
Abstract
Acute tumor lysis syndrome (ATLS) is characterized by severe metabolic abnormalities and organ dysfunction resulting from rapid destruction of neoplastic cells. Metabolic disturbances are thought to be the primary cause of clinical ATLS symptoms, which include renal dysfunction, seizures, and cardiac arrhythmias. The histopathologic lesions associated with organ dysfunction are largely unknown because of the low rate of mortality of ATLS in humans and the few cases of ATLS identified in laboratory animals. Here, we describe histologic, immunohistochemical, and electron microscopic analyses of thirty-one ATLS cases from a cohort of 499 mice that are prone to spontaneous lymphoblastic lymphoma owing to genetic defects in DNA replication fidelity. Seventy-three percent of our cohort died with lymphoblastic lymphoma, and 8% of affected mice died with diffuse microthromboemboli consistent with ATLS. Mice with ATLS had a high spontaneous mortality rate (>50%), a large tumor burden with disseminated disease, and evidence of leukemia. Blood vessels in the lung, kidney, and other organs were occluded by microthromboemboli composed of chromatin, cellular debris, fibrin, platelets, and entrapped erythrocytes and malignant cells. This case series suggests that ATLS can occur at high frequency in mice with disseminated lymphoblastic lymphoma and leads to a high rate of spontaneous death from microthromboemboli.
Keywords
Introduction
Acute tumor lysis syndrome (ATLS) results from massive necrosis or apoptosis of large proliferating tumors and is characterized by marked hyperuricemia, hyperkalemia, and hyperphosphatemia secondary to cell lysis and the metabolism of excessive nucleic acids (Davidson et al. 2004; Hochberg and Cairo 2008). Acute tumor lysis syndrome occurs most commonly in patients with hematologic malignancies; however, ATLS has also been reported with solid malignancies (Davidson et al. 2004; Hochberg and Cairo 2008). Clinically significant ATLS, with end-organ compromise, occurs in approximately 5% of all patients with hematologic malignancies (Hochberg and Cairo 2008) and in up to 25% of high-risk patients, including those with T-cell acute lymphoblastic leukemia and Burkitt’s lymphoma (Coiffier et al. 2008). Acute tumor lysis syndrome also develops secondary to hematopoietic malignancies and solid tumors in veterinary species (LaCarrubba et al. 2006; Radaelli et al. 2009), although the overall incidence is unknown. Life-threatening complications of ATLS include acute renal failure, cardiac arrhythmias, neurologic perturbations, and metabolic acidosis (Davidson et al. 2004; Hochberg and Cairo 2008).
The pathophysiology of ATLS and its associated histopathologic lesions in humans and animals are not well characterized. One known cause of nephropathy in humans is the deposition of urate or calcium phosphate crystals in the kidney tubules (Davidson et al. 2004; Hochberg and Cairo 2008). Seizures and cardiac arrhythmias associated with ATLS are usually attributed to metabolic changes, including hyperkalemia, hyperphosphatemia, and hypocalcemia (Davidson et al. 2004). Similar metabolic derangements have been described in cats, dogs, and a horse with ATLS (Calia et al. 1996; LaCarrubba et al. 2006; Laing and Carter 1988; Mylonakis et al. 2007; Vickery and Thamm 2007), but histopathology has not been routinely reported. A single series of three canine ATLS cases revealed generalized microthrombi in the necropsy specimens of two dogs (Laing and Carter 1988). Additionally, Lovelace et al. describe a single DBA mouse with lymphoblastic lymphoma that exhibited neurologic symptoms associated with multifocal vascular thrombi throughout all tissues; special stains and electron microscopy suggested the microthrombi were composed of DNA and fragmented cellular components, consistent with debris from tumor cell lysis (Lovelace et al. 2003). The authors proposed that symptoms in this mouse may have been secondary to ischemia from mechanical obstruction of the vasculature by thromboemboli. A recent case report described similar microthrombi in one 129/SvEv mouse with acute promyelocytic leukemia that died suddenly during treatment with valproic acid, possibly recapitulating treatment-induced ATLS (Radaelli et al. 2009).
Our case series is extracted from studies looking at the effects of decreased DNA replication fidelity on disease incidence in mice. Defects in DNA polymerase proofreading and mismatch repair increase spontaneous mutation (mutator phenotype) and predispose mice to aggressive lymphoblastic lymphoma (Albertson et al. 2009; Edelmann and Edelmann 2004; Goldsby et al. 2002). In this report we describe a large cohort of animals expressing combinations of DNA polymerase and mismatch repair mutator alleles. Seventy-three percent of these mutator mice developed lymphoblastic lymphoma, and disseminated microthromboemboli were observed in almost 10% of the affected animals. Microthromboemboli were composed of malignant cell debris and associated with tissue compromise and increased risk of spontaneous death. These studies show that ATLS can be common in mice with disseminated lymphoblastic lymphoma, can lead to rapid death, and may be underreported.
A supplemental appendix to this article is published electronically only at http://tpx.sagepub.com/supplemental.
Materials and Methods
Animals and Housing Conditions
Mice used in this study that harbor mismatch repair-deficient alleles have been described previously and are at an elevated risk of lymphoblastic lymphoma: Msh2Δ (Smits et al. 2000), Msh2G674A (Lin et al. 2004), and Mlh1Δ (Edelmann et al. 1999). Mice with error-prone DNA polymerase alleles have also been described previously: Pold1e (Goldsby et al. 2002), Pold1L604G (Venkatesan et al. 2007), and Polee (Albertson et al. 2009). All mice were part of survival studies that have been or will be published elsewhere. Single mutant mice were bred as heterozygotes to fill experimental cohorts, as described previously (Albertson et al. 2009; Goldsby et al. 2002). Mutant cohorts were included in this study if they were at elevated risk of lymphoblastic lymphoma. Cohorts were as follows: C57BL/6J Mlh1Δ/Δ (n = 27), C57BL/6J Msh2Δ/Δ (n = 39), C57BL/6J Msh2G674A/G674A (n = 36), C57BL/6J Pold1e/e (n = 40), 129/SvJ Pold1e/e (n = 37), A/J Pold1e/e (n = 32), and FVB/NJ Pold1e/e (n = 74). Double-mutant mice were generated from double-heterozygous breeding pairs in the C57BL/6J genetic background. Double-mutant cohorts were as follows: Pold1e/e Mlh1+/Δ (n = 34), Pold1e/eMsh2+/Δ (n = 33), Pold1e/eMsh2+/G674A (n = 31), Pold1+/eMlh1 Δ/Δ (n = 38), Pold1+/e Msh2 Δ/Δ (n = 42), Pold1+/eMsh2G674A/G674A (n = 39), Pold1+/L604GMsh2 Δ/Δ (n = 15), Pole+/eMlh1Δ/Δ (n = 9), Pole+/eMsh2Δ/Δ (n = 17), Pole+/eMsh2G674A/G674A (n = 27). Genotyping of animals was performed on genomic DNA purified from ~0.5 cm tail tip segments taken at weaning, as described previously (Albertson et al. 2009; Edelmann et al. 1999; Goldsby et al. 2002; Lin et al. 2004; Venkatesan et al. 2007).
Mice were multiply housed (one to five mice per cage) in ventilated Allentown cages (model MBS75JRHMVX) containing Bed-O-Cob (The Andersons, Maumee, OH) in a non-barrier specific pathogen-free facility with a twelve-hour light/dark cycle at the University of Washington. All life- and health-span studies were conducted with contemporary mutant cohorts and wild-type controls housed together in the same room and cages. Food (irradiated Picolab Rodent Diet 20 #5053; PMI Nutrition International, Brentwood, MO, USA) and reverse-osmosis water were provided ad libitum. All supplies entering animal rooms were autoclaved, and rooms were maintained at 70–74°F, 45–55% humidity, with twenty-eight air changes/hour. Sentinel mice were tested quarterly and were negative for endo- and ectoparasites, mouse hepatitis virus, mouse parvovirus, and rotavirus and tested annually and were negative for Mycoplasma pulmonis, pneumonia virus of mice, reovirus-3, Sendai virus, and Theiler’s murine encephalomyelitis virus. Sentinels were not tested for Helicobacter species or the newly recognized mouse Norovirus. All procedures were approved by the University of Washington Animal Care and Use Committee.
Tumor Analysis
Mice were observed daily from time of weaning. Animals were euthanized by CO2 inhalation when they met one of the following criteria: moribund, visible tumor >1 cm, ulcerating tumor, or dermatitis/self-inflicted wounds that did not respond to treatment with Betadine. Moribund was defined as respiratory distress, decreased activity, or visibly evident loss of weight. Complete necropsies were performed, and brain, kidney, liver, lung, spleen, and any abnormal tissue were taken for histology. In a subset of moribund mice that were euthanized, peripheral blood smears were collected and stained with Wright-Giemsa. White blood cell counts were estimated as an average of ten high-dry (40×) fields × 1600 conversion factor. Tissue samples were fixed in 10% phosphate-buffered formalin, embedded in paraffin, sectioned at 4–5 μm, stained with hematoxylin and eosin, and examined by light microscopy. A board-certified veterinary pathologist (P. M. T.) examined all slides. Tumors were confirmed and identified histologically (Frith et al. 2001), and lymphomas were classified using the Bethesda recommendations (Morse et al. 2002). After routine histological examination, additional sections were stained with Movat’s pentachrome or 4′,6-diamidino-2-phenylindole (DAPI).
Electron Microscopy
Transmission electron microscopy was performed on lung tissue from one mouse for ultrastructural analysis of neoplastic lymphocytes and microthromboemboli. The lung was post-fixed in 2% aqueous OsO4/0.2 M cacodylate buffer (Electron Microscopy Sciences, Hatfield, PA, USA) followed by dehydration and embedding in Epon 812 (Polysciences, Inc., Warrington, PA, USA). Ultrathin sections were then stained with uranyl acetate (Electron Microscopy Sciences), followed by lead citrate (Leica Microsystems, Inc., Bannockburn, IL, USA). Sections were visualized with a JEOL 1230 transmission electron microscope equipped with an 11-megapixel SC1000 ORIUS side-mount Gatan CCD camera.
Immunohistochemistry
Immunohistochemical phenotyping of lymphoma samples was performed by the Fred Hutchinson Cancer Research Center’s Experimental Histopathology Shared Resource. Four-micron sections were cut, deparaffinized, and rehydrated in Dako Wash Buffer (Carpinteria, CA, USA). CD3 antigen retrieval was performed in a Black and Decker steamer for twenty minutes in preheated Trilogy buffer (Cell Marque, Hot Springs, AZ, USA) and cooled for twenty minutes. CD45/B220 antigen retrieval was performed similarly in Target Retrieval solution (pH6, Dako). Slides were rinsed three times in Dako wash buffer, and all subsequent staining steps were performed at room temperature using the Dako Autostainer. Endogenous peroxide activity was blocked using 3% H2O2 for eight minutes, followed by protein blocking. Slides were then blocked in 15% goat serum and 5% mouse serum (Jackson ImmunoResearch, West Grove, PA, USA) in Tris-buffered saline (TBS) containing 1% bovine serum albumin (BSA) for ten minutes. All antibodies were incubated on the tissue for thirty minutes and then washed with wash buffer. CD3 antibody (MCA1477, Serotec) was used at 10 μg/mL, and CD45/B220 antibody (CBL1342 Chemicon) was used at 0.5 μg/mL. Antibodies were detected using biotinylated goat anti-rat (112-065-167, Jackson ImmunoResearch) at 1:200 for thirty minutes followed by Vector Elite ABC. The staining for all slides was visualized with 3,3′-diaminobenzidine (DAB, Dako) for seven minutes, and the sections were counterstained with hematoxylin (Dako) for two minutes. Concentration-matched isotype control slides were run for each tissue sample (Jackson ImmunoResearch Laboratories).
Results
We studied the effect of mutation burden on disease and mortality in genetically engineered mice with error-prone DNA polymerases (Albertson et al. 2009; Goldsby et al. 2002; Venkatesan et al. 2007), loss of mismatch repair (Edelmann and Edelmann 2004), or both. Cohorts of nine to seventy-four mice of each genotype were monitored for spontaneous tumor development and survival. Most cohorts exhibited a high susceptibility to cancer and accelerated mortality compared to wild-type animals. During these survival studies, we noted a high incidence of spontaneous death in mice with disseminated lymphoblastic lymphoma and histopathologic lesions consistent with ATLS (Lovelace et al. 2003). To define the overall incidence of ATLS and further characterize the associated pathologic lesions, we analyzed necropsy and histology data from seventeen cohorts of mutator mice that were at increased risk of developing lymphoblastic lymphoma.
In total, 499 mice were necropsied and had tissue adequate for histologic analysis. Forty-two mice died spontaneously; those remaining were euthanized when moribund (Table 1 and Supplemental Table 1). Lymphoblastic lymphoma was diagnosed in 71% of mice that were euthanized and in 98% of those that died spontaneously (Table 1). Among the 366 mice with lymphoblastic lymphoma, thirty-one (8%) exhibited histologic evidence of ATLS (described below). Fourteen cases of ATLS were identified in animals euthanized and necropsied within minutes of death. Seventeen additional cases were found in mice that expired spontaneously and had tissue adequate for histologic analysis (less than twenty-four hours postmortem); two of these ATLS-associated spontaneous deaths occurred during observation in the laboratory moments prior to necropsy. We found no difference in the severity of histologic lesions between mice that were euthanized and those found dead, indicating that the lesions developed antemortem.
Incidences of Lymphoblastic Lymphoma and ATLS in a Mutator Mouse Cohort

a Mice with informative histology.
b Percentages of euthanized, expired and total mice with informative histology.
c Percentages of euthanized, expired and total mice with lymphoblastic lymphoma (LL).
d Statistically different from moribund mice (p < .0005; Fisher exact test).
Acute tumor lysis syndrome was defined histologically as the presence of microthromboemboli composed of neoplastic cells admixed with abundant dark, basophilic acellular material (Lovelace et al. 2003). These lesions were observed in diverse tissues, including the lung (94%), kidney (61%), liver (23%), brain (16%), and spleen (16%) of affected mice (Supplemental Table 1). All mice with ATLS exhibited highly proliferative, disseminated lymphoma (Figure 1 and Supplemental Table 1) and abundant intravascular lymphoblasts (Figure 1). A thymic or nodal mass was common, as was tumor involvement of the liver, spleen, and kidneys (Figure 1 and Supplemental Table 1). Neoplastic cells were uniformly large and round, with high nuclear-to-cytoplasmic ratios and occasional cytoplasmic vacuoles. Nuclei were central, round to irregular, with a fine chromatin pattern consistent with lymphoblasts. Multifocal areas of lympholysis and necrosis were often observed in thymic and nodal masses (Figure 1D). Neoplastic cells infiltrated the liver, kidneys, and spleen and were arranged in highly cellular sheets that compressed adjacent normal tissues and effaced the normal architecture (Figure 1E and 1F). Evidence of malignant cells in the vasculature of various organs suggested a leukemic component to the disseminated lymphoma (Figures 1E and 1G). Leukemia was verified in peripheral blood taken from euthanized mice with ATLS. Leukemic lymphoblasts were evident in all peripheral blood smears examined (n = 9; Figure 1H and Supplemental Table 1); the average white blood cell count was ~51,000/μL (range = 17,000-90,000; normal range = 4,000-12,000/μL) (Jacoby and Fox 2002).
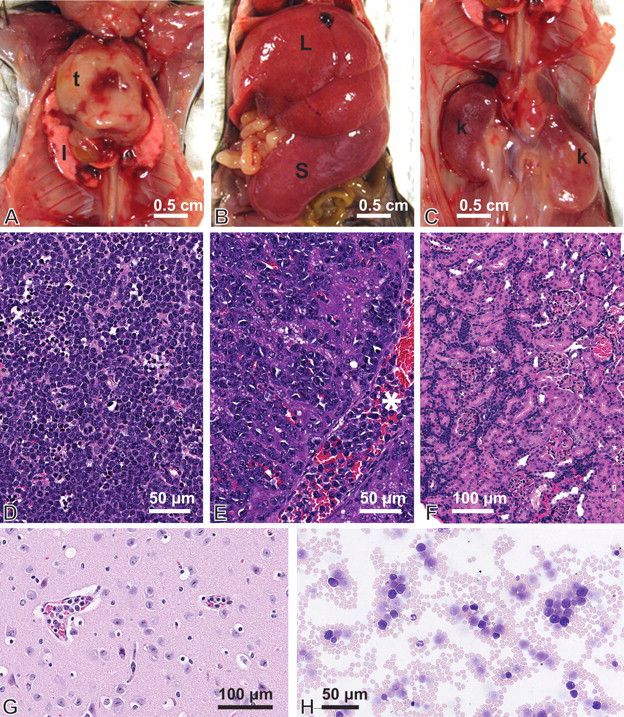
Gross presentation and histology of disseminated lymphoblastic lymphoma. (A) Massive thymic (t) enlargement filling the cranial thoracic cavity. The thymus is markedly enlarged with multifocal hemorrhage (red foci). Similar hemorrhagic foci are present on the lung (1). (B) Markedly enlarged liver (L) and spleen (S) of affected animal. (C) Bilaterally, kidneys (k) are moderately enlarged and mottled. (D) Hematoxylin and eosin (H&E)-stained histologic section of thymic lymphoma with multifocal areas of lympholysis/necrosis. (E) H&E-stained histologic section of liver infiltrated with lymphoma. Normal hepatic architecture is disrupted by sheets of neoplastic lymphocytes that expand the sinusoids and compress the hepatic cords. Note the high numbers of neoplastic cells within the central vein (*). (F) H&E-stained histologic section of kidney infiltrated with lymphoma. The tubules and glomeruli are compressed by accumulations of neoplastic lymphocytes in the interstitium. (G) Intravascular neoplastic lymphoblasts in the brain. (H) Peripheral blood smear stained with Wright-Giemsa. Numerous nucleated cells are observed, including normal neutrophils. Leukemic cells are large and round, with high nuclear-to-cytoplasmic ratios.
Immunohistochemical staining for both T cell (CD3) and B cell (CD45/B220) markers was performed. The majority of tumors were CD45/B220 negative and CD3 positive (faint to strong staining), suggesting T cell lymphoblastic lymphoma (Supplemental Table 1). Five tumors were positive for both CD3 and CD45/B220, and two tumors were CD3 negative and CD45/B220 positive. The absence of a thymic mass in six of the seven mice with CD45/B220-positive lymphomas is consistent with a diagnosis of B cell lymphoma (Supplemental Table 1). Thus, ATLS was associated with both T and B cell lymphoblastic lymphoma.
Microthromboemboli were often found in multiple organs (Supplemental Table 1) and were composed of neoplastic cells admixed with abundant dark, basophilic, acellular material; karyorrhectic and apoptotic debris; platelets; and fibrin (Figure 2 ). The lung was the most common organ involved; twenty-nine of thirty-one animals with ATLS (94%) had microthromboemboli in the pulmonary vasculature (Supplemental Table 1). Alveolar septal vessels were filled with lymphoblasts, and capillaries and veins were markedly distended and occluded by microthromboemboli (Figure 2A–2C). Numerous small- and medium-caliber veins were occluded, and microthromboemboli extended into larger pulmonary veins, where the thrombi partially disintegrated, highlighting the anatomical outflow tract (Figure 2A). Larger thrombi had a tortuous appearance, with a laminated structure resembling lines of Zahn, suggesting the thrombi were exposed to turbulent blood flow (Figure 2B). Additional histochemical staining confirmed that nucleic acids and fibrin were primary components of microthromboemboli (Figure 3 ).
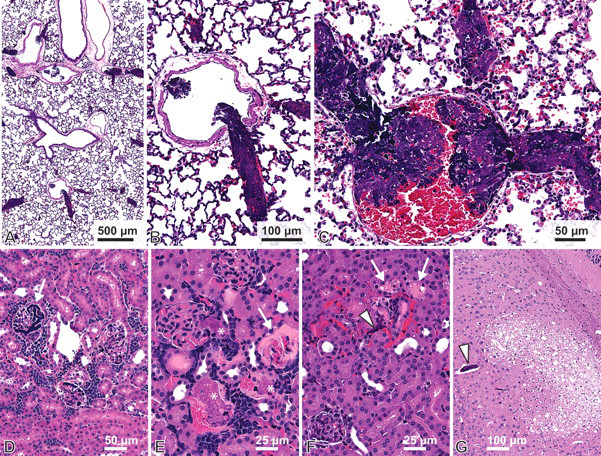
Histologic characterization of microthromboemboli. (A) Hematoxylin and eosin-stained section of lung showing abundant, deeply basophilic microthromboemboli within the vasculature. (B) Higher power of (A). The laminated thrombus is lodged in the outflow tract of a small pulmonary vein into a larger vein. (The cardiac muscle sheath is a normal feature of murine pulmonary veins.) (C) Thrombi are composed of pools of deeply basophilic acellular material, lighter staining material composed of fibrin, entrapped intact neoplastic cells, erythrocytes, and necrotic debris. (D) Deeply basophilic acellular material in the glomerular vessels of an affected kidney (arrow). (E) Foci of fibrinous thrombi (*) and glomerular necrosis (arrow). (F) Renal tubular degeneration and necrosis (arrows) and vascular congestion associated with neoplastic thrombi (arrowhead). (G) Section of cerebrum with locally extensive malacia associated with an occluded vessel (arrowhead).
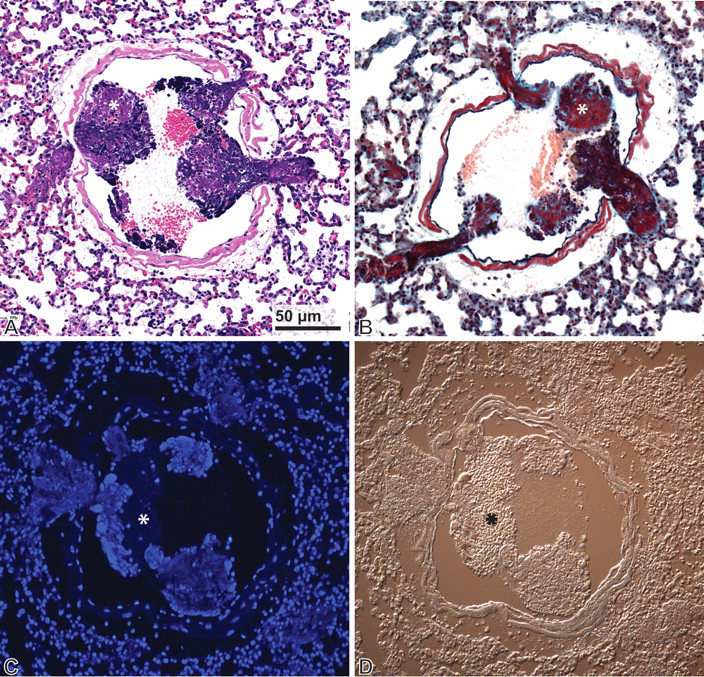
Characterization of microthromboemboli. Sections from the same pulmonary vein were stained with hematoxylin and eosin (A), Movat’s pentachrome (B), and DAPI (C) to confirm the presence of nucleic acids and fibrin. (A) Within a large pulmonary vein, there are numerous thromboemboli lodged in the outflow tracts of smaller veins. Deeply basophilic staining material is admixed with lighter staining material (*) and red blood cells. (B) Fibrin (*) appears deep red, and nuclear debris appears black in the section stained with Movat’s pentachrome. (C) The deeply basophilic material noted on hematoxylin and eosin sections is confirmed as nucleic acid via DAPI staining (bright blue). (D) Differential interference contrast illumination of the same section as (C) to highlight the structural features of the thromboemboli stained with DAPI. The asterisk indicates a region of the thrombus that does not stain with DAPI.
Degenerative lesions consistent with ischemia were often noted adjacent to occluded blood vessels in the kidney and brain (Figure 2D–2G). Renal tubular degeneration occurred in nearly all animals with ATLS and was characterized by numerous cytoplasmic vacuoles, hyaline droplets, and occasional apoptosis or necrosis (Figure 2E and 2F). We did not observe any evidence of crystal deposition in the renal collecting tubules. Tissue compromise was also seen in liver and kidney sections that did not display microthromboemboli. Tissue abnormalities included parenchymal necrosis, hemorrhage, and degeneration and were likely secondary to the large infiltrative tumor burden in those organs or thrombi that did not survive fixation and/or were outside the histologic section.
Electron microscopy was performed on the lung of a single mouse with ATLS. High magnification revealed lymphoblasts within alveolar capillaries (Figure 4). Cells were large, with high nuclear-to-cytoplasmic ratios, central round or irregular nuclei with abundant marginated heterochromatin, and large, irregular nucleoli (Figure 4A and 4B). The cytoplasm was relatively free of organelles, however, mitochondria and ribosomes were observed (Figure 4B, inset). Early and late changes consistent with apoptotic cell death were often noted (Figure 4A and 4C) and cellular debris (ruptured cellular membranes, cytoplasmic fragments, and chromatin) was abundant intravascularly (Figure 4D).
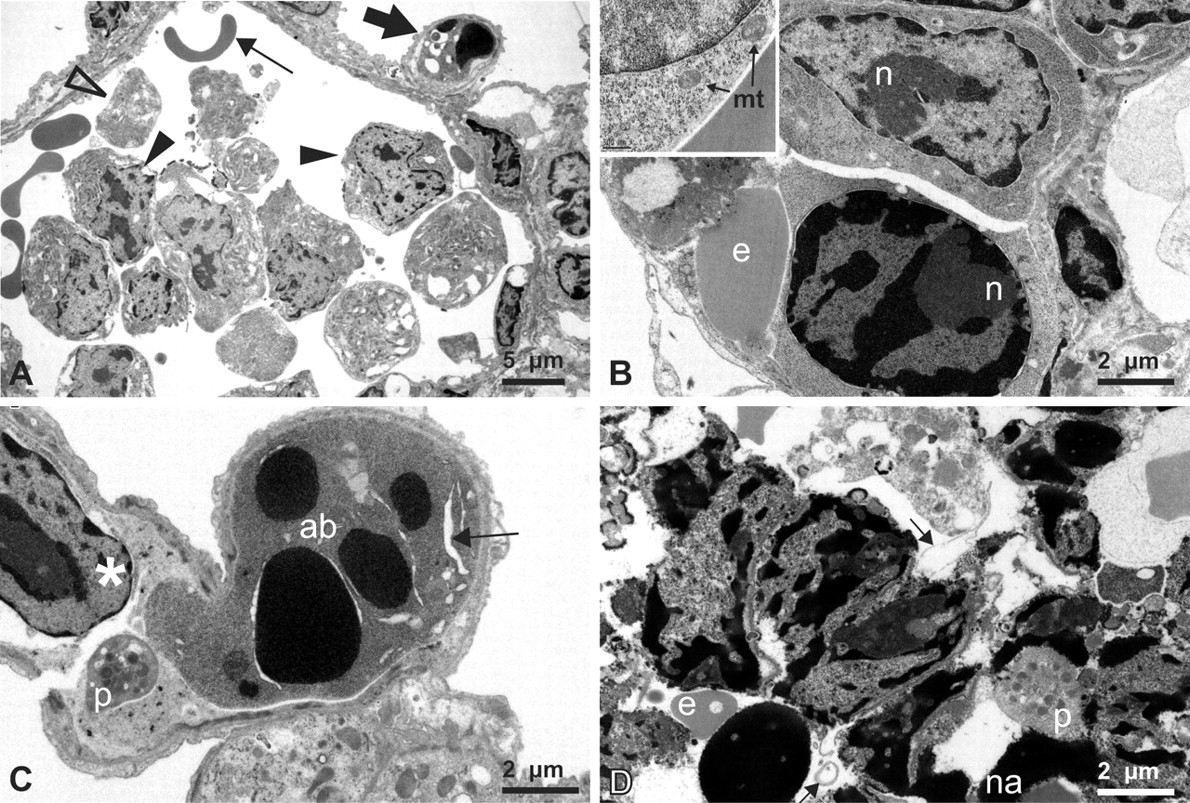
Electron micrographs of malignant lymphoblasts and cell lysis. (A) Pulmonary vessel with abundant intralumenal large nucleated cells (closed arrowheads), partial cell profiles (open arrowhead) and erythrocytes (thin arrow). The small peripheral capillary contains a cell with apoptotic nuclear morphology (thick arrow). (B) High magnification of two lymphoblasts and an erythrocyte (e) within an alveolar capillary. Top cell: a lymphoblast with a large central nucleus; marginated heterochromatin; a large, irregular nucleolus (n); and a high nuclear-to-cytoplasmic ratio. Bottom cell: a lymphoblast with early changes consistent with apoptosis (cytoplasmic shrinkage and nuclear condensation). Inset: cytoplasm is relatively devoid of organelles, with abundant free ribosomes and mitochondria (mt). (C) An apoptotic body (ab) with classic nuclear condensation, blebbing, and evidence of cytoplasmic shrinkage (arrow). Note platelet (p) and lymphoblast (*) also within the capillary. (D) Cellular debris within a pulmonary vein. Ruptured cellular membranes (arrows), electron dense masses (presumptive nucleic acids, na) and platelets (p) are abundant.
Inspection of the microthromboemboli by electron microscopy revealed a collection of debris with varied electron density (Figure 5C and 5D). A large proportion of each thrombus was electron dense, consistent with nucleic acid condensation and the DAPI-positive thrombi detected by fluorescence microscopy (Figure 3C). Cellular fragments were less dense, and neoplastic cells and erythrocytes were commonly trapped in the debris (Figure 5). Large thrombi were laminated and composed of viable and apoptotic lymphoblasts at the margins with central condensed nucleic acid, cytoplasmic fragments, and fibrin; erythrocytes appeared throughout (Figure 5C and 5D). Vessel walls were disrupted, and fibrin was observed within airspaces (Figure 5D). These images suggest that thrombi originate from abundant intravascular lymphoblasts that lyse and block the vessels via buildup of cellular debris, platelets, and fibrin.
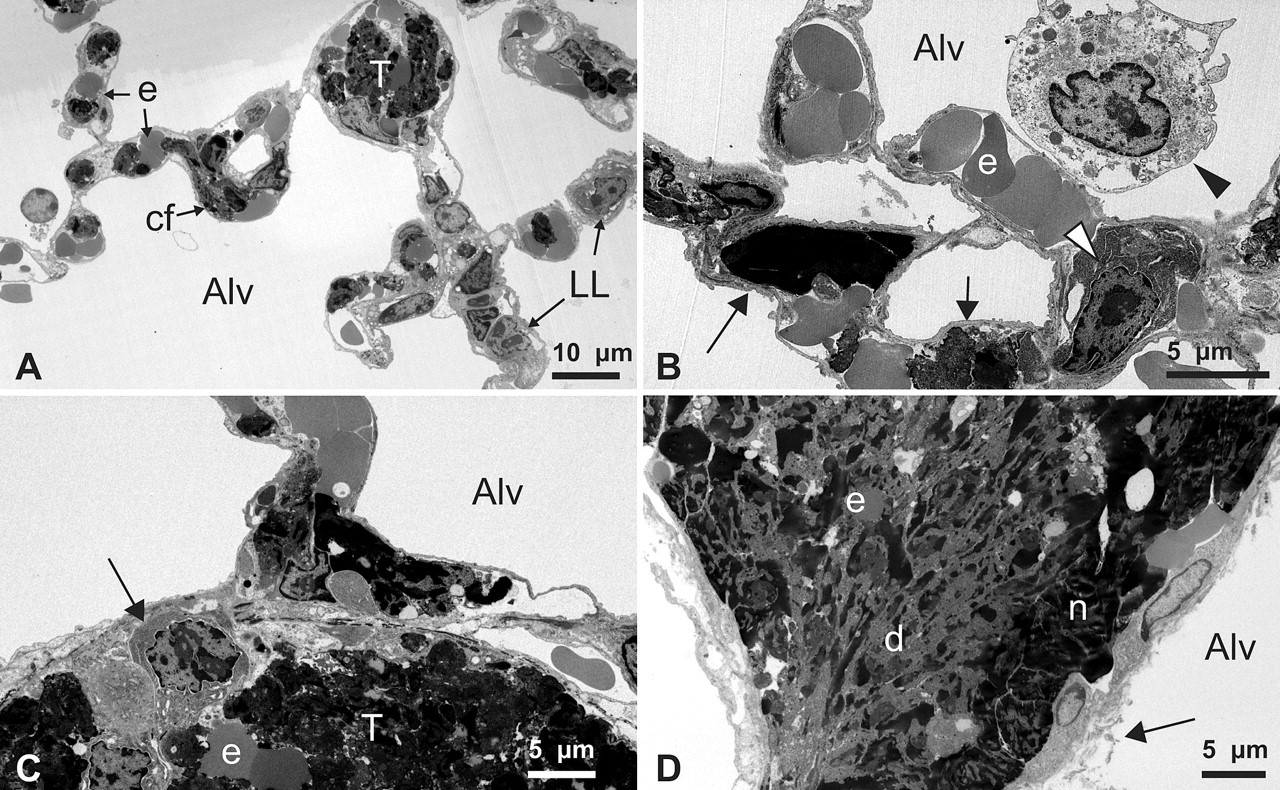
Detailed ultrastructure of pulmonary microthromboemboli. (A) Alveolar septae are expanded by capillaries containing neoplastic lymphoblasts (LL), cell fragments (cf), erythrocytes (e), and a small thrombus (T). Alv, alveolar air space. (B) Higher magnification of alveolar septae with nuclear and cytoplasmic debris (arrows), intact neoplastic lymphoblast (white arrowhead), and deformed and bunched erythrocytes (e). An alveolar macrophage is present within the airspace (black arrowhead). (C) Pulmonary vessel occluded by a thrombus (T) composed of intact (arrow) and lysed neoplastic cells. (D) A large pulmonary thrombus with peripheral margination of degenerate, partially intact nuclei (n) and central layers of cytoplasmic fragments and necrotic debris (d), admixed with fibrin and a few erythrocytic profiles (e). There is also disruption of the endothelium and vessel walls with fibrin strands present within airspaces (arrow).
Discussion
We identified thirty-one cases of ATLS in a cohort of mice highly susceptible to lymphoblastic lymphoma. Acute tumor lysis syndrome cases were identified by the presence of microthromboemboli, lesions similar to those described previously in single case reports (Lovelace et al. 2003; Radaelli et al. 2009). All cases in our series were found in mutator mice with disseminated lymphoblastic lymphoma, primarily of T-cell origin. Furthermore, all mice with ATLS had a high tumor burden and evidence of leukemia. No cases were observed in mice with mature B-cell (pleomorphic/follicular) lymphoma or solid tumors, despite a significant risk of both malignancies in these mouse strains (data not shown). Thus, spontaneous ATLS can be a common complication in mice with mutator phenotypes and disseminated lymphoblastic lymphoma/leukemia.
Lovelace et al. suggested that microthrombi in organs such as the lung or brain may be reflected clinically by the increased respiratory rate or neurological signs they observed in their mouse (Lovelace et al. 2003). Consistent with this hypothesis, we observed parenchymal damage associated with vascular microthromboemboli within the brain, liver, lung, and kidney (Figure 2). The incidence of ATLS was significantly higher in mice found dead (41%) compared to moribund mice that were euthanized (4%; Table 1), suggesting that microthromboemboli likely caused sudden death.
Acute renal tubular and glomerular necrosis was found in most animals that died with ATLS, without any evidence of associated crystals in the renal tubules. Mice have functional urate oxidase, an enzyme not present in humans and other primates (Wu et al. 1989). Urate oxidase metabolizes uric acid, the primary breakdown product of purines, to a more soluble form, allantoin, and theoretically reduces the risk of obstructive nephropathy from uric acid crystals (Wu et al. 1989). Nonetheless, animals with functional urate oxidase develop hyperuricemia in response to tumor lysis (Calia et al. 1996; Laing and Carter 1988; Mylonakis et al. 2007). Thus, endogenous levels of this enzyme can be overwhelmed by excessive DNA metabolism. The absence of renal tubule crystals in our mice suggests that tubular necrosis results from the combination of tumor microthromboemboli and neoplastic infiltrates.
Recent improvements in the treatment and prevention of ATLS in humans have decreased the incidence of renal failure from obstructive nephropathy resulting from urate crystal deposition (Jeha and Pui 2005). Recombinant urate oxidase is efficient at reducing elevated uric acid levels within four hours of treatment (Pui et al. 2001). However, even with effective preventative measures there is a measurable incidence of renal failure in the face of normal uric acid levels (Bosly et al. 2003; Jeha and Pui 2005; Mukherjee et al. 2007; Patte et al. 2001). Therefore, renal failure in humans with ATLS may occur by mechanisms not involving obstructive nephropathy from urate crystals. Malignant infiltration of the renal parenchyma has been hypothesized to contribute to tissue compromise, likely via renal tubule compression (Olgar et al. 2005). However, most kidney involvement with leukemia/lymphoma is clinically silent (Eckman and Lynch 1978; Lundberg et al. 1977; Norris and Wiener 1961). Our study suggests that microthromboemboli may cause end-organ dysfunction in mice. Likewise, a similar mechanism may contribute to renal failure in humans with disseminated lymphoblastic lymphoma or leukemia.
Spontaneous ATLS is presumed to be a rare occurrence, although it has been described in humans and several veterinary species (Hochberg and Cairo 2008; Tosi et al. 2008). Two mechanisms may have induced spontaneous tumor lysis and ATLS in the mice we report here. First, endogenous steroid release owing to the stress of illness could mimic induction chemotherapy in mice with a high tumor burden (Ward et al. 1999). Corticosteroids, used during induction chemotherapy for lymphoma and leukemia, are commonly associated with ATLS in humans (Coiffier et al. 2008; Duzova et al. 2001). Second, intravascular fibrin could shear neoplastic lymphocytes and cause mechanical cell lysis; metabolic abnormalities, microthromboemboli, and spontaneous death may then ensue. Disseminated intravascular coagulation (DIC) results in intravascular fibrinous thrombi and is a well-known complication in patients with neoplasia (Levi 2009). DIC can occur via a variety of mechanisms including circulatory disturbances, neoplastic cell–derived procoagulants, and cytokines (Levi 2009; ten Cate and Falanga 2008). Several veterinary reports of ATLS also describe associated DIC (Mylonakis et al. 2007; Vickery and Thamm 2007). We detected a significant amount of fibrin in the microthrombi observed in our animals (Figure 2E and 3B). However, a definitive diagnosis of DIC based on an abnormal serum coagulation profile was not possible. Both endogenous steroids and DIC may contribute to tumor lysis, ATLS, and spontaneous death in mice with disseminated lymphoblastic lymphoma.
This study suggests that ATLS in mice with lymphoblastic lymphoma can be common, is associated with a high risk of spontaneous death, and may be underreported in the literature. It is likely that metabolic abnormalities consistent with ATLS are present in these animals and precede the development of microthromboemboli. Mouse models of disseminated lymphoblastic lymphoma may provide a powerful tool to investigate the early pathophysiology of ATLS and test the efficacy of preventative measures and treatment strategies.
Footnotes
Acknowledgments
We thank Donovan Anderson, Cammy Mason, and Scott Paulson for technical assistance; Julie Randolph-Habecker for immunohistochemistry; Bobbie Schneider for electron microscopy assistance; Charles Frevert and Brian Johnson for fluorescence imaging; and Diana Lim for illustrations. This work was supported by the National Institutes of Health (R01 ES09927, R01 CA098243, R01 CA111582, and P01 CA77852 to B.D.P.; P01 AG01751 to B.D.P. and P.M.T.), the National Institute of Environmental Health Sciences–sponsored Center for Ecogenetics and Environmental Health (P30 ES07033), and the Comparative Mouse Genomics Center (U01 ES11045) at the University of Washington. T. M. A. was supported by a Young Investigators Award from Seattle Children’s Hospital.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.