
Abstract
The receptor tyrosine kinase receptor (RTK) signaling pathway, mesenchymal-epithelial transition factor (c-Met)/hepatocyte growth factor receptor (HGFR), has been implicated in oncogenesis and is a target of interest in cancer therapy. PF-04254644 is a potent and selective inhibitor of c-Met/HGFR. Wide ligand binding profiling of PF-04254644 revealed a potentially significant interaction with phosphodiesterase (PDE) 3, and follow-up PDE enzyme activity assays confirmed PF-04254644 as a potent inhibitor of PDE3 as well as other PDEs (1, 2, 5, 10, and 11). Clinical observations, laboratory, and echocardiography parameters were recorded in Sprague-Dawley (SD) rats that received PF-04254644 oral dosing for up to seven consecutive days. Toxicological evaluations revealed myocardial degeneration as an adverse event at all tested doses. Echocardiographic evaluations revealed an increase in heart rate (HR) and contractility after the first dose with PF-04254644 and myocardial fibrosis correlated with decreased cardiac function after repeat dosing. A study in telemetry-instrumented rats substantiated that PF-04254644 induced a sustained increased HR and decreased contractility after six days of treatment. Data suggest that the decreased cardiac function and cardiotoxicity are likely due to inhibition of multiple PDEs by PF-04254644.
Introduction
Tyrosine kinases (TKs) are enzymes that catalyze the transfer of phosphate from adenosine triphosphate (ATP) to the tyrosine residues in polypeptides (Krause and Van Etten 2005). These phosphorylated polypeptide products are under tight control and regulate cellular proliferation, function, differentiation, and motility and are expressed at increased levels in cancer cells (Krause and Van Etten 2005). TKs are divided into two main classes: nonreceptor TKs are found in the cytosol, nucleus, and inner surface of the plasma membrane; and receptor TKs (RTKs) are transmembrane proteins with a ligand-binding extracellular domain and a catalytic intracellular kinase domain (Krause and Van Etten 2005). The proof of principle for a TK inhibitor (TKI) as an excellent therapeutic agent for cancer therapy was observed with the success of imatinib mesylate (Gleevec), an inhibitor of the fusion protein Bcr-Abl, which incorporates c-Abl, a nonreceptor TK; and Bcr, a multidomain protein of uncertain origin in chronic myeloid leukemia (Krause and Van Etten 2005). Cardiovascular side effects are well known in cancer patients undergoing treatment with TKIs. This has been recently reviewed by Force et al. (2008) and represents a challenge in the development of new targeted cancer therapeutics. RTK pathway activity associated with cancer cell survival may also perform a similar function in cardiac myocytes, and untoward cardiotoxicity may be unavoidable until downstream RTK pathways that mediate cardiac toxicity are identified (Force et al. 2008). Off-target toxicity associated with RTKIs may be due to their nonselectivity, that is, multitargeted RTKI (Force et al. 2008) or other off-target effects, for example, phosphodiesterase inhibition.
Hepatocyte growth factor (HGF) signaling pathway appears to play an important role in human carcinogenesis and metastasis and is a therapeutic target of interest in anticancer treatment (Stellrecht and Gandhi 2009). The receptor of HGF, hepatocyte growth factor receptor (HGFR), is also known as mesenchymal-epithelial transition factor (c-Met). A small molecule RTK inhibitor (PF-04254644) is an oral, highly potent, selective inhibitor of c-Met/HGFR with anti-tumor and anti-angiogenic activities (Pfizer Inc., data on file; Shen et al. 2008; Zou et al. 2008). PF-04254644, a 6-{(S)-1-[-(6-(1 methyl-1H-pyroazol-4-yl)-[1,2,4] triazolo [4,3-b] pyridazin-3-yl-ethyl}-quinoline (Shen et al. 2008), inhibited c-Met with a low nanomolar potency (8 nM) as well as inhibited tumor growth, phosphorylation of c-Met and downstream signaling molecules including Gab1, Akt, PLCγ, STAT5, and Erk1/2 in xenografted GTL-16 tumors (human gastric carcinoma addiction model) in nude mice (Zou et al. 2008). Routine screening of PF-04254644 for potential off-target pharmacologic effects on more than 200 receptors and kinases revealed a potential interaction with phosphodiesterase 3 (PDE3) (Pfizer Inc., data on file). A full PDE enzyme activity profile on PF-04254644 found that PF-04254644 is not only an inhibitor of PDE3 (IC50<5μM) but also inhibits PDE1A and PDE5 (Ramirez et al. 2008), which are known to play a role in cardiovascular function (Omori and Kotera 2007). PF-04254644 was also found to inhibit PDE1C, PDE10, and PDE11 (Ramirez et al. 2008).
This article reports the nonclinical safety studies conducted in Sprague-Dawley (SD) rats given an oral RTK inhibitor (PF-04254644) potently selective for c-Met/HGFR with known off-target PDE inhibition. The objectives of this work included characterization of systemic exposure, cardiac function, and toxicologic profiles following oral administration of PF-04254644 for up to 7 days in rat preclinical studies and verification of the irreversibility of treatment-induced effects in subsequent rat investigative studies.
Materials and Methods
The off-target pharmacology profile for PF-04254644 and four oral toxicity studies in rats are reported: a single-dose toxicokinetics (TK) study, a 7-day repeat-dose toleration study, a 7-day single-dose investigative study, and a 6-day repeat-dose investigative study. The TK study was conducted in rats to determine the relationship between oral dosing and systemic exposure. The 7-day repeat-dose toleration study in rats with PF-04254644 was conducted to determine systemic and target organ toxicities. The lowest dose selected in this study was based on the predicted therapeutic free plasma concentration in humans, and 2- to 8-fold higher doses were selected to calculate the safety margins over any toxic effects. Investigative studies were conducted to further assess the pathogenesis of the cardiac toxicity by evaluating drug plasma concentrations, temporal changes in myocardial tissue, cardiac function, and serum cardiac troponin I (cTnI) concentrations.
Animals and Husbandry
In all studies, standard procedure and conditions were applied in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All procedures involving laboratory animals were reviewed and approved by the Pfizer Inc. Institutional Animal Care and Use Committee associated with the facility. The single-dose TK study, 7-day repeat-dose toleration study, 7-day single-dose investigative study, and 6-day repeat-dose investigative study were conducted in 7- to 9-week-old male SD rats supplied by Charles River Laboratories, Inc. (Portage, MI).
Drug
PF-04254644, a 6-{(S)-1-[-(6-(1 methyl-1H-pyroazol-4-yl)-[1,2,4] triazolo [4,3-b] pyridazin-3-yl-ethyl}-quinoline (Shen et al. 2008), was supplied by Pfizer Global Research and Development, La Jolla Laboratories (San Diego, CA). All dose levels are represented as the free base equivalent mg/kg dose. PF-04254644 was formulated in 0.5% methylcellulose and given to study animals by oral gavage.
PF-04254644 was assayed using a validated LC-MS/MS method, and toxicokinetic calculations were preformed using the noncompartmental approach with the aid of Watson Bioanalytical LIMS (v7.2.0.03, Thermo Electron Corp, Waltham, MA.)
Off-Target Pharmacology Profile
PF-04254644 was submitted to CEREP BioPrint (Poitiers, France) for in vitro binding and functional assays (68 total) to assess off-target pharmacology as well as screened internally within Pfizer for potential activity against 150 kinases. A PDE enzyme activity assay panel developed at Pfizer utilizing scintillation proximity assay (SPA) technology was used to evaluate PF-04254644 in a 10-point IC50 curve ranging from 1 nM to 10 μM. The assay utilizes recombinant human phosphodiesterase proteins partially purified from insect cells.
Study Design
In the single-dose TK study, 2 male rats per group were given PF-04254644 at 0, 50, 150, or 500 mg/kg/day and euthanized at 24 hrs after dosing. Toxicity was determined by assessing changes in clinical signs and behavior, pretest and posttest body weights. Blood samples for the assessment of systemic exposure to PF-04254644 were collected from all treatment groups at various times on day 1 of the study. Laboratory examinations (hematology and clinical chemistry) were performed at 24 hrs after dosing. Necropsy was performed at the end of treatment; organ weights were recorded, and histopathologic examinations were performed.
In the 7-day repeat-dose toleration study, 5 male rats per group were to be given PF-04254644 at 0, 40, 80, and 320 mg/kg/day for 7 days. Laboratory evaluations (cardiac troponin I [cTnI]), hematology, and clinical chemistry were performed 24 hrs postdose and on day 8. Serum was analyzed for cTnI by a high-sensitivity rodent ELISA-Kit (Life Diagnostics, Inc. West Chester, PA). Blood samples for the assessment of systemic exposure to PF-04254644 were collected from all treatment groups at various times on days 1 and 7 of the study. Necropsy was performed on day 8; organ weights were recorded, and histopathologic examinations were performed. Cardiac function was evaluated in vivo by collecting echocardiography parameters on anesthesized rats during the predose period (baseline values) and on day 6 of the 7-day repeat-dose study. Rats were anesthesized with 1.5% isofluorane (AErrane; Baxter, Inc., Deerfield, IL); underwent hair removal from the thorax by shaving, followed by treatment with a depilatory agent; and then were placed on their left side to collect echocardiographic images in the parasternal long axis, parasternal short axis, and apical four chamber view. Echocardiograms were performed using the Philips SONOS 5500 system equipped with a 15-MHz linear-array transducer (Philips, Andover, MA) (Heyen et al. 2002), and parameters analyzed were end systolic volume (ESV), end diastolic volume (EDV), stroke volume (SV), and cardiac output (CO). Ejection fraction (EF), an indirect measure of cardiac contractility and overall cardiac performance, was reported and calculated by the formula EF = EDV – ESV/EDV × 100%. Cardiac output (SV × HR) was used as an indicator of overall cardiovascular health. All measurements were performed according to the recommendations of the American Society for Echocardiography leading-edge method from three consecutive cardiac cycles.
In the 7-day single-dose investigative study, 12 male SD rats were given 0 or 500 mg/kg dose of PF-04254644. Toxicity was determined by evaluating changes in clinical signs and behavior (at least daily), body weights (pretest and at study termination). Cardiac function was evaluated in vivo by echocardiography on anesthesized rats at baseline predose, 6 hrs and 24 hrs postdose on day 1, as described above. Laboratory evaluations for cTnI and necropsies were performed on 6 rats per group at scheduled time points (2, 8, and 24 hrs post–first dose and on day 7). At necropsy, organ weights were recorded, and the heart was bisected longitudinally through both ventricles immediately adjacent to both atria. Half of the heart was retained in 10% neutral buffered formalin for routine histopathologic evaluation, and the other half was retained in Karnovsky’s fixative (2.5% glutaraldehyde, 4% paraformaldehyde in 0.1M sodium cacodylate buffer with 0.01% calcium chloride, pH = 7.3) for ultrastructural examination. Following fixation in Karnovsky’s, the tissues were postfixed in buffered 1% osmium tetroxide solution and then embedded in epoxy resin after appropriate dehydration. Hearts were sectioned at 0.5 μm thickness using a Leica ultramicrotome; stained with 1% Toluidine Blue, 1% Azure II, and 1% borax; and examined by light microscopy to define areas of interest. Ultrathin sections of 70–90 nm were obtained and mounted on copper grids, stained with 10% aqueous uranyl acetate and 2.5% lead citrate, and examined using a Hitachi 7100 transmission electron microscope at 75 kV. Digital images were captured using an AMT digital camera. Blood samples to assess systemic exposure to PF-04254644 were collected at various times on day 1 and at termination of study.
In the 6-day repeat-dose investigative study, 74 male SD rats were given 0, 40, or 80 mg/kg PF-04254644 or 10.5 mg/kg milrinone, a known PDE3 inhibitor (Earl, Linden, and Weglicki 1986a, 1986b). Rats were segregated into three assessment groups: in vivo cardiovascular analysis including echocardiography and telemetry (n = 28), histo and ultrastructural pathology with cTnI (n = 40), and satellite toxicokinetics (n = 6). Rats intended for in vivo cardiovascular assessment were instrumented with PA-C40 radio telemetry units (Data Sciences Inc., St. Paul, MN) for conscious, unrestricted blood pressure and heart rate measurements (Blasi et al. 2003). Arterial blood pressure and heart rate were measured and analyzed with the DATAQUEST A.R.T. Version 3.0-Gold software (Data Sciences International, St. Paul, MN). The values represent the average of all data points collected from each animal, every 15 min for a 10-sec interval over a 24-hr period. Data were further consolidated and analyzed using 1-hr averages. Echocardiography was performed on anesthetized telemetry instrumented rats as follows: on day 1 at baseline predose, at times of maximal drug concentration (Cmax; 0.5-1 hr postdose for milrinone and 1-2 hrs postdose for PF-04254644), and at 24 hrs; on day 6 prior to dosing (0 hr) and at Cmax. Hearts for histologic and ultrastructural pathology were collected from 3 rats per group at various time points (2, 6, 24, and 144 hrs) postdose and were bisected longitudinally through both ventricles immediately adjacent to both atria. Half of the heart was retained in Karnovsky’s fixative as described above, and the other half of the myocardium was saved in RNA later. Samples of left ventricle, right ventricle, and interventricular septum were retained in Karnovsky’s for ultrastructural examination. The remaining heart tissue was processed for histopathologic examination. Analysis of serum cTn1 was conducted at 2, 6, 24, and 144 hrs postdose.
Statistical Analysis
All blood pressure and heart rate data were analyzed comparing group means using one-way analysis of variance (ANOVA), and multiple comparisons were controlled for using Dunnett’s test to evaluate 1 hr means. The analysis was performed on the raw data (parametric analysis) using a two-tailed test for the planned comparisons between the means of vehicle and treatment groups evaluated as change from vehicle. The alpha = .05 level of significance was used. All data are reported as mean change from vehicle ± standard error of the mean (SEM).
Echocardiography data was used to evaluate whether treatment had effects on the changes of ultrasound parameters from baseline measurements. Mixed effect modeling with appropriate contrasts were carried out at each follow-up time point to determine whether there were any treatment effect at the specific time point. For each time point that demonstrated a significant effect of treatment, follow-up pairwise comparisons against the vehicle were performed. The alpha = .05 level of significance was used.
Results
Off-Target Pharmacology
In a wide ligand binding profile performed at Cerep (Poitiers, France), PF-04254644 inhibited the PDE3 ligand at 98% (10 μM with an IC50 of 170 nM (Table 1 ). Follow-up evaluation of PF-04254644 in an internal PDE assay panel demonstrated strong enzymatic activity at multiple PDE families including 1A, 1C, 2, 3A, 3B, 4, 5, 10, and 11 (Table 2 ). Single-point kinase screening performed internally at Pfizer suggested potential interaction (<10 uM) with Anaplastic Lymphoma Kinase (Alk), Fibroblast Growth Factor Receptor 3 (FGFR3), and Wnt (Wingless Int; data not shown). Follow-up evaluation of ALK, FGFR3, and Wnt in enzymatic and cellular assays suggested IC50 values of 31.7, >10, and 56.7 μM, respectively (Zou et al. 2008).
Select tier 0 and 1 ligand profiling for PF-04254644.
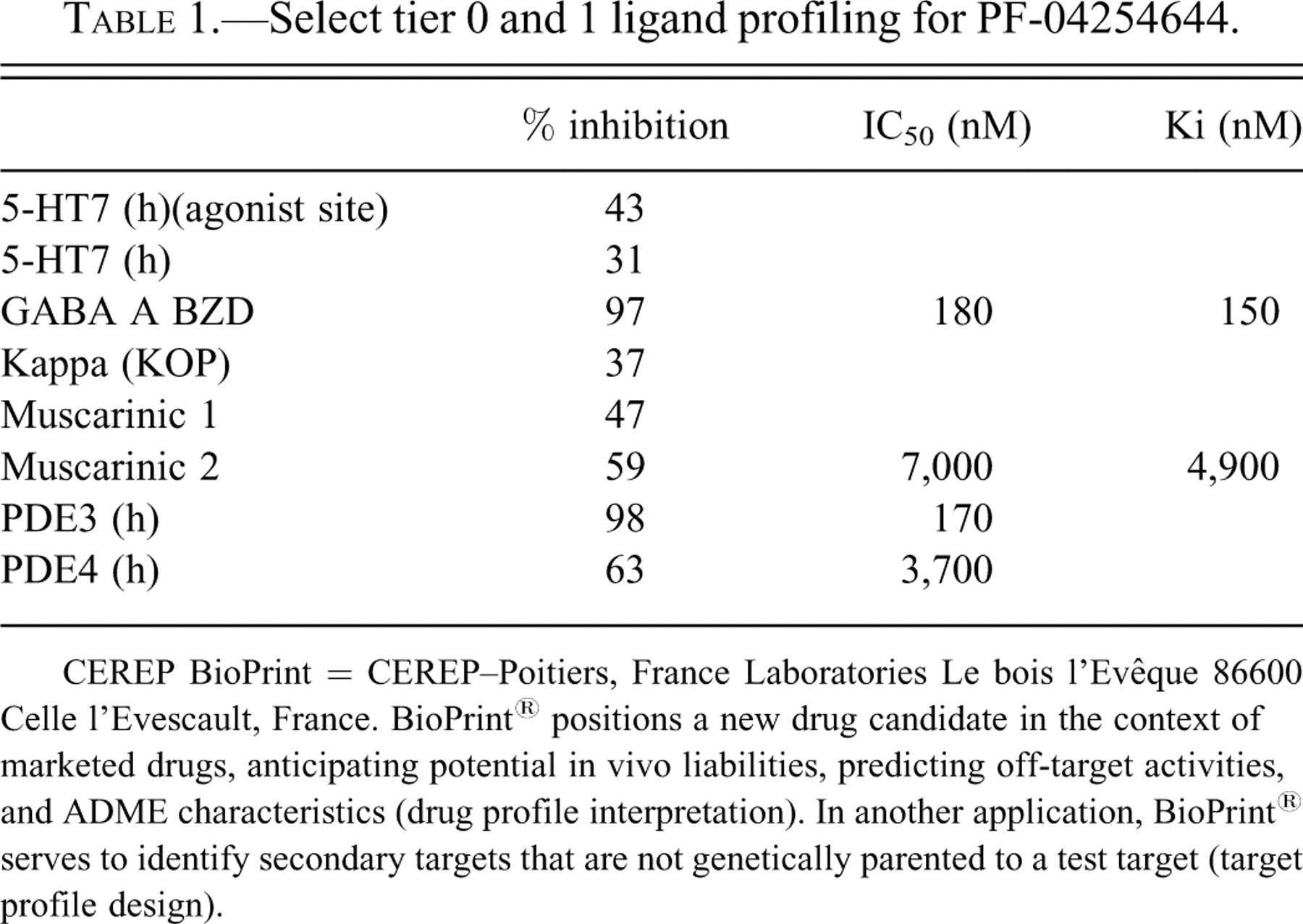
CEREP BioPrint = CEREP–Poitiers, France Laboratories Le bois l’Evêque 86600 Celle l’Evescault, France. BioPrint® positions a new drug candidate in the context of marketed drugs, anticipating potential in vivo liabilities, predicting off-target activities, and ADME characteristics (drug profile interpretation). In another application, BioPrint® serves to identify secondary targets that are not genetically parented to a test target (target profile design).
PDE profile of PF-04254644.
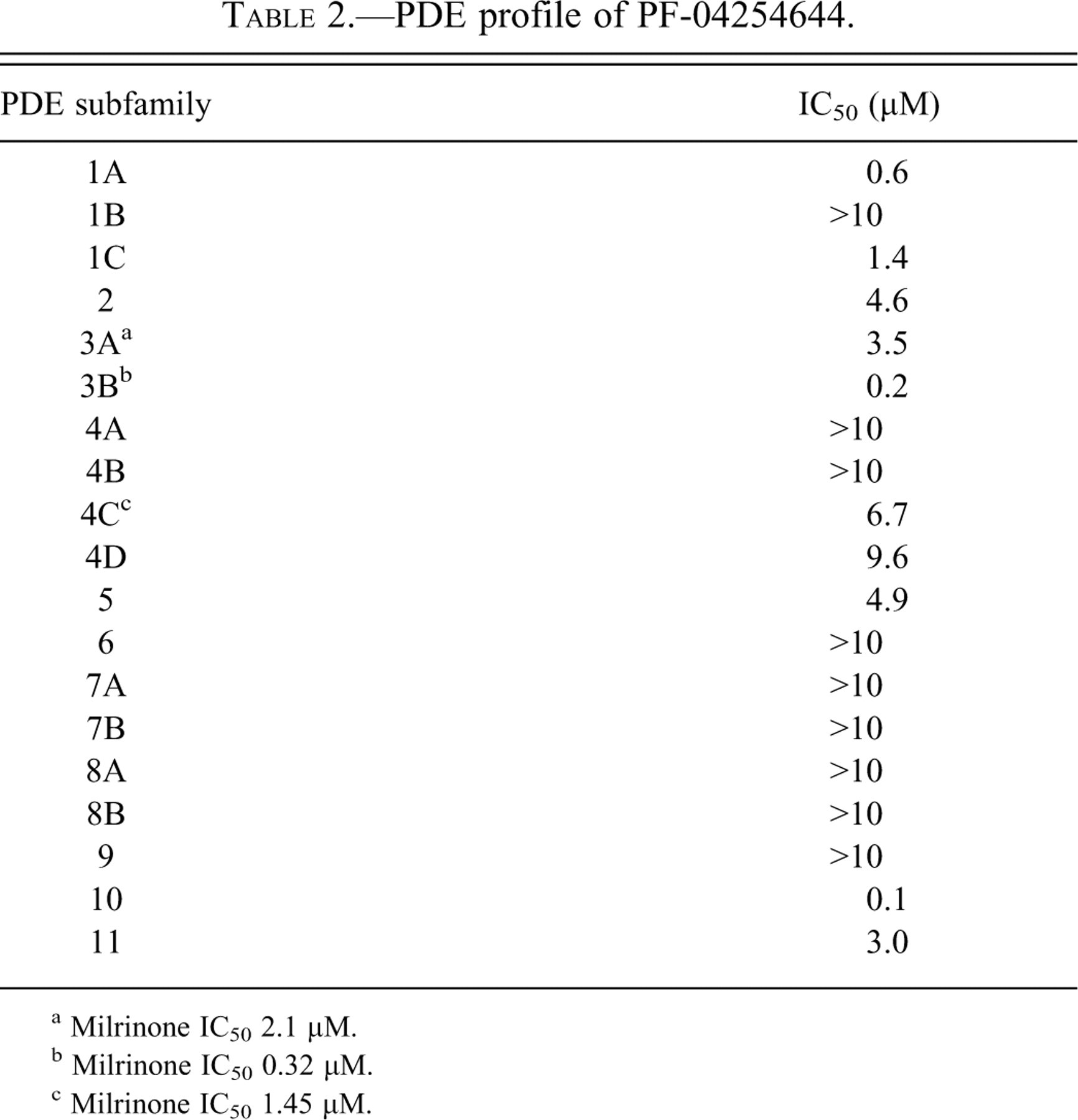
a Milrinone IC50 2.1 μM.
b Milrinone IC50 0.32 μM.
c Milrinone IC50 1.45 μM.
Tolerability
In the 7-day repeat-dose toleration study, treatment-related morbidity occurred in 2 of 5 male rats receiving 320 mg/kg/day PF-04254644. Because of the poor condition, treatment was stopped on day 2, and these animals were euthanized. Other animals in that dose group subsequently received a reduced dose of 160 mg/kg for the remainder of the study.
Clinical Signs, Clinical Chemistries, Hematology, and Organ Weights
Except for the treatment–related morbidity observed in the 7-day repeat-dose study, there were no other PF-04254644-related clinical signs of toxicity, clinical chemistry or hematology findings, macroscopic or organ weight findings in the single-dose TK study, 7-day repeat-dose study, 7-day single-dose investigative study, or 6-day repeat-dose study.
Histopathologic and Electron Microscopic Evaluations
Myocardial changes observed in the single-dose TK, 7-day repeat-dose toleration, 7-day single-dose investigative, and 6-day repeat-dose investigative studies at the microscopic and ultrastructural levels are described below and illustrated in Figures 1 and 2. The grading scheme for the myocardial degeneration detectable at the light microscopic level ranged from minimal to severe and was based on the percentage of myocardium affected. A grade of minimal corresponded to ≤ 10%, mild corresponded to > 10% to ≤ 25%, moderate corresponded to > 25% to ≤ 50% and severe corresponded to >50%.
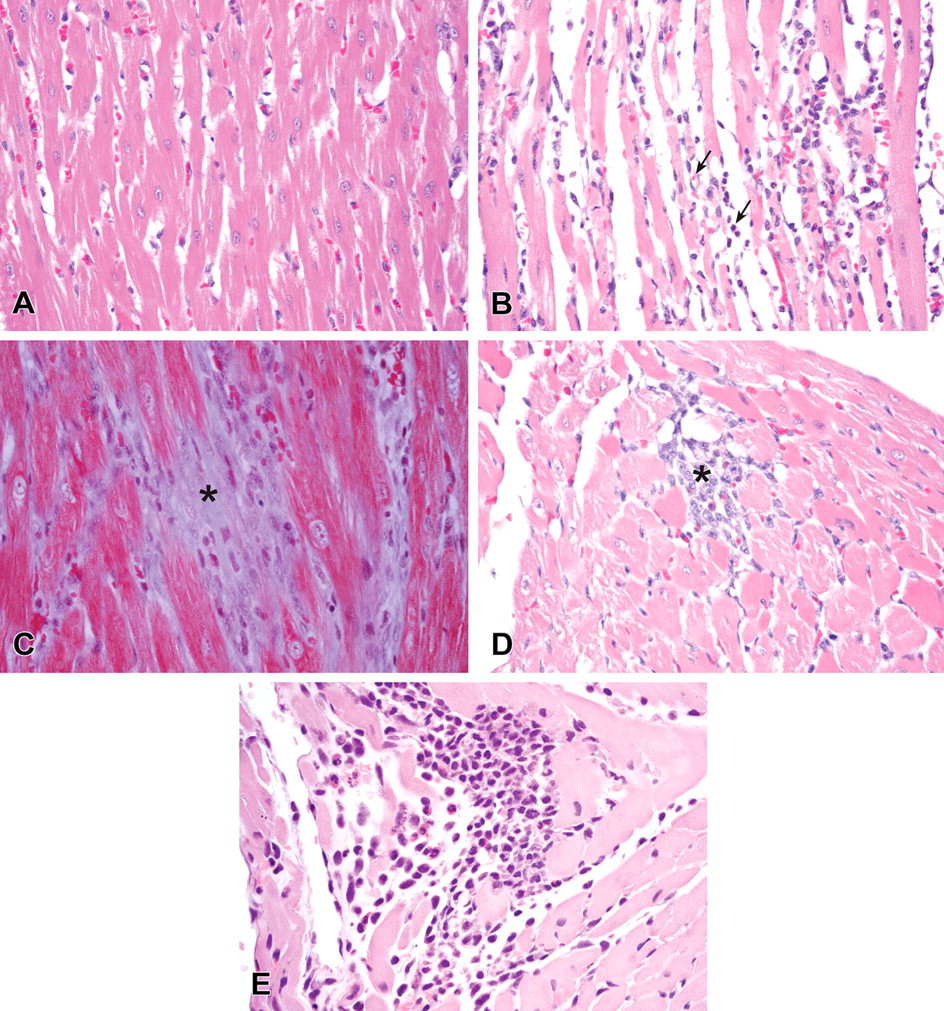
Microscopic findings of hematoxylin and eosin stained or Masson’s Trichrome stained sections of rat myocardial interventricular tissue. (A) Hematoxylin and eosin staining of myocardial sections from a control rat in the TK study (400X). (B) Hematoxylin and eosin staining of myocardium sections from a rat in the TK study with 500 mg/kg PF-04254644 24 hrs after dosing (400X). Moderate myocardial degeneration in the interventricular septum (arrows). (C) Masson’s Trichrome staining of myocardial sections of a rat with 320/160 mg/kg PF-04254644 in the 7-day repeat-dose toleration study after 7 days of dosing (400X). Fibrosis of the myocardial interventricular septum (asterisk). (D) Hematoxylin and eosin staining of myocardial sections of a rat given 80 mg/kg PF-04254644 in the 7-day repeat-dose toleration study after 7 days of dosing (400X). Minimal myocardial degeneration in the interventricular septum (asterisk). (E) Hematoxylin and eosin staining of myocardial tissue taken from a rat 24 hrs after dosing with 10.5 mg/kg milrinone (400X). Note the minimal myocardial degeneration with macrophage and few neutrophil infiltrates.
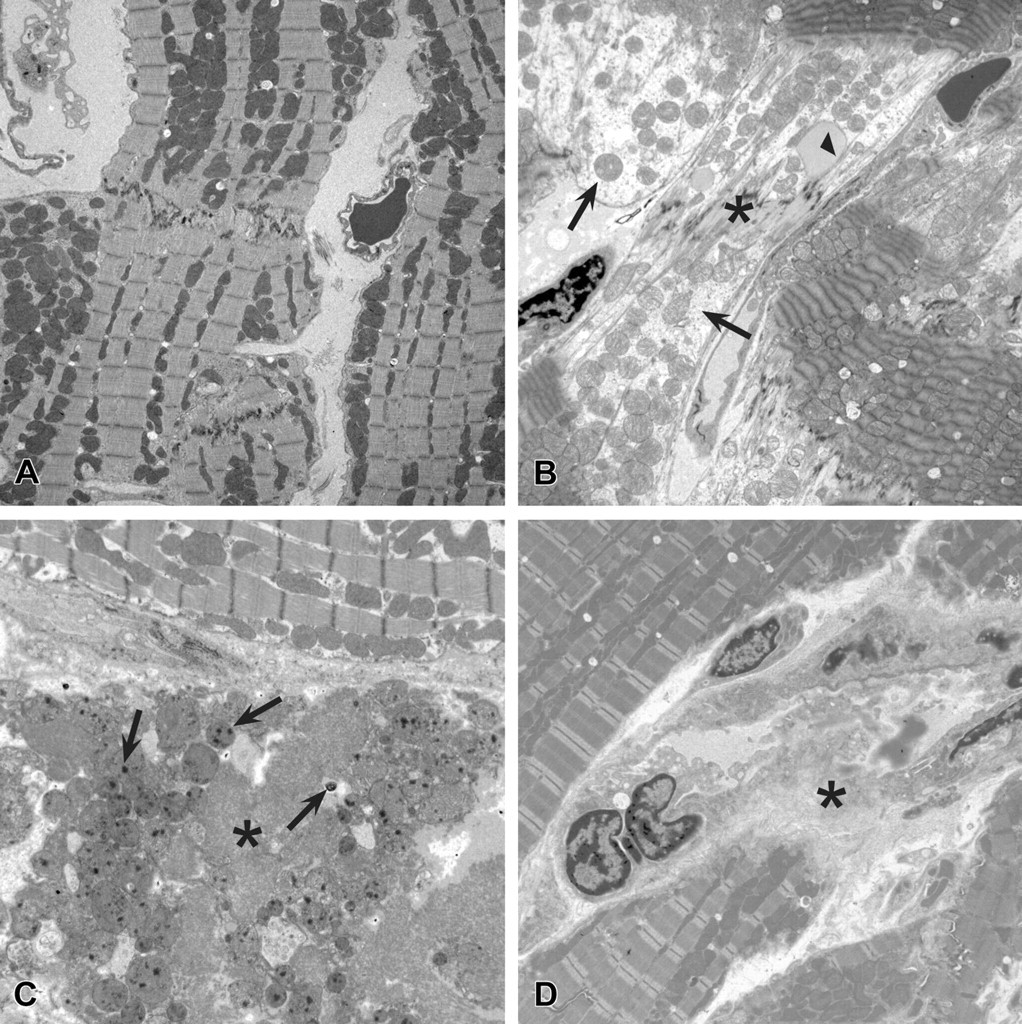
Ultrastructural findings in rat myocardial tissue. (A) Myocardial tissue from a control rat (2,000X). (B) Myocardial tissue from a rat in the 7-day single-dose investigative study 2 hrs after dosing with 500 mg/kg PF-04254644 (2,000X). Note the loss of myofiber actin and myosin filaments and intact mitochondria (arrows), deposition of flocculent material near disrupted intercalated disk (asterisk), and lipid deposition (arrow head). (C) Myocardial tissue from rat in the 7-day single-dose investigative study 8 hrs after dosing with 500 mg/kg PF-04254644 (2,000X). Note the coagulative necrotic myofibers (asterisk) and intramitochondrial densities (arrows). (D) Myocardial tissue from a rat in the 6-day repeat-dose investigative study 24 hrs after dosing with 10.5 mg/kg milrinone (2,000X). Note the coagulative necrosis of myofibers (asterisk) and macrophage infiltration.
In the single-dose TK study at 24 hrs following a dose of ≥150 mg/kg, there was dose-related mild to moderate myocardial degeneration at the apex, interventricular septum, and in the left and right ventricular free walls. The moderate change was characterized by the presence of thin and fragmented hypereosinophilic myofibers with loss of cytoplasmic striations and neutrophils in areas of degeneration (Figure 1). No myocardial changes were observed after a single dose of 50 mg/kg. In the 7-day repeat-dose toleration study, myocardial changes in rats given 320 mg/kg that became morbid were similar to those observed in the TK study and characterized by mild to moderate multifocal areas of degeneration in the apex, interventricular septum, left and right ventricular free walls. After 7 days of repeat dosing with 320/160 mg/kg, fibrosis replaced the degenerative myofibers (Figure 1). Minimal focal myocardial degeneration in the same areas of the heart occurred in 2 of 5 rats given 80 mg/kg PF-04254644 (Figure 1). Minimal myocardial degeneration occurred in 1 of 5 rats given 40 mg/kg. In the 7-day single-dose investigative study, minimal multifocal myocardial degeneration was observed at 2 and 8 hrs after dosing in most rats and progressed to a moderate myocardial degeneration at 24 hrs postdose and myocardial fibrosis on day 7 in all rats given 500 mg/kg. In the 6-day repeat-dose investigative study, no myocardial changes were observed by histopathologic examination at 2 hrs after dosing. However, minimal focal to multifocal myocardial degeneration was observed at 6 hrs postdose in the apex, interventricular septum, and left and right ventricles in all rats given ≥40 mg/kg PF-04254644; this change progressed to fibrosis on day 6. Similar to PF-04254644 at the 40 mg/kg dose level, no myocardial changes were observed in rats given 10.5 mg/kg milrinone at 2 hrs postdose; and minimal multifocal myocardial degeneration occurred in the apex, interventricular septum, left and right ventricular free walls of rats at ≥6 hrs after dosing (Figure 1).
To more accurately assess the onset and reversibility of myocardial changes after a single high dose of 500 mg/kg PF-04254644 in rats, we examined the hearts at the ultrastructural level at various time points. In the 7-day investigative study, myocardial changes in rats given a single 500 mg/kg dose of PF-04254644 were observed as early as 2 hrs postdose and characterized by myofiber lysis with visibly intact mitochondria, disruption of the intercalated disk, and deposition of lipid material (Figure 2). Ultrastructural changes in the heart were more consistently observed than histopathologic changes at 2 hrs postdose. At 8 hrs after dosing, myocardial changes progressed to coagulative necrosis of myofibers with presence of intramitochondrial densities (Figure 2) and were consistently correlated with histopathologic findings. Coagulatively necrotic myofibers and macrophage infiltrates were consistently observed at 24 hrs after dosing by ultrastructural and histopathologic examination. In the 6-day repeat-dose investigative study, myocardial changes in rats given 10.5 mg/kg/day milrinone were undetectable at 2 hrs and minimal at ≥6 hrs postdose and characterized by coagulative necrosis of myofibers with macrophage infiltration (Figure 2).
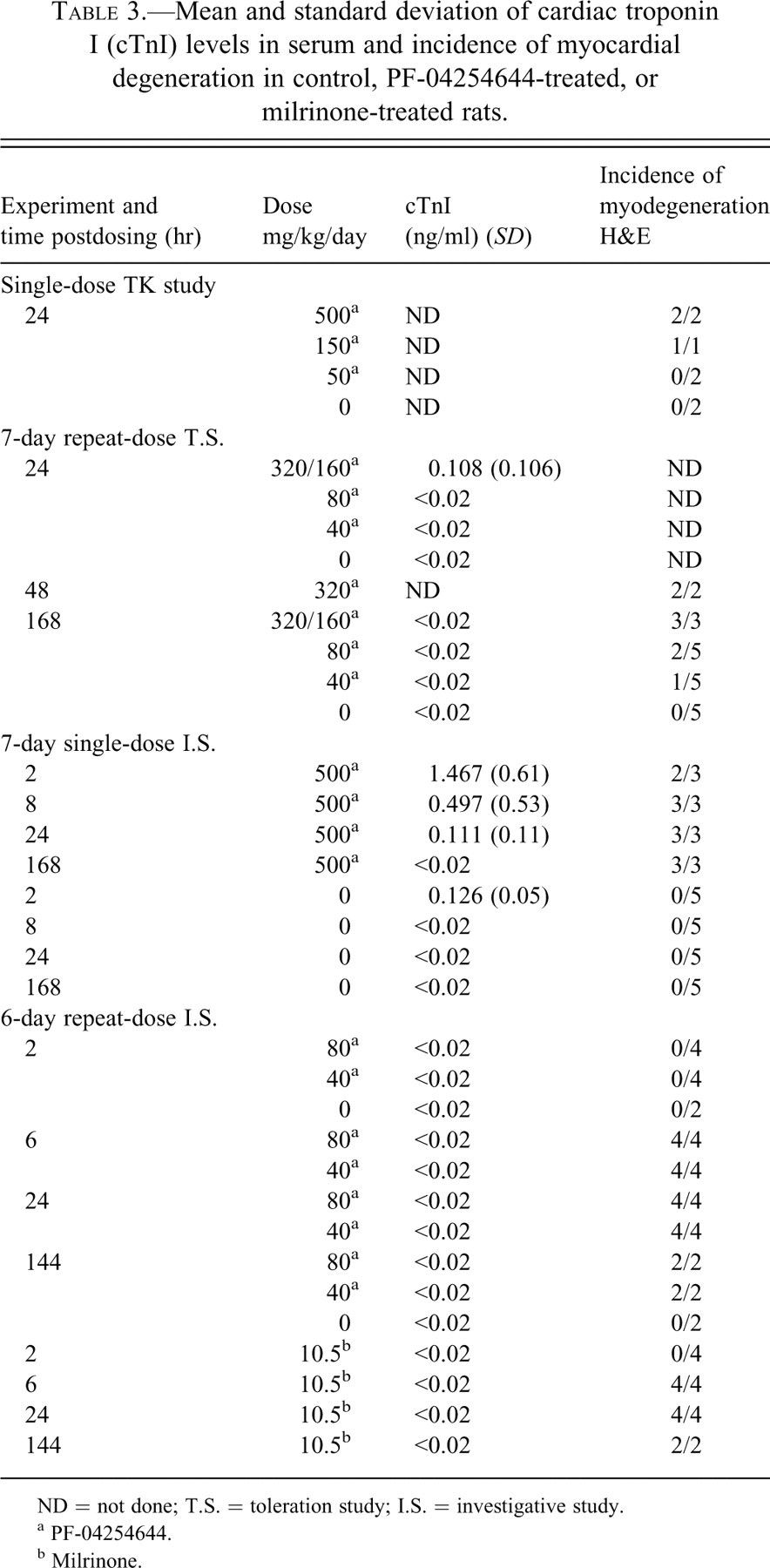
Serum Cardiac Troponin I Levels and Incidence of Myodegeneration
Serum levels of cTnI and the incidence of myodegeneration for the single-dose TK, 7-day repeat-dose toleration, 7-day single-dose investigative, and 6-day repeat-dose investigative studies are summarized in Table 3 . The range of cTnI levels in the serum of vehicle control-treated rats was <0.002 to 0.164 ng/ml. The range of cTnI levels in rats across studies given PF-04254644 was 0.182 to 2.158 ng/ml. The experiment with the most remarkable changes in serum cTnI was the 7-day investigative single-dose study using 500 mg/kg PF-04254644. The serum cTnI levels ranged from 0.564 to 2.158 ng/ml at 2 hrs following a single dose of 500 mg/kg PF-04254644. At the 2-hr time point, the increased cTnI levels were more consistently correlated with findings observed in the myocardium of rats at the ultrastructural level than findings observed in the heart by histopathology on H&E stained sections. At 8 hrs postdose, serum cTnI levels ranged from 0.421 to 1.472 in 4 rats and correlated with mild to moderate myocardial degeneration observed by ultrastructural or histopathologic examination. By 24 hrs, serum cTnI levels ranged from 0.236 to 0.253 ng/ml or returned to background levels and correlated with a mild to moderate myocardial degeneration observed by ultrastructural or histopathologic examination. Comparable cTnI levels at 24 hrs and myocardial changes at 24 to 48 hrs were observed in rats given 500 mg/kg in the 7-day single-dose investigative study or 320 mg/kg in the 7-day repeat-dose toleration study. In the 7-day repeat-dose toleration study, the cTnI level in 1 rat given 320/160 mg/kg was 0.288 ng/ml, while cTnI levels in the other treated rats in that dose group remained within the background range. All rats given 320/160 mg/kg developed a mild to moderate myocardial degeneration on days 2 and 7. Together, these findings suggest that damage to myocardium started early (~2 hrs) and cTnI levels peaked at 2 hrs at doses ≥320 mg/kg PF-04244644. At lower doses (40 and 80 mg/kg) in the 7-day repeat-dose toleration study, cTnI levels were within background levels, although a minimal myodegeneration was observed in 2 of 5 rats given 80 mg/kg after 7 days of dosing (Figure 1) and in 1 of 5 rats given 40 mg/kg PF-4254644 (Table 3). Although cTnI levels were not evaluated in the TK study, a mild to moderate myocardial degeneration (Figure 1) was observed in all rats at 24 hrs after dosing with ≥150 mg/kg PF-04254644. Collectively at higher doses, myocardial changes observed in rats at 24 hrs in the TK and 7-day single-dose investigative studies were of similar severity to those observed at 48 hrs in the 7-day repeat-dose study. In the 6-day repeat-dose investigative study, rats were given lower doses of 40 and 80 mg/kg PF-04254644, and cTnI levels were not increased beyond background levels. However, a minimal focal myocardial degeneration was observed at ≥6 hrs after dosing in all rats given ≥40 mg/kg. Similarly, the dose of milrinone given to rats in this 6-day repeat dose investigative study (10.5 mg/kg) did not increase the levels of cTnI above background values, and a minimal myocardial degeneration was observed in all rats at ≥6 hrs postdose but was undetectable at 2 hrs. In summary, cTnI levels in rats given a higher dose of PF-04254644 (≥320 mg/kg) likely peaked early around 2 hrs after dosing, preceded the development of the maximal myocardial lesion at 24 to 48 hrs, and correlated with the dose-related severity of myocardial injury.
Mean and standard deviation of cardiac troponin I (cTnI) levels in serum and incidence of myocardial degeneration in control, PF-04254644-treated, or milrinone-treated rats.
ND = not done; T.S. = toleration study; I.S. = investigative study.
a PF-04254644.
b Milrinone.
Echocardiography Evaluations
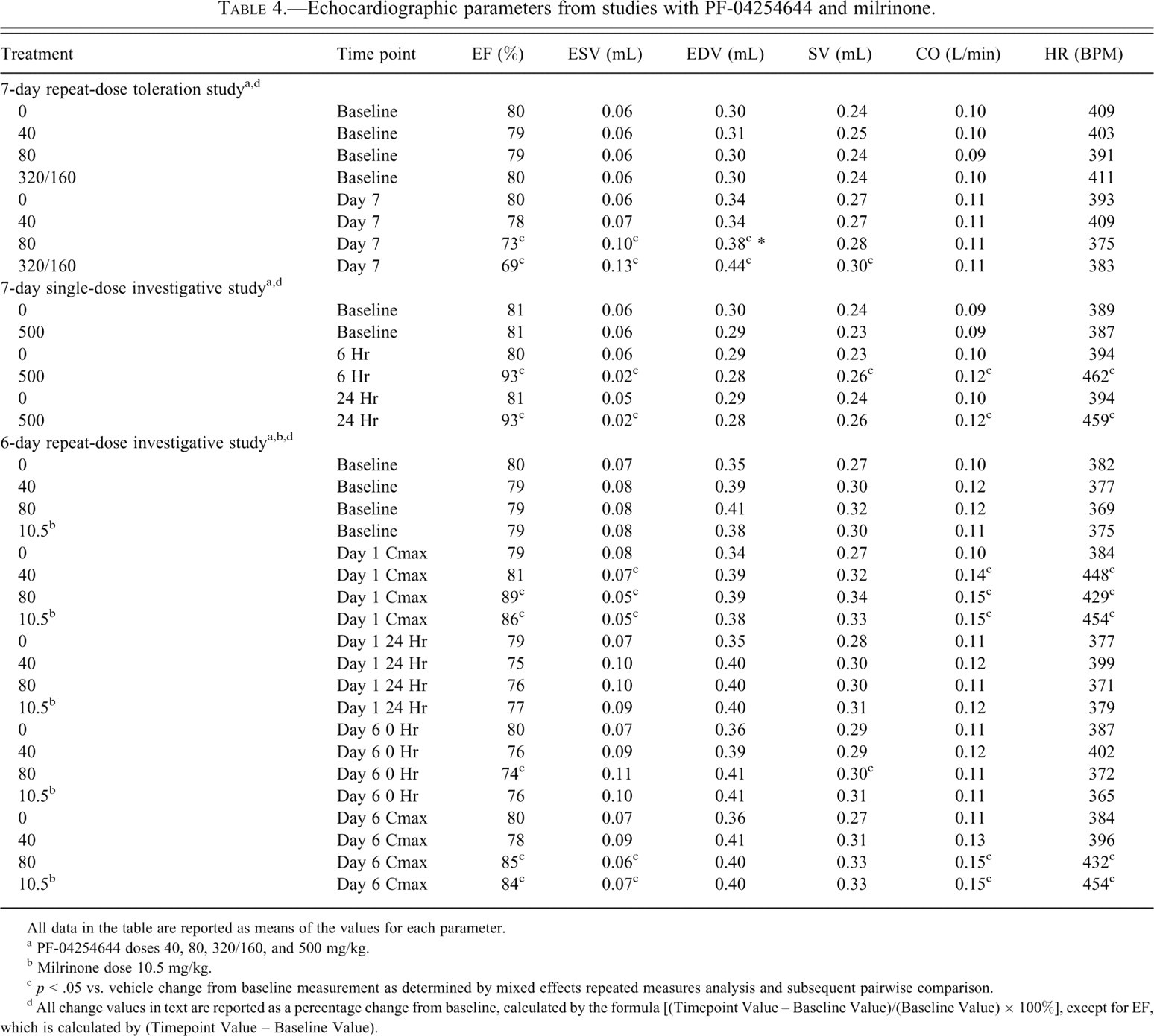
Cardiac functional parameters including EF, ESV, EDV, SV, CO, and HR following treatment with PF-04254644 or milrinone in rats are listed in Table 4 . In the 7-day repeat-dose toleration study, a dose-dependent decrease in cardiac contractility measured by changes in EF and ESV occurred in rats treated with 80 or 320/160 mg/kg. EF was decreased 6% and 11% from baseline, and ESV was increased 67% and 113%, in rats given 80 and 320/160 mg/kg PF-04254644, respectively. Rats treated with 80 or 320/160 mg/kg PF-04254644 had preserved CO because of small increases in diastolic chamber volume. In the 7-day single-dose investigative study, EF was increased by 12% over baseline values at 6 and 24 hrs, and significant increases in HR (19%) and CO (33%) were also observed at 6 and 24 hrs postdose in rats given 500 mg/kg PF-04254644. In the 6-day repeat-dose investigative study, an increase in EF (2% and 10%), CO (17% and 25%), and HR (5% and 17%) was observed at Cmax in rats given 40 and 80 mg/kg PF-04254644, respectively, and all parameters returned to normal or baseline levels 24 hrs postdose. However, a significant decrease in EF (5%) and SV (6%) was evident before dosing with 80 mg/kg PF-04254644 on day 6. In spite of this decreased contractility during time of minimal drug concentrations, treatment with 80 mg/kg PF-04254644 increased EF, CO, and HR (6%, 25%, and 17%, respectively) on day 6 at Cmax. No significant changes in echocardiography parameters occurred on the 6th day of dosing in rats given 40 mg/kg PF-04254644. Milrinone treatment in rats on day 1 induced an increase over baseline in EF (7%), CO (37%), and HR (21%) at Cmax and no significant changes in these parameters at 24 hrs postdose. Similarly, on day 6 at 0 hr before dosing with milrinone, EF and SV were not significantly different from vehicle-treated animals, and EF, CO, and HR increased (5%, 37%, and 21%, respectively) at Cmax. Across all studies, echocardiographic changes in rats given vehicle were consistent with historical data from nontreated and vehicle-treated rats.
Echocardiographic parameters from studies with PF-04254644 and milrinone.
All data in the table are reported as means of the values for each parameter.
a PF-04254644 doses 40, 80, 320/160, and 500 mg/kg.
b Milrinone dose 10.5 mg/kg.
c p < .05 vs. vehicle change from baseline measurement as determined by mixed effects repeated measures analysis and subsequent pairwise comparison.
d All change values in text are reported as a percentage change from baseline, calculated by the formula [(Timepoint Value – Baseline Value)/(Baseline Value) × 100%], except for EF, which is calculated by (Timepoint Value – Baseline Value).
In summary, soon after dosing rats with 500 mg/kg PF-04254644, cardiac contractility measured by EF, CO, and HR as measured by echocardiography R-R interval remained increased over a 24-hr time period. After 7 days of repeat dosing in rats with ≥80 mg/kg PF-04254644, there was an overall reduction in cardiac function with dose-dependent decreases in cardiac contractility and a preserved CO. After a single dose of 80 mg/kg PF-04254644, cardiac contractility and HR increased as early as 3 hrs postdose and returned to normal baseline levels at 24 hrs. However, after repeat dosing and before the last treatment on day 6 (time 0 hr) with 80 mg/kg, there was a significant decrease in cardiac contractility. In spite of this decreased contractility, rats that were given the last dose of 80 mg/kg on day 6 were able to respond with an increase in cardiac contractility (EF), CO, and HR at Cmax. Rats given 40 mg/kg PF-04254644 or 10.5 mg/kg milrinone responded with an increased contractility, HR, and CO at Cmax on day 1 and returned to baseline levels 24 hrs later. On day 6, prior to the last dose (time 0 hr), no changes in cardiac function were detectable in rats given 40 mg/kg PF-04254644 or 10.5 mg/kg milrinone. However, on day 6 at Cmax, rats given milrinone responded with an increased cardiac contractility, HR, and CO; while rats given 40 mg/kg PF-04254644 did not respond with any detectable changes in cardiac function.
Rodent Telemetry
In the 6-day investigative repeat-dose study, HR changes were continuously monitored in rats given 40 or 80 mg/kg PF-04254644 or 10.5 mg/kg milrinone on day 1 through day 6, as shown in Figure 3 . On day 1, an increase in HR or tachycardia (HR change from vehicle ≥25 bpm) was first observed at 2 hrs postdose in rats given 40 or 80 mg/kg PF-04254644 for a duration of 5 hrs. In comparison, tachycardia was first observed 1 hr postdose in rats given milrinone and reached a maximum effect 4 hrs postdose. On day 2, the magnitude and duration of the milrinone-induced tachycardia was greater than that observed with PF-04254644. However, over days 3 to 5, although the initial magnitude of the tachycardia postdose was greater for milrinone than PF-04254644, the overall duration of the PF-04254644-induced tachycardia was longer (at least 8 hrs) than that observed for milrinone (<4 hrs). On day 6, a maximum tachycardia (120 bpm) relative to vehicle controls occurred 1 hr postdose in rats given 80 mg/kg PF-04254644 or 2 hrs postdose in rats given 40 mg/kg and was sustained for 8 hrs. In contrast, milrinone on day 6 induced a maximum tachycardia (100 bpm) relative to vehicle controls that lasted for less than 1 hr and returned to baseline levels in <3 hrs. Therefore over the course of 6 days of repeat dosing in rats, PF-04254644 or milrinone induced a tachycardia (>100 bpm) 1 to 2 hrs after dosing, but the tachycardia (>25 bpm) in rats given PF-04254644 lasted 1 to 6 hrs longer in duration compared with rats given milrinone.
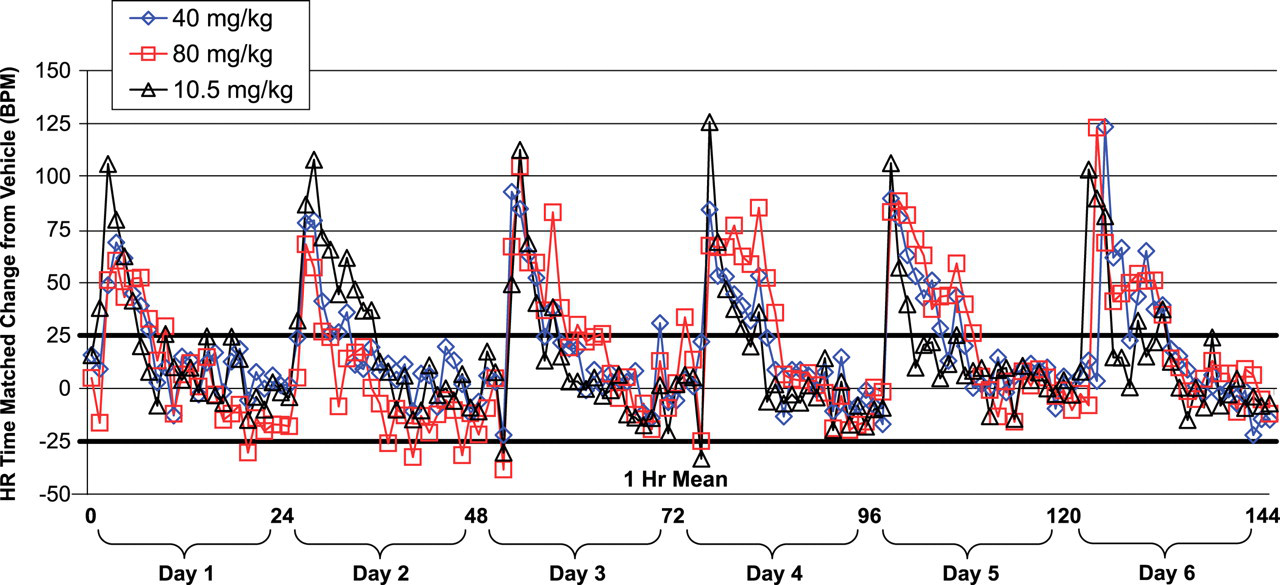
Heart rate profile in telemetry-instrumented rats from the 6-day repeat-dose investigative study. Rats were given 40 or 80 mg/kg PF-04254644 or 10.5 mg/kg milrinone. Heart rate data represented as 1-hr means of time-matched change from rats given vehicle. Dosing occurred at or during hrs 2, 25, 49, 73, 96, and 121. Heavy black bars at 25 and –25 depict baseline HR range prior to dosing for male Sprague Dawley rats aged 7 to 9 weeks.
Systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean blood pressure (MBP) were also monitored for 6 days in rats given 40 or 80 mg/kg PF-04254644 or 10.5 mg/kg milrinone (data not shown). Prior to dosing, BP measurements ranged between –5 mmHg to +5 mmHg of group averages of 118 mmHg SBP, 100 mmHg MBP, and 80 mmHg DBP. The most remarkable drop in blood pressure was observed in rats given milrinone. Over the course of 6 days, a 15-20 mmHg decrease in SBP, DBP, and MBP at time of maximal drug concentration (Cmax) was observed approximately 1 hr postdose and was sustained for 4 to 6 hrs in rats given milrinone. The BP decrease in rats given milrinone was consistent with internal Pfizer data (unpublished data). After 6 hrs of dosing, BP values returned to baseline levels. Upon repeat dosing with milrinone, the decrease in BP diminished to a level similar to PF-04254644-treated rats. The maximal decrease in SBP, DBP, and MBP in rats given 40 or 80 mg/kg PF-04254644 was 8 to 10 mmHg at 1 hr postdose (or Cmax) and was sustained for 4 to 6 hrs and then returned to baseline values after 6 hrs over the 6-day treatment period. With PF-04254644 repeat dosing, the BP values remained within the range of baseline values after 6 hrs of dosing and were consistently higher than baseline BP values observed with milrinone treatment. Therefore, the overall pattern of BP decrease induced by milrinone or PF-04254644 treatment in rats was somewhat similar, but the overall decrease in BP was greater in milrinone-treated rats compared to PF-04254644-treated rats.
Toxicokinetics
Across studies, systemic exposure, as defined by area under the curve (AUC), to PF-04254644 increased with increasing dose, whereas the increase in Cmax was less than dose proportional. Time of maximum plasma concentration (Tmax) for PF-04254644 was in the range of 2 to 4 hrs postdose (data not shown). The ranges for the free plasma concentrations on first and/or last day of dosing were 6.2 to 12 μM in the TK study, 3.1 to 15 μM in the 7-day IVT study, 6.2 to 10.4 μM in the single-dose investigative study, and 2.0 to 3.7 μM in the 6-day repeat-dose investigative study. Except for the 6-day repeat-dose investigative study, the free plasma concentrations of PF-04254644 in rat studies exceeded the IC50 values for PDEs 1A, 1B, 1C, 2, 3A, 3B, 4A, 4B, 4C, 4D, and 5 expressed in cardiac tissue and PDEs 6, 7A, 7B, 8A, 9, 10, and 11 expressed in other tissues (Table 2). In the 6-day repeat-dose investigative study, free plasma concentrations of PF-04254644 exceeded the IC50 values for PDE 1A, 3A, 3B, 10, and 11 (Table 2).
The Tmax for milrinone occurred at 2 hrs postdose (data not shown). The maximal free plasma concentration of milrinone was 2.5 uM and exceeded the IC50 values for PDE3A, 3B, and 4D (Table 2).
Discussion
In rats, PF-04254644 was not well tolerated at doses ≥320 mg/kg, likely because of a dose-related mild to moderate myocardial degeneration observed at 24 to 48 hrs postdose. Clinical signs of morbidity in rats given 320 mg/kg were evident by 48 hrs postdose. Peak serum cTnI levels at 2 hrs postdose in rats given 500 mg/kg PF-04254644 suggests myocardial damage correlated with peak systemic exposure levels at Tmax. Because a minimal myocardial degeneration was observed in most rats given ≥40 mg/kg PF-04254644 and echocardiography and telemetry evaluations confirmed cardiovascular effects at doses ≥40 mg/kg, the no observed adverse effect level (NOAEL) could not be determined from these studies.
Although the mechanism of PF-04254644-induced myocardial toxicity is unknown, further investigation using electron microscopy in single-dose and repeat-dose investigative studies suggested that the PF-04254644-induced myocardial damage was dose- and time-related early impairment of the myofibers and intercalated disks. Within hours of dosing, the loss of cardiac myofibers or myocytolysis and fragmentation of the intercalated disks observed in rats given 500 mg/kg PF-04254644 are features similar to those reported in isoproterenol-induced cardiotoxicity (Dhalla et al. 1992). The myocytolysis and fragmentation of the intercalated disks at higher doses of PF-04254644 could be explained by the excessive force on the myofibers at the apex, possibly due to the cardiovascular effects of increased HR and contractility. Normal appearing mitochondria among fragmented myofibers, lipid deposition, intramitochondrial densities, and disorganized and/or fragmented myofibers were observed at 2 and 8 hrs in rats given PF-04254644 (Fig 2) and have also been observed in rats with catecholamine cardiotoxicity (Dhalla et al. 1992). Intramitochondrial densities representing calcium phosphate or calcium protein complexes suggested increased mitochondrial oxidative activity (Rona 1985). Minimal myocardial changes at lower doses of PF-04254644 (≤80 mg/kg) were similar to those described in rats given lower doses of isoproterenol (Zhang et al. 2008), and these doses correlated with a mild increase in HR (~50 bpm) and, although not reported, presumably contractility in rats given lower doses of isoproterenol (van Steeg et al. 2007). The overlapping ultrastructural findings in PF-04254644-induced cardiotoxicity and catecholamine-induced cardiotoxicity may in part be related to physiological stress of increased HR and contractility driven by shared second messenger mechanisms in the rat cardiovascular system.
Increased contractility of myocardial cells mediated through the accumulation of intracellular cAMP/cGMP can occur either through direct stimulation of beta adrenergic receptors on the heart by catecholamines or through inhibition of cAMP/cGMP breakdown by PDE3 inhibitors, and the subsequent toxic levels of intracellular cAMP/cGMP can lead to myocardial degeneration (Movsesian 1999). In this study, an increase in myocardial contractility determined by echocardiography was observed in rats given PF-04254644 or milrinone, and similar contractility effects have been reported in rats given isoproterenol (Dhalla et al. 1992). Myocardial degeneration in rats induced by milrinone was morphologically similar to isoproterenol (Center for Drug Evaluation and Research 2002) and morphologically similar to PF-04254644 at 24 hrs postdose. The mechanism of myocardial degeneration secondary to the accumulation of cAMP in cardiac myocytes is unknown and may be related to increased oxygen demand, cardiovascular hemodynamics, membrane permeability alterations, free radical oxidation, calcium or fatty acid overload, mitochondrial dysfunction, and induction of apoptosis (Dhalla et al. 1992; Rona 1985). Isoproterenol-induced myocardial degeneration has been characterized as a distinctive infarct-like myocardial necrosis that can be observed at the macroscopic level (Rona 1985), and the most broadly accepted mechanism for this change is increased oxygen demand resulting from a pure vasodilatation-induced tachycardia (Dhalla et al. 1992). Milrinone is a positive inotrope and can increase cardiac contractility directly; however, milrinone also has effects on the peripheral vasculature resulting in arterial and venous dilation. The effects of milrinone are suggested to be through increases in intracellular cAMP and intracellular calcium without an increase myocardial oxygen consumption (Shin et al. 2007). The sustained tachycardia observed in rats given PF-04254644 could lead to an increased oxygen demand in myocardial cells and eventual myocardial hypoxia and myodegeneration. Therefore, the similarity of the myocardial degeneration at the ultrastructural and histopathological levels observed with PF-04254644, milrinone, and reported for isoproterenol in rats suggests that overlapping mechanisms related to intracellular cAMP signaling and subsequent cardiovascular effects (increased HR and contractility) are likely responsible for the toxic phenomenon in the heart.
Although minimal myocardial degeneration was detected in rats given ≤80 mg/kg, cTnI concentrations were not increased above background levels. The possible explanations for the inability to detect increased serum cTnI in rats given lower doses of PF-04254644 may be multifactorial. The sampling time of 2 hrs postdose was likely the appropriate time frame to detect cTnI release based upon cTnI release in rats given higher doses of PF-04254644, which correlated with severity of myocardial injury at 24 hrs after dosing. The assay used to detect release of cTnI may have been of insufficient sensitivity to detect release of cTnI in the presence of minimal myocardial injury. The insensitivity of the cTnI assay may have been further confounded by the wide variation in serum cTnI responses that was observed in several rat experiments where a considerable in-group variation was reported (York et al. 2007).
Recent publications have postulated on the molecular mechanism(s) of cardiotoxicity associated with therapeutic responses in cancer patients undergoing treatment with TK inhibitors. Inhibition of TK pathways targeting c-ABL, HER2, VEGF, and EGFR may carry an inherent cardiovascular risk for developing congestive heart failure (Force et al. 2008; Kerkela et al. 2006). Although several of these kinase targets are not expressed in cardiomyocytes, cardiac liability arises when these RTK inhibitors inhibit unintentional bystander targets that are not essential for killing tumor cells but are involved in cardiomyocyte survival. The common mechanistic theme described with these RTK inhibitor cardiotoxicites is mitochondrial dysfunction and energy depletion (Force et al. 2008; Kerkela et al. 2006). Unlike these RTK inhibitors, PF-04254644 potently inhibited c-Met only with very little activity for other TKs; however, the compound potently inhibited several PDE families with known cardiovascular liabilities. C-Met/HGFR and PDEs have species-specific expression patterns and cofactors that influence downstream regulators. It is conceivable that species-related differences could result in different toxicological manifestations. The cardiotoxic effects observed in rats given PF-04254644 are likely related to the off-target PDE inhibition and not due to inhibition of other kinases.
The screening of PF-04254644 in the PDE enzyme functional assay suggested potent inhibitor activity for multiple members of the PDE family, notably PDE3. PDE3 inhibitors are known vasodilators and positive inotropes with compound-specific profiles in vasodilator and contractility action (Earl, Linden, and Weglicki 1986a; Schlepper, Thormann, and Mitrovic 1989; Sircar et al. 1989). The net effect of PDE3 inhibitors on the cardiovascular system are compound-specific and most likely dependent upon the relative effect of the agent on determinants of myocardial oxygen demand such as heart rate and contractility (Kaufmann and Pargger 1994). In the current study, the cardiovascular profile of milrinone, consisting of increased contractility and tachycardia coupled with a decrease in BP, was consistent with previous publication and unpublished internal data (Desjardins and Cauchy 1995; Ramirez et al. 2008). While there was a similar magnitude of increased contractility in rats given PF-04254644 or milrinone indirectly measured using echocardiography, the decrease in blood pressure was greater in rats given milrinone (data not shown). Furthermore, the tachycardia was sustained for a longer duration upon repeat dosing with PF-04254644; however, this phenomenon may be a direct result of different pharmacokinetic properties of milrinone and PF-04254644 in the rat. These data suggest that the cardiovascular effects in rats following treatment with PF-04254644, while somewhat similar to milrinone, do not fully mimic prototypical PDE3 inhibition in the rat. Consequently, the overall cardiovascular profile of PF-04254644 may be influenced by inhibition of multiple PDE families, notably PDE1 and PDE5 in addition to PDE3. In addition, the possible contribution of inhibition of other PDE families (PDE2, 4C, 4D, 5, 10, 11) to the cardiovascular profile by PF-04254644 cannot be completely ruled out.
Rats treated with PF-04254644 in single- and multiple-dose experiments demonstrated robust and sustained tachycardia most likely driven by PDE inhibition. In addition to PDE3, PDE1 and PDE5 are also inhibited by PF-04254644 (Table 2) and are expressed in the heart and vasculature of several species including rat and human (Bender and Beavo 2006; Lugnier 2006). In vitro models suggest that PDEs 1 through 5 control the spontaneous beating rate of cardiac pacemarker cells by suppressing local calcium releases and that PDE3 is the major family contributing to the total PDE basal activity (Vinogradova et al. 2008). Thus, the off-target inhibition of several PDE families in cardiac pacemarker cells may represent a direct mechanism contributing to tachycardia in addition to any reflexive mechanisms caused by vasodilatation. In the 6-day repeat-dose study, the magnitude and duration of the tachycardia was reflected by the drug plasma concentrations in rats and PDE enzyme IC50 values (Table 4). Plasma drug concentrations of PF-04254644 in rats exceeding the IC50 concentrations of PDE1A, PDE1C, PDE3A, and PDE3B (Table 2) correlated with the time course of effects on cardiovascular parameters, namely, HR and EF (Figure 3 and Table 4). Prolonged inhibition of PDE1A and PDE3B in the current studies may result in the observed cardiovascular profile of increased HR and EF (Pfizer Inc., data on file). Thus, it is plausible that after a single high dose or with repeat dosing at a lower dose of PF-04254644, a combination of increased heart rate and contractility due to the inhibition of several PDE families would contribute to the early cardiac myofiber changes, leading to eventual myocardial degeneration.
The exact pharmacologic mechanism of PF-04254644-induced myocardial degeneration is unknown and may involve multiple signaling pathways and compensatory mechanisms in addition to cAMP and/or cGMP, which in turn can have detrimental effects on the cardiovascular system. In vitro paradigms have suggested that the c-Met signaling pathway may be important in cardiomyocyte protection either directly by blocking cell death from oxidative stress or indirectly from inhibiting apoptotic pathways (Kitta et al. 2003; Ueda et al. 2001). Cardiomyocyte protective kinase signaling pathways downstream of c-Met, including Akt, Mek, and Erk, may also block cell death (Miyamoto, Murphy, and Brown 2009; Wang 2007). Thus, it is conceivable that inhibition of c-Met signaling could exacerbate cardiomyocyte cellular damage. However, current in vivo toxicology data suggest that the mechanism of PF-04254644-mediated cardiotoxicity is not likely directly related to inhibition of the c-Met signaling pathway. Preclinical and clinical results of other propriety potent c-Met inhibitors belonging to a chemical series different from PF-04254644 did not report cardiac toxicity in 1-week rat studies (Pfizer Inc., data on file) or in patients enrolled in phase I/II clinical trials (Eder et al. 2009), respectively. Unlike other potent c-Met inhibitors, PF-04254644 has a unique off-target multi-PDE inhibition profile that is most likely responsible for the observed cardiotoxicity. Additionally, another potent c-Met inhibitor of the same chemical class as PF-04254644, which differed from the structure of PF-04254644 only by the substitution of two fluorine groups for a methyl group on C6, reported an increased HR in dogs at highest doses (Perera et al. 2008), which suggests a possible pharmacophore-related hemodynamic effect. Therefore, whether the pharmacologic mechanism of PF-04254644-related cardiotoxicity involves one or a combination of these effects remains unanswered and is the subject of ongoing investigations. However, the inhibition of c-Met alone without additional PDE inhibition does not result in cardiotoxicity in the rat.
In summary, this article described the cardiovascular effects of PF-04254644 in rat preclinical studies. The effects were considered dose related, and the exact mechanism inducing this change is still unknown. However, myocardial degeneration observed in rats given ≥80 mg/kg PF-04254644 may be due to the off-target PDE inhibitory activity and subsequent induction of increases in HR and cardiac contractility. Ultimately, a variety of mechanisms compensatory and otherwise that converge on either necrotic or apoptotic pathways of cell death may have resulted in the observed myocardial degeneration, and that is the subject of ongoing investigation.
Footnotes
This study was supported by funding from Pfizer, Inc.
Acknowledgments
Special thanks to individuals who directly contributed to this work: Aileen McHarg and Winston Evering for review of the manuscript; Ray Lui, Jingjing Ye, Kirk Kozminski, and Shinji Yamazaki for PK support; Jeri Eades and April Marr for histology assistance; and Darren Samuel and Constance Benedict for graphic arts assistance.