
Abstract
The liver is a target of many inflammatory pathologies of both infectious and noninfectious etiology. As key effectors of the innate immune system, neutrophils are critical for defense against microbial infections but are often the source of profound collateral damage to host tissues during disease states. In this article based on the authors’ presentation at the 2011 Society of Toxicologic Pathology Annual Symposium, they review the molecular mechanisms of neutrophil recruitment to the liver in response to sepsis/endotoxemia, as well as sterile inflammation, and discuss variations in the molecular choreography of neutrophil trafficking in response to these different insults. Furthermore, the authors discuss the functional contributions of neutrophils within the liver microvasculature during severe sepsis, including their contributions to both host defense and organ damage. Given that inappropriate neutrophilic inflammation contributes to the pathogenesis of many liver diseases, a thorough understanding of the molecular mechanisms that regulate the recruitment of neutrophils to the liver, and their functions therein, may reveal new avenues for therapeutic interventions to treat inflammatory liver pathologies.
Introduction
Neutrophils are rapidly recruited to sites of acute inflammation, where they contribute to host defense and tissue healing (Nathan 2006). The essential role of neutrophils in host defense is dramatically illustrated by the profound susceptibility to bacterial and fungal infections resulting from neutropenia or defects in neutrophil trafficking (Lekstrom-Himes and Gallin 2000). These large granular polymorphonuclear leukocytes are uniquely endowed with a high phagocytic capacity as well as a vast arsenal of rapidly synthesized and/or preformed antimicrobial molecules, making them key effectors of innate immune defenses (Segal 2005). At the same time, the potent arsenal of neutrophil weapons functions with poor target specificity and can cause profound damage to host cells and tissues. As such, overexuberant neutrophil recruitment and indiscriminant release of toxic mediators contribute fundamentally to the pathogenesis of many diseases. In particular, many acute and chronic pathologies of the liver are mediated and/or exacerbated by neutrophilic inflammation, such as sepsis and endotoxemia (Jaeschke et al. 1996; Molnar et al. 1997) and ischemia-reperfusion injury (Jaeschke and Farhood 1991; Jaeschke, Farhood, and Smith 1990; Martinez-Mier et al. 2001), as well as alcoholic (Bautista 1997), viral (Takai et al. 2005), autoimmune (Bonder et al. 2004), and toxin- or drug-induced hepatitis (Liu et al. 2006; Ohta et al. 2006). Despite the central role of neutrophils in a variety of liver diseases, the molecular mechanisms that allow these cells to home to the liver, and their functions therein, are not well understood. Given the growing body of clinical evidence supporting the use of antiadhesion molecule therapies (and other modulators of leukocyte recruitment) to treat a variety of inflammatory diseases (Mackay 2008), identifying and characterizing the molecular mechanisms of neutrophil recruitment within the liver may reveal novel therapeutic strategies to combat inflammatory liver pathologies.
Recruitment of neutrophils into inflamed tissues involves a series of molecularly distinct adhesive interactions with the vascular endothelium. The “classical paradigm” of leukocyte recruitment is described in most reviews and textbooks as a multistep cascade of events, involving initial tethering and rolling along the vessel wall (classically in postcapillary venules), followed by firm adhesion and arrest, culminating in transendothelial migration into tissues (Figure 1) (Ley et al. 2007). This classical paradigm of leukocyte recruitment was largely elucidated using simplified in vitro flow chamber experiments in which isolated leukocytes are perfused over a cytokine-activated endothelial monolayer (usually derived from a macro- rather than microvascular source, namely, human umbilical vein) (Smith et al. 1991). Subsequently, these classical recruitment mechanisms were confirmed in vivo using intravital microscopy of thin tissues that were amenable to transillumination (typically, the cremaster muscle, mesentery, or skin) (Ley et al. 1991; von Andrian et al. 1991). These experimental systems also enabled researchers to identify and characterize the cell adhesion molecules that mediate leukocyte-endothelial interactions within inflamed postcapillary venules. Tethering and rolling require adhesion molecules called selectins (primarily E- and P-selectins) that engage in transient low-affinity interactions with the selectin-ligands expressed on leukocytes. Rolling leukocytes then encounter chemokines expressed on the lumenal surface of vascular endothelium, which transmit signals via G-protein coupled receptors (so called “inside-out” signaling) to activate leukocyte integrins (β2 and α4 integrins), leading to firm adhesion (Ley et al. 2007). More recently, mechanistic details of postadhesion events in leukocyte recruitment have begun to emerge, including the observation that adherent cells actively crawl across the endothelial surface in search of optimal sites for transmigration into tissues (Phillipson et al. 2006). Initially, researchers believed that this canonical pathway of cell trafficking controlled leukocyte recruitment throughout the body. However, advances in in vivo imaging techniques now enable the visualization of microvascular beds of internal organs, including the liver, and have revealed that profound variations exist in the mechanisms of leukocyte recruitment in different anatomical sites (Kreisel et al. 2010; Kuligowski, Kitching, and Hickey 2006; Mizgerd et al. 1997; Mizgerd et al. 1996; Petri, Phillipson, and Kubes 2008).
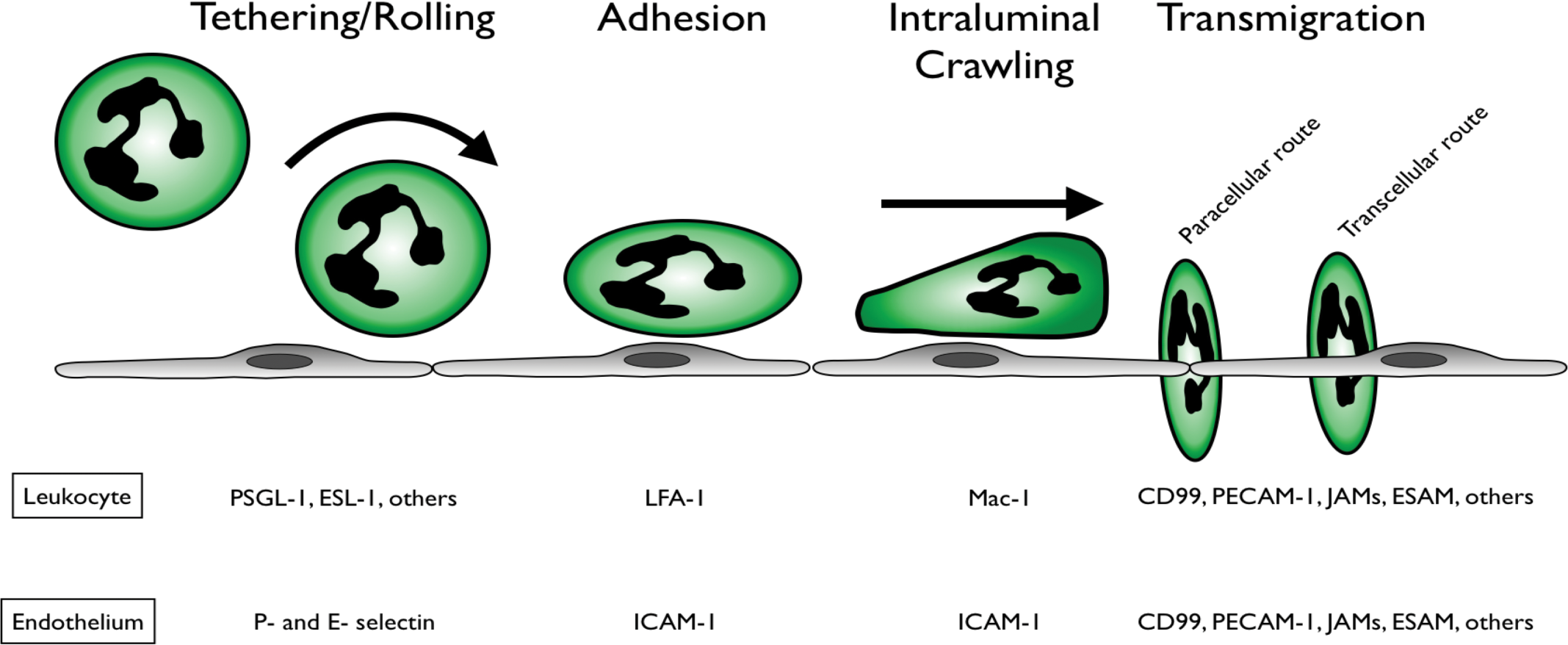
Classical neutrophil recruitment cascade. Within inflamed postcapillary venules, tethering and rolling are mediated by engagement of endothelial selectins with neutrophil selectin-ligands. Stimulation of chemokine receptors on rolling cells transmits “inside-out” signals that activate integrins (LFA-1), resulting in firm adhesion. Intraluminal crawling is supported by neutrophil integrin Mac-1 and is guided by chemokine gradients decorating the surface of the vascular endothelium. Neutrophils exit the venule by transendothelial migration between adjacent endothelial cells (paracellular) or directly through endothelial cells (transcellular).
Studies of hepatic inflammation have revealed that leukocyte trafficking in the liver differs from the aforementioned classical paradigm in three fundamental ways: (1) unlike the postcapillary venules of many tissues, the vast majority of infiltrating leukocytes (>90%) adhere within the capillary-like sinusoids of the liver; (2) a rolling step is not observed within sinusoids, as leukocytes simply tether and immediately adhere; and (3) the adhesion of leukocytes within sinusoids is completely independent of selectins (Lee and Kubes 2008; Wong et al. 1997). In this article based on our presentation at the 2011 Society of Toxicologic Pathology Annual Symposium, we focus on recent studies that have begun to define the molecular choreography of neutrophil recruitment in liver sinusoids and some of the functional roles of neutrophils within the vasculature of the liver during septic as well as sterile inflammation.
Sepsis syndrome, neutrophils, and the liver microvasculature
Sepsis is a syndrome of systemic inflammation in response to infection (Bone, Balk, and Cerra et al., 1992). In its severe form, the inflammatory pathology of sepsis causes shock, multiorgan dysfunction, and death in 20–50% of patients (Dombrovskiy et al., 2007). Equally alarming is the fact that only a single targeted therapy (recombinant activated protein C [rAPC]) is in routine use for the treatment of severe sepsis, while the mainstay of management remains antibiotics and supportive care (Dellinger et al., 2008). The unacceptably high mortality rate and lack of effective therapies can be traced to an incomplete understanding of the inflammatory pathogenesis of sepsis and organ dysfunction. As such, research into the molecular and cellular pathogenesis of sepsis is needed to better understand this complex syndrome and to identify effective therapies.
The innate immune system and neutrophils in particular play a central role in the inflammatory pathology and multiple organ dysfunction of severe sepsis (Brown et al. 2006). A consistent finding in both animal models of sepsis and hyperinflammatory sepsis in humans is that, regardless of the initial site of infection, activated neutrophils become sequestered in the expansive capillary networks of internal organs, such as the liver and lungs, where neutrophil-mediated damage and organ dysfunction ensue (Andonegui et al. 2009; Brown et al. 2006; Ye et al. 2008). Within the septic liver, infiltration by neutrophils contributes to significant hepatocellular damage, vascular hypoperfusion, and ultimately, organ dysfunction (Dhainaut et al. 2001). While fulminate liver failure is a relatively rare complication of sepsis, inflammatory liver damage and hepatic dysfunction are very common in severe sepsis and represent underappreciated contributors to disease progression and mortality (Dhainaut et al. 2001; Kortgen et al. 2009; Martin et al. 2003). In addition, the liver impacts other organs as a source of many acute-phase proteins and other inflammatory mediators. Therefore, understanding the molecular mechanisms of neutrophil recruitment within the liver may reveal new therapeutic strategies to prevent immune-mediated organ dysfunction during severe sepsis.
Studies aiming to identify the molecular mechanism (or mechanisms) of adhesion within liver sinusoids during sepsis/endotoxemia have consistently concluded that classical adhesion molecules are not involved. Initially, selectins, β2 integrins, and α4 integrins were interrogated for a role in neutrophil recruitment to the endotoxemic liver. Using antibody blockade as well as selectin-deficient mice, Wong et al. (1997) demonstrated that neutrophil sequestration in sinusoids proceeded unabated in the absence of functional E-, P-, or L-selectins. Next, β2 integrins (CD18) were investigated based on their well-documented role in firm adhesion within other organs. In addition, it had previously been demonstrated that neutrophils could utilize β2 integrins to mediate all steps in the recruitment cascade (tethering, rolling, and adhesion) in the splanchnic circulation when shear rate was reduced by 50% (similar to the shear encountered in the slow-flowing sinusoids) (Gaboury and Kubes 1994). Nevertheless, numerous studies have observed that neutrophil recruitment to the septic/endotoxemic liver is unaffected by blockade or genetic deficiency of β2 integrins or their endothelial ligand intercellular adhesion molecule 1 (ICAM-1) (Jaeschke et al. 1996; McDonald et al. 2008; Menezes et al. 2009; Wong et al. 1997). Finally, while α4 integrin/vascular CAM 1 (VCAM-1) interactions are not generally thought to support neutrophil recruitment to inflamed tissues, neutrophils from patients with severe sepsis have up-regulated expression of α4 integrin and can adhere to VCAM-1 in vitro (Ibbotson et al. 2001). However, like the β2 integrins, studies using neutralizing antibodies have found that α4 integrins are dispensable for neutrophil adhesion within the liver sinusoids (Fox-Robichaud and Kubes 2000; McDonald et al. 2008).
These observations led many researchers to adopt the default hypothesis that activated neutrophils simply become physically trapped in the narrow lumens of inflamed liver sinusoids (and pulmonary capillaries) as a consequence of the “cytokine storm” in acute sepsis (Bonder et al. 2004; Fox-Robichaud and Kubes 2000; Jaeschke et al. 1996). This hypothesis was supported by in vitro evidence that neutrophils activated by bacterial lipopolysaccharide (LPS) or N-formylated peptides became rigid (less deformable) as a result of cytoskeletal reorganization, resulting in an inability to pass through 5 µm pores that were easily traversed by resting neutrophils (Worthen et al. 1989). However, the physical trapping hypothesis was initially challenged by observations from intravital microscopy studies that showed sinusoids remaining patent and perfused despite the presence of many adherent neutrophils, implying that the cells were not mechanically wedged in the vessel lumen (Fox-Robichaud and Kubes, 2000; Wong et al. 1997). In addition, a number of studies demonstrated that specific adhesion molecules mediate the recruitment of other leukocytes within inflamed sinusoids in mouse models of hepatitis—α4 integrin (TH1), VAP-1 (TH2), and β2 integrins (CD8+) (Bonder et al. 2005; John and Crispe 2004). More recently, the physical trapping hypothesis has been directly challenged by studies that have discovered active molecular mechanisms of neutrophil and monocyte adhesion in the inflamed liver during sepsis/endotoxemia (Bonder et al. 2005; John and Crispe 2004; McDonald et al. 2008; Menezes et al. 2009; Shi et al. 2010).
Given that classical adhesion molecules were dispensable for sinusoidal neutrophil sequestration, we conducted a screening assay for surface expression levels of various endothelial adhesion molecules in vivo. Unexpectedly, we observed that the nonclassical adhesion molecule hyaluronan (HA) was expressed at disproportionately high levels on the luminal surface of the liver sinusoidal endothelium (McDonald et al. 2008). Using a model of endotoxemia, it was found that removing HA from the sinusoidal endothelium or blocking its interaction with CD44 (the principle receptor for HA) dramatically reduced neutrophil recruitment within inflamed liver sinusoids (McDonald et al. 2008). Subsequently, it was found that CD44-HA interactions also mediate neutrophil recruitment to the liver during Escherichia coli sepsis, as well as monocyte recruitment to foci of hepatic Listeria monocytogenes infection (Menezes et al. 2009; Shi et al. 2010). From these studies, it has become clear that during sepsis, neutrophil sequestration within the liver microvasculature is not a passive event but instead is an actively coordinated event mediated by a unique molecular adhesion mechanism within liver sinusoids.
Neutrophils and Intravascular Immunity in the Liver During Sepsis
The reasons for this seemingly inappropriate and injurious sequestration of neutrophils in the liver (and lungs) during sepsis are incompletely understood. It has been hypothesized that this represents a clever immune evasion strategy by bacteria to distract neutrophils away from the focus of infection by misdirecting them to the capillary networks of the liver and lungs. Indeed, simplified models of sepsis such as endotoxemia induced by injection of bacterial LPS cause neutrophils to accumulate in the liver sinusoids and pulmonary capillaries, indicating that the stimulus for sequestration in these organs is indeed provided by the pathogen. Furthermore, signaling via Toll-like receptor 4 (TLR4, the receptor for LPS) is responsible for this observed misdirection of neutrophils during endotoxemia and gram-negative sepsis (Andonegui et al. 2009). Even in models of polymicrobial sepsis where multiple TLR- and non-TLR ligands are released, signaling via TLRs remains the dominant pathway causing neutrophil sequestration within the lungs and liver, as well as inhibiting their ability to home to peripheral sites of infection through a phenomenon termed “neutrophil paralysis” (Alves-Filho et al. 2006; Alves-Filho et al. 2009).
However, the demonstration that neutrophil recruitment to septic liver sinusoids is an actively orchestrated event rather than passive physical trapping suggests the possibility that neutrophils are intentionally directed to the liver as part of a coordinated host defense strategy. Indeed, the liver is a primary site for clearance of bacteria and bacterial products from the blood and is uniquely endowed with a vast population of resident intravascular macrophages (Kupffer cells) that efficiently capture and kill bacteria that enter the hepatic circulation (Benacerraf et al. 1959; Crispe 2009). However, a number of pathogens including E. coli, Klebsiella pneumoniae, Salmonella typhimurium, and trypanosome parasites can be cleared effectively after intravenous inoculation in animals that lack Kupffer cells, indicating that additional cell types contribute to bacterial eradication in the liver (Gregory and Wing 2002). Specifically, a number of studies have suggested that neutrophils contribute to the clearance of circulating pathogens within the liver (Clark et al. 2007; Gregory, Sagnimeni, and Wing 1996; Gregory and Wing 2002). We hypothesize that sequestration of neutrophils within liver sinusoids enhances trapping of bacteria from the bloodstream during sepsis, limiting the spread of infection to distant organs, yet this concerted attempt to defend the host occurs at the expense of damage to the liver.
Clearance of bacteria from the bloodstream is a challenging task for the immune system, especially during severe sepsis when the number of bacteria in the bloodstream may outnumber phagocytes in the body. It stands to reason that the phagocytic capacity of neutrophils and macrophages may easily be overwhelmed in the setting of septic bacteremia. Interestingly, Brinkmann and colleagues recently discovered a novel neutrophil effector mechanism, termed “neutrophil extracellular traps” (NETs), that enables these cells to capture and kill microbes in a manner that is much more efficient than pursuing and engulfing individual bacteria. NETs are webs of decondensed chromatin decorated with antimicrobial granule proteins that are expelled from the nucleus of activated neutrophils (Brinkmann et al. 2004). In vitro, NET release by neutrophils can be stimulated by a wide variety of microorganisms and/or inflammatory mediators through both cell death–dependent and –independent pathways and have been shown to effectively trap and kill gram-positive and gram-negative bacteria as well as pathogenic fungi and parasites in vitro (Papayannopoulos and Zychlinsky 2009; Pilsczek et al. 2010). More recently, studies investigating the functions of NETs in vivo have revealed that NETs are critical for containing bacterial infections in models of necrotizing fasciitis and bacterial pneumonia (Beiter et al. 2006; Buchanan et al. 2006; Li et al. 2010).
A putative role for NETs in sepsis was first reported by our laboratory with the demonstration that a novel collaboration between neutrophils and activated platelets (stimulated with LPS or serum from septic patients) caused NETs release in vitro (Clark et al. 2007). We hypothesize that, in vivo, neutrophils recruited to the sinusoids of the liver during sepsis may be similarly activated to cast NETs into the bloodstream and capture circulating bacteria. Using a simplified model of bacterial trapping (endotoxemia followed by intravenous infusion of E. coli), we observed that neutrophil accumulation in liver sinusoids enhanced hepatic clearance of circulating E. coli and that this bacterial trapping is likely dependent on the release of NETs within these vessels (Clark et al. 2007). However, further studies are needed to clearly demonstrate the presence of intravascular NETs within liver sinusoids, as well as a definitive role for NETs in the defense against bacterial dissemination during sepsis. Preliminary observations from our laboratory using spinning-disk confocal intravital microscopy of the liver have revealed the presence of intravascular NETs in sinusoids during endotoxemia and gram-negative sepsis, as well as a role for these structures in controlling severe bacterial infections by preventing the spread of bacteria to distant organs (McDonald and Kubes, unpublished observations).
In addition to their putative antibacterial functions, NETs released within the liver sinusoids may also contribute to hepatic damage and vascular dysfunction. NETs are covered with potent antimicrobial substances (proteases and cationic proteins) that may also be directly toxic to host cells (reviewed by Papayannopoulos and Zychlinsky 2009). Interestingly, a recent report by Esmon and colleagues revealed that histones, a major component of NETs, are primary mediators of mortality in sepsis due to their cytotoxicity against host cells (Xu et al. 2009). Immunoneutralization of histones protected against organ dysfunction and mortality in polymicrobial sepsis in mice (Xu et al. 2009). Furthermore, Esmon and colleagues demonstrated that rAPC cleaves histones and neutralizes their cytotoxicity, revealing a potential link between histones/NETs and the mechanism of action of the only disease-modifying therapy for severe sepsis (rAPC) (Xu et al. 2009). In addition, NETs may contribute to hepatic vascular dysfunction by activating the coagulation pathway and thrombus formation in liver sinusoids. Neutrophil serine proteases and histones (both major components of NETs) have recently been shown to initiate microvascular coagulation in vivo, possibly contributing to the development of disseminated intravascular coagulation, a feared complication of severe sepsis (Massberg et al. 2010). Therefore, the generation of intravascular NETs during sepsis is likely a double-edged sword, contributing to antimicrobial defenses at the expense of damage to host organs.
Neutrophil recruitment to sterile inflammation in the liver
In addition to infectious insults, sterile tissue injury and cell necrosis can induce profound neutrophilic inflammation. In this setting, neutrophils are not functioning as antimicrobial effectors but instead are charged with the task of clearing debris and initiating the wound-healing process (Nathan 2006). Indeed, neutrophils are emerging as central orchestrators of resolution and restitution following tissue injury and may even contribute to the avoidance of autoimmunity following sterile inflammation (Christoffersson et al. 2010; Soehnlein and Lindbom 2010; Yang et al. 2010). However, overexuberant or prolonged neutrophil infiltration can exacerbate tissue injury, leading to disease. Within the liver, inappropriate inflammation in response to sterile tissue injury contributes to the pathogenesis of many noninfectious diseases, including drug- or toxin-induced liver injury, ischemia-reperfusion injury, autoimmunity, alcoholic steatohepatitis, and others (Bautista 1997; Bonder et al. 2004; Jaeschke and Farhood 1991; Jaeschke, Farhood, and Smith 1990; Liu et al. 2006; Ohta et al. 2006). Although sterile inflammation is a well-known phenomenon, the mechanisms that initiate and regulate the innate immune response to cell death have only recently begun to unfold (Chen and Nuñez 2010; Kono and Rock 2008).
Tissue injury and necrotic cell death result in the liberation of many endogenous damage-associated molecular pattern molecules (DAMPs, also known as danger signals, alarmins) that stimulate the sterile inflammatory response. An explosion of recent research has focused on defining the nature of these endogenous inflammatory signals; this research has been reviewed in depth elsewhere (Chen and Nuñez 2010; Kono and Rock 2008). The list of DAMPs that can activate sterile inflammation is continuously expanding, but in general DAMPs can be dichotomized into two categories: (1) intracellular substances released passively from necrotic cells or actively from stressed cells (including HMGB-1, ATP, monosodium urate, and others) or (2) extracellular substances that become “activated” to a proinflammatory state following tissue injury (including short-fragment HA, biglycan, collagen fragments, and others) (reviewed by Kono and Rock, 2008). The multitude of DAMPs released and/or generated at sites of sterile injury yields a complex environment of inflammatory mediators that must be detected by the innate immune system and translated into guidance signals to direct neutrophil recruitment to the site of tissue damage.
Using a model of focal heptatic necrosis induced by localized thermal injury to the surface of the liver (Figure 2), we observed that neutrophil recruitment to sites of sterile inflammation differed dramatically from that induced by septic inflammatory stimuli. In contrast to endotoxemia/sepsis where neutrophils adhere within sinusoids and remain stationary, neutrophils responding to sterile inflammation were found to be highly motile within the sinusoids (McDonald et al. 2010; Menezes et al. 2009). Specifically, adherent neutrophils migrated intravascularly through the sinusoid channels toward the focus of tissue necrosis, ultimately infiltrating directly into the area of damage (Figure 2) (McDonald et al. 2010). These observations suggested that sterile liver injury not only is a potent inflammatory stimulus but also clearly generates an intravascular gradient of chemoattractant signals that guide neutrophils to migrate precisely into the area of injury. Indeed, numerous DAMPs can stimulate the production of CXC chemokines that are critical for neutrophil recruitment in various models of sterile inflammation (Chen and Nuñez 2010; Eigenbrod et al. 2008; McDonald et al. 2010). Consistent with these observations, chemokine ligands of CXCR2 (MIP-2 and KC, homologues of human IL-8) were expressed on the luminal surface of liver sinusoids around sites of necrosis, forming a chemokine gradient that guided neutrophil migration in the direction of the injury (McDonald et al. 2010). However, the chemokine gradient was observed to end 100–200 µm from the injury border, yet neutrophils continued to migrate past this point and penetrate directly into the area of tissue necrosis, indicating that additional chemoattractants are involved (McDonald et al. 2010). Two recent in vivo imaging studies have clearly demonstrated that injured cells themselves can attract neutrophils, suggesting the existence of chemoattractant DAMPs released following necrosis (McDonald et al. 2010; Ng et al. 2011). Interestingly, in vitro neutrophil chemotaxis assays have revealed that much of the chemoattractive potential of dead cells resides within the mitochondria (Zhang et al. 2010; Pittman and Kubes, unpublished observations). Given their evolutionary origin as bacterial endosymbionts, mitochondria have retained a number of prokaryotic signatures, including the use of N-formylated methionine for the initiation of protein translation (Galper 1974). As such, like their bacterial counterparts, mitochondrial N-formyl peptides are potent neutrophil chemoattractants in vitro and in vivo (Carp 1982; Zhang et al. 2010). Consistent with the hypothesis that neutrophil attraction to necrotic cells is mediated by the release of mitochondrial N-formyl peptides, the antibody blockade or genetic deficiency of formyl peptide receptor 1, or defects in downstream signaling, inhibits neutrophil migration toward tissue necrosis in vitro and in vivo (McDonald et al. 2010; Ng et al. 2011).
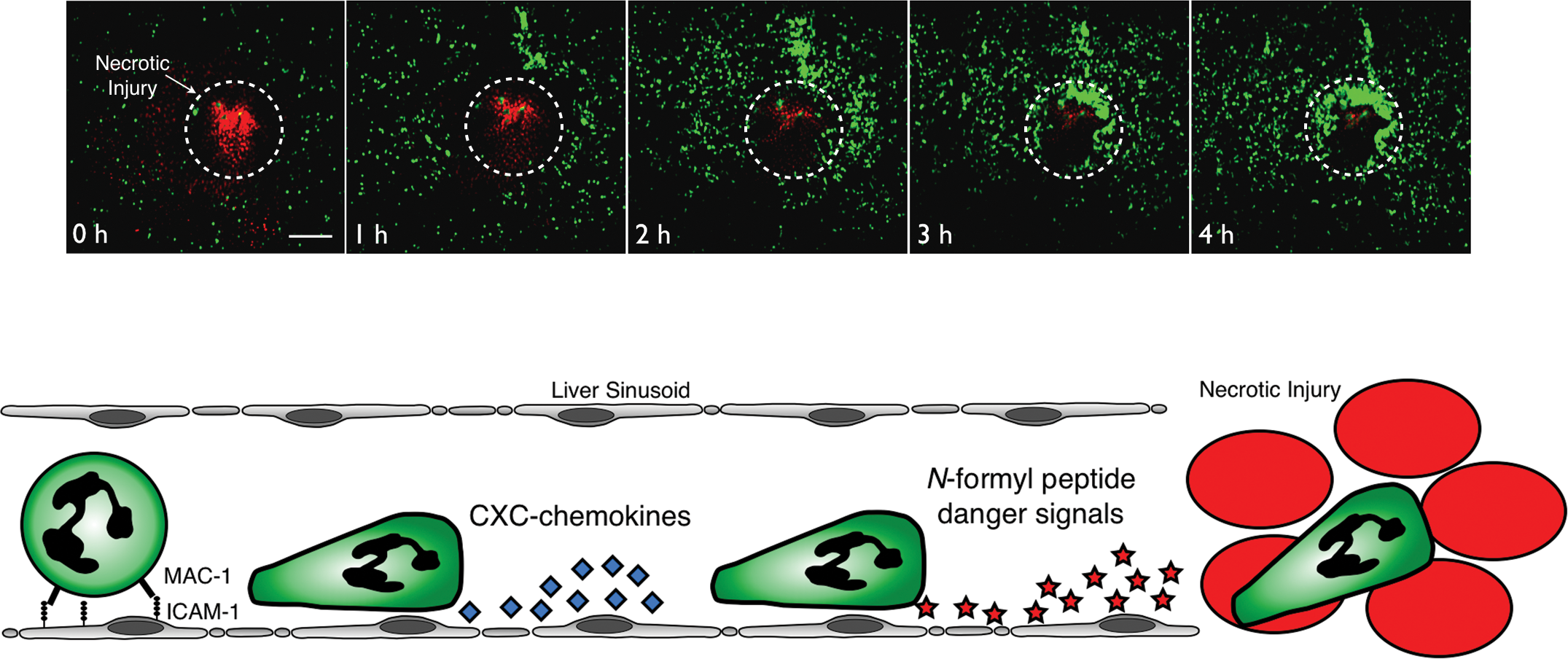
Neutrophil recruitment to sites of sterile inflammation in the liver. Around sites of focal hepatic necrosis, neutrophil adhesion to sinusoidal endothelium is mediated by integrins (Mac-1) binding to endothelial ICAM-1. Subsequently, neutrophils migrate through the intravascular channels toward the site of injury, ultimately penetrating directly into the area of damage. Initially, neutrophil chemotaxis is directed by an intravascular gradient of CXCR2 ligands that line the surface of the sinusoidal endothelium. Next, as neutrophils move closer to the injury, chemoattactant danger signals emanating from the necrotic tissue predominate (such as mitochondrial N-formyl peptides), thereby attracting the neutrophils to migrate directly into the focus of tissue injury.
In addition to their intravascular motility in response to liver injury, another key difference displayed by neutrophils responding to sterile versus septic inflammation is the use of different adhesion molecules for recruitment. While CD44 has been found to mediate the recruitment of neutrophils to the liver in response to septic/infectious stimuli, emerging evidence indicates that neutrophil infiltration at sites of sterile inflammation requires a different mechanism of adhesion. We observed that neutrophil recruitment to sites of focal hepatic necrosis was independent of CD44 but instead required adhesive interactions between αMβ2 integrin (Mac-1) and its endothelial ligand ICAM-1 (Figure 2) (McDonald et al., 2010). Furthermore, Liu and colleagues (2006) reported that neutrophil recruitment to the liver following acetaminophen-induced hepatocellular necrosis was dramatically reduced in the absence of ICAM-1. The molecular mechanisms that underlie this differential adhesion molecule usage between septic and sterile liver inflammation have been explored by Menezes et al. using a simplified model of focal fMLP-induced inflammation and investigating the effects of superimposing this stimulus in the setting of sepsis/endotoxemia. Applying an fMLP-soaked filter paper to the surface of the liver resulted in recruitment of neutrophils by Mac-1-ICAM-1-dependent adhesion and crawling, similar to focal necrosis (Menezes et al., 2009). However, in mice treated with LPS (or rendered septic by intraperitoneal E. coli infection), neutrophil adhesion around the fMLP filter was now CD44 dependent, and the adherent neutrophils remained stationary and failed to crawl toward the filter. Surprisingly, if the same experiment was performed in IL-10-/- mice, neutrophils in liver sinusoids reverted back to the use of Mac-1 for firm adhesion. It was found that high levels of IL-10 produced locally within the liver caused down-regulation of Mac-1 surface expression on neutrophils, causing CD44 to become the dominant adhesion receptor in the setting of sepsis/endotoxemia. Taken together, these findings suggested that CD44-dependent adhesion within the septic liver is the result of IL-10 production and down-regulation of Mac-1, whereas Mac-1 expression remains high during sterile inflammation enabling engagement with ICAM-1 (Figure 3) (Menezes et al. 2009). It remains to be determined whether the immobility of neutrophils during sepsis/endotoxemia is solely the result of Mac-1 down-regulation or whether this is due to the sepsis-induced “paralysis” of neutrophil chemotactic machinery that has been suggested by a number of studies (Alves-Filho et al. 2006; Alves-Filho et al. 2009; Khan et al. 2005).
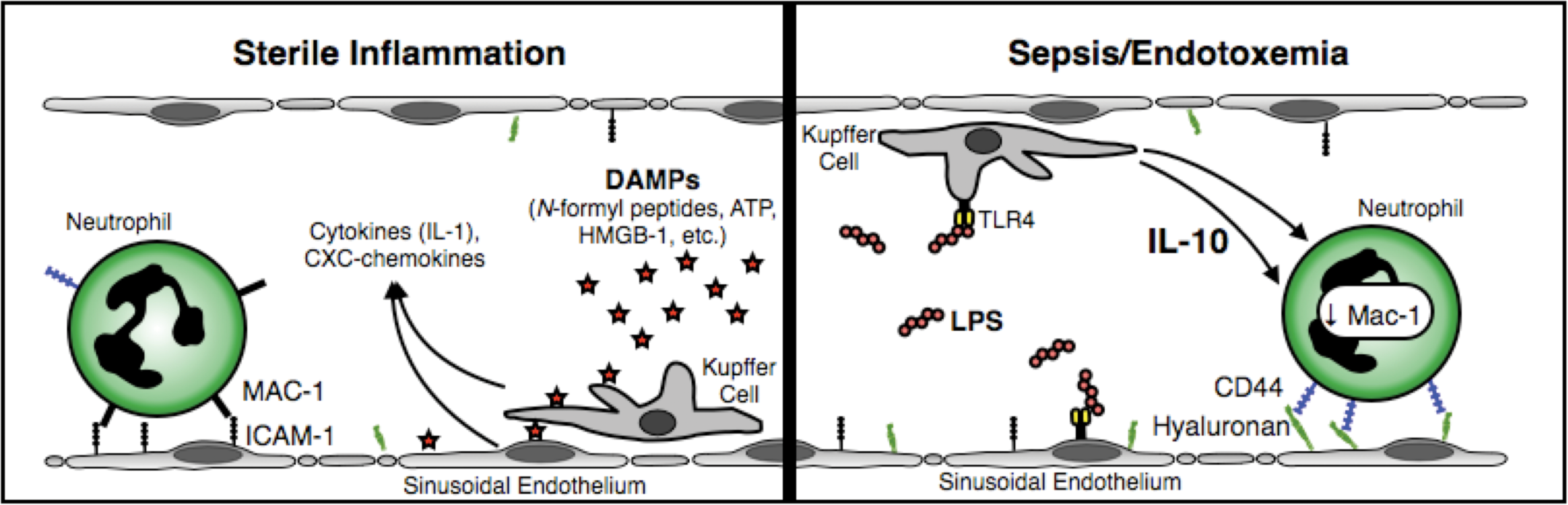
Different adhesion mechanisms mediate neutrophil recruitment to the liver during sepsis/endotoxemia and sterile inflammation. Tissue injury and cell necrosis result in the release of damage-associated molecular pattern molecules (DAMPs) that stimulate local sentinel cells in the liver microvasculature (Kupffer cells, endothelium) to produce proinflammatory cytokines (such as IL-1β) as well as CXC-chemokines. These mediators choreograph neutrophil recruitment via conventional β2 integrin- (Mac-1) dependent adhesion and intravascular crawling. However, during endotoxemia or gram-negative sepsis, high levels of bacterial lipopolyssacharide (LPS) in the blood stimulate Kupffer cells, sinusoidal endothelium, and other local hepatic sentinel cells to produce large amounts of the anti-inflammatory cytokine IL-10. Neutrophils passing through the hepatic circulation are exposed to high levels of IL-10, resulting in downregulation of Mac-1 surface expression, yielding CD44 as the dominant adhesion molecule for recruitment within the sinusoids.
Conclusions
Neutrophils play critical roles in innate immune responses within the liver. The intravascular events in neutrophil trafficking within the microcirculation of the liver are choreographed by different molecular mechanisms, depending on the nature of the inflammatory insult, enabling versatility in neutrophil function in the setting of sepsis versus sterile inflammation. During sepsis, neutrophils are actively recruited to the liver by a precisely coordinated molecular mechanism involving adhesive interactions between neutrophil CD44 and endothelial HA. We hypothesize that this active sequestration of neutrophils within the liver microcirulation represents a coordinated host response involving the release of NETs to protect against hematogenous dissemination of bacteria. Given that NETs harbor various cytotoxic molecules such as histones and proteases, it has been suggested that NETs may be a potential therapeutic target in sepsis and other infectious diseases. However, given their putative role in antibacterial defenses, caution is warranted, as therapeutic inactivation of NETs to lessen tissue pathology may come at the expense of worsening bacterial dissemination. In the setting of sterile inflammation, neutrophil recruitment to sites of hepatocellular necrosis proceeds using an alternate adhesion mechanism involving neutrophil Mac-1 and endothelial ICAM-1. Furthermore, neutrophils responding to sterile inflammation are highly motile and chemotax toward areas of tissue necrosis along gradients of CXC-chemokines followed by formyl peptide receptor–dependent danger signals released from necrotic cells (likely mitochondrial formylated peptides). We believe that this multistep intravascular recruitment cascade enables focusing of the innate immune response on the existing area of damage, thereby sparing the surrounding healthy tissue from the potentially injurious effects of neutrophilic inflammation. Further studies are needed to investigate whether other tissues employ a similar recruitment strategy or whether this intravascular neutrophil response is unique to the architecture of the liver sinusoids.
Footnotes
Abbreviations
Acknowledgements
The authors thank Dr. Björn Petri for critically reviewing this manuscript. Work in the authors’ laboratory is supported by Canadian Institutes for Health Research operating grants and group grant, as well as the Canadian Foundation for Innovation. Braedon McDonald is supported by an MD/PhD studentship from the Alberta Heritage Foundation for Medical Research (Alberta Innovates Health Solutions). Paul Kubes is an Alberta Heritage Foundation for Medical Research Scientist and the Snyder Chair in Critical Care Medicine.
The author(s) declared no potential conflicts of interests with respect to the authorship and/or publication of this article. The author(s) declared the following financial support in regards to the authorship of this article: The authors will receive a $400 honorarium for the contribution of this article provided by the Society of Toxicologic Pathology. This in no way affected the content of the manuscript.