
Abstract
In simple terms, inflammation can be defined as a beneficial, nonspecific response of tissues to injury that generally leads to restoration of normal structure and function. In this concept, resolution of the inflammatory response, once it has achieved its protective and pro-immunogenic functions, becomes a critical determinant of what might be considered the paradox of inflammation. On one hand, inflammation is essential to resolve tissue injury and maintain homeostasis. On the other, inflammation is a key participant in the great majority of human diseases. Accordingly, to achieve complete resolution of inflammation, it is necessary to both turn off inflammatory mediator production and inflammatory cell accumulation and to remove inflammatory cells and debris without initiating an autoimmune response. Much of this process involves key activities of the mononuclear phagocyte series of cells, including resident and recruited macrophages. Recognition of activated and dying acute inflammatory cells by mononuclear phagocytes has been shown to (a) enhance macropinocytic activity for removal of debris, (b) enhance uptake of the effete inflammatory cells themselves, (c) induce inflammosuppressive and immunosuppressive mediators such as TGFβ and IL-10 that can down-regulate and limit proinflammatory mediator production, and (d) induce production of growth factors for tissue cells that may play key roles in tissue repair. Defects in these highly regulated processes are associated with persistent inflammation and/or autoimmunity in overaggressive resolution mechanisms such as nonresolving fibrosis or persistent tissue destruction as in emphysema.
Introduction
Resurging interest in inflammation, for example as participating in obesity, cardiovascular disease, or cancer, has led to a focus on the cytokines, chemokines, and other inflammatory mediators that orchestrate the process (Libby 2002; Nathan 2002; Tracey 2007; Hotamisligil 2006) to an extent that often downplays the critical role played by the cells that both induce and respond to these mediators. Thus, we argue that while a cytokine theory of disease is important to recognize, the balance between the cellular and humoral elements should always be kept in mind: an interesting recapitulation of similar arguments in the concepts of immunity that were prevalent a century ago (Mechnikov 1908). Likewise, the term regulation is all too often unclear since it could imply either an enhancing or inhibitory function or, as is often the case, a vague and uncharacterized double meaning. Here we will focus on the role of cells in suppressing the inflammatory response, leading to its normal resolution; as in the words of the eighteenth-century Scottish physician, William Cullen, “If an inflammation is to be cured while the state and texture of the part remain entire, the disease is said to be terminated by Resolution.” A rationale for this perspective is that an understanding of normal resolution in a protective and self-limited inflammatory response can provide insight into, and eventual therapeutic interventional opportunities toward, the persistent inflammatory processes that contribute to so many human diseases. A brief consideration of these processes, and especially their consequences, will therefore be the subject of this article. Rather than providing a comprehensive review (for which, see Serhan and Savill 2005; Barton 2008; Han and Ulevitch 2005; Luster, Alon, and von Andrian 2005), we will alternatively raise a few general points and, more importantly, identify areas needing more intensive investigation. Additionally, given our own areas of interest, the focus will be on processes and diseases of the respiratory tract. However, we suggest that while there clearly will be important tissue-specific elements to inflammation and its resolution, the concepts discussed herein will have broad applicability to resolution of inflammation of other organ systems and sites.
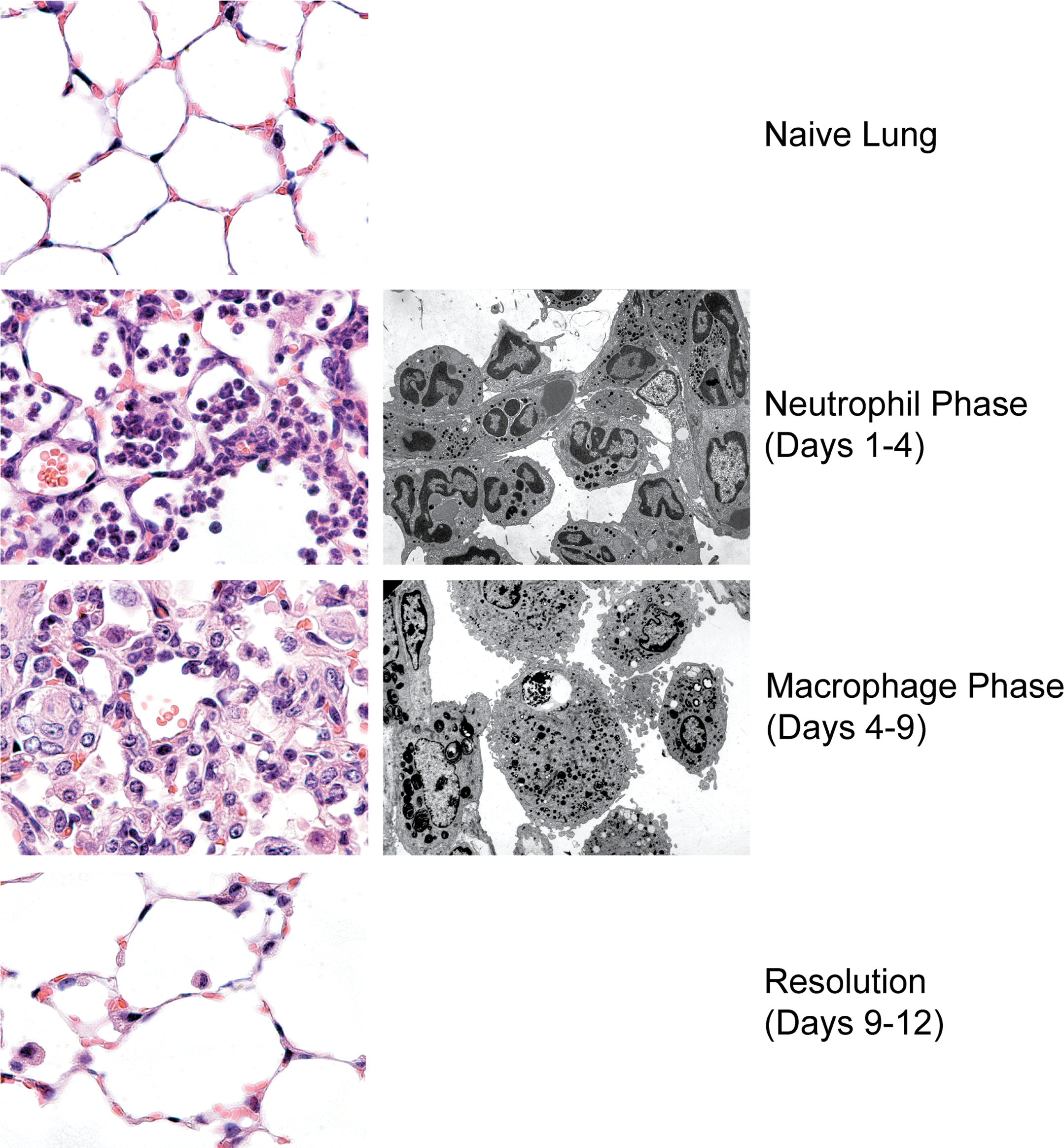
Sequence of histological changes during an acute, self-limited inflammatory process. The left panels depict the murine lung parenchyma prior to injury (top), during the neutrophilic and macrophage phases (middle panels), and during resolution (bottom) following inflammation induced with bacterial lipopolysaccharide (20μg). The right panels depict equivalent electron micrographs of the neutrophil and macrophage stages.
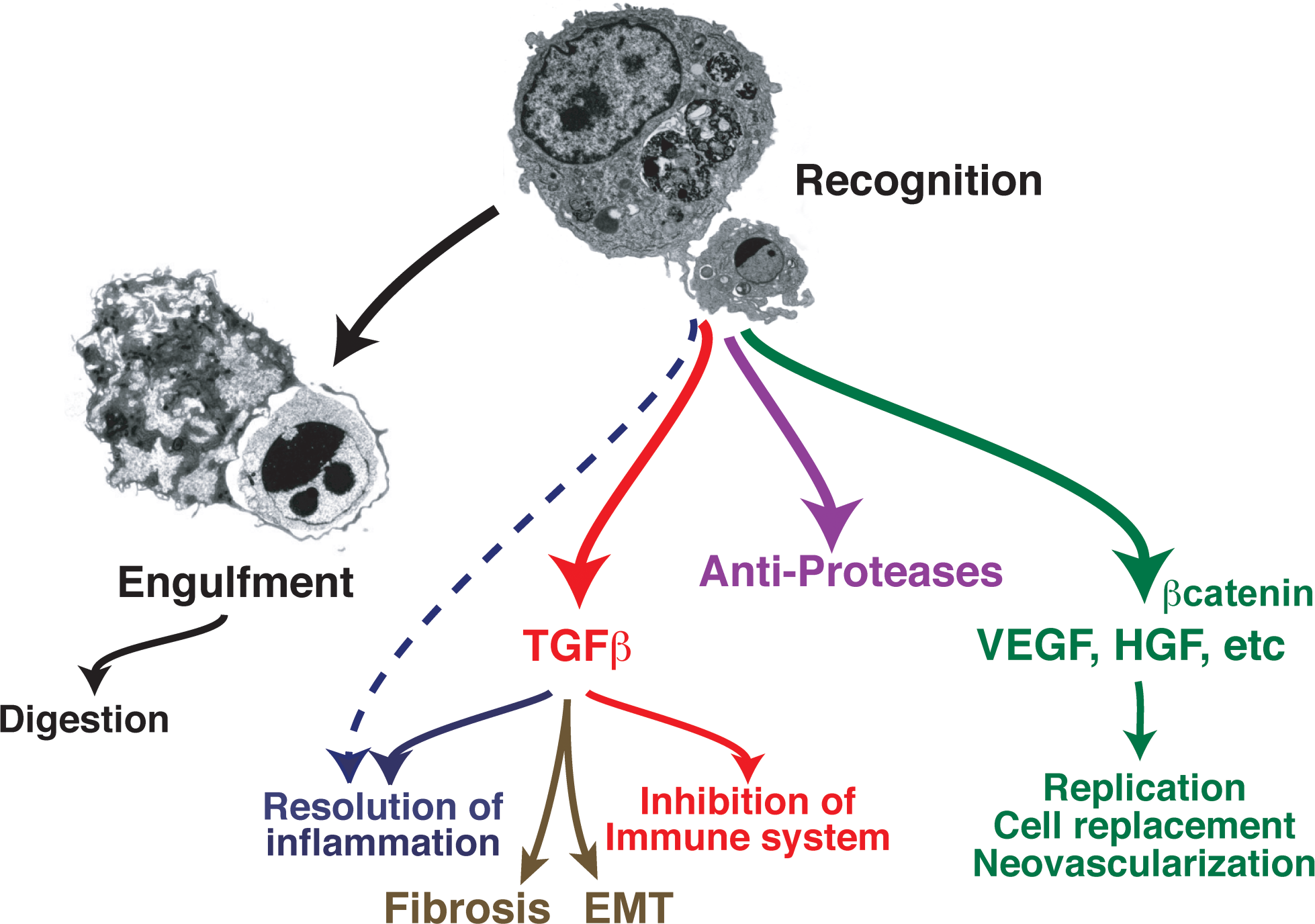
Responses of macrophages to apoptotic cells that may participate in resolution of inflammation.
Resolution of Inflammation
As a response of the naïve tissue to insult, pro-inflammatory mediators are produced either from endogenous leukocytes (macrophages, monocytes, dendritic cells, or lymphocytes) and/or from the tissue cells themselves. These mediators are the key orchestrators of the inflammatory response, initiating recruitment of neutrophils and later monocytes and lymphocytes to areas of injury and inducing the systemic responses well known to accompany classical inflammation (e.g., changes in blood flow and temperature). However, pro-inflammatory mediator production in the self-limited response is soon turned off, in part presumably due to cessation of the initial stimulus consequent on the protective functions of the inflammatory response itself—namely, removal of the invading pathogen (PAMPs), removal of the damaged tissue (DAMPs), and so on. In addition, a wave of anti-inflammatory mediators occurs later in the response. Removal of any tissue debris, and in particular of the recruited inflammatory leukocytes, accompanies this inflammosuppressive effect and is in fact one of the mechanisms for its induction.
Removal of Neutrophils as a Paradigm for Clearance of Inflammatory Cells
In an acute, self-limited inflammatory reaction (Figure 1), recruited neutrophils are quickly cleared from the lesion as they undergo programmed cell death (PCD), often generalized as “apoptosis” (though strictly speaking, apoptosis represents but a subset of a number of forms of PCD; Kroemer et al. 2009; Fink and Cookson 2005; Okada and Mak 2004). Recognition of these apoptotic cells by phagocytes leads not only to their engulfment and rapid digestion but also to a host of additional responses that are involved in the overall resolution (see the following). Uptake of apoptotic neutrophils in vivo is highly efficient, to the degree that detection of dying neutrophils in injured tissues is normally difficult (Scott et al. 2001; Matute-Bello et al. 1997), even during the clearance phase of acute neutrophilic inflammation, as, for example, in bacterial pneumonia (Dockrell et al. 2001, 2003). Accordingly, we have suggested that histologic detection of apoptotic neutrophils should trigger consideration of either a massive wave of apoptosis and/or a defect in the clearance mechanisms (see the following).
The primary cells involved in apoptotic cell clearance are macrophages, although dendritic cells are also important players and may thereby contribute to either immunosuppressive or autoimmune sequelae. In addition, it has also become apparent that tissue cells such as endothelial cells, fibroblasts, and epithelial cells can also have, or develop, the ability to recognize and ingest apoptotic cells (Aguilar et al. 1994; Monks et al. 2005; Petrovski et al. 2011; Suzuki, Takeda, and Farbman 1996; Svensson et al. 1999; Vandivier et al. 2009). The recognition and uptake mechanisms for apoptotic cells are highly evolutionarily conserved and differ from more classical types of phagocytosis (as mediated by antibody or complement) in both physical uptake mechanisms and intracellular signaling pathways. As such, we have given this unique form of phagocytosis the name efferocytosis (from effero; to carry to the grave; to bury; deCathelineau and Henson 2003).
Ligands on cells undergoing PCD and the receptors that recognize them are many—the processes appear to be highly redundant (see Table 1 and Gardai et al. 2006; Grimsley and Ravichandran 2003; Savill et al. 2002; Savill, Gregory, and Haslett 2003). Of the ligands expressed by apoptotic cells, phosphatidylserine (PS) carries particular importance. PS is normally confined to the inner cell membrane, but is rapidly exposed on the cell surface during early apoptosis. Many direct receptors for PS have been identified, as well as a number of soluble molecules that can tether PS to phagocyte surface receptors and act as “bridge molecules” or opsonins (Bratton and Henson 2008). Here for simplicity we will group these together as “PS recognition structures” or PSRS. Intriguingly, activated neutrophils, as well as other leukocytes, may also expose PS on their cell surfaces when they become activated, though in this case the exposure is transient rather than permanent as in apoptosis. Nonetheless, it has become apparent that such transient PS exposure may itself be enough to trigger some recognition and removal before the cells actually die (Marguet et al. 1999; Callahan, Williamson, and Schlegel 2000; Frasch et al. 2008).
Apoptotic cell ligands and their phagocytic receptors.

Another general ligand that becomes exposed and/or altered on apoptotic cells is calreticulin, a protein normally found in the endoplasmic reticulum. A critical receptor for calreticulin is LDL receptor related protein-1 (LRP-1). The collectin family of pattern recognition molecules (e.g., mannose binding lectin, surfactant proteins A and D, C1q, and other related proteins) also recognizes surface changes on apoptotic cells (Palaniyar et al. 2004; Palaniyar, Nadesalingam, and Reid 2003; Jensen et al. 2007) and can act through LRP-1.
Macropinocytosis and Removal of Debris
The process of efferocytosis appears to be a modified form of macropinocytosis that is dependent on the low molecular weight, Rho-family GTPase, Rac. Uptake of the apoptotic cell is accompanied by simultaneous ingestion of surrounding fluid (deCathelineau and Henson 2003; Bratton and Henson 2008), and in fact, the soluble “bridge molecules” mentioned previously can themselves enhance macropinocytosis (Hoffmann et al. 2001; Somersan and Bhardwaj). This then may contribute importantly to the clearance of debris, fluid phase constituents, and other leftovers from the inflammatory response. Intriguingly, some of the collectin family members, including the Ficolins, are able to recognize DNA and chromatin (Palaniyar et al. 2004; Palaniyar, Nadesalingam, and Reid 2003; Jensen et al. 2007), thereby providing a mechanism for removal of these classes of cell debris, including perhaps the neutrophil extracellular NETS that may have been generated in the course of the protective stages of the inflammatory process (see the article in this symposium by McDonald et al.).
What Happens to the Macrophages?
True resolution of inflammation requires subsequent removal of the macrophages. Mechanisms for this may be tissue specific, although in a fundamental sense the two main modes of macrophage removal include excretion from the body or apoptosis and their own subsequent efferocytosis. In the lung, some of the inflammatory macrophages may exit the body via the mucociliary escalator to be coughed up and swallowed. However, clear evidence of apoptosis and local removal exists (Janssen et al. 2011), and we suggest that this is the dominant fate of macrophages in the lungs, and perhaps at other inflammatory sites in the body as well. In the peritoneum, migration of macrophages to the local lymph nodes has been reported (Cao et al. 2005; Bellingan et al. 1996, 2002). Even in this case, the eventual fate of the macrophage is still likely apoptosis and ingestion since there is no evidence for necrotic death or excretion of macrophages from the blood stream or lymphatics into the lungs or GI tract for external disposal.
Inflammosuppressive and Immunosuppressive Consequences of Apoptotic Cell Recognition
The previous comments emphasize the importance of clearing cells and debris produced as inevitable consequences of the inflammatory response in order to allow injured tissues to recover and return to normal structure and function. However, inflammation itself also has to be halted for resolution to occur. In other words, following correction of the initiating event (i.e., removal of pathogens), the production of proinflammatory chemokines and cytokines needs to be turned off. The protective functions of inflammation may be assumed to contribute to the first of these steps by removing the stimulus, thereby decreasing its induction of pro-inflammatory mediators. However, it has also become apparent that recognition of apoptotic inflammatory cells by phagocytes not only results in apoptotic cell removal, but also actively induces the generation of anti-inflammatory (inflammosuppressive) mediators, such as TGFβ and IL-10, and a spectrum of lipid mediators with anti-inflammatory properties (Fadok et al. 1998; Freire-de-Lima et al. 2006). In other words, removal of apoptotic cells is generally actively inflammosuppressive, and in fact is also able to actively suppress the innate and adaptive immune response (Savill et al. 2002; Huynh, Fadok, and Henson 2002; Savill 1997). The pathways primarily responsible for these resolution-inducing effects appear to involve the recognition of phosphatidylserine on apoptotic cells, although the precise set of receptors and/or bridge molecules that mediate these effects is not entirely clear. Likely candidates include the TAM family of receptor tyrosine kinases, whose PS-recognizing bridge molecule ligands are GAS6 and Protein S (Scott et al. 2001; Ishimoto et al. 2000); however, it is likely that other receptors are also involved.
Many of the inflammosuppressive and immunosuppressive effects of efferocytosis appear to result from the aforementioned induction of anti-inflammatory mediators, such as TGFβ. However, a number of studies also suggest that efferocytosis turns off production of pro-inflammatory mediators, for example by blockade of the NFκB pathways (Amarilyo et al. 2010; Cvetanovic and Ucker 2004). Likely relevant to these processes is the treatment regimen termed extracorporeal photopheresis (ECP) that has been proposed as a therapeutic strategy for patients with chronic inflammatory diseases (Babic 2008; Chiesa-Fuxench and Gonzalea-Chavez 2010; Szodoray et al. 2010). In this method, leukocytes are removed from the patient, treated ex vivo with the light-sensitive dye psoralen and light, and then reinfused. One can reasonably infer that these cells undergo apoptosis, providing a source of PS-exposing cells, that when recognized and ingested by phagocytes induce immunosuppression. However, it is also important to note that a number of studies have also suggested pro-inflammatory effects deriving from the presence of apoptotic cells (as well as pro-immunogenic consequences of apoptotic cell ingestion). A number of explanations for this dichotomy include the presence on apoptotic cells of ligands that, unlike PS, are potentially pro-inflammatory (Peter et al. 2010). In this context, it is important to note that the processes involved represent a delicate balance between pro-inflammatory effects of dying cells, especially when they undergo secondary necrosis, versus the strong anti-inflammatory effect seen normally during resolution of acute inflammatory events. To no surprise (as noted in the following), defects in apoptotic cell recognition and removal and/or changes in this balance can contribute to persistent inflammation and autoimmunity.
Regenerative Consequences of Apoptotic Cell Recognition
Although less well studied for the signaling pathways involved, apoptotic cell recognition can also induce macrophages to synthesize and secrete growth-maintenance factors (e.g., VEGF and HGF; Golpon et al. 2004; Morimoto et al. 2001), which exert potent effects on parenchymal tissue cells such as endothelial cells and epithelial cells (see Fig. 2). This raises the general concept that cell removal can lead to restoration of tissue cells, to maintain the homeostatic balance. We believe that this may be a more extensive story than has yet been defined and is well worth further investigation.
As noted earlier, one of the anti-inflammatory and anti-immunogenic molecules induced by recognition of apoptotic cells is TGFβ. Since this multi-potent mediator also plays an important role in fibrotic processes, it too represents a two-edged sword in the broad question of inflammation resolution. An intriguing study of apoptotic cell contribution to lung injury and repair demonstrated that blockade of apoptosis early led to increased severity and death, whereas blockade later in the process led to decrease in fibroproliferative sequelae (Douglas et al. 2006). This supports the active role played by apoptosis (presumably of the inflammatory cells) in both resolution and potentially later in generation of fibrosis in circumstances when complete restoration of tissue structure cannot be achieved. The source of fibroblasts and myofibroblasts for such “abnormal” reparative process is not clear, but differentiation of fibroblasts and epithelial-mesenchymal transition (EMT) are both candidates and are both driven by TGFβ. It is also worth noting that fibroblast and myofibroblast accumulation/development are normal processes of wound healing in other tissues, such as the skin. Here this fibrotic response can, for the most part, resolve later to restore normal function, though in other tissues functional impairment is all too often the consequence.
Consequences of Defective or Inefficient Inflammatory Cell Removal
Not surprisingly for such an important and highly regulated set of processes, defective and/or imbalanced responses to apoptotic cells can lead to a variety of pathologic responses. As such, defects in apoptotic cell clearance are beginning to be recognized as participants in a multitude of diseases. Here we will mention two such examples.
Systemic defects in apoptotic clearance mechanisms have been widely demonstrated in autoimmune diseases, such as systemic lupus erythematosus (Ren et al. 2003; Gaipl et al. 2005, 2007; Baumann et al. 2002; Herrmann et al. 1998) where increased numbers of apoptotic cells can be detected in the circulation (Ren et al. 2003; Courtney et al. 1999). Similar findings have been reported in mice prone to autoimmunity (Scott et al. 2001; Hanayama et al. 2004; Kim et al. 2003; Ling, Pi, and Holoshitz 2007; Botto et al. 1998; Heidari et al. 2006). These observations have led to the concept that secondary necrosis of dying cells may release intracellular constituents to the immune system as a critical source of autoantigens. Clearly, this would only be a small part of the overall story of autoimmunity and there are many other changes in the broader aspects of the immune system that also participate in the pathogenesis. However, in the light of increasing evidence of apoptotic cell recognition leading to elements of immunosuppression, one might speculate that defects in apoptotic cell clearance may contribute more to the autoimmune process than merely a supply of potential antigen.
Chronic obstructive pulmonary disease (COPD), which is characterized by severe changes to the airways (chronic bronchitis) and permanent loss of alveoli (emphysema), is another example of a disease that may be associated with defective recognition and removal of apoptotic and damaged cells. Once again, the starting point for such a concept was the detection of apoptotic cells within the parenchyma of the lungs from COPD patients (Kasahara et al. 2001). As noted earlier, such detection immediately raises the likelihood of significant ongoing cell death and especially, given the normal efficiency with which such cells are removed, of an accompanying defect of the clearance processes. Such a defect in apoptotic cell removal has been shown (Hodge et al. 2003, 2006, 2005, 2007; Morimoto, Janssen, Fessler, McPhillips, et al. 2006; Morimoto, Janssen, Fesser, Xiao, et al. 2006; Richens et al. 2009). The major etiologic agent for COPD is cigarette smoke, which both injures the lungs and induces persistent inflammation. However, only a proportion of smokers in fact develop the disease. This leads to the suspicion that one contributory element to disease development might indeed be an underlying propensity for inefficient apoptotic cell clearance, a concept that is supported by finding that blood monocytes matured into macrophages in vitro still show a defect in apoptotic cell uptake if taken from patients with COPD (R. W. Vandivier, unpublished data). Intriguingly, both inflammation and the presence of apoptotic cells have been shown to persist years after the patient has stopped smoking, leading investigators to propose a secondary and self-sustaining process in the lungs, probably autoimmune in nature (Lee et al. 2007; Aoshiba and Nagai 2009; Curtis, Freeman, and Hogg 2007; Taraseviciene-Stewart et al. 2006). Once again, the defective apoptotic cell recognition and uptake can be shown in patients after smoking cessation (Hodge et al. 2005), supporting the presence of an intrinsic defect and a possible link to autoimmunity.
Since the capacity for apoptotic cell uptake by phagocytes is programmable, deficiencies in this capacity may be induced by the local environment or may arise from intrinsic genetic or epigenetic factors. Studies of genetic polymorphisms in phagocytic receptors or uptake mechanisms are in their infancy but provide fertile areas for investigation. In addition, it has become apparent that a variety of stimuli can exogenously reduce macrophage efferocytic capacity. Thus, macrophages “programmed” toward a more inflammatory state (in murine systems sometimes termed M1 polarization) are less efficient at efferocytosis compared with cells programmed toward a reparative state, the latter showing up-regulation of receptors associated with apoptotic cell recognition (Gordon and Martinez 2010; Varin et al. 2010). In addition, stimulation of the phagocytes to increase the ratio of Rho:Rac activation (Rho is inhibitory) can induce immediate suppression of apoptotic cell uptake (Erwig et al. 1999; Tosello-Trampont, Nakada-Tsukui, and Ravichandran 2003), and if the imbalance is more persistent, could contribute to some of the effects seen, for example in COPD or other chronic inflammatory states. This latter effect may in some circumstances be mitigated by inhibitors of Rho kinase as a key downstream mediator of the Rho effect (Morimoto, Janssen, Fessler, McPhillips, et al. 2006; Richens et al. 2009; Janssen et al. 2008). This has led to suggestions for using statins (HMG-CoA reductase inhibitors) for enhancing apoptotic cell clearance via their effect on Rho activation through preventing its prenylation (Morimoto, Janssen, Fessler, McPhillips, et al. 2006; Morimoto, Janssen, Fessler, Xiao, et al. 2006; Gordon and Martinez 2010; Varin et al. 2010; Vandivier, Henson, and Douglas 2006).
Conclusions
This discussion has focused on roles played by macrophages in resolution of acute inflammatory responses, in particular the removal of the inflammatory cells and debris and promotion of various forms of repair. However, it is important to note that structural cells of the tissues also play key roles in the resolution of inflammation and exhibit similar properties (up to and including ingestion of apoptotic cells) that were here described for macrophages. In addition, for reasons of limited cell numbers, while dendritic cells probably do not contribute to the bulk of clean-up in inflammation, their participation in the immunologic sequelae are of substantial importance. The arena for discussion of these concepts has been the lung, but while this is certainly a unique organ that has direct exposure to the outside environment, we suggest that the essential elements of macrophage participation in resolution of inflammation are equally applicable to other tissues and organs. Most importantly we propose that increasing our knowledge of normal resolution processes will have a direct future impact on our ability to deliberately enhance the resolution of chronic inflammation and thereby mediate an earlier return to normal tissue structure and function.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by HL68864, HL88138, and HL81151.