
Abstract
With the growing number of genomic data in public repositories, efficient search methodologies have become a basic need to reach the relevant genomic data. However, this need cannot be fulfilled with the current repositories because they offer a limited search option which is a lexical matching of textual descriptions or metadata of the experiments. This technique is insufficient to get the required information needed to detect similarities between experiments within a large data collection. Due to the limitation of the existing repositories, in this study, we develop a text-based experiment retrieval framework by using both lexical and semantic similarity approaches to find similarities between experiments, and their retrieval performance was compared. This study is the first attempt to use text-driven semantic analysis approaches for developing a retrieval framework for experiments. An empirical study was conducted on a large textual description of Arabidopsis microarray experiments from the Gene Expression Omnibus database. In the proposed model, Jaccard similarity was used as a lexical similarity approach; Latent Semantic Analysis, Probabilistic Latent Semantic Analysis and Latent Dirichlet allocation were used as semantic similarity approaches to detect similarities between the textual descriptions of the experiments. According to the experimental results, relevant experiments can be retrieved successfully by text-driven semantic similarity approaches compared with the lexical similarity approach.
Keywords
1. Introduction
Over recent years, with the help of developments in computational biology, the accumulation of genomic data has been increasing rapidly. The genomic data are stored in various kinds of data formats such as sequences, networks and experimental measurements. Gene Expression Omnibus (GEO) [1], ArrayExpress [2], GenBank [3] and Arabidopsis Information Resource (TAIR) [4] are widely used public data repositories that contain high-throughput biological experiments. Accessing and organising these experiments is a significant task for researchers to obtain hypotheses from the retrieved information. They also need efficient and fast access tools that make their task easy, so recently the development of such retrieval methods has been of interest to researchers.
Experimental studies are stored in data repositories with the metadata information, also known as textual annotations, which contain brief information about the experiment such as organism name, author name, laboratory design and free-style descriptions about the experimental setup. They can be considered as text documents associated with the related experiment. To search for an experiment from a data repository, users generally use metadata information because current data repositories only provide metadata-based search using lexical matching or similarity of textual annotations of the experiments within large data collections. In addition, when searching through a database, logical operators such as AND and OR can be used to compound the values of the attributes. Although such a search is easily implemented, it has major limitations like preventing the conformability of the user when he or she demands a semantic query. Besides this search option, the keyword-based search can be used to search for an experiment. It can provide conformability by using text descriptions through the search, but sometimes text annotations do not have enough information about the experiment. Moreover, lexical similarity does not consider polysemy and synonymy of the terms or words that always exist in the words of natural language; however, the semantic similarity between texts can be found by considering the synonyms and polysemy of each term in the texts. Therefore, semantic-based search is a more powerful technique to overcome current searching limitations.
To date, retrieving relevant experiments has been addressed by different studies focusing on various data formats such as complementary DNA (cDNA) microarrays [5,6], time-series microarrays [7] and metagenome-sequencing samples [8]. There are studies that use query-by-example retrieval in which an abstract content is constructed to represent each experiment within the data collection and then retrieved relevant experiments by using similarity between obtained abstract contents to overcome the limitations of the lexical metadata-based retrieval [9]. In the study by Şener and Oğul [10], a content-based retrieval framework was proposed by using a dataset of Arabidopsis microarrays from the GEO database. They represented each experiment by a fingerprint as the content of the experiment, then an overlap score was calculated between these fingerprints, and the Jaccard coefficient was used to determine the gene set similarity of the experiments which is defined as the relevant information of the compared experiments. It was stated in their study that similarities between experiments can be successfully detected by the proposed framework. In addition to this study, Açıcı et al. [11] proposed a computational framework for detecting similarities between microRNA (miRNA) experiments. A normal-uniform mixture model was adopted to detect differentially expressed miRNAs; then, binarised real-valued fingerprints were obtained using a rank-based threshold. They also introduced an efficient similarity metric to find similarities between obtained fingerprints of the experiments. An open-source platform, called miRWalk, was developed to predict miRNA-binding sites of known genes of species including human, mouse, rat, etc. They combined different predictive algorithms to improve target prediction, and miRNA-target gene interaction can be visualised via a network graph [12]. In addition, there is a study in which an R package and web application were developed for retrieving information from the repository of miRNA sequences and annotation data [13]. With the developed retrieval systems, a user can query information related to the name, accession, sequence, species, version and family information of a miRNAs. In another study conducted by Şener and Oğul [14], a computational content-based framework was developed for finding relevant experiments from the data collection. In the model, different fingerprinting strategies were used and an empirical study was conducted on the microarray experiments of Arabidopsis Thaliana. According to the results, the relevant experiments can be successfully detected by the proposed content-based retrieval framework. Besides this study, Sener et al. [15] developed a retrieval framework for whole-metagenome sequencing sample retrieval. In their study, different fingerprinting approaches were used to get a representative fingerprint for the content of the sequencing samples. They also applied feature extraction and selection methods to reduce the computational complexity of the proposed system. Furthermore, there are also software tools that are applicable for content-based retrieval focusing on extracting signatures based on differentially expressed genes [16–19]. CellMontage [16] was developed as a tool to be used for searching an experiment within a data collection. Spearman’s rank correlation coefficient was used to find similarities between differentially expressed profiles of the compared experiments. Moreover, ProfileChaser proposed by Engreitz et al. [17] is one of the most widely used tools based on differential expression to construct fingerprints of the experiments. SPIEDw [18] is another search engine in which a gene list with their expression values is taken as a query in the given data repository and relevant experiments are retrieved having similar expression values to the query experiment. In addition, SEEK [19] is a query-based search engine developed for transcriptomic data collection mostly including microarray and sequencing experiments. Besides these software tools, a web portal [20] is also available for visual identification of associations between signatures and searching for similar experiments using signatures within the data collections.
In this article, unlike the current studies and approaches, we propose a text-based experiment retrieval framework for genomic databases using lexical and semantic similarity approaches. An experimental study was conducted on textual information of microarray experiment collection from the GEO database. This study, to the best of our knowledge, is the first attempt to apply semantic analysis approaches to textual information of gene expression experiments to find similarities between experiments from data collection. According to the experimental results, the proposed system has been successful in detecting similarities between experiments. Moreover, it can easily be adaptable to the different types of genomic data.
The article is structured as follows: ‘Introduction’, ‘Methods’, ‘Results and Discussion’ and ‘Conclusion’.
2. Methods
2.1. Retrieval framework
The proposed framework aims to find similarities between experiments within a given data collection (Figure 1). To do so, metadata or textual descriptions of each experiment in the collection are taken as input for the retrieval methods. Lexical similarity and semantic similarity approaches were used as retrieval methods in the model. To adapt these methods to our study, textual descriptions of experiments are represented as documents, and words in the related description are represented as the terms. Retrieved experiments are listed based on the similarity score between the query experiment and other experiments in the data collection.
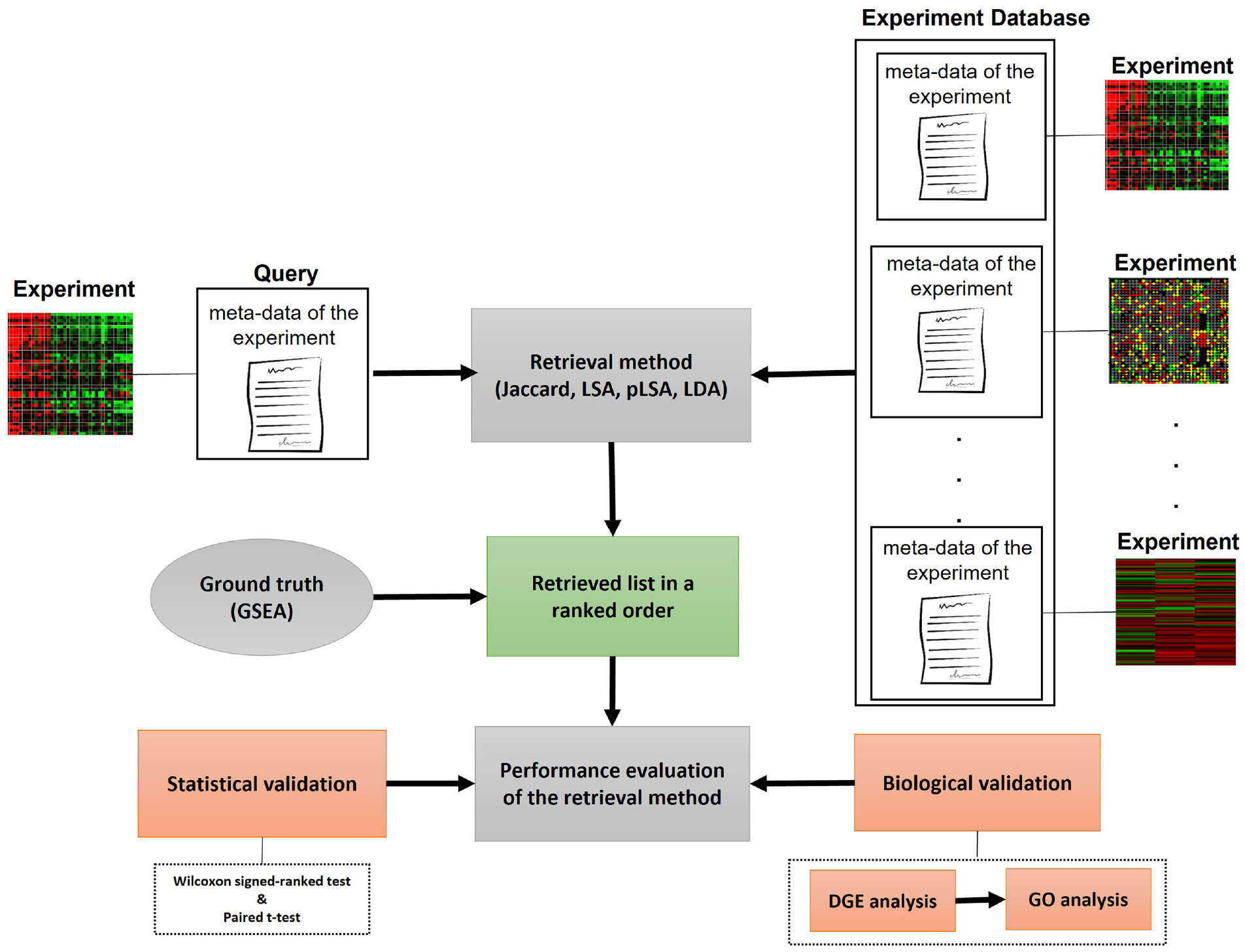
Overview of the proposed framework.
2.2. Jaccard similarity
Finding similarity between a set of documents within a data collection depends on a lexical match or similarity between words in the user’s query document and the ones in the rest of the documents. Lexical similarity is defined as a measure to show how similar a set of words also called documents. There are two main approaches for finding similarity lexically between compared sets of words such as character-based and term-based similarity approaches [21]. Jaccard similarity is one of the term-based approaches defined as the intersection of two sets of words divided by a union of them [22]. Let A and B be two documents to be compared, the Jaccard similarity between them is calculated using formula (1). The similarity score ranges between 0 and 1, where 0 represents no match, and 1 represents the perfect match between the compared documents

2.3. Latent Semantic Analysis
Latent Semantic Analysis (LSA) is a corpus-based semantic similarity method, which is widely used for detecting relationships between documents and terms. It was introduced by Landauer et al. [23] to give a key insight by reducing the dimension in information retrieval problems. There are four main steps in LSA, such as the following:
Creating a term-document matrix; this matrix represents a collection of documents in which rows represent words and columns represent sets of words. The word frequency across the documents is stored in the cell of the matrices. In LSA, bag-of-words representation is used because the order of words is unimportant.
Transformation term-document matrix; after obtaining the raw term frequency matrix, the matrix is transformed using inverse document frequency or entropy-based score.
Applying Singular Value Decomposition (SVD); SVD is performed to get the k-largest singular values. Each document and term are represented by a k-dimensional vector.
Retrieval in the reduced space; similarities are computed among a set of documents in the reduced dimension. The cosine distance is used to calculate the angle between the term and document vectors.
2.4. Probabilistic latent semantic analysis
Probabilistic latent semantic analysis (pLSA) is a technique from the category of topic model, and it was developed by Hofmann [24] in 1999. Unlike LSA, which is derived from linear algebra techniques and uses occurrence tables, pLSA is based on a mixture model decomposition. It starts with an aspect model, which interrelates co-occurrence data and an unobserved class variable. The data in the model are represented by three sets of variables such as documents, words and topics that can be represented as follows:
A document is defined as
A word is defined as
A topic is defined as
A generative process is performed such as first, a document


2.5. Latent Dirichlet allocation
Latent Dirichlet allocation (LDA) is a generative probabilistic topic model, which aims to detect latent topics defined as a natural group of the documents in the data collection. LDA is an unsupervised machine learning algorithm introduced by Blei et al. [26]. It assumes that a document is a bag-of-words, and they consist of more than one topic based on the words in the related document. Basic terms are defined, such as the following:
A word is the basic unit of the vocabulary. A vector
A document is the set of
There are
To adapt the LDA model to our study, were represented as documents. Furthermore, there are

3. Results and discussion
3.1. Dataset
In this study, we used the same dataset and ground truth as the study of Şener and Oğul [14]. Overall, 120 Arabidopsis time-course experiments from GEO were used in the retrieval model. Each experiment has textual annotations or descriptions of the experimental study. Figure 2 shows an example of an experiment used in the study. As shown in the figure, detailed descriptions are given with status, title, organism, experiment type, summary and overall design. We used ‘Summary’ and ‘Overall design’ experiment descriptions to detect similarities between compared experiments. Preprocessing steps were applied to transform data in the applicable format. In the preprocessing steps, stop words and punctuation marks were eliminated, abbreviations and numbers that did not make sense to the user were removed from the dataset and stemming word was used as the main dataset. All preprocessed data can be accessed from the link below.

An example experiment.
https://github.com/dygdedesener/data/blob/main/dataset_text_based_exp_retrieval.xlsx.
3.2. Experimental setup and evaluation
A retrieval framework based on the lexical and semantic similarity for descriptions of microarray experiments was proposed to get relevant experiments within the data collection. Textual descriptions of each experiment were used as a query, and a ranked list was obtained from the collection. The retrieved experiment with a high similarity score is expected to be more likely relevant to the query experiment.
As shown in Figure 3, relevance information between the experiments was defined by ground truth which describes whether compared experiments have true relevance or not. In our study, as mentioned previously, we used the same ground truth as the study of Şener and Oğul [14]. In the study, they performed Gene Set Enrichment Analysis (GSEA) [27] to set an acceptable ground truth for the compared experiments. The idea behind using GSEA as ground truth is that biologically relevant experiments should have common enriched gene sets because similar behaviour can be detected in response to an environmental factor during the experiment. Jaccard index (formula (1)) was used to calculate gene set–based similarity. A threshold was defined to define relevant information of the experiments. Let us assume that there are two distinct gene sets, called A and B, obtained from two compared experiments, then the Jaccard index is calculated as the ratio of their common gene sets divided by all gene sets. Setting the ground truth by a threshold was done by assuming a Gaussian distribution of all experiment pairs, the threshold was set as 0.35. The threshold was found by summing the mean of all values in the dataset and the standard deviation of the data. If two compared experiments have a Jaccard index value greater than the threshold, they are called relevant or otherwise irrelevant experiments.
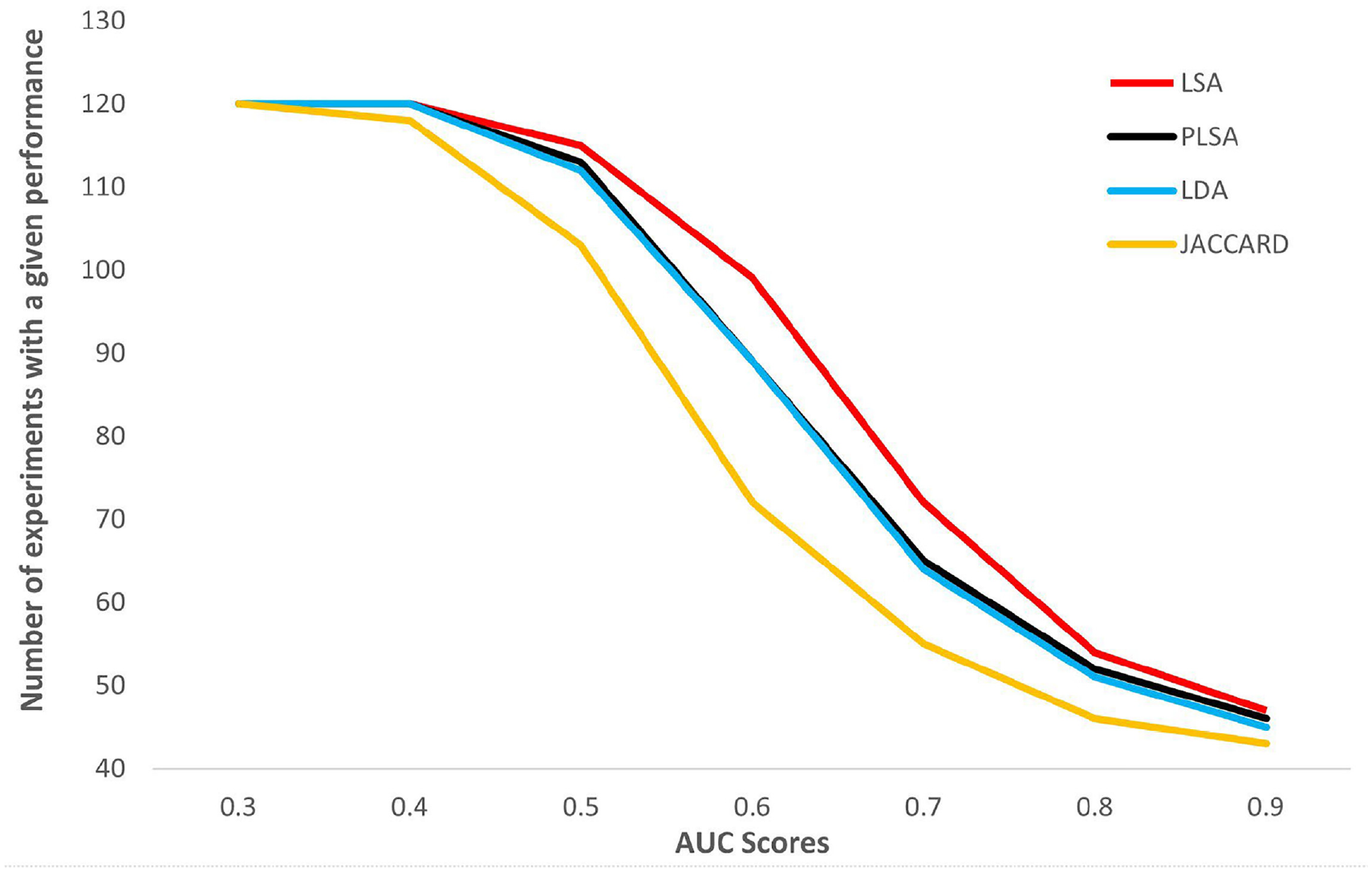
Comparison of retrieval performance of similarity approaches.
After obtaining relevant experiments based on the similarity approaches, biological validation of the experimental results should be performed to get evidence for the results. To do so, Gene Ontology (GO) analysis was performed to get biological validation for selected query experiments and the experiments retrieved by the system [28]. We first applied Differential Expression Analysis to get upregulated and downregulated gene lists for each experiment using DEBrowser Tool [29]. The analysis was performed using the DESeq2 R package [30] with the criteria absolute fold change > 2 and p-value < 0.05. Then, GO analysis results were obtained for each experiment with a software tool g:Profiler [31]. This analysis returned GO terms that belong to the Biological Processes (BP), Molecular Function (MF) and Cellular Component (CC) for the query and the retrieved experiment. In our study, GO terms with BP ontology were used for identifying common GO terms between the query experiment and the retrieved experiment. Having at least one common GO term between compared experiments is proof that they are also biologically similar experiments.
3.3. Empirical results
The retrieval task of the system was conducted by taking each experiment as a query within the experiment collection in the proposed framework. A ranked list was obtained for each query experiment based on the similarity scores generated for compared experiments. The expected result is getting higher ranks for true relevance experiments while lower ranks for other experiments.
Receiver Operating Characteristic (ROC) curves were used for evaluating the system performance. In the curve, the x-axis represents the false positive rate (FPR) (formula (5)), and the y-axis represents true positive rate (TPR) (formula (6)). In the given formulas, TN is true negatives and FN is false negatives. For a ranked retrieved list, a positive experiment means a relevant experiment and a negative experiment means an irrelevant experiment. For each query experiment, a ranked listed based on the similarity scores is obtained; thereafter, an ROC curve is created by thresholding a test set. An area under curve (AUC) score for the query is calculated by the area under the ROC curve associated with it and the average AUC of all query experiments is calculated for the similarity approach. Instead of giving an average AUC score for each method, the number of experiments that have AUC scores equal to or greater than the given score was used for the visualisation of the performance of the methods. The better retrieval performance is observed by getting a higher average AUC value


Jaccard similarity was used for finding lexical similarity, while LSA, PLSA and LDA methods were used for finding semantic similarity between textual descriptions of experiments. The retrieval performance of these approaches, given in Figure 3, was compared by plotting AUC scores across the number of experiments, which has a given score or a better score than the related score. The high curve shows an effective retrieval performance. As can be seen from the figure, biologically relevant experiments can be detected successfully by the LSA method for many queries. It can be clearly seen that an average AUC of larger than 0.7 can be achieved by two out of three of the experiments. In addition, the average AUC score for the LSA method is 0.79, while 0.77 for LDA, 0.77 for pLSA and 0.73 for the Jaccard similarity (lexical similarity). Besides this, we also compared the performance of the proposed framework with the study of Şener and Oğul [14], which is the retrieval framework based on the content similarity of the same microarray experiments used in our study. In their study, they achieved an average AUC score of 0.74, while in our study we got an average AUC score of 0.79 by the LSA method. This means that a retrieval framework based on the semantic similarity of textual descriptions of the experiments has become more successful than a content-based retrieval model in detecting similarities between experiments.
To investigate how the dimensionality of LSA vectors affects the retrieval performance, we performed the method using different vector dimensions. Figure 4 indicates the effect of the dimension on the retrieval performance of the LSA method. We simply computed the average AUC scores for different numbers of dimensions. As shown in the figure, the optimal vector dimension is 5 since the highest average AUC score was obtained using this vector dimension; hence, the optimal vector dimension is selected as 5 for the LSA method.
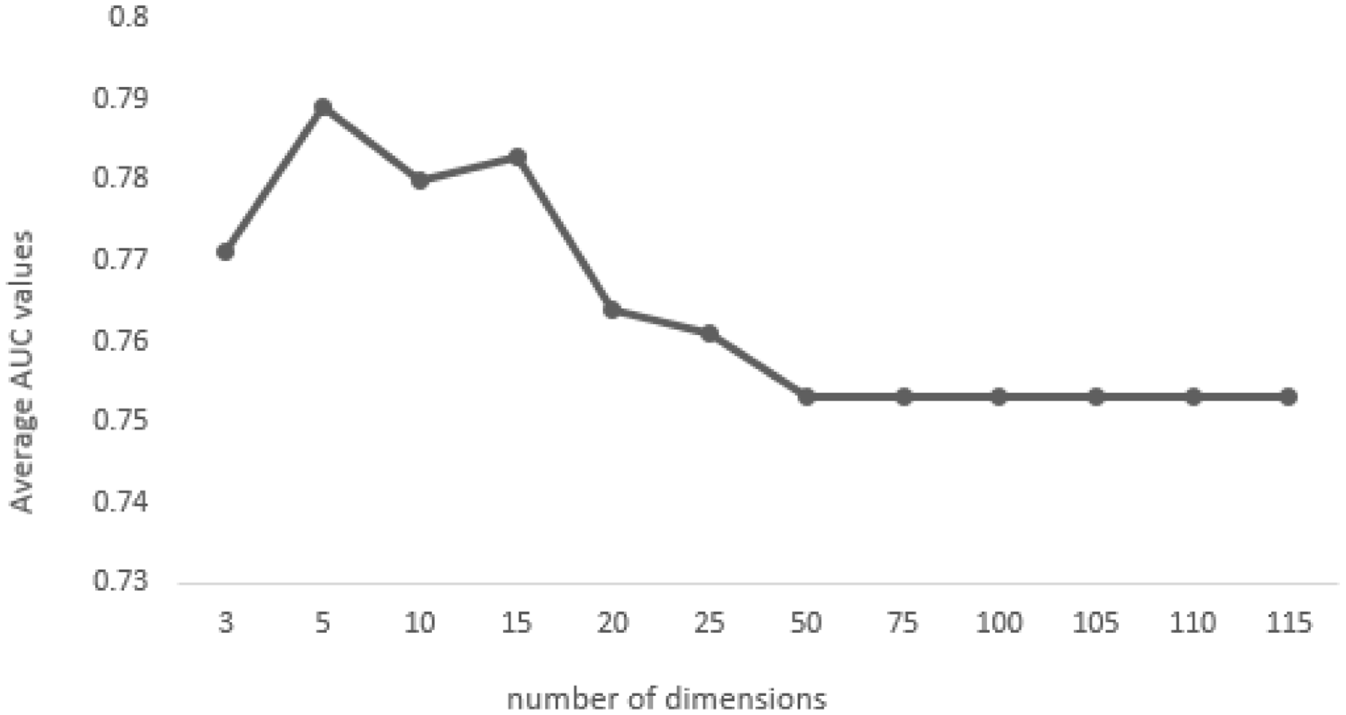
Effect of the number of dimensions in the LSA method.
After observing the performance of the retrieval methods, we applied a statistical significance test to determine whether the performance difference between the most successful method and the other methods was significant. To do so, a paired t-test and a non-parametric Wilcoxon signed-rank test were performed to get a difference between pairwise AUC scores of the used methods. We got p-values that are below 0.05 in both tests, except for the Wilcoxon result for LSA versus pLSA. We got a p-value of 5.83734E−11 and 2.34E−11 for Jaccard, 0.054 and 0.018 for pLSA and finally 0.003 and 0.009 for the LDA method for Wilcoxon and paired t-test, respectively (Table 1). The results justify that achievement in finding relevant experiments using the LSA method in terms of AUC scores is statistically significant.
Significance tests for the pairwise comparisons of used methods.

LSA: Latent Semantic Analysis; pLSA: Probabilistic Latent Semantic Analysis; LDA: Latent Dirichlet allocation.
In addition to the indirect evaluation based on the gene set similarity, the retrieval ability of the proposed system was evaluated by discovering textual annotations of the experiments manually. For this, two query experiments were selected as sample query experiments to observe the relevance between their annotations. The selection criteria for those experiments are that except for itself at least two relevant experiments should be retrieved from the dataset with a high similarity score. The first sample query experiment was performed to observe the expression data from xylem-pole pericycle cells of Arabidopsis roots undergoing lateral root initiation [32]. In the experiment, transcript profiling was used on sorted pericycle cells undergoing lateral root initiation to define the receptor-like kinase ACR4 of Arabidopsis as a key factor. They also used FACS on Arabidopsis roots containing a green fluorescent protein (GFP) marker for xylem-pole pericycle cells at different time points including the arrest of G1-to-S transition, auxin signalling, and progression through G1-to-S and G2-to-M transition or asymmetric cell division. The accession number of this experiment is GSE6349. The second sample query experiment was performed to observe volatiles of certain rhizobacteria’s growth inhibitory effects on Arabidopsis thaliana. The accession number is GSE35325-2 in which ‘-’ indicates the experiment number in the same GEO entry.
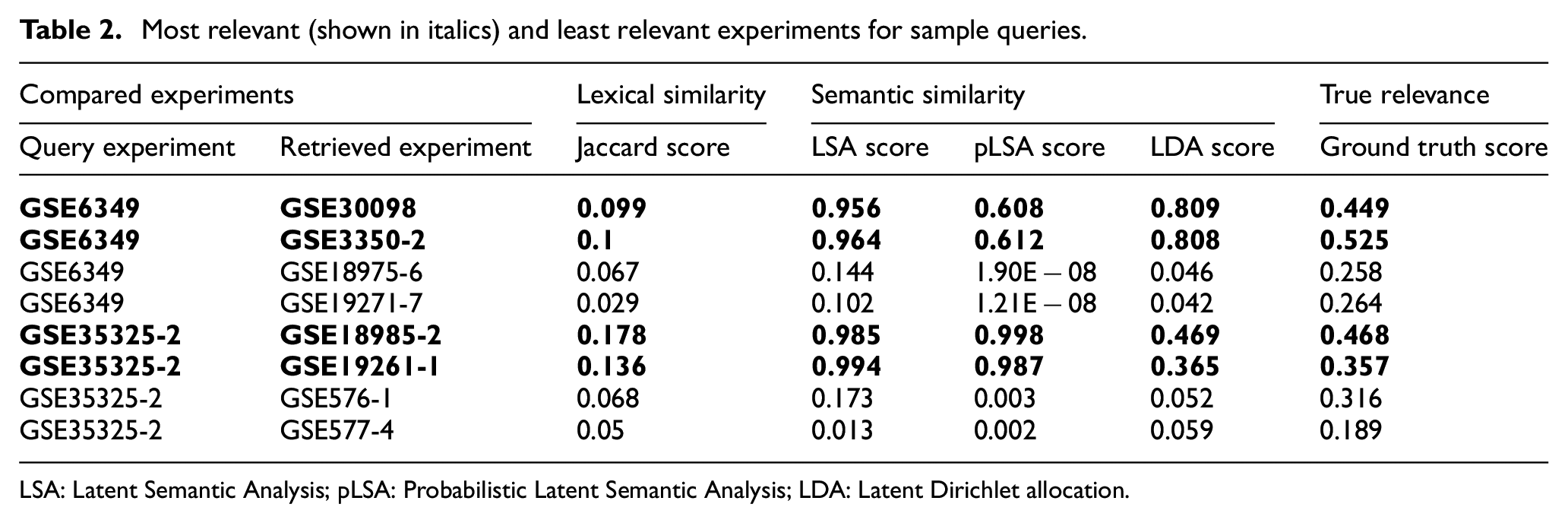
Table 2 gives the retrieval results of selected sample query experiments. The two relevant experiments (given in bold) and two irrelevant experiments retrieved by the system with their lexical and semantic similarity scores are given in the table. The ground truth score obtained by gene-set-based similarity, known as true relevance, is also given in the same table. The results are strong evidence that semantic similarity approaches have a significant correlation between the true relevance of the retrieved relevant and irrelevant experiments. According to the results, lexical similarity cannot succeed in detecting similarities between experiments because low similarity scores were obtained even for relevant experiments.
Most relevant (shown in italics) and least relevant experiments for sample queries.
LSA: Latent Semantic Analysis; pLSA: Probabilistic Latent Semantic Analysis; LDA: Latent Dirichlet allocation.
For the first query experiment, GSE30098 is the most relevant experiment retrieved by the system. This experiment is about the expression analysis of Arabidopsis plant roots during sulphur deficiency. The experiment was conducted to understand the link between development and stress in Arabidopsis root by using genome-wide assays [33]. The most notable similarity between the query and this experiment is that they were developed for observing plant root development processes at different time periods and in response to environmental factors. The second most relevant experiment, named as GSE3350-2, is the study of mechanisms behind auxin-induced cell division by using lateral root imitation [34]. The similarity between the experiments is that they are conducted for investigating auxin hormone and cell division. We can also clearly state that the proposed system can infer the relevance between experiments with the same objective and using the same stimulus.
For the second query experiment, numbered GSE35325-2, the most relevant experiment is GSE18985-2, which identifies early gibberellin (GA) responsive genes in the roots of an Arabidopsis GA-deficient mutant. The most notable similarity between experiments is that the effects of growth inhibitory bacteria and a supportive hormone were observed in these experiments. The second most relevant experiment was GSE19261-1, which shows an interesting relevance. The retrieved experiment was conducted to observe the effects of light on the growth process of the plant, whereas the query experiment also presents the results of a study on the inhibitory effect of the volatile of a certain bacterium on plant growth. Evidently, both the query and retrieved experiment are the results of studies about observing plant growth for the same objective with a different stimulus.
The relevance between selected query experiments and retrieved experiments can also be observed by GO analysis. We observed that four common GO terms and related p-values are enriched for both the query and relevant experiments given in Tables 3 and 4. As can be shown from the tables, the relevant experiments have common GO terms with low p-values, which are the main criteria for evaluating GO analysis results.
Common GO terms enriched for query experiment (GSE63349) and the first relevant experiment (GSE30098).

GO: Gene Ontology.
Common GO terms enriched for query experiment (GSE35325-2) and the first relevant experiment (GSE18985-2).

GO: Gene Ontology.
4. Conclusion
With the exponential growth of genomic data in public repositories, efficient search methodologies to get relevant data from the repositories have become a significant need for researchers. Current data repositories provide only metadata-based searching approaches that mainly use lexical matching of textual descriptions of the experiments. This technique fails to retrieve relevant experiments from large data collections. Owing to the existing limitation of current data repositories’ searching options, in this study, we aim to develop a retrieval framework using semantic similarity approaches in data repositories. Similarities between experiments were found by using both lexical and semantic similarity approaches and their retrieval performance was compared. In the proposed model, Jaccard similarity was used as a lexical similarity approach, and LSA, pLSA and LDA were used as semantic similarity approaches to detect similarities between the textual descriptions of the experiments.
We used textual annotations of GEO experimental datasets to test the performance of the proposed system. This study, to the best of our knowledge, is the first attempt to use semantic similarity approaches for detecting similarities between textual descriptions of microarray experiments. Evaluation of the proposed system was done by the ROC performance of the similarity approaches. It has been observed that for most of the experiments, high ROC scores were obtained. To perform an experimental evaluation of the results, we selected sample query experiments and compared their textual annotations with the retrieved ones manually. It is clearly seen that the inference by the system can be proved with a biological justification of the results. According to the experimental results, the relevant experiments can be retrieved successfully by semantic similarity approaches compared with the lexical similarity approach. Although text-driven semantic similarity approaches have been successful in finding similarities between experiments in our study, they have some drawbacks. Topic models lack interpretable embeddings because the generated static topics are not known and they are hidden variables of the documents. Besides, the models are constructed in a bag-of-words representation assuming terms in a text can be exchangeable, sentence structure is not modelled which might cause some limitations to represent information hidden in the sentence structure of a text. In addition to this, LSA involves SVD which increases the computational cost of the system when the number of experiments has been increased in the dataset collection.
In conclusion, text-driven semantic similarity approaches have promised a successful result in retrieving relevant experiments and they provide us to get biological similarity between the query experiment and the retrieved experiments. We also expect that the proposed system will be the key insight into future studies that goes beyond simple metadata-based approaches. The results also encourage us to use data mining techniques for retrieving relevant experiments from the genomic data collections. Finally, the proposed retrieval framework is expected to be applicable not only to the microarray experiments but also to other types of genomic data contexts that have textual annotations.
Footnotes
Author’s Note
Duygu Dede Sxener is also affiliated to Department of Computer Engineering, Baskent University, Ankara, Turkey.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.