
Abstract
Microglia are the histiocytes of the central nervous system. These long-lived cells undergo very little turnover in normal physiological states; however, in pathological conditions, increased egress from the bone marrow and chemoattractive signals in the brain can substantially modulate the indigenous population. Although they were initially conceived of as “resting” cells, recent data suggest that they would be more aptly described as “surveillance” cells. Microglia are specifically adapted to sense various types of danger and differentially react with a classical or alternative reparative response. Our understanding of macrophage function has shifted away from focusing on cell lineage to a more systems-based biology of gene networks accomplishing the detoxification and immune functions. With our greater appreciation of microglial involvement in the innate immune response, we have entered a new era in which the modulation of microglia can be proposed as a means of modulating neurological disease.
Keywords
Introduction
Microglia are the tissue-based macrophages or histiocytes of the central nervous system (CNS). Beginning with Ivan Mechnikov’s in vivo observations in starfish penetrated by rose thorns, the entire concept of innate defense has undergone several paradigm shifts over the past century. From the initial observations, phagocytosis was front and center a hallmark of an innate defense. The initial concept of the body’s macrophages was embodied in the term “reticuloendothelial system,” emphasizing the close relationship between myeloid and endothelial cells (for review, see Ross 2010; Gordon 2007). This concept was founded on the principle that the body needed a form of detoxification and immunity that was initially accomplished by cells distributed throughout the body. Over the decades, the discovery of multipotent stem cells and hematopoietic lineages gave way to the new paradigm of the “mononuclear phagocyte system,” a concept that embodied the idea of terminally differentiated cells that can undergo further differentiation with activation and basically accomplish the same detoxification and early inflammatory response. More recently, our understanding of macrophage function has shifted away from focusing on cell lineage to a more systems-based biology of gene networks accomplishing the detoxification and immune functions. In this current paradigm, genetic programs are executed through populations of cells, while transcriptional regulation of gene expression in individual cell elements within a population follows a more probabilistic nature (Hume 2000; Ross 2010).
All organ systems possess tissue-based macrophages called histiocytes that are best thought of as local sentinels for various insults. Although capable of carrying out numerous inflammatory functions, in their natural “quiescent” state, their inflammatory capabilities are fully repressed. Extending well beyond the lymphoid system, histiocytes populate all organ systems, constituting up to 15% of an organ’s cells. Known by a wide variety of names in different organs, they are sentinels for the entire gamut of potential insults.
Histiocytes of the CNS are referred to as microglia and are the key innate immune cell in the CNS. Although all eukaryotic cells possess intracellular and extracellular molecules capable of sensing pathogenic signals, macrophages and microglia are notable for particular specialized abilities to detect danger signals. Once activated, these macrophage elements have the added capacity to differentiate down different pathways depending on the particular eliciting stimulus (Ross 2010).
Founded in historical static observations of silver-stained sections, later followed by numerous immunohistochemical studies, our concept of “resting” microglia has undergone a revolution in recent years. New dynamic in vivo images recently acquired in the living brain (Davalos et al. 2005; Nimmerjahn, Kirchhoff, and Helmchen 2005) are highly reminiscent of the studies performed by Mechnikov before the turn of the century. In vivo imaging of microglia in the rodent brain suggests that the term “resting” perhaps is best dropped in favor of the descriptor “surveillance.” Imaging live fluorescently tagged microglia has shown that these “resting” cells in fact exhibit very dynamic cell processes that project and retract extensively throughout the neuropil in a fashion highly suggestive of sampling or surveying the local environment (Nimmerjahn, Kirchhoff, and Helmchen 2005). This “resting” state of surveillance with extension of cytoplasmic processes is highly activated upon insult (Davalos et al. 2005).
The concept of innate immunity has evolved from our understanding of inflammation. But innate immunity is not a single response, as implied by the term “inflammation”; rather it is “designed” to sense the type of insult and appropriately tailor the cell’s response to the particular threat. Sensing the type of insult directs the innate immune response down different paths. The innate immune response to microbes and other insults is a very early and rapid defense that in vertebrates is later influential in tuning the adaptive immune response. Although tuning the later host adaptive immunity is a key function of the innate immune response, shaping adaptive immunity is not discussed further in this review. Instead, we focus on the initial phases of host response to CNS damage in which microglia play a key role.
Innate Immune Response Detector Molecules
Recognition of nonself and altered or stressed-self structures is central to innate immune stimulation. Pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) are present in a broad range of tissue insults and are detected by, and initiate, the innate immune response. Cellular receptors that recognize PAMPs and DAMPs have been intensely studied in the past fifteen years (for review, see Ross 2010; Takeuchi and Akira 2010; Gordon 2007). Although expressed on membranes and intracellularly by most cells, the receptors are particularly characteristic of neutrophils, macrophages, and dendritic cells. One class of these receptors is referred to as Toll-like receptors (TLRs) because of the similarity to Toll molecules of Drosophila that are key to establishing the dorsal and ventral axis during Drosophila development. Ten TLRs have been identified in humans (Ross 2010; Takeuchi and Akira 2010; Mogensen 2009). All TLRs have an extracellular or intraluminal ligand-binding domain consisting of leucine-rich repeat regions capable of detecting specific PAMPs or DAMPs, which is connected to a Toll/interleukin (IL)–1 receptor cytoplasmic domain (Fig. 1 ). TLRs are cell membrane proteins, but they are not limited to the cell surface. Rather, TLRs are present in different and specific membrane compartments of the cell. Because different TLRs detect different PAMPs, it is logical that they would have a differential distribution on select membranes surfaces. TLRs on the surface of the cell detect surface pathogens, whereas TLRs on internal membranes (e.g., lysosomes) are tuned to detecting pathogens that preferentially occur in endosomal compartments. Irrespective of the membrane distribution, intracellular signaling occurs through the Toll/IL-1 receptor cytoplasmic domain through recruitment of adaptor proteins, eventually leading to the activation of protein kinases, followed by the activation of transcription factors and eventual gene transcription (e.g., IL-1β and IL-18; Fig. 1). Activation of these genetic programs allows the cells to participate in various gene networks of the immune system.
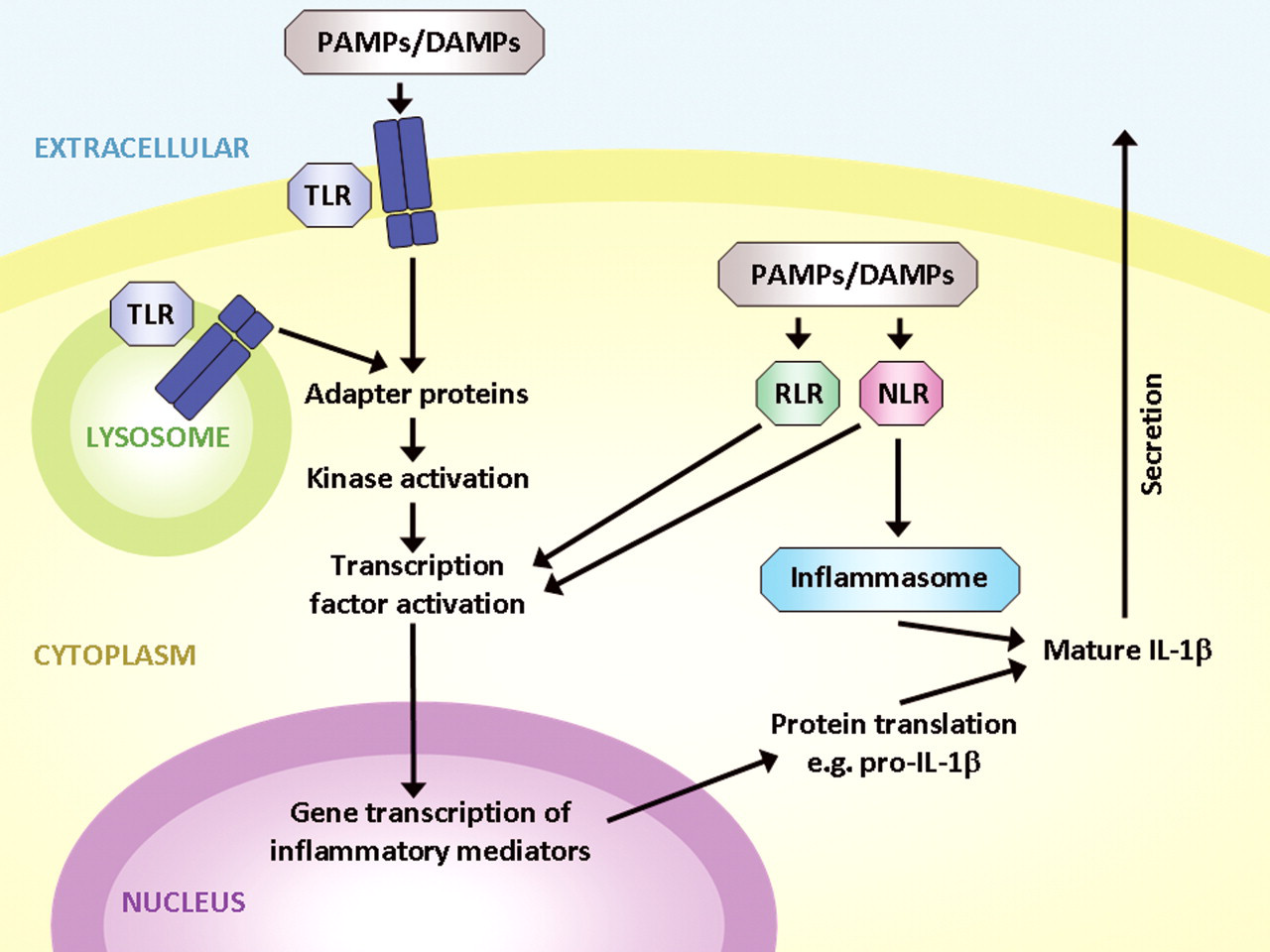
Schematic of innate immune response: pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) are detected on the surface of cells or within endosomal compartments by Toll-like receptors (TLRs). Binding to TLRs leads to homodimerization or heterodimerization and the recruitment of adapter proteins. Subsequent kinase activation leads to the activation of transcription factors that mediate new gene transcription of inflammatory mediators (e.g., pro forms of interleukin [IL]–1β). The additional stimulation of nuclear oligomerization domain–like receptors (NLRs) and retinoic acid–inducible gene–1–like receptors (RLRs) promotes both additional gene transcription and the activation of caspase-1 activity in inflammasomes that activate cytokines and lead to their secretion.
In the brain, all TLRs have been shown to be expressed on microglial cells, in addition to limited expression of some TLRs on neurons and other glial cells. TLR signaling plays a significant role not only in the innate immune response to CNS infections but also in inflammatory and demyelinating disorders, including multiple sclerosis, ischemic injury, and neurodegeneration (Aravalli, Peterson, and Lokensgard 2007; Lehnardt 2010).
TLRs are not the only sensing component of the innate immune response. Other surface receptors, such as carbohydrate receptors (e.g., the mannose receptor, cluster of differentiation [CD] 206) or oxidized lipoprotein detectors (e.g., scavenger receptors, CD68) have the capacity to detect microbial carbohydrates or altered lipoproteins, respectively. Additionally, there are cytoplasmic receptors that are also key components of the innate immune response, such as retinoic acid–inducible gene–1–like receptors and nuclear oligomerization domain–like receptors (NLRs). Unlike TLRs, they are found exclusively within the cytoplasm and recognize cytosolic viruses and bacterial products. Like TLRs, however, they contain leucine-rich repeats capable of binding many different ligands (e.g., peptidoglycans). After binding specific ligands, both types of receptors promote gene transcription. In addition, NLRs recruit cytoplasmic proteins to form an inflammasome capable of activating caspase-1. Caspase-1 then cleaves cellular proteins such as those transcribed by TLR activation (e.g., IL-1β and IL-18) to their active forms that upon secretion contribute to the inflammatory cascade.
Beyond the TLRs and NLRs, macrophages also possess PAMP receptors that do not signal but have an immediate effect (Ross 2010), for example, enabling pathogen binding and subsequent internalization. Such immediate effectors include scavenger receptors (CD36) and C-type lectin domain receptors (the mannose receptor, CD206; dendritic cell–specific intercellular adhesion molecule–3–grabbing nonintegrin, CD209; complement receptor; immunoglobulin receptor; etc.).
In summary, rather than being considered “resting” tissue macrophages, histiocytes are more appropriately considered in terms of their innate immune function as tissue sentinels. They are physiologically active, constantly surveying the local environment. They are particularly endowed with receptors to detect PAMPs and DAMPs. They possess both immediate effectors and program effectors capable of specific activation eliciting specific genetic programs. Because different pathogens lead to different activations, it would not be appropriate to think of macrophages as a single type of responder cell; rather, it would be more appropriate to consider them terminally differentiated cells with the capacity for a spectrum of different responses.
Microglia Origin
Although macrophages were first described in the late nineteenth century in starfish, microglia were first recognized by Nissl in 1899 and formally described by del Rio Hortega in 1932 as resting ramified cells using silver carbonate staining (del Rio-Hortega 1932; Barron 1995). The origin of mammalian microglia has been studied and debated for more than a century. Current evidence suggests that in mammals, early microglia are derived from yolk-sac progenitors that migrate into the CNS around the second trimester (Ransohoff and Perry 2009; Chan, Kohsaka, and Rezaie 2007). Myeloid progenitors continue to enter the CNS throughout development switching from a liver origin to bone marrow origin at about the time of birth. By birth, a substantial complement of microglia are present in the brain, the result of a combination of migration and local proliferation. There is a fair amount of regional variation in overall density of the microglial cells, ranging from 5% in the cortex and corpus callosum up to 12% in the substantia nigra of the murine brain (Lawson et al. 1990) and from 0.5% in gray matter areas of cerebellum and cerebral cortex to 16.6% in the pons and medulla in the nondiseased human brain (Mittelbronn et al. 2001). Overall, there is significantly higher microglial density in white versus gray matter.
Microglial Turnover
What are the origin and magnitude of microglial turnover in normal and pathological states? For more than a century, this issue has been so extensively studied that the number of reviews and position papers on microglia turnover may exceed the number of microglia in the brain. With the vast differences between species' life spans, it is not surprising that no single half-life of microglia can be given. Also, there is substantial heterogeneity between different populations of microglia within an individual organism. The general consensus is that perivascular microglia are fairly dynamic compared with parenchymal microglia. In the unperturbed state, most investigators would accept an estimate of months to years for the half-life of perivascular microglia and years to lifetime half-life of parenchymal microglia. But in pathological states, microglia turnover can be robust, presumably commensurate with the need for new cells.
A comprehensive review of the origin and turnover of microglia can be found in recent publications (Soulet and Rivest 2008; Ransohoff and Perry 2009). These authors have compared myriad different murine experimental systems labeling monocytes—parabiosis models, chimeric mouse models, models with and without irradiation, and models with and without immunosuppression—and attempted to derive a conclusion regarding the origin of brain microglia. Although it might be too early to claim that a consensus has been reached, a fairly uniform perspective has developed. In normal and pathological states following full-body irradiation with shielding of the CNS, most studies have shown that there is limited or no infiltration of bone marrow–derived mononuclear cells into lesions such as cuprizone-induced demyelination or facial nerve transection. The hypothesis here is that despite activation of local microglia, there is an absence of key migration signals to attract circulating monocytes. Parabiosis experiments have generated results that could be interpreted differently. However, it appears that two events are necessary for microglia turnover: progenitor cells need to egress from the bone marrow and be directed into the CNS. Limitations of microglia turnover from the periphery appear to be related to the absence of key signals like chemokine (C-C motif) ligand 2, which are critical for migration induction. Perhaps a corollary of these conclusions is that microglia normally do not turnover. This new perspective places more emphasis on local proliferation of either microglia or microglial-like stem cells for the normal physiology of the CNS (Geissmann et al. 2010).
A conclusion from cell turnover experiments in general is that most tissues have long-lived histiocytes. The mechanisms controlling the replacement of these resident macrophages are for the most part unknown, and perhaps normal physiological renewal is limited to local stem cell proliferation. Such a conclusion is tempered by the possibility of species-specific macrophage behavior. Additionally, injury and repair can be associated with repopulation of the tissue by new histiocytes, but whether those histiocytes adopt the original phenotype or a new phenotype is open to future study.
Microglial Turnover in Pathological States
In general, there are two types of experimental paradigms for studying microglia increase in pathological states. There are models of disease without gross disturbance of the blood-brain barrier such as facial nerve axotomy or severance of the entorhinal pathway. Alternatively, there are numerous experimental models in which the blood-brain barrier is breached either physically or immunologically, such as by a direct stab wound or experimental allergic encephalomyelitis.
In the entorhinal-dentate perforant pathway lesion model, there is physical sectioning of the perforant pathway that is spatially separated from the site of microglial activation (Prang, Del Turco, and Kapfhammer 2001). In this model, microglia respond to an anterograde axonal and terminal degeneration that is believed to occur in the absence of any overt blood-brain barrier breakdown. Microglia proliferate in the initial two to three days after sectioning producing an abundant CD34-positive cell population that declines by day 5. But even without overt blood-brain damage, there is a question of whether bone marrow–derived cells begin to migrate at a later date. Morphologic and phenotypic changes in the course of microglial activation have been nicely reviewed (Ladeby et al. 2005). Proliferation, migration, differentiation, and normalization of microglial numbers run a variable time course dependent on the nature of the injury and host physiology.
Microglia Function
Numerous physiological and pathological functions have been attributed to microglia in addition to their role in the innate immune response (see above). During embryogenesis, the nervous system is notable for massive developmental apoptosis, with twice as many neurons being generated than are retained in the mature organism. Microglia are thought to be important not only in the final phagocytosis, but they may also promote the death of developing neurons (Ransohoff and Perry 2009; Marin-Teva et al. 2004; Bessis et al. 2007). Experimental systems such as the facial nerve transection model have suggested that microglia are active at synaptic stripping in pathological processes (Trapp et al. 2007; Wake et al. 2009). From such findings, it has been conjectured that microglia may be important in physiological synaptic remodeling. Moreover, recent observations indicate a role for microglia in regulating developmental synaptogenesis as well as modulating synaptic activity (Bessis et al. 2007; Coull et al. 2005). Microglia have also been recognized as regulators or promoters of adult neurogenesis in the dentate gyrus and subventricular zone (Battista et al. 2006; Choi et al. 2008; Walton et al. 2006).
Beyond scavenging apoptotic bodies, microglia have the ability to scavenge extracellular molecules such as amyloid (see below), and much attention is focused on the possible neurotoxic and beneficial effects of microglia in neurodegenerative diseases (Weiner and Frenkel 2006; Hickman, Allison, and El Khoury 2008; Rogers et al. 2002; Ransohoff and Perry 2009). Neuroinflammation associated with microglial response may also contribute to the neurotoxicity of environmental agents and neurotoxicants; however, very few studies are reported in this field of toxicology, and most rely on in vitro systems. Most of the work has focused on heavy metals and the organometal trimethyltin. The results of these studies point to significant diversity and heterogeneity of the microglial response (Harry and Kraft 2008). More recently, microglia have also been shown to internalize and be activated by gold nanoparticles and thus may play an important role in the CNS response in the emerging field of nanotherapeutics and nanodiagnostics (Hutter et al. 2010). Last, in addition to all of the innate immune response functions described above, microglia are the only known endogenous CNS cells that are capable of presenting antigen to T cells participating in adaptive immune responses.
Types of Microglia
Beginning with a common precursor cell, a variety of environmental cues produce heterogeneous states of macrophage and microglial activation. Our concept of macrophage activation states has undergone significant changes over the past decade with the recognition of alternative activation states. Following this discovery, macrophage differentiation states were initially divided into a simple two-state system, classical (M1) versus alternative (M2), but as we learn more about microglial function this binary system has begun to crack and expand, and it appears more likely that a wide spectrum of differentiation states rather than two discrete states is closer to the truth (Gordon and Martinez 2010; Mosser and Edwards 2008; Martinez, Helming, and Gordon 2009; Gordon 2003). The topic is even further complicated by the apparent substantial discrepancy between characterizations of macrophages from different species (Martinez et al. 2006; Martinez, Helming, and Gordon 2009). Differentiation markers and responses in the two most commonly studied macrophage systems, human primate and murine, are not consistent, resulting in substantial confusion in comparison between the systems. Given the limited overlap between the murine and human systems, significant caution must be recommended when attempting to extrapolate data from one species to the other. Nevertheless, it is convenient from a heuristic perspective to consider the simplified two-state system, as long as it is remembered that this is only a simplification.
In general, the M1 proinflammatory state is a defensive state in which oxidative metabolites and proinflammatory cytokines are produced by macrophages stimulated by a diverse set of ligands, including interferon-γ, lipopolysaccharide (LPS), and other TLR activators. Engagement of the respective receptors leads to intracellular signaling through adapter proteins such as MyD88 and others. Activation of the adapter proteins leads to activation of kinases, phosphorylation of transcription factors, and eventual genetic program induction. Proinflammatory genes, including interferon-γ, tumor necrosis factor–α, IL-1β, IL-18, chemokines, and proteases, are subsequently produced. This proinflammatory activation is suppressed by glucocorticoids. Further activation of the M1 pathway occurs by assembly of the NLR inflammasome and caspase-1 activation, leading to conversion of proforms of IL-1β and IL-18 into secreted active forms. Functionally, the M1 activation pathway is important for helper T cell type 1 responses, killing of intracellular pathogens, and mediating tumor resistance.
In contrast, alternative activation (M2) is more of a reparative, matrix remodeling, and anti-inflammatory response on the part of macrophages stimulated by IL-4, IL-13, and other agonists. On the basis of the activating stimulus, alternative activation can be subdivided into different categories with associated functional differences. However, the field is still evolving, and different nomenclatures are in use, adding to the difficulty in interpreting and comparing the findings; for example, the state of acquired deactivation is either labeled as a distinct third macrophage activation state or included under the umbrella term “alternative activation.” Overall, alternative activation is associated with a variety of functions spanning—helper T cell type 2 responses (allergy and immunoregulation), killing and encapsulation of parasites, tumor promotion, matrix deposition, and tissue remodeling—and includes the production of suppressive inflammatory cytokines (e.g., IL-10) and membrane metalloproteases 1 and 12.
Classical (M1) and alternative (M2) activation are associated with distinct gene expression profiles, with, at least in the murine system, well-established gene and also protein signatures that have been successfully used to assess macrophage activation states in vivo. The most widely used markers are inducible nitric oxide synthase for M1 activation and Ym1, mannose receptor (CD206), and arginase 1 for M2 polarization and represent an important new tool for assessing macrophage and microglial phenotype and function. Unfortunately, the majority of these markers are not differentially regulated or not expressed at all in human cells, and human-specific gene signatures have only recently begun to be developed. Establishing reproducible and widely accepted signatures is not only important to describe activation states in vivo but even more crucial to assess if macrophage or microglial phenotypes and functions in (transgenic) mouse disease models truly mimic what is happening in human diseases (e.g., Alzheimer’s disease [AD]).
Despite evidence indicating a separation of macrophage activation states, there is also evidence emerging to suggest that there can be transitions from one state to another. In the spinal cord injury model (Kigerl et al. 2009), M1 activation is induced rapidly and persistently. Supernatants of such activated cells are toxic to dorsal root ganglion neurons, leading to short branched neurite extension in vitro. It appears that, in vivo, this classical activation is slowly down-regulated, with transient activation of a more M2-like profile over a period of three to seven days. This transition and intermingling of activation pathways suggests that chronic inflammation may in fact be a mixture of both classically and alternatively activated cells and that in a given disease or injury condition, the balance between these concurrent pathways may determine the protective or deleterious role of microglial activation (Li et al. 2007; Hanisch and Kettenmann 2007; Schwartz et al. 2006).
Once activated, it is critical for host survival that the inflammatory process and macrophages in particular be turned off. Numerous pathways have been identified to down-regulate classical activation, including the ubiquitination of surface TLRs, the production of suppressor cytokines, production of microribonucleic acids to decrease cellular transcription, the up-regulation of receptor antagonists, and the release of receptors for cytokines to decrease their overall signaling. An intriguingly brain-specific interaction between the CD200 molecule on neurons and CD200 receptor on microglia leads to inhibitory signaling. Genetic knockout experiments have demonstrated the importance of this means of turning off macrophages in experimental models such as experimental allergic encephalomyelitis and LPS administration (Hoek et al. 2000; Koning et al. 2007). The interactions between CD200 and CD200R as well as other ligand-receptor pairs (e.g., CD47 and CD172a/SIRPα, CX3CL1 and CX3CR1) are also in part responsible for the remarkably down-regulated phenotype of microglia in the healthy CNS compared with other tissue macrophages. Interruption of this signaling by loss of ligands due to impairment of neuronal integrity may constitute a type of danger signal and can subsequently lead to microglial activation (Ransohoff and Perry 2009; Hanisch and Kettenmann 2007; Frank et al. 2006; Wang et al. 2007).
Microglial Markers
It is clear from the above discussion that finding “a” marker for microglia is not an appropriately framed question. In the surveillance state, most important cellular markers are repressed, more so in microglia than in other macrophages, suggesting that microglial phenotype is regulated by the CNS microenvironment (Ransohoff and Perry 2009). Nevertheless, these surveillance histiocytes can be labeled immunohistochemically. Unfortunately, although there are many available labels, species heterogeneity requires careful selection. “Resting” macrophages notably have receptors for colony-stimulating factor 1 (CD115) (Hume 2006); however, this molecule is not abundant, is rapidly internalized with activation, and is present on other CNS cells. A new and particularly robust marker for macrophages is the ionized calcium-binding adapter molecule–1 that demarcates macrophages in multiple mammalian species in the resting as well as, more robustly, in the activated state. Ionized calcium-binding adapter molecule–1 and the lysosomal marker CD68 are currently among the most widely used “general” microglia and macrophage markers (Fig. 2 ). Major histocompatibility complex type 2 molecules, complement receptors, and scavenger receptors are expressed mostly on selectively activated macrophage elements. In mice, the epidermal growth factor module containing mucin-like hormone receptor 1 has a specific antibody (F4/80) that labels well but does not work well in human tissues, because of the presence of different isoforms and wider expression on other leukocytes (Hume 2006). Experimental studies have used transgenic mice in which one copy of the CXCR1 gene has been replaced by the gene for green fluorescent protein to highlight microglial cells (Davalos et al. 2005; Jung et al. 2000; Nimmerjahn, Kirchhoff, and Helmchen 2005). Innate immune receptors such as TLRs and NLRs, discussed above, are all expressed in macrophages; however, possibly because of the low number of molecules present in individual cells, they have not been widely used to mark macrophages. Downstream elements of the innate immune response including transcription factors are all present in activated cells, but even then, they are not abundant. The mRNA for newly expressed genes (e.g., interferon-γ) are excellent targets of in situ hybridization probes (Fig. 3 ), but the protein gene products tend to be soluble and thus not readily localized in traditional preparations.
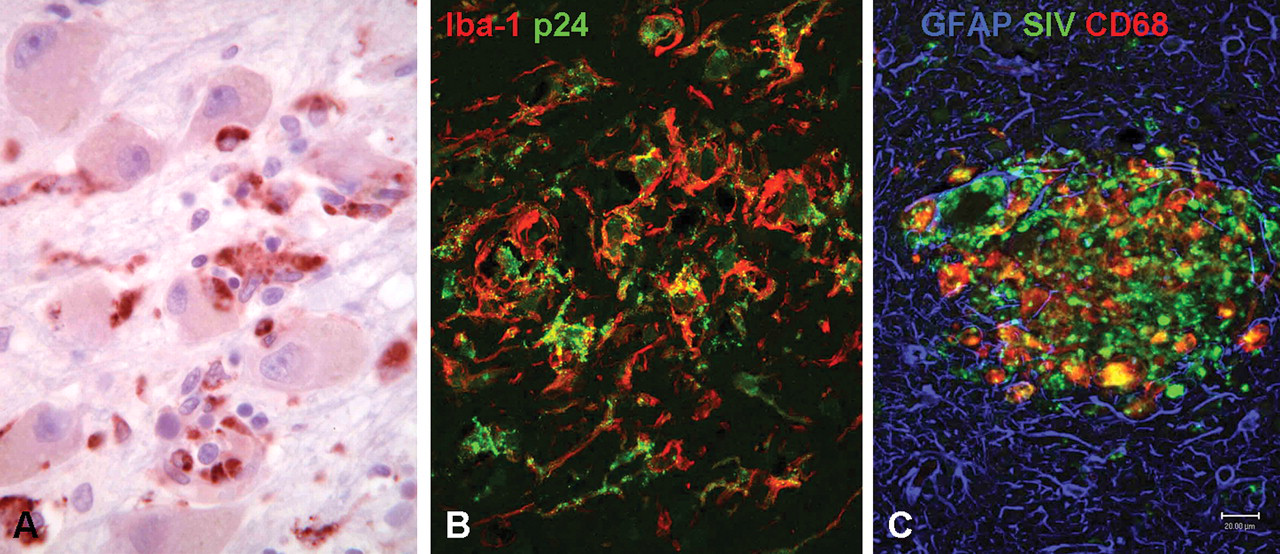
Microglia are intimately involved in the central nervous system’s innate immune response to virus infection. (A) Lateral geniculate nucleus of a patient with West Nile virus encephalitis immunostained for the macrophage/microglia marker cluster of differentiation (CD) 68 shows large infected neurons undergoing phagocytosis by CD68-positive macrophages (red). (B) Brain of human immunodeficiency virus (HIV)–infected patient with acquired immunodeficiency syndrome with encephalitis double immunostained for the macrophage/microglia marker ionized calcium-binding adapter molecule–1 (Iba-1) (red) and for HIV p24 (green) demonstrating nodules of infected or activated macrophages/microglia. (C) Brain of simian immunodeficiency virus (SIV)–infected macaque with encephalitis triple immunostained for CD68 (red), the astrocyte marker glial fibrillary acid protein (GFAP) (blue), and SIV (green) highlights a microglial nodule.
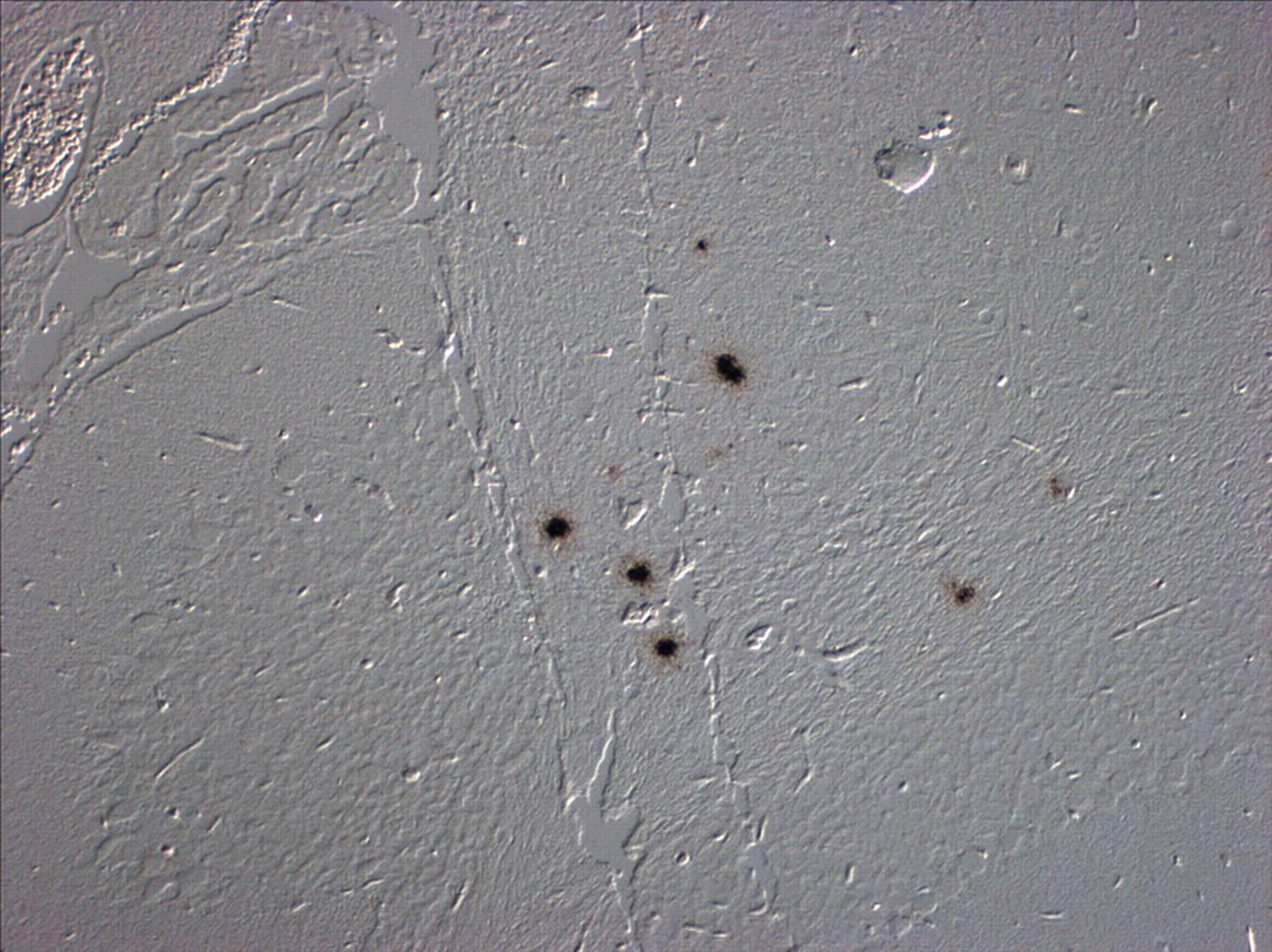
Differential interference contrast of brain of a mouse infected with H5N1 influenza virus after in situ hybridization for interferon-γ and autoradiography. Clusters of black emulsion grains demarcate cells that are transcribing interferon-γ mRNA consistent with activation of innate immune response.
In conclusion, there is no ideal single marker for tissue macrophages. Although there is a broad spectrum of potential markers (including plant lectins that robustly label macrophages of various species), few of these accurately display the nature of macrophage activation. Certainly, multiple CD markers closely aligned with function might be helpful, but these are usually most useful in flow cytometry when used as a panel and have limited use in situ. Even if there were a universal microglial marker, its use would be limited, because in the case of microglia (Soulet and Rivest 2008), the number of cells is not as important as their functional differentiation. Given their overall prevalence in the CNS, finding a certain density of microglia is almost meaningless, because the activation state of the cell will determine much of its physiological functions.
In Vivo Imaging of Microglial Dynamics
There is a critical need to monitor the activation of microglia in a dynamic way in living animals. As described above, two-photon microscopy has been successful in rodents, but a less invasive technique (i.e., one not requiring removal of portions of the skull) and one capable of imaging throughout the brain would substantially advance the field. We and others have attempted to develop positron emission tomography (PET) to quantitatively assess microglial activation during neuroinflammation in human and nonhuman primates (Banati et al. 1999; Cagnin et al. 2001a; Venneti et al. 2004; Venneti et al. 2009; Venneti, Wang, and Wiley 2008). PET is quite expensive but very sensitive and in theory quantitative. Fundamental to the success of PET is the availability of specific ligands to bind receptors of interest. Given the abundance of potential receptor targets on macrophages, it is a bit surprising that as late as 2010, there are very limited numbers of radioactive ligands that can be used to tag tissue histiocytes in general and CNS microglia specifically. Perhaps the most commonly used receptor for monitoring brain microglia to date is the peripheral benzodiazepine receptor (PBDR). This molecule in the external membrane of mitochondria is of unknown function though associated with the permeability transition pore (McEnery et al. 1992). Given its limited and historical connection to benzodiazepine binding, the receptor name has been changed recently to translocator protein–18 (Papadopoulos and Lecanu 2009). A group of radiolabeled ligands to the PBDR has been used in PET experiments to document the activation of CNS macrophages. In part, this technique is possible because in the resting state, the brain shows very little evidence of PBDR expression and an equally low amount of receptor binding. With disease, the overall binding of PBDR ligands increases, and attempts have been made to use alterations in binding kinetics to follow microglial activation.
We have used the nonhuman primate model of lentiviral infections (simian immunodeficiency virus encephalitis) as a model system to assess the utility of PET in detecting and assessing microglial activation. Although initially promising in identifying and distinguishing encephalitic from nonencephalitic animals, follow-up experiments have drawn into question the sensitivity of PBDR ligands for quantitatively detecting changes in microglial activation in various disease processes. We have attempted to extend these nonhuman primate studies into human diseases and encountered similar difficulties with overall sensitivity (Wiley et al. 2006, 2009). In studies of patients with acquired immune deficiency syndrome with minor cognitive impairment and studies of aged individuals with minor cognitive impairment or frank AD (Fig. 4 ), we have not been able to detect microglial activation using PBDR ligands, despite their initial promise in the nonhuman primate models. Although others have shown some positive results in comparisons of groups of individuals (Banati et al. 2000; Cagnin et al. 2001b), on an animal-to-animal or human-to-human basis, the PET studies suggest that new and more sensitive ligands need to be synthesized and tested before PET will be a useful in vivo methodology for following activation of microglia in living animals (Venneti, Wiley, and Kofler 2009).
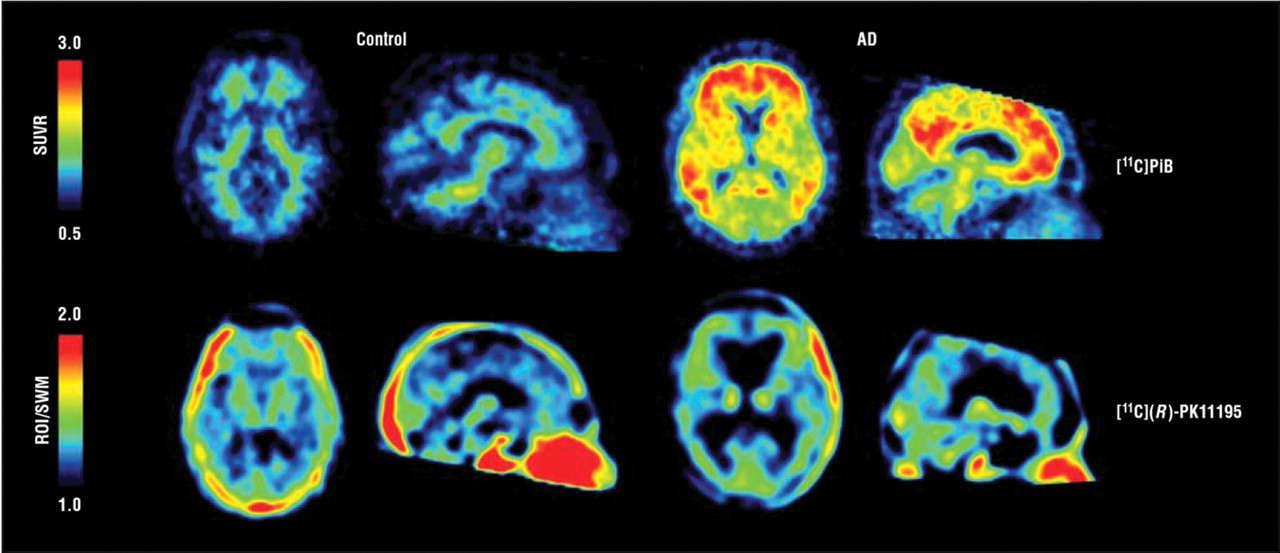
Transaxial and sagittal parametric positron emission tomographic images of the brains of a patient with Alzheimer’s disease (AD) (right) and an age-matched control (left) showing (top) relative abundance of β-amyloid binding ([11C]PiB) and (bottom) relative abundance of macrophages ([11C](R)-PK11195). Areas of significantly elevated [11C]PiB retention are evident in the patient with AD in a manner consistent with the known pattern of amyloid deposition in AD. No similar pattern is noted in the [11C](R)-PK11195 images, suggesting inadequate sensitivity of the PK probe to detect microglial activation associated with AD. (Reproduced with permission from Wiley et al. 2009.)
Microglial Therapeutics
With our improved understanding of microglial physiology and pathology, a new field of microglial therapeutics has emerged. This field is premised on the ability to pharmacologically manipulate existing microglia, such as switching their classical and alternative activation status or replacing defective or senescent microglia in storage disorders and degenerative diseases such as AD. The previously described turnover experiments have suggested that for therapeutic purposes, some sort of a preconditioning stimulus to elicit the ingress of new microglial elements would vastly improve replacement of brain microglia. Although irradiation is a known preconditioning stimulus, other less toxic approaches are possible.
Bone marrow transplantation with CD34 myeloid progenitor cells has been attempted in a variety of genetic storage disorders. Such studies have suggested that a limited number (1%) of CNS microglia can be of bone marrow origin within three weeks of transplantation. One such disease for which microglial therapeutics has been attempted is X-linked adrenoleukodystrophy (Cartier et al. 2009). In this peroxisomal disorder, male patients develop a severe leukoencephalopathy secondary to impairment of peroxisomal oxidation of very long chained fatty acids and their subsequent accumulation within the tissues. Made famous by the movie Lorenzo’s Oil, both animal models and human patients have been studied using bone marrow transplantation. Given the integrity of the blood-brain barrier, enzyme replacement therapies for the most part have not been effective in the treatment of leukodystrophies. However, treated in its early stages with bone marrow transplantation, there is definitely a therapeutic effect. Why does this work? Although it is clearly possible for slow engraftment of the brain to occur, in most leukodystrophies, the enzymatic defect is intracellular, thus requiring cell contact for effective spread. In the case of adrenoleukodystrophy, the peroxisomal transporter is not an enzyme and certainly would not be expected to be secreted, yet nevertheless the transplantation is effective, possibly through an entirely separate mechanism such as modifying the neuroinflammatory response.
Given the success in these very rare genetic disorders, it is not surprising that microglial therapeutics has been approached for more common disorders such as neurodegenerative diseases, including AD. AD clearly seems to be associated with an innate immune response in that the hypothetically toxic β-amyloid (Aβ) fragments can interact with microglial receptor for advanced glycation end products receptors, complement receptors, and CD36 (Heneka and O’Banion 2007; Hickman, Allison, and El Khoury 2008; Rogers et al. 2002). Whether Aβ itself is a toxin or whether its presence leads to the secretion of soluble factors is unknown. In vivo, AD is characterized by the presence of activated microglia associated with the pathological hallmark of plaques and tangles. Transgenic mouse models have indicated that these plaques have abundant microglia and astrocytes highly reminiscent of the human disease (Fig. 5 ). Bone marrow transplantation experiments have suggested that microglia associated with these plaques may have a bone marrow origin (Simard et al. 2006). However, as mentioned above and extensively discussed in the literature, the precise impact of this microglial activation on the disease process is still uncertain and both detrimental (e.g., release of proinflammatory and neurotoxic mediators) as well as beneficial effects (e.g., amyloid phagocytosis) are postulated (Hickman, Allison, and El Khoury 2008; Ransohoff and Perry 2009; Rogers et al. 2002; Weiner and Frenkel 2006).
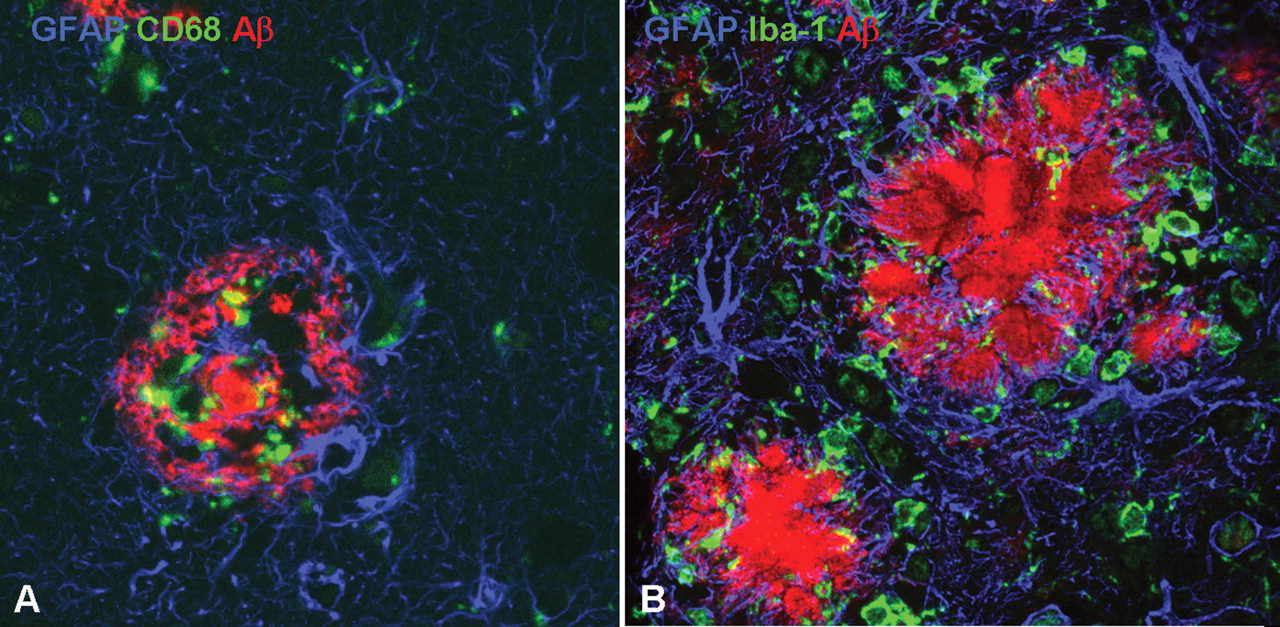
Microglia are essential components of the central nervous system’s innate immune response for clearing putative neurotoxic β-amyloid (Aβ) fibrils. (A) Brain of a patient with Alzheimer’s disease triple immunostained for glial fibrillary acid protein (GFAP) (blue), cluster of differentiation (CD) 68 (green), and Aβ (red). A dense amyloid core plaque (red) is infiltrated by activated macrophages (green) and a surrounding halo of reactive astrocytes (blue). (B) Brain of a transgenic mouse over expressing Aβ triple immunostained for GFAP (blue), ionized calcium-binding adapter molecule–1 (Iba-1) (green), and Aβ (red). A dense amyloid core plaque (red) is surrounded by activated macrophages (green) and reactive astrocytes (blue).
Although it appears that under normal circumstances, the phagocytic capability of microglia is insufficient to remove the amyloid accumulating in AD, several studies have demonstrated dramatic increases in amyloid removal following various interventions (e.g., LPS or interferon-γ injection) or with concomitant diseases leading to microglial activation (DiCarlo et al. 2001; Frenkel et al. 2005; Akiyama and McGeer 2004; Akiyama et al. 1996; Wisniewski, Barcikowska, and Kida 1991). Perhaps one of the most exciting therapeutic approaches in the treatment of AD has been immunization to Aβ (Bard et al. 2000; Buttini et al. 2005; DeMattos et al. 2001; Janus et al. 2000; Schenk et al. 1999; Wilcock et al. 2004; Lemere et al. 2004). After active or passive immunization, there is ample evidence of increased microglial activity with internalization of amyloid and decrease of plaque load in the brain (Bacskai et al. 2001; Wilcock et al. 2004), with an overall switch from an inflammatory to a phagocytic profile (Morgan et al. 2005). However, other microglia-independent mechanisms are also involved in plaque clearance, such as a direct effect of antibodies on Aβ leading to dissolution of amyloid fibrils or neutralization of A-oligomers and capturing of amyloid in the peripheral circulation, resulting in net efflux from the brain (Das et al. 2003; Bacskai et al. 2002; Carty et al. 2006; Klyubin et al. 2008; Solomon et al. 1997; DeMattos et al. 2001; Boche and Nicoll 2008; Weiner and Frenkel 2006; Wilcock et al. 2003, 2004). Results from the first human active immunization trial suggested mild beneficial effects in some clinical outcome measures with slowing of cognitive performance decline in antibody responders (Gilman et al. 2005; Hock et al. 2003). However, in a subset of patients, meningoencephalitis occurred as an adverse event, which has been attributed mainly to an unwanted activation of T-cells (Gelinas et al. 2004; Weiner and Frenkel 2006), but other mechanisms have also been suggested, such as overexuberant microglial activation prompted by opsonization of Aβ and alterations in fluid balance in the brain triggered by interaction of antibody-Aβ immune complexes and the cerebral vasculature (Boche and Nicoll 2008). Currently, several human clinical trials are under way using passive and active immunization approaches with newly developed vaccines that were specifically designed to minimize the risk for unwanted T-cell activation.
Normal aging is associated with an overall more proinflammatory state as well as a decline in both innate and adaptive immune functions, a process termed immunosenescence (Richartz et al. 2005). Analogously, microglial senescence in either normal or pathological aging may be associated with microglial dysfunction and possibly the formation of a distinct chronic neuroinflammatory state. Dystrophic microglia with nonramified atrophic or fragmented tortuous processes have been seen with aging and have been associated with telomere shortening and decreased telomerase activity (Flanary et al. 2007; Streit 2004). A shift toward a proinflammatory state possibly due to decreased CD200 expression by neurons or increased interferon-γ may contribute to this microglial dysfunction (Frank et al. 2006). General functional impairment such as decreased phagocytosis or decreased amyloid clearance can be associated with either increased neurotoxicity or the formation of neurofibrillary tangles (Hickman, Allison, and El Khoury 2008; Richartz et al. 2005; Streit et al. 2008).
Conclusions
In conclusion, microglia are key innate immunity cells of the CNS. They are specifically adapted to sense various types of danger and differentially respond with a classical or alternative reparative response. With our improved understanding of these activations and responses, we have entered a new era in which the modulation of microglia can be proposed as a means of modulating neurological disease. Such modulation could be carried out by simple pharmacological manipulation of microglial elements, more complicated genetic therapy, or even the overall replacement of senescent microglia by bone marrow transplantation. In any event, our greater appreciation of microglial involvement in the innate immune response has opened new vistas in the approach to neurological therapies.