
Abstract
Hepatic drug-metabolizing enzyme (DME) induction is an adaptive response associated with changes in preclinical species; this response can include increases in liver weight, hepatocellular hyperplasia and hypertrophy, and upregulated tissue expression of DMEs. Effects of DME induction on clinical pathology markers of hepatobiliary injury and function in animals as well as humans are not well established. This component of a multipart review of the comparative pathology of xenobiotically mediated induction of hepatic metabolizing enzymes reviews pertinent data from retrospective and prospective preclinical and clinical studies. Particular attention is given to studies with confirmation of DME induction and concurrent evaluation of liver and/or serum hepatobiliary marker enzyme activities and histopathology. These results collectively indicate that in the rat, when histologic findings are limited to hepatocellular hypertrophy, DME induction is not expected to be associated with consistent or substantive changes in serum or plasma activity of hepatobiliary marker enzymes such as alanine aminotransferase, alkaline phosphatase, and gamma glutamyltransferase. In the dog and the monkey, published studies also do not demonstrate a consistent relationship across DME-inducing agents and changes in these clinical pathology parameters. However, increased liver alkaline phosphatase or gamma glutamyltransferase activity in dogs treated with phenobarbital or corticosteroids suggests that direct or indirect induction of select hepatobiliary injury markers can occur both in the absence of liver injury and independently of induction of DME activity. Although correlations between tissue and serum levels of these hepatobiliary markers are limited and inconsistent, increases in serum/plasma activities that are substantial or involve changes in other markers generally reflect hepatobiliary insult rather than DME induction. Extrahepatic effects, including disruption of the hypothalamic-pituitary-thyroid axis, can also occur as a direct outcome of hepatic DME induction in humans and animals. Importantly, hepatic DME induction and associated changes in preclinical species are not necessarily predictive of the occurrence, magnitude, or enzyme induction profile in humans.
Keywords
Introduction
The biotransformation and elimination of lipophilic substances of endogenous or foreign origin is a critical activity performed by living organisms to guard against injury caused by intracellular accumulation of potential toxicants. In animals and man, biotransformation or xenobiotic metabolizing enzyme systems are present in most if not all tissues, with the highest concentration found in liver. Expression of these enzymes is influenced by a range of factors including diet, sex, age, environmental exposures, and most importantly, endobiotics and xenobiotics, including drugs and chemicals. Although drug-metabolizing enzyme (DME) systems are generally present at low levels in the liver, they can be rapidly and reversibly upregulated in response to endogenous or exogenous stimuli, a process known as enzyme induction (Dickins 2004; Xu et al. 2005). An extensive review of DME systems is beyond the scope of this article, however, additional information is available in the introduction to this series (Botts et al. 2010) and in several excellent reviews in the literature (Dickins 2004; Graham and Lake 2008; Handschin and Meyer 2003).
Although changes in liver morphology and hepatic DME, including CYP450 activities, in response to microsomal enzyme induction are well characterized in preclinical species, the effects of microsomal enzyme induction on clinical pathology markers of hepatic injury and function are less commonly described. Increased alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and/or gamma glutamyltransferase (GGT) activities in liver parenchyma have been described in association with drug-induced CYP450 induction in the rat, the dog, and the human. However, reports of increased ALT, AST, ALP, or GGT activity in serum or plasma and their association with DME induction are conflicting in these species. Therefore, the objective of this review is to summarize information available in the literature on the effects of hepatic DME induction on clinical pathology parameters in these preclinical species and in humans. Additionally, select endocrine changes associated with DME induction will also be reviewed.
Hepatic Effects of DME Induction in Animals
Following administration of xenobiotics, enzyme induction is first observed in the liver because of its anatomical location, function, and high concentration of DMEs. Centrilobular hepatocellular hypertrophy, the most common histological change associated with enzyme induction in animals, is an effect considered adaptive and not injurious (Greaves 2007; Schulte-Hermann 1974). However, this adaptive response can be overcome following intense or prolonged induction stimuli, leading to hepatocellular degeneration, necrosis, or proliferation (Williams and Iatropoulos 2002). Hepatobiliary injury can also be an indirect consequence of enzyme induction, resulting from increased formation of highly reactive intermediates that cause oxidative injury or formation of adducts that covalently bind cellular proteins, nucleotides, or DNA/RNA (Klaunig et al. 1998). A more comprehensive review of the morphologic consequences of hepatic microsomal enzyme induction is provided in an accompanying article in this series (Maronpot et al. in press).
Commonly Used Biomarkers for Monitoring Hepatic Effects
Noninvasive monitoring for liver injury is routinely performed by measurement of hepatobiliary markers in plasma or serum. This paper will focus on the activities of ALT, ALP, and GGT, as these enzymes are generally liver associated and often reported to be altered in association with microsomal enzyme induction. Furthermore, these enzymes are recommended as indicators of hepatocellular (ALT activity) and hepatobiliary (ALP and GGT activities) injury in preclinical studies (Boone et al. 2005; Weingand et al. 1996). Glutamate dehydrogenase (GLDH) and α-glutathione S-transferase (αGST) have also been shown to be sensitive indicators of hepatocellular injury, however, use of these markers is less common, and data from prospective studies with prototypic inducer compounds are more limited (Giffen et al. 2002; O’Brien et al. 2002). Aspartate aminotransferase is also used as a marker of liver injury, particularly for sequential monitoring of hepatic injury, where declining values in serial measurements reflect recovery from hepatic injury owing to its shorter circulating half-life than ALT (Meyer and Harvey 2004). Aspartate aminotransferase is generally considered to be less sensitive and less specific for liver (Center 2007; Loeb 1999) and will not be discussed further for preclinical species.
Alanine aminotransferase is present in the highest concentrations in periportal hepatocytes and catalyzes a crucial reaction in gluconeogenesis and the urea cycle. Because of its predominantly cytosolic location, ALT can be rapidly released into the serum following irreversible hepatocellular injury or necrosis. Alanine aminotransferase release also occurs following reversible hepatocellular injury, generally thought to occur via cytoplasmic blebbing (Lemasters et al. 1990). Distinguishing among these conditions is generally not possible based on circulating ALT activity alone. However, reversible or less extensive injury is generally associated with changes of smaller magnitude than irreversible or widespread cellular injury (Lassen 2004; Solter 2005). Increases in serum ALT activity are not liver specific, as increased serum ALT has been reported following strenuous exercise or severe muscle necrosis in dogs and restraint in cynomolgus monkeys (Center 2007; Landi et al. 1990; Solter 2005; Valentine et al. 1990). In the future, separate analysis of the two isoforms of ALT in serum may provide a novel method for better discrimination between tissue origin, severity of hepatic injury, and mechanism of increase, as ALT1 and 2 isoenzymes with unique subcellular and tissue localizations and patterns of response to metabolic or xenobiotic stimuli are described in rodents, dogs, and humans (Glinghammar et al. 2009; Goldstein et al. 2007; Jadhao et al. 2004; Lindblom et al. 2007; Liu et al. 2009; Rajamohan et al. 2006; Thulin et al. 2008; Yang et al. 2009).
In animals, increases in serum ALP activity are most commonly associated with hepatobiliary or bone changes. Unlike ALT, ALP is a membranous enzyme located primarily on the canalicular surface attached by a glycosyl phosphatidyl inositol (GPI) anchor. In cholestatic disease, ALP release is mediated by endogenous phospholipases in association with increased serum concentrations of bile acids. Upregulated ALP expression in the absence of biochemical or morphologic evidence of cholestasis has also been observed on canalicular and basolateral surfaces of hepatocytes and biliary epithelia. Alkaline phosphatase activity is also not liver specific. In the rat, the intestinal isoform is the predominant ALP isoform in serum, with increases in serum ALP activity following food consumption and decreased activity associated with fasting (Center 2007; Hoffmann et al. 1999; Solter 2005). In the dog, increased serum ALP activity can also result from induction of a dog-specific, glucocorticoid-induced ALP isoenzyme in response to increases in endogenous or exogenous corticosteroid hormones (Solter et al. 1994).
The liver is the primary source of serum GGT activity, where the enzyme is a membrane-bound protein found on hepatocytes and biliary epithelia that plays a critical role in cyclic regeneration of amino acids from extracellular glutathione for synthesis of intracellular glutathione. As such, GGT plays a pivotal role in cellular detoxification. Similar to ALP, increased serum GGT activity during cholestasis is almost certainly a result of the release of membrane-bound GGT in part or wholly by bile acids. Increased GGT activity therefore can occur in animals as a result of both cholestasis and biliary epithelial injury. Gamma glutamyltransferase is considered less sensitive than ALP for detection of cholestasis in both the dog and the rat (Bain 2003; Center 2007; Leonard et al. 1984; Loeb 1999).
Overview of Hepatobiliary Biomarker Changes Associated with Prototypic Hepatic DME Induction in Preclinical Species
Reports associating induction of microsomal DMEs with increased liver or blood ALT, ALP, or GGT activities are available in the preclinical literature. However, this review revealed extensive differences in both study design and analytic methodologies across studies included in these reports. One of the goals of this article is to provide a retrospective evaluation of data from rat and dog studies of prototypic inducers where concurrent assessment of hepatic DME induction and liver and serum or plasma ALT, ALP, and/or GGT activities were reported. Retrospective studies of preclinical toxicology data from rats, dogs, and monkeys will also be reviewed. Additionally, a summary of findings for rats given prototypic inducer compounds as part of a recent prospective hepatotoxicogenomics project—including confirmation of DME induction, liver histopathology, and changes in ALT, ALP, and GGT activity—will be presented. Effects of DME, especially phase II enzyme induction on the thyroid-hypothalamic-pituitary axis, will also be discussed.
Corticosteroid-mediated Induction of Hepatobiliary Biomarker Enzyme Activities in the Rat and the Dog
The finding of marked increases in liver ALT activity in rats given cortisol acetate has long been considered as evidence that ALT is an inducible enzyme (Haining 1970; Rosen et al. 1959). Dexamethasone, a more potent glucocorticoid than cortisol and a potent inducer of CYP3A in the rat (Lake et al. 1998; Martignoni et al. 2004), is also associated with increases in hepatic and serum ALT activities in rats (Jackson et al. 2008; O’Brien et al. 2002). In these studies, increases in serum ALT activities consistently exceeded increases in hepatic ALT activities. Importantly, histopathologic findings were limited to hepatic glycogen deposition with minimal to mild multifocal areas of hepatocellular necrosis (Jackson et al. 2008). Because liver histologic findings were of mild severity and disproportionate relative to the large magnitude of concurrent serum ALT increases, pharmacologically mediated induction of hepatic gluconeogenesis was considered as a potential mechanism for these findings (Jackson et al. 2008). This mechanism was considered particularly relevant, as glucocorticoids are known to play a central role in the metabolic response to stress via induction of hepatic gluconeogenesis, primarily via upregulation of phosphoenolpyruvate carboxykinase (PEPCK) (Jin et al. 2004; Sapolsky et al. 2000).
In another study, in which male Sprague Dawley rats were given increasing doses of dexamethasone (0.01 to 300 mg/kg/day) for four days (Ennulat et al. 2010), hepatic expression of CYP3A3 and ALT1 mRNA (but not hepatic ALT activity) were evaluated in parallel with clinical chemistry and liver histopathology. In this study, progressive, dose-responsive increases in serum ALT activity were observed and greatly exceeded increases in liver ALT1 and CYP3A3 mRNA expression, both of which peaked at approximately three-fold of control means at doses ≥30 mg/kg/day (Figure 1). Hepatocellular single-cell necrosis of minimal severity was observed in rats given ≥3 mg/kg/day, with no increase in severity or incidence at doses ≥30 mg/kg/day. These findings are consistent with previous studies of glucocorticoids, where the magnitude of increases in serum ALT activities were larger than expected when based on the minimal severity of liver histopathologic changes (Jackson et al. 2008). Furthermore, comparable increases in serum ALT activity occurred in rats both with and without evidence of hepatocellular necrosis. Therefore, the progressive, dose-responsive increase in serum ALT activity in the absence of a dose-responsive increase in either hepatocellular necrosis or ALT1 or CYP3A3 expression suggest a post-transcriptional mechanism for the increase in serum ALT activities. Although dexamethasone is a known inducer of CYP3A in the rat, the data from these studies (Ennulat et al. 2010; Jackson et al. 2008) support that glucocorticoid-mediated induction of liver and/or serum ALT activities in these rats is more consistent with direct pharmacological or hepatotoxic events rather than changes associated with CYP450 induction.
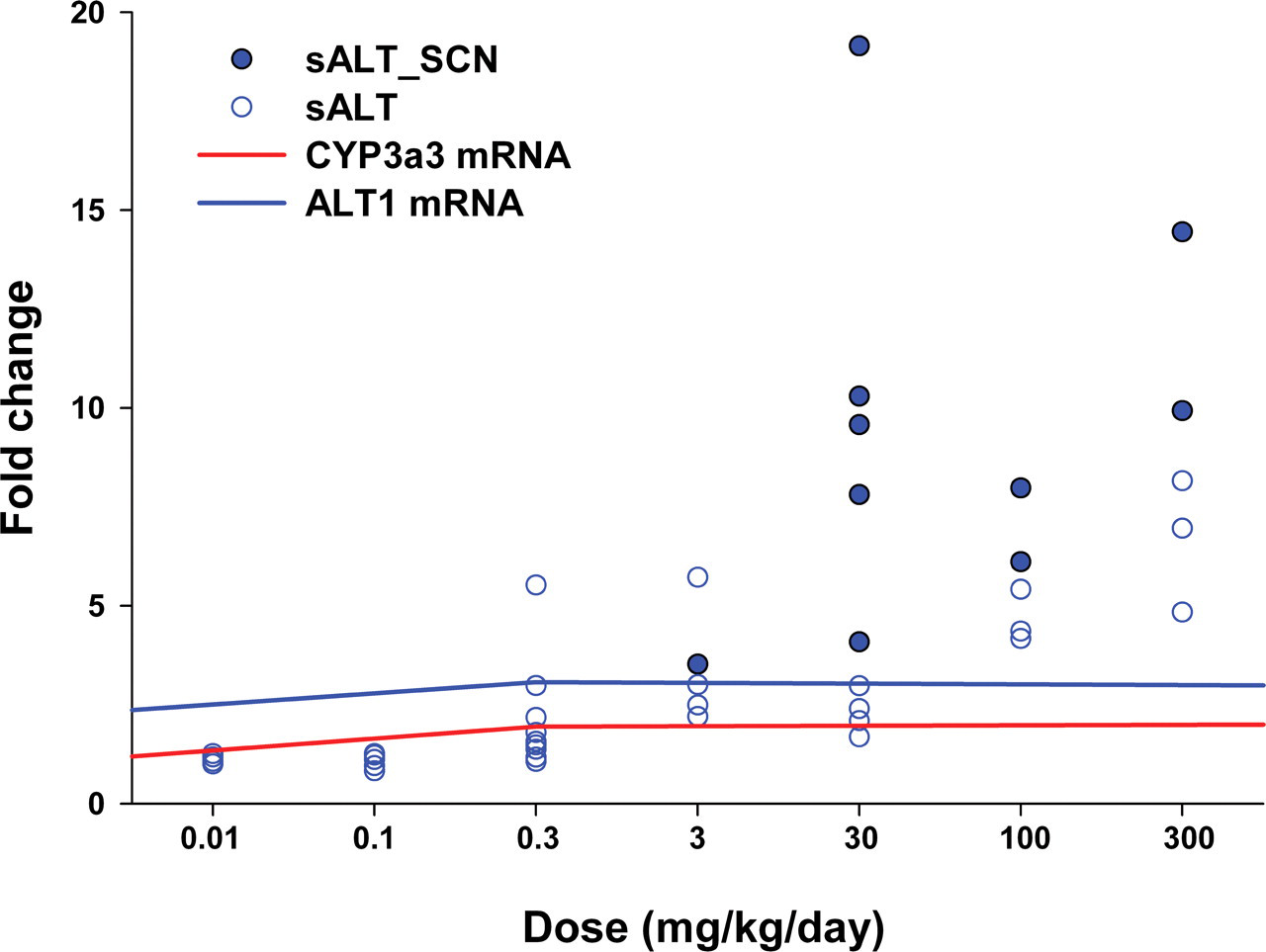
Serum alanine aminotransferase (sALT) and hepatic CYP3a3 and ALT1 mRNA changes in male Sprague Dawley rats given ascending doses of dexamethasone for four days. Mean hepatic CYP3a3 and ALT1 mRNA expression and individual rat serum ALT activities are normalized to concurrent control mean. For serum ALT values, open circles represent rats without microscopic evidence of hepatocellular necrosis, and solid circles represent rats with minimal hepatocellular single-cell necrosis.
The hepatic response of the dog to corticosteroid administration differs from that of other animals, particularly the rat and the monkey, in the following ways: (1) dexamethasone has no effect or reduces CYP activity in dog hepatocytes in vitro (Jayyosi et al. 1996; Lu and Li 2001); (2) the dog is also unique in expression of a corticosteroid-inducible isoenzyme of ALP (CIALP) in addition to the liver- and bone-specific ALP isoenzymes found in the circulation of the dog and other species (Hoffmann et al. 1999). In dogs given prednisone, increases in hepatic ALP and GGT, but not ALT, activities are seen, often along with increases in serum ALP, GGT, and ALT activities (Rutgers et al. 1995; Solter et al. 1994). Increased serum activity of the liver ALP isoenzyme (LALP) can occur following a single dose of prednisone in the dog (Wiedmeyer et al. 2002a); however, significant increases in serum CIALP activity do not occur before seven to ten days of prednisone administration (Solter et al. 1994; Wiedmeyer et al. 2002b). These differences are related in part to different regulatory mechanisms, as the onset of hepatic CIALP mRNA expression is delayed in comparison to LALP mRNA (Wiedmeyer et al. 2002b). Corticosteroid-mediated induction and release of both ALP and GGT into serum in the dog occurs independently of cholestasis, as both hepatic and serum bile acid concentrations are unchanged in dogs given prednisone (Solter et al. 1994). Although these increases in hepatic and serum ALP and GGT activities are attributed to induction, the increased serum ALT activities in these studies may be related to hepatotoxicity, because increases of hepatic ALT were not seen. Although histopathology was not performed in these studies, other studies of dogs given comparable doses of prednisone consistently demonstrate hepatocellular glycogenosis and hypertrophy with focal hepatocellular necrosis (Fittschen and Bellamy 1984; Rutgers et al. 1995). Therefore, corticosteroid-mediated induction of ALP and GGT in dogs is a unique, pharmacologically mediated event that is not related to cholestasis or induction of hepatic DME activity.
Hepatobiliary Biomarker Findings in Association with Prototypic Hepatic Enzyme-inducing Agents in the Rat and the Dog
Our current understanding of the relationship between liver and serum hepatobiliary biomarker enzyme activities and DME induction in rats and dogs is based on numerous, yet widely varied published studies of prototypical DME inducers in these species (Tables 1–4). In rodents, the most common inducers evaluated were phenobarbital (PB), ethanol, and PPARα agonists (Tables 1 and 3). In dog, hepatic metabolizing enzyme induction was most commonly reported for anticonvulsant drugs such as PB, phenytoin (PHE), or primidone (PRI, Table 2).
Changes in liver parameters in rats treated with various drugs associated with hepatic drug-metabolizing enzyme induction

Abbreviation: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GGT, gamma glutamyltransferase; NM, not measured.
a Fold change compared to concurrent controls.
b Homogenate, per g liver weight unless otherwise specified.
c Plasma membrane fraction.
d Microsomal fraction.
Changes in liver parameters in dogs treated with various drugs associated with hepatic drug-metabolizing enzyme induction.
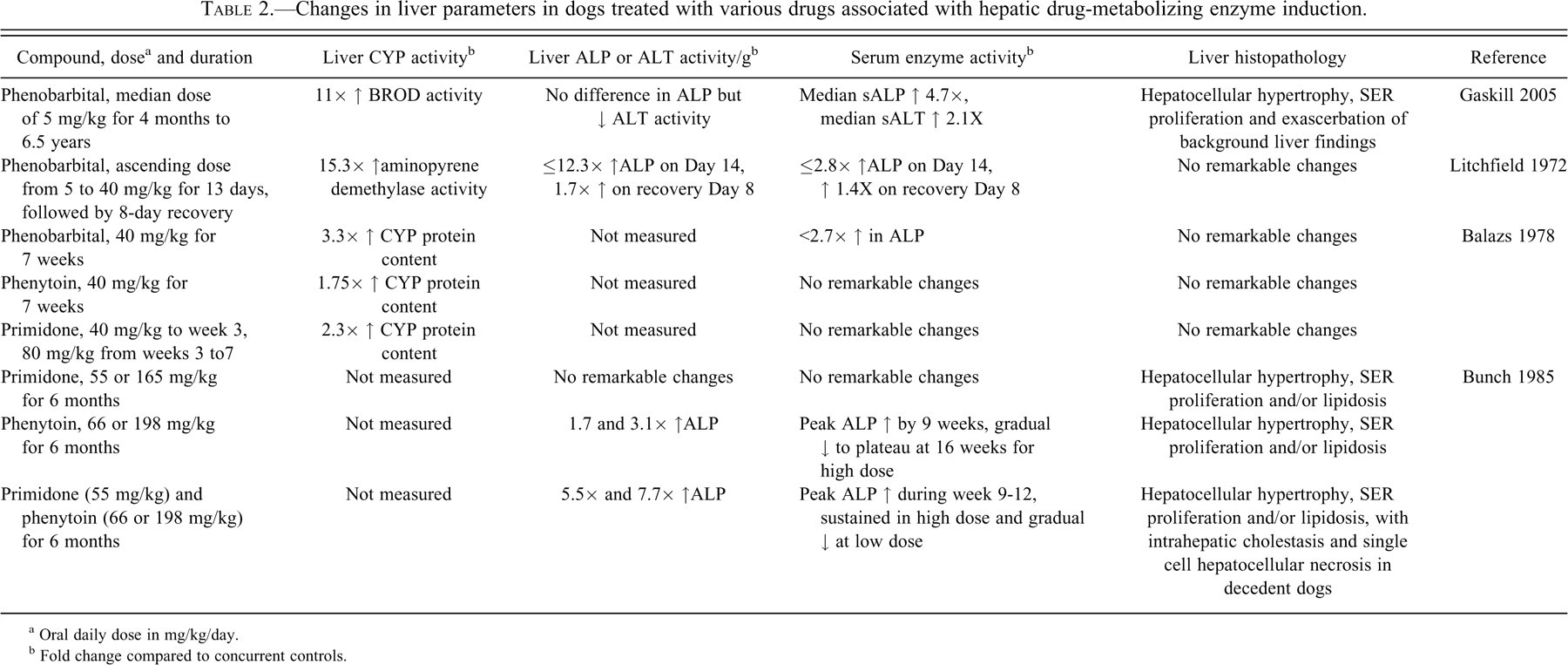
a Oral daily dose in mg/kg/day.
b Fold change compared to concurrent controls.
Changes in liver parameters in rat models of chronic ethanol consumption.

Abbreviation: LDC; Lieber DeCarli diet, CHO; carbohydrate.
a Fold change compared with concurrent controls.
b Homogenate.
c Ethanol.
Pathology findings and CYP gene expression changes in rats given prototypic drug-metabolizing enzyme inducer compounds
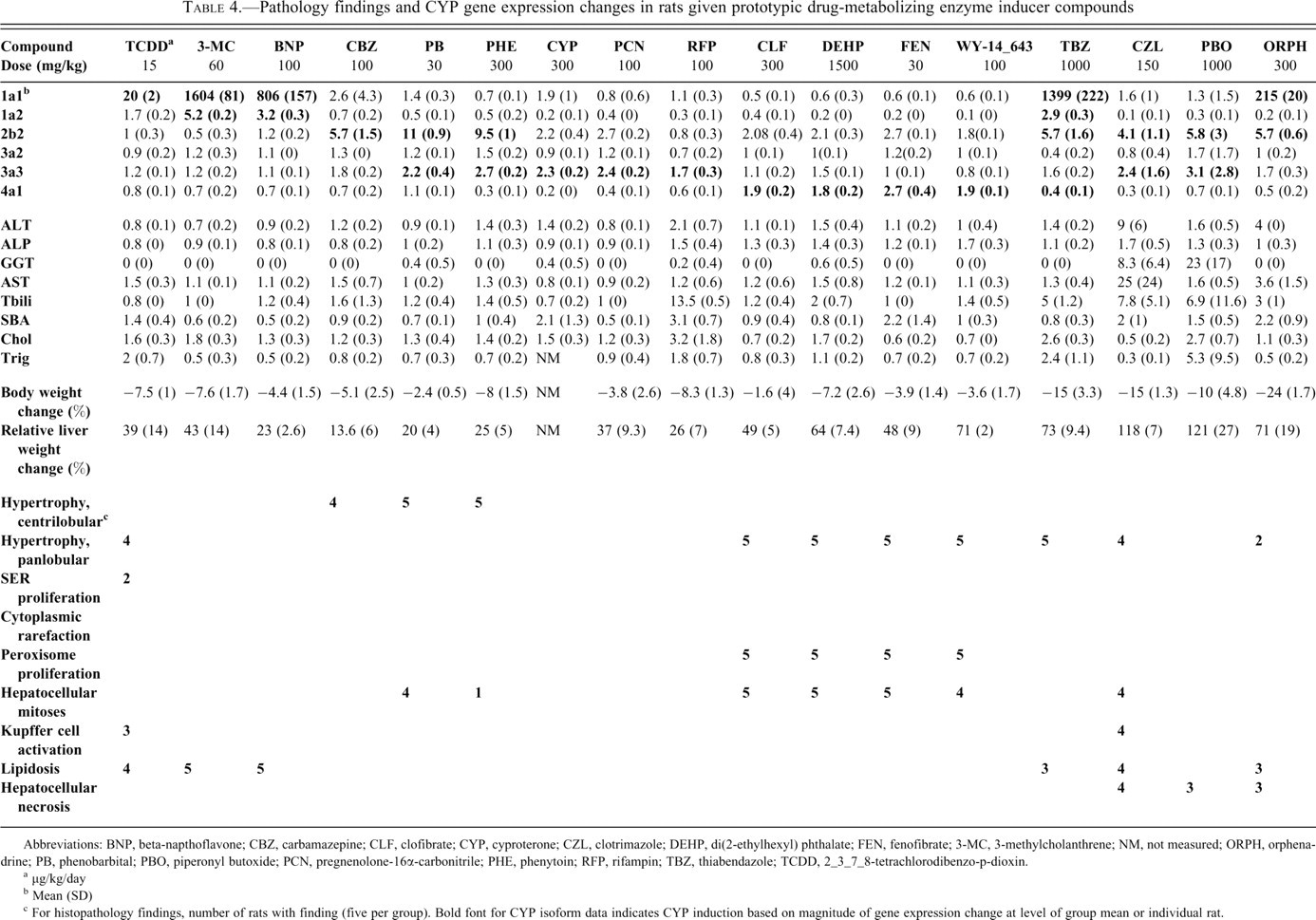
Abbreviations: BNP, beta-napthoflavone; CBZ, carbamazepine; CLF, clofibrate; CYP, cyproterone; CZL, clotrimazole; DEHP, di(2-ethylhexyl) phthalate; FEN, fenofibrate; 3-MC, 3-methylcholanthrene; NM, not measured; ORPH, orphenadrine; PB, phenobarbital; PBO, piperonyl butoxide; PCN, pregnenolone-16α-carbonitrile; PHE, phenytoin; RFP, rifampin; TBZ, thiabendazole; TCDD, 2_3_7_8-tetrachlorodibenzo-p-dioxin.
a μg/kg/day
b Mean (SD)
c For histopathology findings, number of rats with finding (five per group). Bold font for CYP isoform data indicates CYP induction based on magnitude of gene expression change at level of group mean or individual rat.
There are rare reports in which PB was shown to minimally increase hepatic ALT activity in the rat (Boll et al. 1998). However, effects on serum activity are unknown, as concurrent serum ALT activity was not evaluated, and to our knowledge, hepatic ALT activities have not been reported in repeat-dose studies in rats given PB. In contrast, rats given comparable doses of PB for up to five days exhibit increased liver GGT activity in association with hepatic DME induction without concomitant increases in serum GGT activity (Goldberg et al. 1981; Ratanasavanh et al. 1979; Roomi and Goldberg 1981). Notably, the magnitude of hepatic GGT increase was different between these three studies and was related to whether the whole liver or the hepatic plasma membrane fraction, the predominant location of liver GGT, was evaluated. The lack of concordance between liver and serum GGT activities is not surprising, given the low baseline activity (Lahrichi et al. 1982) and exceptionally short circulating half-life of GGT in the rat (Boyd 1983; Huseby 1992; Loeb 1999).
In the dog, induction of hepatic DME activity with increasing doses of PB was associated with increases in both hepatic and serum ALP activities (Litchfield and Conning 1972). Similar increases in hepatic and serum ALP activities were noted in dogs given comparable doses of PB for up to nine weeks (Balazs et al. 1978; Unakami et al. 1987). In contrast, in a prospective study of dogs given therapeutic doses of PB for up to six years, no effect on hepatic ALP or ALT activities were observed, despite increases in serum ALP and ALT activities and confirmation of hepatic DME induction by benzyloxyreso-rufin-O-dealkylase activity as a measure of CYP2B11, and CYP2B immunoblotting in liver homogenate (Gaskill et al. 2005). The cause of this discrepancy is unclear, but it may be related to methodologic differences in tissue extraction techniques, as whole homogenates rather than membrane fractions were evaluated in the latter study (Gaskill et al. 2005). In dogs given PHE alone or in combination with PRI, induction of hepatic DME activity was associated with dose-responsive increases in both hepatic and serum ALP activities (Bunch et al. 1985); however, serum ALP activities declined to baseline levels over time. For all of these studies, the predominant histologic findings at study termination were hepatocellular hypertrophy and SER proliferation, with exacerbation of spontaneous or background hepatic pathology in dogs given the lower pharmacologic doses of PB, and hepatocellular necrosis and intrahepatic cholestasis in the decedent dogs given PRI and PHE in combination. Based on the lack of prominent hepatobiliary pathology in all but dogs given the combination PHE/PRI, a relationship between hepatic metabolizing enzyme induction and increases in serum and/or liver ALP activities in the dogs in these studies cannot be excluded. Furthermore, although hepatic ALP activity in dogs given PB or PHE alone or in combination appears to not correlate with serum ALP activities, the disproportionate changes in liver and serum hepatobiliary marker enzyme activities in these studies do not exclude the potential for serum and hepatic levels to both reflect induction. Rather, differences in serum and hepatic enzyme activities may be related to methodologic differences in tissue extraction or to differences between serum and tissue half-life, as rate of clearance is a major determinant of serum activity of hepatobiliary marker enzymes (Boyd 1983).
Other microsomal enzyme inducers have also been studied for effects on liver and/or serum hepatobiliary marker enzyme activity. Fibrate hypolidemics and other PPARα agonists are potent inducers of CYP4A in the rat (Peterson et al. 2004; Sabzevari et al. 1996). Minimal increases in serum ALT and/or ALP activity were seen in rats given WY-14643, WAY-144112, and clofibrate and were associated with increased hepatic ALP, but not ALT, activities for WY-14643 and WAY-14112 (Kramer et al. 2003; O’Brien et al. 2002; Peterson et al. 2004). Importantly, increases in enzyme activities for these PPARα agonists were accompanied only by centrilobular hypertrophy. Chronic administration of aminopyrine, a nonspecific CYP substrate in rats, also causes CYP450 induction, based on increases in both microsomal enzyme activities and protein content, and was associated with an up to eight-fold increase of hepatic GGT activity, and an approximately three-fold increase in serum GGT activity (Satoh et al. 1982). As above, histologic findings in this study were limited to hepatocellular hypertrophy. These examples are difficult to further interpret in terms of relationship between DME induction and serum hepatobiliary marker enzyme activity because of differences in CYP enzyme induction profile and/or mechanism of induction, and lack of corroborative information from other studies.
Chronic ethanol consumption in rodents and man is commonly associated with induction of CYP2E1 and increases in serum and liver GGT activities (Roberts et al. 1995; Teschke et al. 1983). Despite extensive methodological differences between rat chronic ethanol consumption models, particularly with regard to ethanol dosage and dietary carbohydrate content, increased hepatic GGT activities consistently paralleled increases in serum GGT activities (Table 3). In contrast, increases in hepatic and/or serum ALT activities were less commonly observed, and no effects on liver ALP activities were reported despite sporadic mild increases in serum ALP activities. Although induction of hepatic DME activity was not assessed in these rat chronic ethanol consumption studies, in comparable rat dietary models of chronic ethanol consumption, ethanol was shown to cause up to a 2.5-fold increase in CYP2E1 protein (Roberts et al. 1995). When histopathology was assessed, microscopic findings were limited to lipid accumulation and increased membrane GGT activity in centrilobular hepatocytes (Misslbeck et al. 1986), or panlobular hepatocellular lipidosis in association with increased hepatic CYP2E1 protein (Oh et al. 1998). These data suggest that hepatic GGT induction can be associated with CYP induction and increases in serum GGT activity in the rat.
In summary, these studies of prototypic inducer compounds in the rat and the dog demonstrate that the relationship between induction of hepatic DME activity and increases in liver or serum hepatobiliary marker enzyme activities is both compound and species-specific.
Retrospective Analyses of Rat, Dog, and Monkey Toxicology Study Data
Retrospective analyses of toxicology studies on new chemical entities provide an additional resource for evaluation of the relationship between hepatic microsomal enzyme induction and clinical pathology parameters in rats, dogs, and monkeys. For each toxicology study, end points evaluated included liver weight and histopathology, clinical chemistry, and hepatic CYP450 content and activity. Importantly, hepatic hepatobiliary marker enzyme activity was not assessed, as this is not a standard end point in preclinical toxicology studies used for human risk assessment (Boone et al. 2005; Weingand et al. 1996).
For the rat, data were reviewed from toxicology studies in male and/or female Fischer or Sprague Dawley rats given ten new chemical entities from five different therapeutic classes for approximately two weeks to three months (Amacher et al. 1998). Centrilobular or panlobular hepatocellular hypertrophy was associated with increases in absolute liver weights of >20%. However, there was no relationship between the magnitude of liver weight increase or hepatocellular hypertrophy and the degree of CYP450 induction. Importantly, CYP450 induction, hepatocellular hypertrophy, and increased absolute liver weights, in the absence of other histologic findings, were not associated with changes in serum ALT activity or other measured serum hepatic enzymes. For the compounds used in these studies, the results indicated that CYP450 induction and secondary hepatic adaptive changes were not associated with changes in serum indicators of hepatic injury in the rat.
In the retrospective analyses of preclinical toxicology study data for candidate or investigative drugs in the dog (Amacher et al. 2001) and monkey (Amacher et al. 2006), increased microsomal enzyme activity, consistent with hepatic DME induction, was seen in one or both sexes for eight of nine compounds from five different pharmacological classes in the dog, and for nine of ten compounds from four different pharmacological classes in the monkey. Microsomal enzyme induction in both species was variably associated with hepatocellular hypertrophy and relative liver weight increases, but no changes in clinical chemistry parameters or histopathology findings indicative of hepatotoxicity were seen in either species.
Prospective Evaluation of the Association between CYP Induction and Hepatobiliary Marker Changes in the Rat
Data collected from rats given prototypic inducer compounds as part of a recent hepatotoxicogenomics project were also evaluated to assess the relationship between DME induction and changes in serum ALT, ALP, and GGT activity (Ennulat et al. 2010). In this study, male Sprague Dawley rats (N = 254) were given seventeen prototypic inducer compounds for four days by oral gavage (Table 4). Study end points collected on Day 5 included histopathology, clinical pathology, body and relative liver weight, and CYP450 gene expression. Data were normalized across studies to concurrent control mean, such that individual values for treated animals were expressed as a fold change of the control mean for liver weight, CYP450 mRNA expression, and serum ALT and ALP activity. Absolute rather than normalized values were assessed for GGT because of the low level of GGT activity in the rat (Huseby et al. 1992; Loeb 1999). CYP450 gene thresholds used to identify rats with DME induction were ≥ five-fold upregulation of CYP450 1A1 or 2B2 and ≥ two-fold upregulation of 1A2, 3A2, or 4A1 mRNA. Based on these criteria, enzyme induction was present in all seventeen compounds. Microscopic findings consisted primarily of hepatocellular hypertrophy and lipid or glycogen accumulation. Peroxisome proliferation and increased hepatocellular mitoses were observed for several of the PPARα agonists, Kupffer cell activation was noted in rats given 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin and clotrimazole, and a dose-responsive increase in hepatocellular single-cell necrosis was identified in rats given clotrimazole, piperonyl butoxide, and orphenadrine. With the exception of rats given treatments which caused necrosis, minimal (<1.5-fold) increases in ALT, ALP, and GGT activities were the most common serum clinical chemistry findings. Rats given clotrimazole, piperonyl butoxide, and orphenadrine had concurrent dose-responsive increases in total bilirubin and bile acids of up to eight- and 2.2-fold, respectively.
Because of the strong association between ALT changes and hepatocellular necrosis, changes in serum ALT, ALP, and GGT activity were evaluated for all seventeen inducer compounds, and then separately for the three compounds that caused necrosis and the fourteen inducer compounds that did not elicit necrosis (Figure 2). Mean serum ALT, ALP, and GGT activities were only minimally increased in treated rats relative to controls, however, the greatest increases and variability of these enzymes was seen in rats given the inducer compounds that also caused hepatocellular necrosis. When the compounds that caused necrosis were excluded, ALT and GGT and, to a lesser extent, ALP activities decreased to levels comparable to those seen in control rats. These findings provide further support that hepatic DME induction in the absence of hepatocellular degeneration or necrosis is not associated with noteworthy increases in serum ALT, ALP, or GGT activities in the rat.
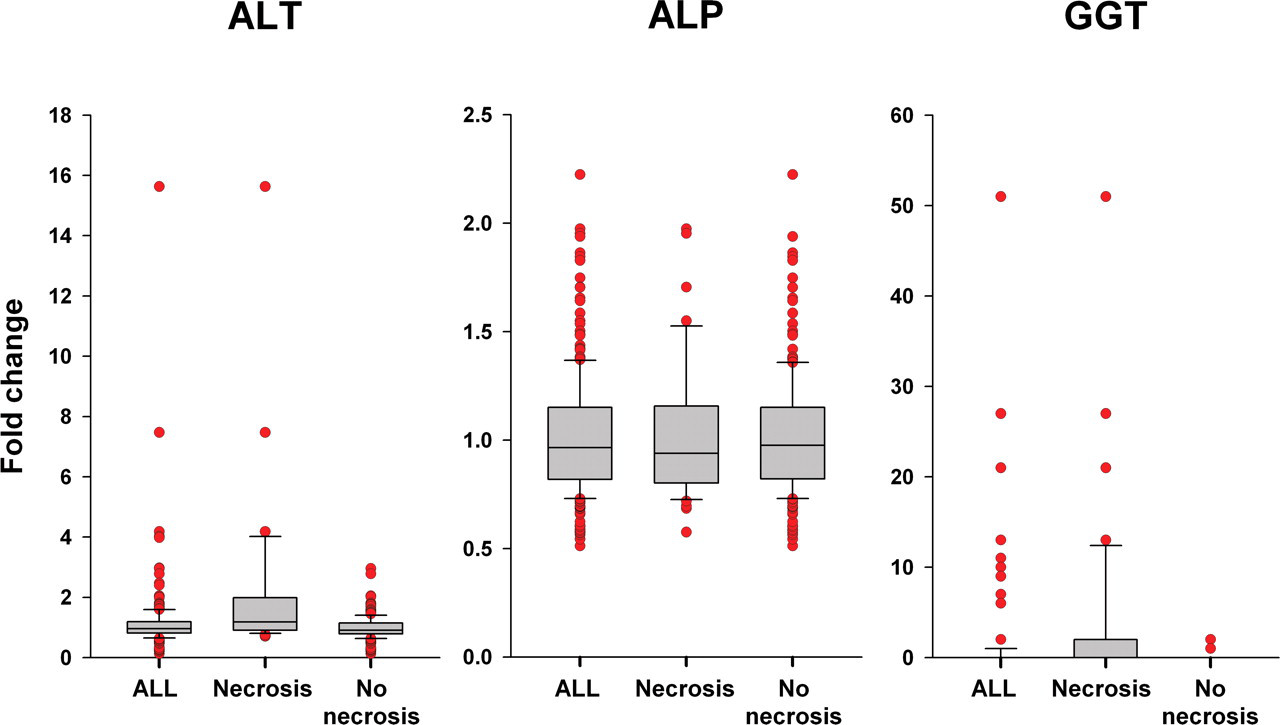
Serum alanine aminotransferase, alkaline phosphatase, and gamma glutamyltransferase changes for prototypic DME inducer compounds in the presence and absence of hepatocellular necrosis. Boxplots of individual animal fold change data for alanine aminotransferase, alkaline phosphatase, and gamma glutamyltransferase from left to right for all animals (N = 17 compounds, <244 rats), animals given inducer compounds that caused necrosis (n<42 rats given clotrimazole, piperonyl butoxide or orphenadrine), and animals given prototypic inducer compounds that did not cause necrosis (n = 15, <202 rats). The top, bottom, and line through the middle of the box correspond to the 75th percentile (top quartile), 25th percentile (bottom quartile), and 50th percentile (median), respectively. The whiskers extend to the 10th percentile and 90th percentiles, and outlier values are represented by red circles.
Effects of Enzyme Induction on Endogenous Thyroid Hormones in Preclinical Species
In rodents and other species, alteration of the hypothalamus-pituitary-thyroid axis can be a secondary consequence of enzyme induction. Most notably, PB administration in rats has been associated with decreased thyroxine (T4) and tri-iodothyronine (T3) owing to induction of liver UDP-glucuronyltransferase (UGT) and accelerated biliary excretion (O’Connor et al. 1999). Decreases in functional T4 and T3 levels were further associated with compensatory increases in circulating thyroid-stimulating hormone (TSH) concentration and increased thyroid gland weights following four weeks of PB administration (O’Connor et al. 1999). These findings have been replicated in numerous comparable studies in Sprague Dawley rats (Barter and Klaassen 1994; Capen 1997; Hood et al. 2003, Hood, Hashimi et al. 1999; Hood, Liu et al. 1999; McClain et al. 1989). Development of thyroid follicular hypertrophy, hyperplasia, and progression to neoplasia occured with chronic PB administration in rats (Capen 1997; McClain et al. 1989; Meek et al. 2003); however, similar patterns of thyroid hormone changes without thyroid tumorigenesis were seen in rats given other microsomal enzyme inducing compounds including 3-methylcholanthrene and pregnenolone-16-α-carbonitrile (Barter and Klaassen 1994; De Sandro et al. 1992; Saito et al. 1991).
The Sprague Dawley rat appears to be unique in the development of thyroid tumors with chronic PB administration. Mice and other laboratory rat strains chronically given PB do not develop thyroid neoplasia (Kato et al. 2005; Kato et al. 2008; Lecureux et al. 2009; Meek et al. 2003), presumably owing to negligible effects on TSH and thyroid follicular cell proliferation following chronic PB administration (Hood et al. 2003; Viollon-Abadie et al. 1999). Thyroid tumorigenesis is also not reported in dogs, which develop similar thyroid hormone changes with chronic PB administration (Gaskill et al. 1999). Further, thyroid carcinogenesis has not been seen clinically in patients on chronic PB regimens (Capen 1997; Meek et al. 2003).
The marked differences in thyroid tumor susceptibility between different animal species and rat strains treated with PB is probably related to differences in thyroid hormone homeostatic mechanisms, as well as the specific DME changes that influence thyroid function. Notably, although induction of T4- and T3-UGTs has been the most well-studied mechanism proposed to explain thyroid hormonal effects of PB in rats, additional factors affecting thyroid hormone metabolism may be linked to hepatic DME induction, including induction of sulfotransferases and hepatic T4 and T3 transporters, inhibition of deiodinases, and differences in circulating half-life of thyroid hormones or binding proteins (Kato et al. 2005; Kato et al. 2008; Lecureux et al. 2009; McClain et al. 1988; McClain 1995; Qatanani et al. 2005).
Conclusions on DME Induction and Clinical Pathology Biomarkers in Preclinical Species
Induction of DME activity can be associated with upregulation of liver activities of the hepatobiliary biomarker enzymes ALT, ALP, and GGT in rats, but the pattern and magnitude of increase of these enzymes in both the liver and the serum vary greatly between different compound classes in the rat. Additionally, the discrimination between whether increases in these hepatobiliary biomarkers in the serum is owing to upregulation within the liver or release from hepatocytes following liver injury requires both concurrent assessment of liver histopathology with clinical chemistry and consideration of marker biology and compound pharmacology. Based on data from published rat studies and in particular, results of the prospective study with prototypic inducer compounds, the weight of evidence suggests that in the absence of liver histopathologic findings other than hypertrophy, CYP induction alone is not associated with changes in serum or plasma activity of hepatobiliary marker enzymes in the rat.
In nonrodents, based on retrospective evaluation of data from dog and monkey preclinical toxicology studies in which hepatic DME induction was confirmed, there is also no consistent relationship between hepatic microsomal induction and hepatotoxicity, as reflected in changes in clinical chemistry parameters. However, induction of liver ALP activity in both phenobarbital- or corticosteroid-treated dogs suggests that release of serum liver enzymes can occur independently of liver injury or induction of DME activity.
Hepatic Metabolic Enzyme Induction and Associated Clinical Pathology Changes in Humans
In contrast with published preclinical information, clinical reports on DME induction have focused predominantly on effects on pharmacokinetics properties, drug–drug interactions, and bioactivation specific to a xenobiotic rather than relationship to liver pathology. Among clinical investigations of enzyme induction that have included effects on routine laboratory findings, reports with comparative liver histologic or functional end points are particularly limited. Thus, interpretation of the significance of the clinical pathology findings in humans exposed to prototypic DME-inducing agents has been largely based on empirical and indirect observations. These reports on associations between DME induction and human clinical pathology findings have predominantly focused on CYP families 1–3, the phase I enzymes responsible for metabolism of a majority of marketed drugs (Dresser et al. 2000; Tanaka 1998). The phase II UGT enzymes, which are responsible for the majority of drug conjugation reactions, have also received attention for influencing human clinical pathology (Evans and Relling 1999). Administration of prototypical agents that induce these DMEs and associated alterations in laboratory biomarkers thus forms the basis for the current understanding of the relationship between induction and clinical pathology results in humans. As with preclinical studies, collective results of these published clinical reports have generally not supported an effect on routine laboratory end points that is directly or consistently linked with phase I and/or II DME induction.
Hepatobiliary Biomarker Findings in Association with Prototypic Hepatic-inducing Agents in Humans
The drugs that have been most commonly studied clinically for the relationship between DME and laboratory findings are the early generation antiepileptics (Table 5 ), which include PB, PHE, PRI, and carbamazepine (CBZ) (Perucca et al. 1984). Clinical consequences of DME induction with the anti-mycobacterial rifampicin are also described in the literature. Although these agents induce a variety of human CYPs, they share the potential of profound induction of CYP3A4, rendering CYP3A4 a particularly common focus of studies on clinical effects of DME induction (Mohutsky et al. 2010). Extrapolation of findings related to the CYP3A subfamily from humans to animals is problematic. For example, CYP3A4 is highly inducible through PXR ligand binding (Hewitt et al. 2007; Jones et al. 2000) and only weakly induced through CAR in humans (Faucette et al. 2006), in contrast with induction of CYP3A by both PXR and CAR in rodents (Graham and Lake 2008). Even within human populations, CYP3A4 inductive responses vary widely (Hewitt et al. 2007; Wilkinson 1996). Beyond inter-individual differences in exposure to inducers, age, obesity, and even exercise level may affect CYP3A4 activity (Anderson 2008; Kotlyar and Carson 1999; Meijer et al. 2001). Acute-phase inflammatory reactants can profoundly inhibit the extent of CYP3A4 induction (Cooper et al. 2008; Jover et al. 2002). These variables likely contribute to a greater extent to the conflicting information on clinical pathology findings with DME induction in man than in traditional preclinical studies in comparatively homogeneous laboratory animal species with comparable CYP-inducing agents.
Serum or plasma hepatobiliary marker enzyme changes in adults and children given anticonvulsants.
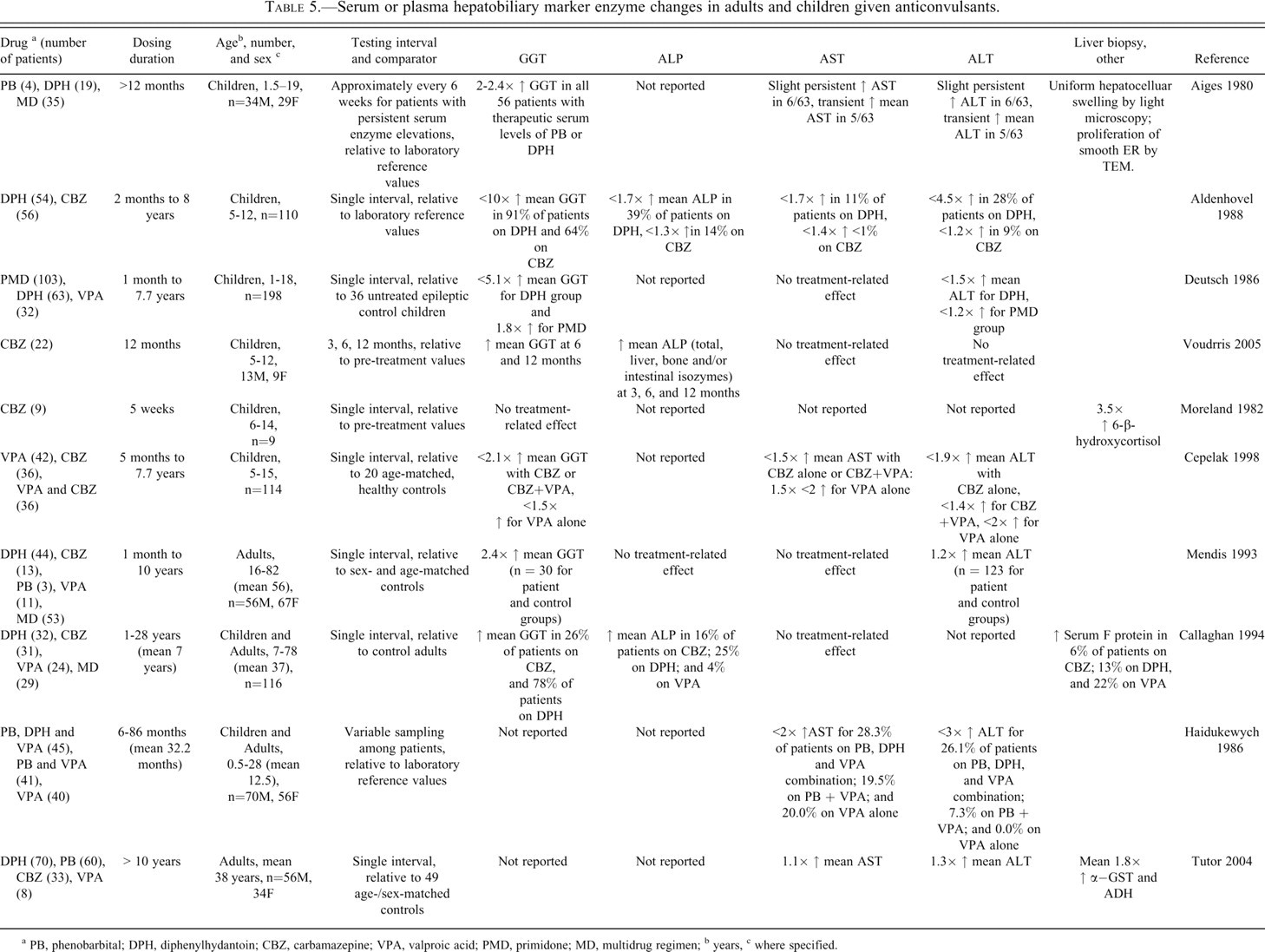
a PB, phenobarbital; DPH, diphenylhydantoin; CBZ, carbamazepine; VPA, valproic acid; PMD, primidone; MD, multidrug regimen;
b years,
c where specified.
A majority of clinical studies reporting clinical pathology alterations with CYP3A4-inducing antiepileptics have involved children. Children are considered particularly susceptible to DME induction, partly because of a greater liver-to-body weight ratio and differences with adults in metabolic and protein binding capacity, and renal and hepatic function (Ginsberg et al. 2004; Kennedy 2008; Moreland et al. 1982). Hormonal changes with puberty are thought to further influence CYP3A4 regulation (Kennedy, 2008). In an exemplary study of children receiving long-term PB and/or PHE (Aiges et al. 1980), increased activity of one or more routinely measured serum hepatobiliary marker enzymes was reported in all subjects. In particular, greater GGT activity was seen in all of the patients, whereas increases in ALP, ALT, and/or AST activity occurred in a far lower proportion. Other studies of children or infants of nursing mothers given these antiepileptics have similarly shown increases, most consistently in serum GGT, and more variably in other serum hepatobiliary marker enzymes (Bartels et al. 1975; de Wolff et al. 1982; Frey et al. 2002; Merlob et al. 1992). The incidence of the serum enzyme increases among immature subjects across these studies tended to be greater than that reported in adults on comparable regimens of these antiepileptics (Table 5). However, the predominance of GGT increases over other laboratory changes, and the magnitude of change in all of the serum enzymes evaluated (typically ≤ three-fold reference values in the absence of other findings supportive of hepatic injury) were generally similar between children and adults in these reports (Table 5).
The predominance of increases in GGT over changes in other serum enzymes with administration of potent CYP3A4 inducers has also been supported by findings in humans given rifampicin (Bartels et al. 1975; Ellis et al. 1979; Perucca et al. 1988). Further, correlation analyses between serum enzymes in a few of these clinical studies have demonstrated no association between GGT and ALP and only an occasional association between GGT and ALT (Acheampong-Mensah 1976; Deutsch et al. 1986; Haidukewych and John 1986; Sano et al. 1981; Tutor et al. 1988). However, DME induction as a direct cause of GGT increases is contradicted by the wide variability in serum GGT activity among human subjects receiving these inducing agents, and lack of correlation between patients' serum GGT activities and DME induction based on urinary excretion of D-glucaric acid or antipyrine clearance (Frezza et al. 1989; Heinemeyer et al. 1986; Hermida et al. 2002; Hildebrandt et al. 1975; Ohnhaus and Studer 1983). In addition, weak to no correlation between serum GGT activity and drug dose or plasma drug concentration has been reported in those clinical studies that comparatively evaluated these end points (Aldenhövel 1988; Braide and Davies 1987; Cepelak et al. 1986; Sano et al. 1981). Collectively, these findings suggest that these increases in serum activity of GGT, as well as other routine hepatobiliary marker enzymes, were not directly related to the DME induction potential of the drugs.
Hepatobiliary Biomarker Findings in Association with (CYP2E1-inducing) Ethanol Consumption in Humans
Human CYP2E1 is of particular clinical importance because of its ready induction by ethanol consumption and its role in the metabolism of well-known, potentially hepatotoxic xenobiotics (Meskar et al. 2001; Mohutsky et al. in press). In contrast with CYP3A4, CYP2E1 is generally comparable between man and laboratory animals in both ligand profile and induction potential (Martignoni et al. 2006). However, CYP2E1 is responsible for metabolism of only 2% to 4% of pharmaceutical agents (Kalra 2007) and shows prominent polymorphism. In humans, CYP2E1 induction has been the purported cause of altered serum hepatobiliary biomarkers, particularly GGT, in heavy alcohol consumers without detectable liver disease. Increased serum GGT has long been considered a sensitive measure of alcohol intake and is even proposed as a marker of alcohol abuse (Borg 1996; Gheno and Mazzei 1983). However, variation in serum GGT response to alcohol is prominent with age, sex, and ethnic influences that are distinct from those known to affect CYP2E1 activity (Braide and Davies 1987; Lucas et al. 1995; Whitfield 2001). In addition, several studies have shown that the magnitude of GGT activity in alcohol-consuming subjects correlated with other circulating hepatobiliary biomarkers and/or the severity of morphologic hepatic injury (Alatalo et al. 2009; Banciu et al. 1983; Conigrave et al. 2002; Daeppen et al. 1999; Salaspuro 1987; Whitfield et al. 1981). These latter findings suggest that prominent increases in GGT, especially when accompanied by other hepatobiliary marker changes, likely reflect true hepatic injury in alcoholics. Serum GGT in subjects with moderate to heavy alcohol consumption has shown particularly close correlation with AST, and serum AST has been significantly linked with hepatic pathology based on biopsy (de Lédinghen et al. 2004; Gomes et al. 2005; Kazemi-Shirazi et al. 2008; Teschke et al. 1983; Vădan et al. 2003) or computed tomography (Allaway et al. 1988). Notably, serum ALT has traditionally been considered less reliable than AST in patients with potential nutritional deficiencies such as chronic alcoholism because of the role of vitamin B6 as cofactor for ALT (Moh and Watson 1989). A comparable pattern of serum enzyme change and corresponding histologic findings has also been found in some rat chronic ethanol consumption studies, with more pronounced serum GGT increases in comparison with AST and ALT in the rodents (Devi et al. 2009).
Gamma Glutamyltransferase Increases in Association with Prototypic Hepatic-inducing Agents in Humans—The Role of Glutathione Consumption
The relatively common occurrence of increases in GGT activity in association with DME-inducing drugs and ethanol has led to further research on the causes of the serum enzyme change in man. Increased circulating GGT at the lower levels seen in these clinical studies is now generally considered to reflect increased tissue glutathione (GSH) consumption. In support of this hypothesis, alcohol-consuming patients have been shown to have significantly lower plasma GSH that is inversely correlated to circulating GGT activity (Whitfield 2001). Induction of CYP2E1 may also increase GSH tissue demand due to oxidative stress (Lu and Cedarbaum 2008). Enhanced potential for increased GSH consumption and ROS generation have also recently been reported in children given CBZ, DPH, or PB (Aycicek and Iscan 2007). Notably in human alcoholics, the increases in serum or plasma GGT activity have been consistently associated with much less of a change in hepatic tissue GGT activity (Ishii et al. 1986; Ivanov et al. 1980; Selinger et al. 1982; Teschke et al. 1983; Yamauchi et al. 1984). This result is presumed partly due to the specific hepatic tissue components analyzed, and contributions from extrahepatic tissues to blood GGT activity (Ikeda and Taniguchi 2005; Whitfield 2001). Still, the contrast between circulating and hepatic GGT activity suggests that serum values with alcohol consumption may reflect enzyme release into circulation in excess of retention in hepatic tissue (Hauge et al. 1998; Seitz et al. 1985), consistent with the extracellular GGT-mediated catabolism of GSH (Chikhi, et al. 1999; Ikeda and Taniguchi 2005). In summary, serum or tissue GGT increases with administration of DME-inducing xenobiotics in humans is now generally considered an adaptive response that is only indirectly linked with DME induction (Zhang et al. 2005).
Hormone Effects Associated with Prototypic Hepatic Enzyme-inducing Agents in Humans
The potential for DME induction to cause endocrine disruption has long been a focus of clinical studies. The CYP3A and CYP2 enzymes are particularly responsible for metabolism of multiple steroid hormones, and CYP3A4 has been specifically implicated in causing human endocrine disruption because of its role in metabolizing progesterone, estradiol, testosterone, and cortisol (Anderson 2004; Sarlis and Gourgiotis 2005). Phase II enzymes additionally conjugate several nonsteroidal and some steroidal hormones; the UGT1A family is particularly responsible for conjugation of thyroid and estrogenic hormones (Kato et al. 2008; Mackenzie et al. 2000; Takahashi et al. 2008), and the UGT2B family has a significant role in glucuronidation of human androgens (Barbier and Bélanger 2003). Drug-metabolizing enzyme induction has also been suspected of altering other endogenous human products, including circulating insulin, glucose, lipids, bilirubin, and vitamins D, K, B12, and folate (Benedetti et al. 2005; Lahtela 1986; Moreau et al. 2008; Podvinec et al. 2004; Putignano et al. 1998; Roth et al. 2008; Xie et al. 2003; You 2004). In this section, only clinical pathology linking DME induction to human thyroid and reproductive hormone levels will be discussed owing to limited comparable information on these other effects with laboratory animals.
Endogenous Thyroid Hormone Effects Associated with Prototypic Hepatic Enzyme-inducing Agents in Humans
Effects of DME-inducing agents on circulating thyroid hormone levels have been reported in a few human studies. In particular, the UGT1A enzymes facilitate T4 excretion and are induced by a wide variety of agents that activate human PXR, CAR, and/or AhR (Xie et al. 2003; Kato et al. 2008). Accordingly, decreased serum T4 and free T4 (fT4) in volunteers given rifampicin alone or with PB or antipyrine for fourteen days were attributed to increased UGT-mediated T4 turnover (Ohnhaus and Studer 1983). Similarly, decreased fT4 and increased thyroid gland volume were reported in volunteers after one month of dosing with rifampicin (Christensen et al. 1989). Comparable effects on serum thyroid hormone profiles have also been described in patients given DPH, PB, CBZ, and/or oxcarbazepine (Chin and Schussler 1968; Connel et al. 1984; Finucane and Griffiths 1976; Hansen et al. 1974; Hirfanoglu et al. 2007; Liewendahl et al. 1985; Tanaka et al. 1987; Vainionpää et al. 2004). Notably, T3 was not comparably affected by UGT-inducing agents in these studies, a finding which has been attributed to primary metabolism of T3 through sulfation and deiodination rather than glucuronidation in humans, in contrast with rats (Benedetti et al. 2005; Findlay et al. 2000; Visser et al. 1993). More significantly, no notable increase in TSH was reported in most of these clinical studies (Apeland et al. 2006; Benedetti et al. 2005; Isojärvi et al. 2001; Verrotti et al. 2009). Because of this lack of TSH change or clinical thyroid illness, the collective human literature suggests that little to no effect on thyroid function occurs in otherwise healthy subjects given DME-inducing agents. However, in persons with underlying thyroid conditions, increased incidence of hypothyroidism has been infrequently reported in association with phase II enzyme-inducing therapies (Takasu et al. 2006). As noted previously, prominent differences in thyroid hormone biology and induction of specific UGTs potentially accounts for much of the difference between rodent and human susceptibility to thyroid hormone disruption associated with DME-inducing agents (Kato et al. 2005; Kester et al. 2003; Wu and Farelly 2006).
Endogenous Reproductive Hormone Effects Associated with Prototypic Hepatic Enzyme-inducing Agents in Humans
In contrast with preclinical drug safety studies, in published clinical studies, reproductive hormone effects have received greater attention than thyroid hormone changes in association with DME induction. Induction of phase I oxidative hydroxylation and/or phase II conjugation reactions may facilitate steroid hormone inactivation or excretion (You 2004). In addition to the well-studied example of rifampin on estradiol-containing contraceptive metabolism in women (Isojärvi 2008; Mohutsky et al. 2009; Thorneycroft et al. 2006), increased endogenous estradiol metabolism has been a proposed clinical outcome of CYP3A4 induction, in general (Yu et al. 2005). Clinical antiepileptic regimens have been most often linked with reproductive hormone disruption through co-induction of aromatase (CYP19), responsible for conversion of testosterone to estradiol (Isojärvi, 2008; Herzog et al. 1991). Reproductive hormone alterations have also been attributed to DME induction via AhR- and PPAR-γ–activating agents (Nakanishi 2008). Animal models are variable in their relevance for study of these reproductive effects (Taubøll et al. 2008). Rationale for the use of nonhuman primates, specifically cynomolgus monkeys, as models for the effects of DME induction on steroid reproductive hormone phase II metabolism has been presented (Barbier and Bélanger 2003; Nishimura et al. 2008).
Conclusions on DME Induction and Clinical Pathology Biomarkers in Humans
As in preclinical species, toxicity associated with DME induction in man has been based largely on the effects of a few prototypic inducing agents. In general, administration of CYP3A4- or CYP2E1-inducing agents has been associated with increased serum or plasma activity of GGT most commonly, and ALP, AST, and ALT less often. However, as noted in animal studies, these findings have been inconsistent between studies and generally show no or only poor association with direct measures of DME induction. Rather, downstream effects of ROS formation, as well as idiosyncratic or pharmacologically mediated hepatotoxicity, more likely influence these blood chemistry changes. Increases in serum GGT may particularly represent an adaptive response to xenobiotic-related oxidative stress. Drug-metabolizing enzyme–inducing agents have also been reported to influence human endocrine patterns. Although the overall mechanisms for DME induction are similar between humans and laboratory animals, the CYP induction profile and response to specific inducers in preclinical species are limited in predictability of the response in humans.
Overall Summary
For both laboratory animals and man, collective preclinical and clinical study data generally do not support a consistent effect of DME on traditional clinical laboratory parameters. However, this review paper does support the following findings: in the rat, in the absence of findings other than hepatocellular hypertrophy, DME induction alone is not expected to be associated with increases in serum or plasma activities of ALT, ALP, or GGT. In the dog and the monkey, there is also no consistent relationship between administration of DME-inducing agents and changes in these clinical pathology parameters in the preclinical literature. However, in dogs given phenobarbital or corticosteroids, increased liver ALP or GGT activities suggest that direct or indirect induction of select hepatobiliary injury markers can occur both in the absence of liver injury and independent of DME induction. Correlations between tissue and serum activities of ALT, ALP, or GGT are limited and inconsistent, and increases in serum/plasma activities that are substantial or involve changes in other hepatobiliary markers generally reflect hepatobiliary insult rather than DME induction. Most notably, hepatic DME induction and associated changes in preclinical species are not necessarily predictive of the occurrence, magnitude, or enzyme induction profile in humans.
Footnotes
The views expressed in this article are the personal views of the authors and do not necessarily represent the policies, positions, or opinions of their respective organizations.