity that are critical for first-line host defense. Many pathogen-associated molecular patterns activate both complement and TLRs, and recent studies in animal models have revealed a marked synergistic interaction between the two systems. In mice deficient in a membrane complement regulator, prototypical TLR ligands such as LPS, zymosan, and polyI:C caused increased systemic complement activation, which in turn led to a profound elevation of proinflammatory cytokine biosynthesis. This phenotype required interaction between complement and TLRs because complement activation alone by cobra venom factor without TLR engagement did not lead to appreciable cytokine production. The regulatory effect of complement on TLR signaling was mediated by the anaphylatoxins C5a and C3a through a receptor-dependent mechanism and involved increased mitogen-activated protein kinase and nuclear factor κB activation. The crosstalk between complement and TLRs may also impact adaptive immunity, for example, the differentiation of T helper 17 (Th-17) cells. Given that excessive activation of either TLR or complement has been associated with inflammatory tissue injury as occurs in sepsis and autoimmune diseases, the new insight on complement and TLR crosstalk may have therapeutic implications.
Complement
Complement is a major component of innate immunity that plays a crucial role in host defense (Dunkelberger and Song 2010a). It is composed of over thirty proteins in the plasma and on the cell surface. Complement is activated in a cascade of proteolytic reactions via three different pathways: the classical, alternative, and mannose-binding lectin (MBL) pathways (Fig. 1). The classical pathway is triggered by antibody-antigen complexes and requires the participation of C1, C2, and C4. Activation of the classical pathway generates a C3-cleaving enzyme complex called the classical pathway C3 convertase, C4b2a. The MBL pathway is activated upon recognition of specific sugar molecules on microbial surfaces by lectins. It uses MBL-associated serine proteases (MASPS) as well as C2 and C4 to generate the same C3 convertase (C4b2a) as in the case of classical pathway of complement activation. The alternative pathway is thought to be constitutively active at a low level due to spontaneous hydrolysis of C3. Hydrolyzed C3 interacts with factor B and factor D to initiate the alternative pathway and subsequently generates the alternative pathway C3 convertase C3bBb. The latter enzyme complex, which is also formed ultimately as a result of classical pathway and MBL pathway complement activation, is self-perpetuating and works to rapidly amplify complement activation, regardless of the pathway by which complement activation is initiated. Newly formed C3bBb is a labile complex, and its half-life is extended by the plasma protein properdin. Recent studies have shown that properdin could also play a role in the initiation of alternative pathway complement activation, likely in an activating surface- and animal species–specific manner (Kemper, Atkinson, and Hourcade 2010; Hourcase 2006; Spitzer et al. 2007; Kimura et al. 2008; Agarwal et al. 2010; Ferreira, Cortes, and Pangburn 2010).
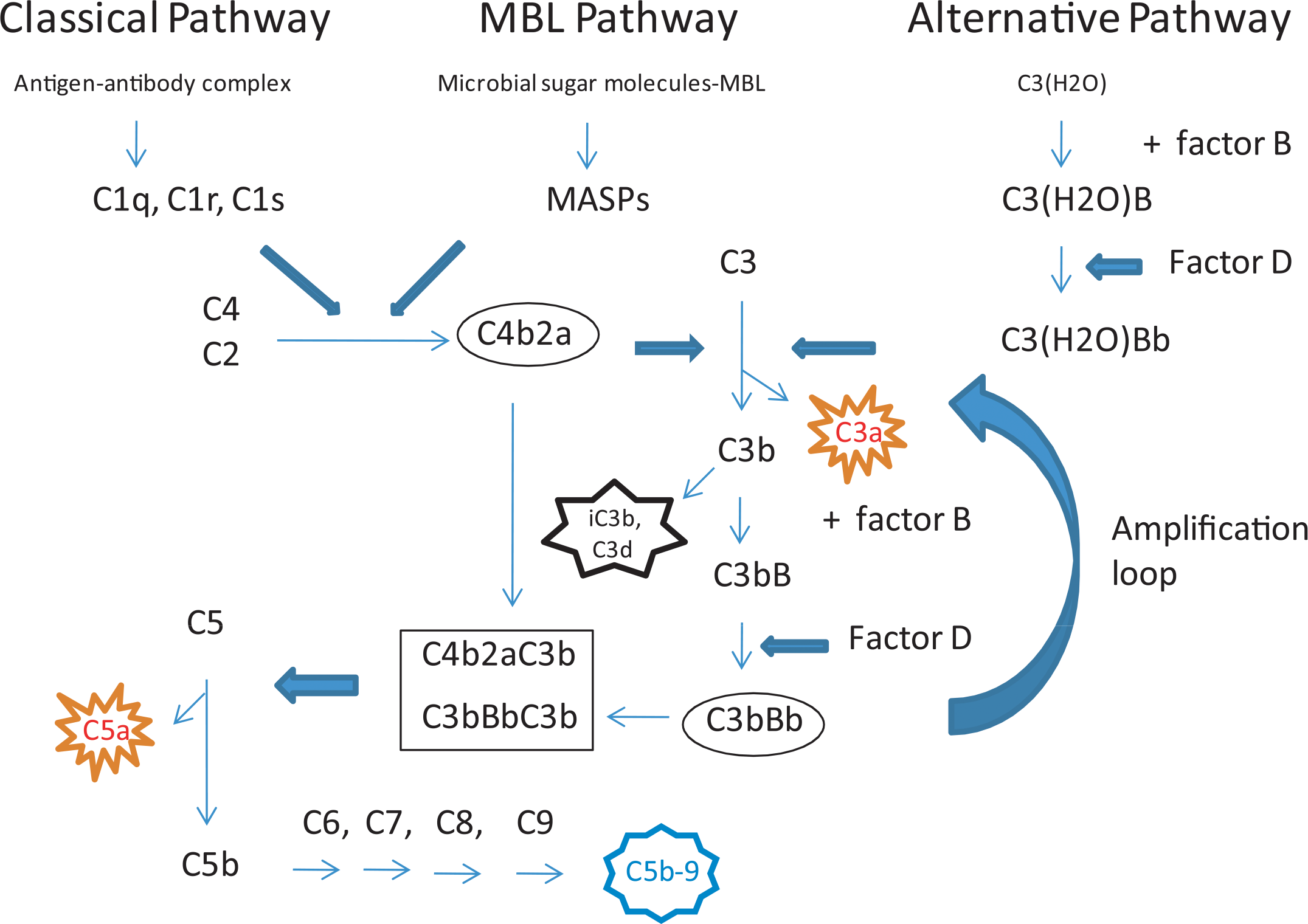
Complement activation cascade. Complement can be activated by three pathways in a cascade of proteolytic reactions. All pathways converge at the C3 cleavage step from which an alternative pathway amplification loop involving C3bBb is engaged. Proteolytic enzyme activity is indicated by block arrows. The C3 convertases are indicated by an oval box, and C5 convertases are indicated by a rectangular box. Effector functions of activated complement are indicated by star boxes (orange for anaphylatoxins, black for opsonization by activated C3 fragments, and blue for membrane attack complex).
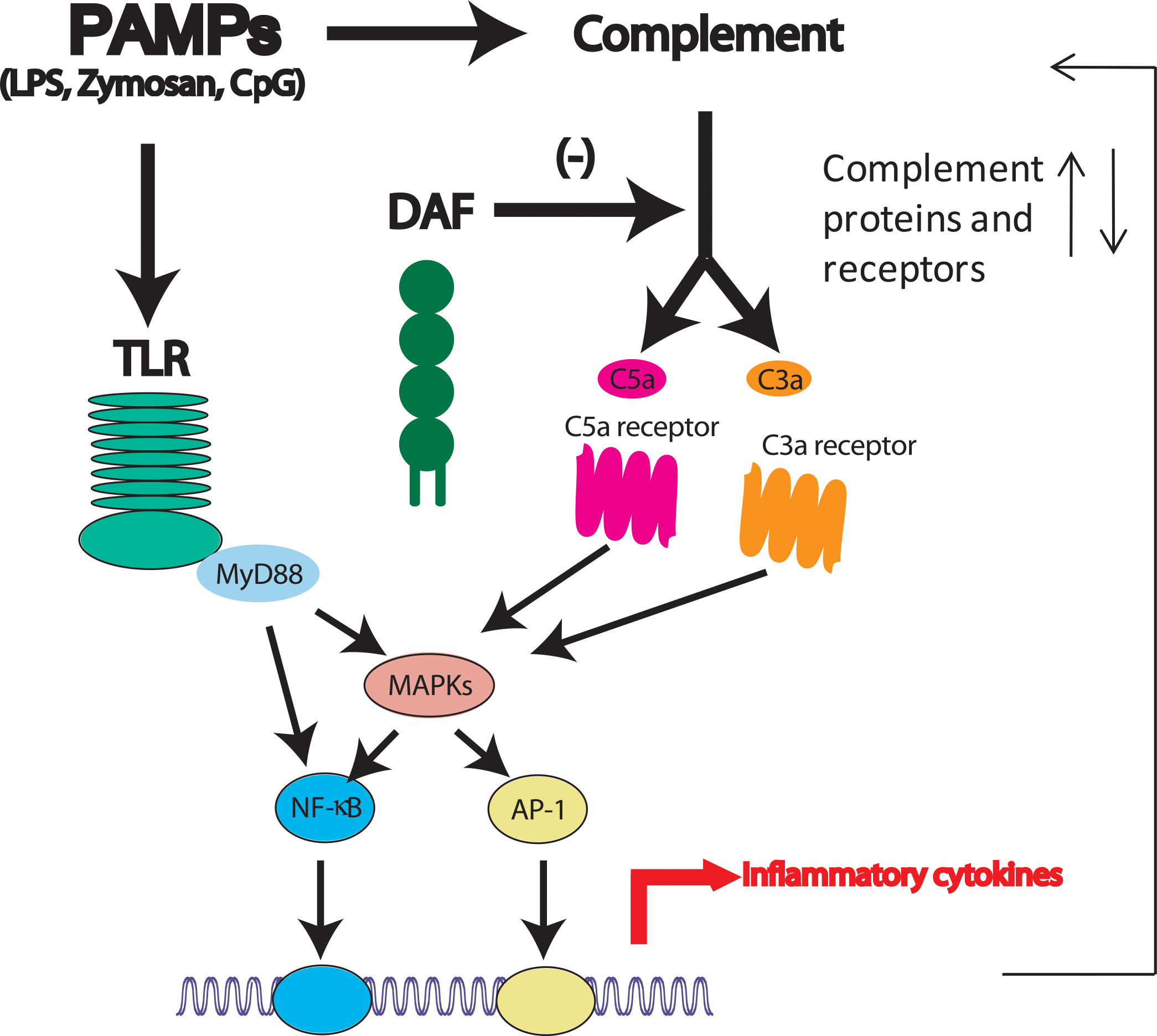
Interaction between complement and the TLR pathways. Pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), zymosan, and CpGs can activate both pathways. Activated complement regulates Toll-like receptor (TLR) signaling through the activities of G protein-coupled anaphylatoxin receptors C5aR and C3aR, intracellular signaling molecules such as MAPKs, and transcriptional factors such as NF-kB. In the absence of the complement regulatory protein decay-accelerating factor (DAF), complement activation and its effect on TLR signaling is amplified. The absence of DAF may be mimicked by strong complement activators such as cobra venom factor (CVF) or pathological conditions such as sepsis. On the other hand, TLR activation may also regulate complement protein biosynthesis and receptor expression (modified from Zhang et al 2007).
Activated complement achieves its biological effect by promoting inflammation through the generation of C3a and C5a, referred to as anaphylatoxins, by opsonization of target cells or activating surfaces and by formation of the membrane attack complex (Dunkelberger and Song 2010a). Apart from these effector functions, complement also regulates adaptive immune responses (Dunkelberger and Song 2010b). For example, complement is well known to be a natural adjuvant for B cell activation and antibody production (Carroll 2000, 2004; Fearon and Carroll 2000). This effect on B cell biology is achieved through multiple mechanisms, including lowering the threshold of B cell receptor activation, facilitating antigen retention on follicular dendritic cells, and promoting memory B cell survival (Fearon and Carroll 2000; Holers 2005). Recent evidence has suggested that complement also plays a regulatory role in T cell immune responses, although the mechanisms by which complement exerts this effect remain to be fully delineated (Dunkelberger and Song 2010b). Most evidence supports a prominent role of the anaphylatoxins, working through their cognate G protein coupled receptors, in regulating the function and behavior of antigen-presenting cells, either directly or indirectly (Dunkelberger and Song 2010b). Finally, complement is also known to play a role in “waste disposal,” namely, in the processing and removal of apoptotic cells and immune complexes (Walport 2001a, 2001b). This hypothesis is supported by the clinical observation that deficiency of the early complement components, particularly of the classical pathways, such as C1q, C4, and C2, is a high-penetrance risk factor for developing systemic lupus erythematosus (SLE) (Walport 2001a, 2001b). Furthermore, mice deficient in C1q and C4 also developed autoimmune diseases resembling human SLE with characteristic findings such as glomerulonephritis and appearance of high titer autoantibodies (Botto et al. 1998; Chen, Koralov, and Kelsoe 2000; Paul et al. 2002).
While the pro-inflammatory and cytolytic activities of complement are needed to provide necessary host defense against pathogenic infections, excessive and uncontrolled complement activation has the potential to cause severe tissue injury. This is especially true considering that the alternative pathway is constitutively active and has the ability to undergo self-amplification. To prevent these undesirable consequences, host cells use a number of fluid-phase and membrane-bound regulators that function to inhibit complement activation on autologous cells (Kim and Song 2006). The membrane-bound complement regulators include decay-accelerating factor (DAF), membrane cofactor protein (MCP), complement receptor-1 (CR1), and CD59. Fluid-phase regulators include factor H, C4-binding protein, and factor I. Except CD59, which is an inhibitor of membrane attack complex formation in the terminal stage of complement activation, all the aforementioned proteins work by inhibiting the activity of C3 and C5 convertases. Inhibition of C3 convertase activity is achieved by one of two ways: (1) promoting the dissociation (“decay acceleration”) of the two partners in the C3 convertases, namely, C4b and C2a in C4b2a and C3b and Bb in C3bBb; this activity is provided by DAF, CR1, and factor H; and (2) proteolytic degradation of C4b and C3b by factor I. The activity of factor I requires C4-binding protein (for C4b cleavage) or fH, MCP, CR1 (for C3b cleavage) as a cofactor. The latter activity is referred to as cofactor activity by the concerned complement regulators (Kim and Song 2006).
Under normal physiological conditions, a fine balance between complement activation and regulation is maintained. Not surprisingly, when this delicate balance is disturbed, there are significant consequences to human health. On the one hand, genetic deficiencies of key complement components will render the affected individuals susceptible to opportunistic infections (Walport 2001a, 2001b), and on the other, dysfunctional complement regulators can result in uncontrolled complement activation and attack of self tissues, leading to autoimmune and inflammatory diseases (Kim and Song 2006). For example, deficiency of DAF and CD59 on hematopoietic stem cells causes the human hematological disorder paroxysmal nocturnal hematoglobinutia, which is characterized by sensitivity of red blood cells to autologous complement attack and lysis mediated by the alternative pathway (Parker 1996). Mutations in factor H and MCP have been found to be associated with atypical hemolytic uremic syndrome and C3 glomerulopathy (Fremeaux-Bacchi et al. 2006; Le Quintrec et al. 2010; Kavanagh, Richards, and Atkinson 2008). The importance of membrane complement regulators has also been demonstrated by studies of knockout mice lacking key complement regulators. Gene deletion of factor H resulted in uncontrolled systemic complement activation via the alternative pathway and secondary C3 and factor B deficiency, leading to age-dependent C3 glomerulopathy with features resembling human membranoproliferative glomerulonephritis (Pickering et al. 2002). In our own studies of DAF knockout (DAF–/–) mice, we showed that mice deficient in DAF displayed increased sensitivity to anti-GBM glomerulonephritis and renal ischemia reperfusion injury (Sogabe et al. 2001; Yamada et al. 2004). Furthermore, DAF deficiency significantly exacerbated the autoimmune disease phenotype of MRL/lpr mice (Miwa et al. 2002). Thus, while mice in a MRL/lpr mouse colony maintained at our animal facility generally had low incidence of dermatitis—and when they did, their skin lesions were usually very small—MRL/lpr mice deficient in DAF developed severe skin inflammation, often with large open lesions, at high prevalence (Miwa et al. 2002). This phenotype was shown to reflect increased complement-dependent tissue injury in the absence of DAF, as further deletion of the C3 gene in DAF-deficient MRL/lpr mice rescued the exacerbated dermatitis phenotype (Miwa et al. 2007).
Toll-Like Receptors
Unlike soluble complement proteins that act as fluid-phase sentinels of pathogen infection, mammalian Toll-like receptors (TLRs) are a group of structurally related molecules that are expressed on the cell surface and serve as pattern recognition receptors (PRRs) to detect the presence of microbial infection (Takeda, Kaisho, and Akira 2003; Medzhitov 2001; Modlin 2002). TLRs were initially identified based on homology to the Drosophila Toll receptor (Rock et al. 1998). Mutant Drosophila carrying loss-of-function mutations in the Toll receptor were highly susceptible to fungi infection and unable to produce an anti-fungal peptide (Lemaitre et al. 1996; Hoffman 2003). This provided the first evidence for the existence of innate cellular receptors that can sense and respond to microbial infection. Subsequently, a human homolog of Toll was identified and shown to be capable of inducing the production of inflammatory cytokines and costimulatory molecules (Medzhitov, Preston-Hurlburt, and Janeway 1997). Later, a loss-of-function mutation of the murine homolog of the human Toll was identified in lipopolysaccharide (LPS)-hyporesponsive mice (Poltorak et al. 1998; Hoshino et al. 1999). These studies have established that Toll or Toll-like receptors are evolutionarily conserved pattern recognition receptors that can sense microbial pathogens and trigger direct innate immune responses in the host.
More than ten members of TLRs have been identified in humans and mice (Akira, Uematsu, and Takeuchi 2006; Pasare and Medzhitov 2003a; Takeda 2005). These TLRs have been found to recognize different microbial and, in some cases, endogenous ligands. For example, TLR4 can be activated by LPS from gram-negative bacteria, heat shock proteins (Poltorak et al. 1998; Hoshino et al. 1999; Takeda 2005; Ohashi et al. 2000), and the anti-cancer drug taxol (Kawasaki, Gomi, and Nishijima 2001; Kawasake et al. 2000; Takeuchi and Akira 2001; Byrd-Leifer et al. 2001). TLR2 can be activated by the yeast cell wall component zymosan and lipoteichoic acid from gram-positive bacteria (Takeuchi and Akira 2001; Underhill et al. 1999; Schwandner et al. 1999; Takeuchi et al. 1999; Volman, Hendriks, and Goris 2005). TLR3 is activated by double-stranded RNAs from viruses, and TLR9 recognizes CpG DNA motifs present in viruses and bacteria (Alexopoulou et al. 2001; Akira and Takeda 2004; Hemmi et al. 2000). Available evidence indicates that TLRs involved in the recognition of molecular structures unique to bacterial and fungal cells (TLR1, TLR2, TLR4, TLR5, and TLR6) are localized to the plasma membrane and can be recruited to phagosomes, whereas TLRs that recognize viral and bacterial nucleic acids (TLR3, TLR7, and TLR9) are localized in intracellular compartments (Takeda, Kaisho, and Akira 2003; Takeda and Akira 2005).
Structurally, TLRs contain extracellular leucine-rich repeats responsible for ligand binding, transmembrane domains, and cytoplasmic Toll/interleukin-1 receptor (TIR) domains required for initiating intracellular signaling (Kawai and Akira 2006). Ligand-mediated activation of TLRs leads to receptor dimerization, recruitment of adaptor molecules, and activation of various protein kinases (Akira and Takeda 2004; Kawai and Akira 2006). These events culminate in the activation of nuclear factor-kB (NF-κB), activation protein-1 (AP-1), and interferon regulatory factor 3 (INF3), transcription factors that regulate the expression of inflammatory cytokines IL-6 (IL-8), TNF-α, IL-1β, and IL-12, as well as type I interferons- IFN-β and multiple forms of IFN-α (Akira and Takeda 2004; Kawai and Akira 2006). One of the key adaptor molecules that have been shown to be critical for TLR signaling is MyD88 (Kawai and Akira 2006; Hayashi et al. 2001; Hemmi et al. 2002; Kawai et al. 2001; Hacker et al. 2000; Schnare et al. 2000). MyD88 is required for the activation of all TLRs except TLR3, which signals via a MyD88-independent pathway (Akira and Takeda 2004; Kawai and Akira 2006). The MyD88-dependent pathway primarily controls the production of inflammatory cytokines whereas the MyD88-independent pathway regulates the production of anti-viral interferons (Akira and Takeda 2004; Kawai and Akira 2006). Notably, both MyD88-dependent and -independent pathways can be operative in a single TLR (e.g., TLR4), and either pathway is sufficient to cause co-stimulatory molecule (CD40, CD80, CD86) and MHCII expression on antigen-presenting cells (APCs) (Akira and Takeda 2004; Kaisho et al. 2001).
In addition to producing inflammatory cytokines and anti-viral interferons, activation of TLRs also elicits adaptive immune responses (Pasare and Medzhitov 2004; Iwasaki and Medzhitov 2004; Akira, Takeda, and Kaisho 2001; Barton and Medzhitov 2002). A key mechanism in the regulation of adaptive immunity by TLRs is the up-regulation of co-stimulatory molecules on APCs (Iwasaki and Medzhitov 2004). Of interest, MyD88-deficient mouse APCs were still capable of expressing co-stimulatory molecules in response to TLR ligation (Kaisho et al. 2001; Akira, Takeda, and Kaisho 2001), yet these mice displayed profound defects in adaptive immune responses (Kaisho et al. 2001; Prinz et al. 2006). This suggested that both inflammatory cytokines and co-stimulatory molecules are necessary for generating optimal adaptive immune responses. It has been demonstrated in an in vitro T cell proliferation assay that TLR-induced inflammatory cytokines, of which IL-6 was shown to be an essential component, functioned to reverse the inhibitory effect of T regulatory cells (Treg) during T cell priming (Pasare and Medzhitov 2003b, 2004). More recently, IL-6 has been established as a master cytokine that, together with TGF-β, drives the differentiation of a new class of IL-17-producing T helper cells referred to as Th-17 cells (Veldhoen et al. 2006; Bettelli et al. 2006). Th-17 cells are highly pro-inflammatory and are now widely recognized as playing a critical role in a variety of autoimmune diseases (Steinman 2007; Bettelli, Oukka, and Kuchroo 2007). In addition to IL-6, TNF-α and IL-1β as well as inhibition of IFN-γ and IL-12 have been shown to promote Th-17 differentiation (Veldhoen et al. 2006; Bettelli et al. 2006; Steinman 2007; Bettelli, Oukka, and Kuchroo 2007). It was also demonstrated that IL-6 drives Th-17 cell differentiation at least in part through up-regulation of IL-23R on and IL-21 production by Th-17 cells, thus generating a paracrine and autocrine amplification loop in Th-17 cell differentiation (Zhou et al. 2007; Nurieva et al. 2007; Korn et al. 2007).
Crosstalk between Complement and TLRs
Given that both the complement system and TLRs are co-evolved to detect and respond to pathogenic infections, it is not surprising that many microbial components and signature molecules are recognized by both forms of innate immunity. For example, LPS is the prototypical ligand for TLR4 and a well-known complement activator (Bjornson and Bjornson 1977; Morrison and Kline 1977). Zymosan, a component of yeast cell wall, is a strong alternative pathway complement activator as well as a ligand for TLR2/6 (Volman, Hendriks, and Goris 2005). Until recently, the potential interaction between the two innate immune systems, when activated coincidentally, has not received much attention despite the fact that the biology of complement and TLRs has each been studied extensively. Using DAF–/– mice, we have characterized a striking interaction between complement and TLR signaling in vivo (Figure 2; Zhang et al. 2007). The realization of a synergistic regulation of TLR response by complement in this model was somewhat fortuitous, and experiments that led to this conclusion were catalyzed by the unexpected observation that LPS injection into DAF–/– mice produced marked elevated levels of pro-inflammatory cytokines than it did in normal mice (Zhang et al. 2007). In fact, our expectation of the initial experiment had been exactly the opposite based on published reports that DAF was an LPS-binding protein (Heine et al. 2001; El-Aamalouti et al. 1997, 1999). We hypothesized that DAF, a glycosylphosphatidylinositol (GPI)-anchored membrane protein, may function as an LPS coreceptor to facilitate TLR4 signaling. A similar role has been established for CD14, also a GPI-anchored membrane protein whose deficiency in mice compromised LPS-induced TLR4 activation (Haziot et al. 1996). Thus, had our original hypothesis been correct, we would have observed decreased LPS-dependent cytokine production mediated by TLR4 signaling. Instead, we found LPS-treated DAF–/– mice had significantly elevated plasma IL-6, TNF-α, and IL-1β levels compared with wild-type (WT) mice. On the other hand, plasma IL-12 level in DAF–/– mice was lower than that of WT mice (Zhang et al. 2007).
Enhanced production of pro-inflammatory cytokines in DAF–/– mice occurred at the transcriptional level as evidenced by increased IL-6 mRNA synthesis in the spleen, lung, and fat tissues of DAF–/– mice (Zhang et al. 2007). The difference in IL-6 mRNA levels between WT and DAF–/– mice was detectible as early as one hour after LPS injection. Because LPS is a well-known complement activator and DAF inhibits complement activation, we wondered if the phenotype of DAF–/– mice was related to increased complement activation. To test this possibility, we investigated the LPS responses of DAF–/– mice that were deficient in C3. This experiment showed that the phenotype of DAF–/– mice was indeed dependent on complement. Thus, unlike DAF–/– mice, DAF–/– C3–/– mice had normal plasma levels of IL-6, TNF-α, IL-1β, and IL-12 after LPS challenge (Zhang et al. 2007). Measurement of activated C3 fragments in the plasma after LPS challenge confirmed that DAF–/– mice were more sensitive to LPS-induced complement activation. To directly demonstrate that enhanced complement activation was responsible for increased production of cytokines in DAF–/– mice after LPS challenge, WT mice were simultaneously challenged with LPS and cobra venom factor (CVF), a potent complement activator. Significantly, while CVF treatment alone did not induce appreciable amount of IL-6 or IL-12 production in WT mice, it synergized with LPS to cause a dramatic increase in IL-6 production while profoundly suppressing IL-12 biosynthesis (Zhang et al. 2007). This finding thus recapitulated the phenotype of LPS-treated DAF–/– mice and provided further support for a regulatory effect of complement on TLR4 signaling.
Several interesting questions arose from these initial observations regarding the mechanism of action of complement and the extent to which TLR-complement interactions occurred. Subsequent experiments were therefore designed to address these questions. By using receptor antagonists and gene knockout mice, it was established that the regulatory effect of complement on TLR4-induced cytokine production in LPS-treated DAF–/– mice or LPS/CVF cotreated WT mice was mediated mainly by the C5a receptor (C5aR) and, to a lesser extent, the C3a receptor (C3aR) (Zhang et al. 2007). Further analysis also revealed that the hyper-LPS response phenotype of DAF–/– mice was correlated with increased NF-kB and mitogen-activated protein kinase activation in the spleens of LPS-treated mice. It was notable that while complement enhanced LPS-induced IL-6, TNF-α, and IL-1β production, it inhibited IL-12 production in the same experiments (Zhang et al. 2007). The finding that complement suppressed TLR4-mediated IL-12 production both in DAF–/– and WT mice was consistent with previous works showing a negative regulation by C5a on IL-12 and IL-23 biosynthesis in murine peritoneal macrophages (Hawlisch et al. 2005). These results together implied a differential regulatory mechanism by complement on IL-12 than those operative in the regulation of the proinflammatory cytokines such as IL-6 and TNF-α. It was demonstrated that the inhibitory effect of complement on IL-12 production was at least partially mediated by IL-10. As it turned out, in addition to IL-6, TNF-α, and IL-1β, complement also synergized with LPS to enhance IL-10 production both in vivo and in isolated peritoneal macrophages (Zhang et al. 2007). Complement-mediated inhibition of IL-12 biosynthesis in vivo was attenuated in IL-10 knockout mice, and antibody blocking of IL-10 in cultured peritoneal macrophages also blunted the inhibitory effect of C5a on LPS-induced IL-12 production (Zhang et al. 2007). In other studies using pharmacological approaches, it was found that the inhibitory effect of C5a on IL-12 biosynthesis in peritoneal macrophages was mediated by a phosphoinositol-3 kinase (PI3K)-dependent mechanism (Hawlisch et al. 2005). However, the involvement of PI3K in this process was questioned by a subsequent study showing that the inhibitory effect of C5a on IL-12 production in murine peritoneal macrophages was still observed in PI3K knockout mice (Okazaki et al. 2011). It is possible that redundant mechanisms exist, and in the absence of PI3K, other pathways may have compensated for the lack of PI3K-mediated signaling events.
Another question arising from the observed regulatory effect of complement on TLR4 signaling is whether complement also exerts a similar effect on the signaling pathways of other TLRs. We addressed this issue by testing the sensitivity of DAF–/– mice to several TLR ligands, including zymosan (for TLR2/6) and CpG (for TLR9), and by evaluating the sensitivity of WT mice to coincidental stimulation of the concerned TLR ligands and CVF. These experiments showed that complement could also impact the signaling of other TLRs, but the effect on specific cytokine production may not be identical to that observed in the case of TLR4 activation (Zhang et al. 2007). Thus, while coincidental complement activation augmented IL-6 and suppressed IL-12 production induced by zymosan, it similarly suppressed IL-12 production but had no effect on IL-6 biosynthesis after mice were challenged with CpG (Zhang et al. 2007). Furthermore, in contrast to what has been observed in the regulation of TLR4 signaling by complement where C5aR seemed to play a major role, the suppressive effect of complement on CpG-triggered IL-12 production appeared to be mediated mainly by C3aR rather than by C5aR (Zhang et al. 2007).
Interaction between complement and TLRs can occur both ways. While, as described previously, the complement anaphylatoxins augmented TLR-induced proinflammatory cytokine production, other studies have demonstrated that TLR activation could also increase complement protein synthesis and effector function. For example, in the RAW264.7 murine macrophage cell line, LPS treatment dramatically increased complement factor B gene transcription and protein synthesis (Kaczorowski et al. 2010). A similar effect of LPS on factor B production could be observed in thioglycollate-elicited mouse peritoneal macrophages. This effect by LPS was TLR4-dependent and was mediated by TIR-domain-containing adapter-inducing interferon-β (TRIF), Jun N-terminal kinase (JNK), and NF-kB (Kaczorowski et al. 2010). A screen of other TLR ligands showed that polyI:C, a double-strand RNA analog, and a ligand for TLR3 and cytosolic double-stranded RNA receptors also stimulated the up-regulation of factor B. The effect of polyI:C appeared to be mediated by the cytosolic receptors rather than by TLR3 but was also dependent on JNK and NF-kB (Kaczorowski et al. 2010). In another recent study, an example was provided of a marked positive modulatory effect of TLR activation on cell sensitivity to C5a in vitro and in vivo. In this model, pre-exposure of peripheral blood mononuclear cells and whole blood to various TLR ligands or bacteria enhanced C5a-mediated pro-inflammatory responses (Raby et al. 2011). This effect was dependent on TLR4 but did not involve up-regulation of C5aR. Instead, TLR activation was found to reduce the activity of C5L2, a second C5a receptor that in some studies has been described as a decoy receptor to negatively modulate C5aR function (Raby et al. 2011). The TLR-induced hypersensitivity to C5a was mimicked by C5L2 blockade and was abolished in C5L2 knockout mice. Furthermore, TLR-dependent suppression of C5L2 activity could be traced to reduced C5L2 expression (Raby et al. 2011).
Because cytokines produced by antigen-presenting cells in response to TLR activation play critical roles in T cell differentiation, we investigated whether the synergistic interaction between complement and TLRs might also have consequences for T cell immune responses. Specifically, the inflammatory cytokine IL-6 has been shown to be a key driving force for Th-17 cell polarization. Using an in vitro mouse T cell activation system stimulated by plate-bound anti-CD3 and soluble anti-CD28 antibodies, we showed that T cells activated and cultured in the presence of 5% serum from mice that had been cotreated with LPS and CVF or C5a contained a higher percentage of Th-17 effector cells than T cells activated in the presence of sera from mice treated with LPS, CVF, or C5a alone (Fang et al. 2009). This effect was critically dependent on IL-6 in the mouse serum as addition of an IL-6-neutralizing antibody prevented the differentiation of Th-17 cells (Fang et al. 2009). Furthermore, serum from C3–/– or C5aR–/– mice cotreated with LPS and C5a did not show enhanced activity in polarizing Th-17 cells, confirming that a higher serum IL-6 level, produced by TLR4 and complement interaction in a C5aR-dependent manner, was responsible for the observed effect on Th-17 differentiation (Fang et al. 2009). Collectively, these results demonstrated that complement and TLR interaction may not only enhance the host innate immune response but also their adaptive immunity to pathogen infection. On the other hand, the interaction between the two innate immune components may also be exploited by certain pathogens to subvert the host immune attack as has been detailed in a recent study (Wang et al. 2010). In this example, the periodontal pathogen Porphyromonas gingivalis produced a protease with C5 convertase activity that generated C5a without forming the harmful membrane attack complex (Wang et al. 2010). The C5a, through ligation with its congate receptor C5aR on macrophages, then synergized with the TLR2 pathway to increase cAMP production, which in turn inhibited nitric oxide release, thus promoting the survival of P. gingivalis in the host (Wang et al. 2010).
Concluding Remarks
In summary, complement and TLRs are two important components of innate immunity. Although each has been studied extensively as a separate entity, given the fact that many microbial ligands and signature molecules are capable of activating both, it is likely that pathogen infection will simultaneously trigger both pathways in vivo. In some situations, endogenous ligands derived from damaged or altered self may also cause coincidental activation of the two pathways. Crosstalk between the two systems to enhance the immediate immune reaction to pathogen infection and to better prime the host in mounting a more robust adaptive immune response has obvious advantages from an evolutionary point of view. In other conditions, however, synergistic amplification of the inflammation response may be detrimental. Understanding the mechanisms and intricacies of such interactions may help to devise novel therapeutic strategies to prevent or treat overwhelming and life-threatening inflammatory conditions such as septic shock and trauma-induced systemic inflammation syndrome. Such knowledge may also benefit the effort to prevent infectious diseases by developing more effective vaccines or by specifically targeting immune subversion tactics of certain pathogens.
Footnotes
Acknowledgment
I thank Dr. Takashi Miwa for help in manuscript preparation.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work in the author’s laboratory has been supported by NIH grants GM092108, AI44970, AI85596, and AI49344.