
Abstract

Occupational Intakes of Radionuclides: Part 2
Approved by the Commission in November 2013
Abstract–The 2007 Recommendations of the International Commission on Radiological Protection (ICRP, 2007) introduced changes that affect the calculation of effective dose, and implied a revision of the dose coefficients for internal exposure, published previously in the Publication 30 series (ICRP, 1979, 1980, 1981, 1988b) and Publication 68 (ICRP, 1994b). In addition, new data are available that support an update of the radionuclide-specific information given in Publications 54 and 78 (ICRP, 1988a, 1997b) for the design of monitoring programmes and retrospective assessment of occupational internal doses. Provision of new biokinetic models, dose coefficients, monitoring methods, and bioassay data was performed by Committee 2, Task Group 21 on Internal Dosimetry, and Task Group 4 on Dose Calculations. A new series, the Occupational Intakes of Radionuclides (OIR) series, will replace the Publication 30 series and Publications 54, 68, and 78. Part 1 of the OIR series has been issued (ICRP, 2015), and describes the assessment of internal occupational exposure to radionuclides, biokinetic and dosimetric models, methods of individual and workplace monitoring, and general aspects of retrospective dose assessment. The following publications in the OIR series (Parts 2–5) will provide data on individual elements and their radioisotopes, including information on chemical forms encountered in the workplace; a list of principal radioisotopes and their physical half-lives and decay modes; the parameter values of the reference biokinetic model; and data on monitoring techniques for the radioisotopes encountered most commonly in workplaces. Reviews of data on inhalation, ingestion, and systemic biokinetics are also provided for most of the elements. Dosimetric data provided in the printed publications of the OIR series include tables of committed effective dose per intake (Sv per Bq intake) for inhalation and ingestion, tables of committed effective dose per content (Sv per Bq measurement) for inhalation, and graphs of retention and excretion data per Bq intake for inhalation. These data are provided for all absorption types and for the most common isotope(s) of each element. The electronic annex that accompanies the OIR series of reports contains a comprehensive set of committed effective and equivalent dose coefficients, committed effective dose per content functions, and reference bioassay functions. Data are provided for inhalation, ingestion, and direct input to blood. The present publication provides the above data for the following elements: hydrogen (H), carbon (C), phosphorus (P), sulphur (S), calcium (Ca), iron (Fe), cobalt (Co), zinc (Zn), strontium (Sr), yttrium (Y), zirconium (Zr), niobium (Nb), molybdenum (Mo), and technetium (Tc).
Keywords: Occupational exposure; Internal dose assessment; Biokinetic and dosimetric models; Bioassay interpretation
AUTHORS ON BEHALF OF ICRP
F. PAQUET, M.R. BAILEY, R.W. LEGGETT,
J. LIPSZTEIN, T.P. FELL, T. SMITH, D. NOSSKE,
K.F. ECKERMAN, V. BERKOVSKI, E. ANSOBORLO,
A. GIUSSANI, W.E. BOLCH, J.D. HARRISON
PREFACE
Publication 130 (ICRP, 2015) was the first in a series of ‘Occupational Intakes of Radionuclides’ (OIR) publications replacing the Publication 30 series (ICRP, 1979, 1980, 1981, 1988b) and Publication 68 (ICRP, 1994b) to provide revised dose coefficients for occupational intakes of radionuclides by inhalation and ingestion. It provided an introduction to the series of reports, and included sections on control of occupational exposures, biokinetic and dosimetric models, monitoring methods, monitoring programmes, and retrospective dose assessment.
The current publication, the second in the OIR series, is the first to provide data on individual elements and their radioisotopes, including information on chemical forms encountered in the workplace, a list of principal radioisotopes and their physical half-lives and decay modes, the parameter values of the reference biokinetic models, and data on monitoring techniques for the radio-isotopes most commonly encountered in workplaces. For most of the elements, reviews of data on inhalation, ingestion and systemic biokinetics are also provided.
Dosimetric data provided in the printed reports of the series include tables of committed effective dose per intake (Sv per Bq intake) for inhalation and ingestion, tables of committed effective dose per content (Sv per Bq measurement) for inhalation, and graphs of retention and excretion data per Bq intake for inhalation. These data are provided for all absorption types and for the most common isotope(s) of each element section.
The electronic annex that accompanies this series of reports contains a comprehensive set of committed effective and equivalent dose coefficients, committed effective dose per content functions, and reference bioassay functions for inhalation, ingestion and for direct input to the blood.
The new biokinetic and dosimetric models, dose coefficients and bioassay data presented and used in this OIR series of reports supersede those applied in the Publication 30 series, the first volumes of which were published almost 40 years ago. Since that time, ICRP has made modifications to the radiation and tissue weighting factors used in the calculation of effective dose (Publications 60 and 103), updated some characteristics of the Reference male and female (Publication 89), updated radionuclide decay data (Publication 107), adopted new anthropomorphic phantoms (Publication 110) and revised biokinetic models for inhalation, ingestion and systemic distribution of radionuclides (Publication 130 and this report). All of these changes ensure that the ICRP dose coefficients make appropriate use of scientific knowledge and reduce the uncertainties associated with the calculation of doses after internal contamination.
This report provides data for the following elements: Hydrogen (H), Carbon (C), Phosphorus (P), Sulphur (S), Calcium (Ca), Iron (Fe), Cobalt (Co), Zinc (Zn), Strontium (Sr), Yttrium (Y), Zirconium (Zr), Niobium (Nb), Molybdenum (Mo) and Technetium (Tc). Subsequent reports will provide data for most of the other elements.
An important question is whether the improvements made to biokinetic and dosimetric models have substantial impacts on the numerical values of dose coefficients. An analysis of the data shows that, for inhalation of reference forms of radionuclides (aerosols of 5μm, type F, M or S), the vast majority of new dose coefficients are slightly lower (within a factor 2) than those published in the Publication 30 series. In some very rare cases (14C monoxide, 14C dioxide, 59Fe Type F, 90Sr Type S, 60Co Type S), dose coefficients have increased by a factor of about 2 because of the revision of the biokinetic models and a better description of radionuclide retention and distribution in tissues. For ingestion, the new dose coefficients are similar to or a few percent lower than the previous dose coefficients. The most significant decrease is for moderately soluble forms of Yttrium-90, where the dose coefficient is now lower by a factor of 5. In this specific case, the older coefficient could be seen as very conservative.
It is reassuring that differences between the old and the new data are mostly small, confirming that the protection of workers was already reliably based on existing data. The increased sophistication and realism of the new biokinetic and dosimetric models gives us additional confidence in the data provided and contributes to reductions in uncertainties. It also means that they are readily applied to the interpretation of bioassay data. It should also be noted that the new data in the OIR series also extend the existing data sets, providing specific coefficients for isotopes and chemical forms that were not described previously, contributing to improvements in exposure and dose assessments and the protection of workers. Furthermore, this series provides physiologically based biokinetic models than can be used for applications other than radiation protection, including in toxicology, pharmacology and medicine.
Three Task Groups participated to the completion of this report. INDOS and DOCAL were involved until 2014 and then were replaced by IDC, newly created in 2014.






The membership of Committee 2 was:


The work of the authors was aided by significant contributions from G. Etherington, E. Blanchardon, D. Melo, L. Bertelli, D. Gregoratto, D.W. Jokisch, G. Ratia and all the INDOS, DOCAL and IDC members.
References
1. INTRODUCTION
(1) This publication is the second part of a series aimed at providing revised dose coefficients for occupational intakes of radionuclides (OIR) by inhalation and ingestion. It also presents radionuclide-specific information for the design and planning of monitoring programmes, and retrospective assessment of occupational internal doses. (2) The OIR series replaces the Publication 30 series (ICRP, 1979, 1980, 1981, 1988b), and Publications 54, 68, and 78 (ICRP, 1988a, 1994b, 1997). The revised dose coefficients, dose per content values, and reference bioassay functions have been calculated using the Publication 100 (ICRP, 2006) Human Alimentary Tract Model, and a revision of the Publication 66 (ICRP, 1994a) Human Respiratory Tract Model (HRTM) which takes account of more recent data. The revisions made to the HRTM are described in Part 1 of the OIR series (ICRP, 2015). Revisions have also been made to many models for the systemic biokinetics of radionuclides, making them more physiologically realistic representations of uptake and retention in organs and tissues, and excretion.
1.1. Methodology used in the OIR series
(3) The general methodology for producing the biokinetic and dosimetric models is given in Part 1 of the OIR series (ICRP, 2015). Part 1 also contains the Glossary for the OIR series. For each element, detailed reviews of the literature were undertaken to identify experimental studies and human contamination cases that provide information to quantify absorption to blood from the respiratory and alimentary tracts, and the biokinetics following systemic uptake. These reviews, and the analyses of the data obtained from them, are summarised in each element section. (4) In the case of inhalation, chemical forms are usually addressed in order of decreasing solubility in the lungs. Where information was available, HRTM absorption parameter values were derived from experimental data from both in-vivo and in-vitro studies. For in-vitro studies, estimation of the dissolution parameter values [rapidly dissolved fraction (fr), rapid and slow dissolution rates (sr and ss)] was usually straightforward. However, for in-vivo studies, simulation modelling was often needed to derive them from the data available, typically retention in organs and excretion in urine and faeces [for further information, see Supporting Guidance 3 (ICRP, 2002)]. (5) In some recent publications, the authors derived HRTM parameter values; if so, they are reported. In most cases, parameter values were derived by the ICRP Task Group (INDOS or IDC) members and their colleagues. This is indicated in the text by wording such as ‘analysis carried out here …’; the first such occurrence for each element is given as ‘analysis carried out here (i.e. by the Task Group) …’. (6) Material-specific rates of absorption have been adopted (and dose coefficients and bioassay functions provided for them in the accompanying electronic annex) for a limited number of selected materials, i.e. those for which:
there are in-vivo data from which specific parameter values can be derived; results from different studies are consistent; it was considered that occupational exposure to the material is likely; and the specific parameter values are sufficiently different from default Type F, M, or S parameter values to justify providing additional specific dose coefficients and bioassay functions. (7) Other materials were assigned to default HRTM absorption types using the criteria described in Publication 71 (ICRP, 1995) and Supporting Guidance 3 (ICRP, 2002) for making such assignments using experimental data. Type M is assumed for particulate forms of most elements ‘by default’, i.e. in the absence of such information. A material is assigned to Type F if the amount absorbed into blood by 30 d after intake is greater than the amount absorbed over the same period from a hypothetical material with a constant absorption rate corresponding to a half-time of 10 d, under identical conditions. Similarly, a material is assigned to Type S if the amount absorbed into blood by 180 d is less than the amount absorbed over the same period from a hypothetical material with a constant rate of absorption to blood of 0.001 d−1 (extrapolation was used in some cases, as indicated in the text). For studies where it was possible to apply the criteria, a statement is made to the effect that results ‘are consistent with’ (or ‘give’) assignment to Type F (M or S). For studies where the results point towards a particular type but there was insufficient information to apply the criteria, a statement is made to the effect that the results ‘indicate’ or ‘suggest’ Type F (M or S) behaviour. (8) Assignments are not made here on the basis of the known solubility of chemical forms in aqueous media, because this is not considered to be a reliable guide to absorption from the respiratory tract (ICRP, 1994a, Section E.2.2.1). If it is considered appropriate in a particular situation, it would need to be carried out with caution. In practice, it might well be possible to assign a radionuclide to which workers have been exposed to an absorption type without knowing its chemical form (e.g. from environmental and/or bioassay measurements). These could include in-vitro dissolution tests on air filters or swabs, in-vivo measurements (chest compared with whole body), or excretion measurements (urine compared with faeces). Nevertheless, for each element, a default absorption type is recommended for use in the absence of information on which the exposure material can be assigned to Type F, M, or S. For most elements, Type M is recommended by default. (9) For soluble (Type F) forms of each element, estimates are made of the overall rate of absorption from the respiratory tract to blood where information is available. In general, this results from dissolution of the deposited material, and also transfer through lining fluids and epithelium into blood. Nevertheless, for simplicity, this is usually represented by the rapid dissolution rate (sr) (see ICRP, 2015, Section 3.2.3). Due to the wide range of estimated values of sr, element-specific values are adopted in the OIR series for those elements for which estimates could be made. Justification of the value chosen for an element is given in the subsection headed ‘Rapid dissolution rate for element’. (10) For some elements, a significant fraction of the dissolved material is absorbed slowly. In some cases, this can be represented by formation of particulate material (which is subject to clearance by particle transport). In others, some dissolved material appears to be attached to lung structural components, and removed only by absorption to blood. To represent the latter type of time-dependent uptake, it is assumed that a fraction of the dissolved material is retained in the ‘bound’ state (fb), from which it goes into blood at a rate sb. Evidence for retention in the bound state, rather than by transformation into particulate material, may be in one or more forms, such as systemic uptake rather than faecal clearance of the retained material, slower clearance than for insoluble particles deposited in the same region of the respiratory tract, or autoradiography showing diffuse rather than focal retention of activity. (11) The bound state was included in the HRTM mainly to take account of slow clearance of dissolved materials from the alveolar-interstitial region. Applying the same bound state parameter values in all regions could lead, unintentionally, to high calculated doses to the bronchial (BB) and bronchiolar (bb) regions. Hence, in the OIR series, it is assumed that for those elements for which a bound state is adopted (fb > 0), it is only applied in the conducting airways (ET2, BB, and bb regions) if there is supporting experimental evidence. Justification of the values chosen for an element is given in the subsection headed ‘Extent of binding of element to the respiratory tract’.
1.2. Data presented in the OIR series
(12) Data presented in the OIR series are in a standard format for each element and its radioisotopes. Each element section provides information on chemical forms encountered in the workplace; principal radioisotopes, and their physical half-lives and decay modes; reviews of data on inhalation, ingestion, and systemic biokinetics; the structure and parameter values for the systemic biokinetic model; monitoring techniques; and detection limits typically achieved in a practical monitoring programme. The detection limits presented in this publication were derived from a compilation of data from laboratories in Europe, Asia, North America, and South America that perform routine monitoring of the specified radionuclide. The sensitivity of the measurements depends on the technique, the counting time, and other factors. For example, in-vivo detection limits depend on the detection system (type, quality, and number of detectors), counting geometry, and shielding and design of the installation. These details are outside the scope of this publication. (13) Dosimetric data are provided in the printed publications of the OIR series and in the electronic annex. The methodology for dose calculation is described in OIR Part 1 (ICRP, 2015) and in Publication 133 (ICRP, 2016). Due to the amount of data to be provided, the printed publications provide tables and graphs restricted to tables of committed effective dose per intake (Sv per Bq intake) for inhalation and ingestion, tables of committed effective dose per content (Sv per Bq measurements) for inhalation, and graphs of retention and excretion data per Bq intake for inhalation. (14) Data in the printed publications are provided for all absorption types of the most common isotope(s) and for an activity median aerodynamic diameter (AMAD) (refer to Glossary from Part 1 (ICRP, 2015)) of 5 µm. In cases for which sufficient information is available (principally for actinide elements), lung absorption is specified for different chemical forms, and dose coefficients and bioassay data are calculated accordingly. The dose coefficients and dose per content values presented in the OIR series are given for a reference worker at light work (ICRP, 2015). (15) The electronic annex that accompanies the OIR series contains a comprehensive set of committed effective and equivalent dose coefficients, dose per content functions, and reference bioassay functions for almost all radionuclides included in Publication 107 (ICRP, 2008) that have half-lives equal to or greater than 10 min, and for other selected radionuclides. Data are provided for a range of physicochemical forms and for aerosols with median sizes ranging from an activity median thermodynamic diameter (AMTD) (refer to Glossary from Part 1 (ICRP, 2015)) of 0.001 µm to an AMAD of 20 µm. Data for ingestion and injection (i.e. direct entry to blood) are provided to allow the interpretation of bioassay data for cases of inadvertent ingestion (e.g. of material on contaminated skin) or rapid absorption through intact or damaged skin (injection). (16) The dose coefficients and other radionuclide-specific data are provided as a set of data files that can be accessed by the user directly or by using the accompanying data viewer (see Annex A). The data viewer permits rapid navigation of the dataset and visualisation of the data in tabulated and graphical formats, such as graphs of the time series of dose per content coefficients, or predicted activity content per dose (Bq Sv–1) as a function of time after intake. Graphical presentations of decay chains and nuclear decay data from Publication 107 (ICRP, 2008) are also included. (17) The present publication, Part 2 of the OIR series, provides the data above on the following elements: hydrogen (H), carbon (C), phosphorus (P), sulphur (S), calcium (Ca), iron (Fe), cobalt (Co), zinc (Zn), strontium (Sr), yttrium (Y), zirconium (Zr), niobium (Nb), molybdenum (Mo), and technetium (Tc). Subsequent parts will provide data for most of the other elements.
1.3. References
2. HYDROGEN (Z = 1)
2.1. Chemical forms in the workplace
(18) Hydrogen is a non-metallic element that occurs mainly in oxidation states -I and I. Hydrogen is able to react chemically with most other elements. Tritium (3H) is a radioisotope of hydrogen. It is found in industry in a variety of chemical forms, including hydrogen gas (elemental tritium), tritiated water (HTO), methane, metal tritide, luminising compounds, tritium-contaminated pump oils, and a variety of organic compounds used in biomedical or other research. Tritium is an important fuel for controlled nuclear fusion in both magnetic and inertial confinement fusion reactor designs. Table 2.1 shows the isotopes of hydrogen addressed in this publication. Isotopes of hydrogen addressed in this publication. B-, Beta-minus decay
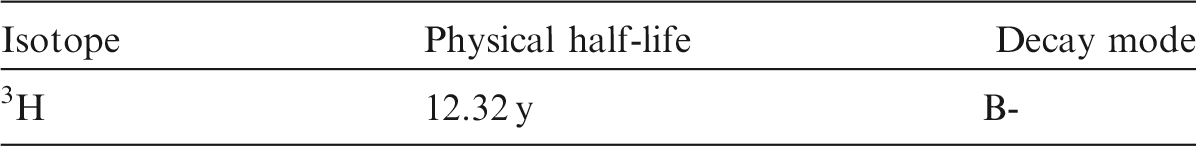
2.2. Routes of intake
2.2.1. Inhalation
(19) Extensive information on absorption of tritium from the respiratory tract is available from occupational exposures, and from human volunteer studies with inhaled tritium gas and tritiated water. Information is also available from experimental studies of tritiated organic compounds and particulate forms (mainly metal tritides and luminous compounds) in rats and in vitro.
2.2.1.1. Gases and vapours
(20) Absorption parameter values and types, and associated fA values for gas and vapour forms of hydrogen (tritium) are given in Table 2.2, and for particulate forms in Table 2.3. Exposures to gas or vapour forms of tritium are more common than exposures to particulate forms, and it is therefore recommended in the OIR series that gas/vapour form is assumed in the absence of information (ICRP, 2002a).
(a) Tritiated water (HTO)
(21) Pinson and Langham (1957) demonstrated that inhaled HTO is translocated to blood almost completely and instantaneously, and then distributes uniformly throughout the body without changing chemical form. For HTO, it is therefore assumed here that there is 100% deposition in the respiratory tract with instantaneous (Type V) absorption (Table 2.2). (22) Absorption through skin can add significantly to uptake during unprotected exposure to airborne HTO, and should be accounted for in workplace control. Publication 30 (ICRP, 1979) noted that Osborne (1966) had shown that exposure to an atmosphere contaminated by tritiated water at a concentration of C Bq m−3 results in the absorption of 10−2 C Bq min−1 through intact skin. Based on the Reference Man breathing rate of 0.02 m3 min−1 (ICRP, 1975), the inhalation rate of tritiated water was calculated to be 2 × 10−2 C Bq min−1, and it was assumed that this was all absorbed into body fluids. The total rate of absorption of tritiated water into body fluids was therefore calculated to be 3 × 10−2 C Bq min−1. Uptake of tritium through skin is not included in the inhalation dose coefficient for tritiated water given in this publication. Nevertheless, urine bioassay measurements, which are the basis for most tritium dose assessments, represent the body water concentration from all routes of intake, and therefore reflect skin absorption as well as inhalation of airborne tritium. ET1, anterior nasal passage; ET2, posterior nasal passage, pharynx, and larynx; BB, bronchial; bb, bronchiolar; AI, alveolar-interstitial. For intake of these forms of tritium, the systemic model for HTO is applied to absorbed activity. For tritium in unspecified gas or vapour form (including unspecified organic vapours), the default option for gases and vapours is recommended: 100% total deposition in the respiratory tract, default distribution between regions,¶ and Type F absorption. Percentage deposited refers to how much of the material in the inhaled air remains in the body after exhalation. Almost all inhaled gas molecules contact airway surfaces, but usually return to the air unless they dissolve in, or react with, the surface lining. In the case of tritium gas and methane, a small fraction is absorbed into body fluids; of that, a fraction is metabolised and the rest is subsequently exhaled. Since instantaneous absorption to blood is assumed, calculations can be performed assuming direct injection into blood, and the regional deposition does not need to be considered. However, for completeness, the default distribution is assumed.¶ Default distribution between regions (20% ET2, 10% BB, 20% bb, and 50% AI). Not applicable for Type V absorption because all activity deposited in the respiratory tract is absorbed instantaneously. Absorption parameter values for inhaled particulate forms of tritium and ingested tritium.* The systemic model for tritiated water is applied to intake of all forms of tritium other than biogenic tritiated organic compounds, for which the systemic model for organically bound tritium is applied. It is assumed that the bound state can be neglected for tritium, i.e. fb = 0.0. The value of sr for Type F forms of hydrogen (100 d−1) is element-specific. The values for Types M and S (3 d−1) are the general default values. See text for summary of information on which parameter values are based, and on ranges of parameter values observed for individual materials. For biogenic organic compounds, Type F default parameter values are used for absorption from the respiratory and alimentary tracts, but a specific systemic model, organically bound tritium, is used for absorbed tritium. Materials (e.g. ‘glass fragments’) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of tritium (1.0). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract.


(b) Tritium gas (elemental tritium, HT)
(23) Publication 30 (ICRP, 1979) identified tritium in the form of hydrogen gas as one of two gases (the other being 37Ar) for which exposure is dominated by irradiation of the lung (rather than the skin), because the emissions have insufficient energy to reach the basal layer of the skin. However, as described in Publication 68 (ICRP, 1994, Annex A), on the assumption that 0.01% of inhaled HT is absorbed and converted to HTO (see below), the estimated effective dose from absorbed HT is several times higher than that due to irradiation of the lung from gas within it. That conclusion remains applicable, and therefore dose coefficients are calculated here for tritium in the form of hydrogen gas, based on its absorption. (24) Studies in which human volunteers inhaled tritium gas (composed of 93% HT) showed that approximately 1% of the inhaled HT dissolved in body fluids and tissues, and approximately 1% of the dissolved HT (i.e. ∼0.01% of the inhaled HT) was subsequently converted to HTO in the gut and the rest was exhaled (Peterman et al., 1985a,b). For further information, see Section 2.2.3.1. These results appear to accord with the data of Pinson and Langham (1957). For HT, it is therefore assumed here that there is 0.01% effective deposition in the respiratory tract with instantaneous (Type V) absorption and conversion to HTO (Table 2.2). In occupational exposure conditions, HT in air is always accompanied by HTO vapour, and the latter dominates with regard to human exposure.
(c) Tritiated methane, CH4–xTx
(25) The dosimetric implications of inhaling methane gas were examined by Phipps et al. (1990). They made the conservative assumption that 1% of the methane was metabolised, based on observations by Dougherty et al. (1967) which indicated that approximately 0.3% of methane infused into sheep was converted to carbon dioxide. Carlisle et al. (2005) and Didychuk et al. (2014) investigated the extent of oxidation and organic fixation of 3H and 14C following inhalation of a mixture of 3H- and 14C-labelled methane by rats. In a pilot study, Carlisle et al. (2005) measured retention of activity in skin, liver, brain, and carcass at 1 and 24 h after 4 h of exposure. They estimated that uptake was approximately 0.1% of intake. Approximately 70% of 3H retained in liver and 10% of 3H retained in other tissues was organically bound. In a more comprehensive study using the same methods, Didychuk et al. (2014) followed retention in these tissues up to 14 d after exposure. They estimated that uptake was approximately 0.3% of intake for both 3H and 14C. For tritiated methane, it is assumed here that there is 0.3% effective deposition in the respiratory tract with instantaneous (Type V) absorption (Table 2.2). It is also assumed here that the absorbed tritium follows the systemic model for HTO.
(d) Unspecified organic vapours
(26) Organic solvents (e.g. benzene, toluene) are widely used in technological processes for production and application of tritium luminous compounds and other organic compounds. Solvent vapours labelled with tritium due to contact with tritiated high-activity compounds can be released into the working-room air. Volatile organic compounds have a wide range of solubility in body fluids, and their systemic behaviour after absorption is likely to depend on the chemical form inhaled (see Section 3.2). Smith et al. (1983) reported a wide range of urinary excretion patterns in workers following inhalation of a variety of tritiated organic compounds, but deposition fractions and absorption rates were not reported. In the absence of specific information, the default option for gases and vapours is taken, which is likely to be conservative. For tritium in unspecified organic forms, it is assumed here that there is 100% deposition in the respiratory tract (with default regional distribution, Table 2.2) and Type F absorption (Table 2.2). (27) Following inhalation by workers, the solvent vapours are absorbed into the blood and metabolise in the body as foreign compounds (Park, 1968; Balonov et al., 1984). A substantial fraction of tritium from those compounds is excreted rapidly from the body in urine and faeces; the other fraction is retained for some days in liver or kidney, and the rest catabolises to HTO. It is assumed here that the absorbed tritium follows the systemic model for HTO, which is also likely to be conservative.
2.2.1.2. Particulate materials (liquid and solid)
(28) Tritium can be released into the work environment in particulate form, and several studies of the dissolution of solid tritiated compounds have been conducted. Due to the low energy of the tritium beta emissions, self-absorption within particles can reduce doses significantly, even for particles as small as 1 µm diameter. For erbium tritide (ErT3-x), Kropf et al. (1998) calculated that the fraction of beta energy that escapes is in the range 0.5–0.1 for particle diameters in the range 1–5 µm. (29) Cheng et al. (1997), Inkret et al. (2001), and Zhou and Cheng (2003) demonstrated that tritium is released from metal tritides into simulated lung fluids as HTO. It is assumed here that for inhalation of inorganic particulate material, as well as ‘non-biogenic’ organic particulate material, the biokinetics of tritium absorbed into body fluids follows that of HTO.
(a) Biogenic tritiated organic compounds
(30) A wide variety of ‘biogenic’ tritiated organic compounds are used in biomedical research and in the pharmaceutical industry. These are generally precursors of biological macromolecules, such as labelled glucose, amino acids, or nucleosides. They are water soluble and are expected to be readily absorbed into blood following inhalation; Type F is assumed here. Their systemic behaviour varies from one compound to another, but they typically show a longer retention time than internally deposited HTO. A biokinetic model developed for application to organically bound tritium (OBT) is applied in this publication to tritium absorbed to blood after intake of biogenic tritiated organic compounds. OBT refers here to carbon-bound tritium formed in living systems through natural biological processes. OBT is distinguished here from ‘foreign’ or ‘non-biogenic’ tritiated organic compounds, which are typically excreted much more rapidly than biogenic organic compounds and are assigned the systemic model for HTO in this publication. In Table 2.3, biogenic organic compounds are presented as having ‘specific parameter values’, although they are assigned Type F parameter values for absorption from the respiratory and alimentary tracts to emphasise the difference in systemic model from that used for other particulate Type F forms of tritium.
(b) Tritiated pump oil
(31) Hydrocarbon mineral oils in vacuum pumps used in 3H-handling facilities can contain significant amounts of 3H, and give rise to airborne 3H contamination in both vapour and aerosol forms. In a pilot study, the biokinetics of 3H were followed for 30 d after intratracheal instillation of tritiated pump oil into rats (Trivedi and Cheng, 2000). At 3 d and 28 d after administration, the concentration of 3H in the lungs fell to approximately 4% and 0.7%, respectively, of that at 30 min. Up to 3 d, most of the 3H remaining in the body was in the lungs, while from 5 d onwards, most of it was in the carcass. Approximately 30% of the initial lung deposit (ILD) was excreted in urine and faeces, and the authors inferred that most of the rest (∼70% ILD) had been exhaled. Most of the urinary excretion occurred in the first few days. Estimation of absorption parameter values would be difficult and was not attempted here (i.e. by the Task Group). Furthermore, the behaviour may have been affected by the large mass administered (∼100 mg), and its form (a viscous oil) would have limited its dispersion in the lungs. The results suggest fast or moderate absorption (Type F or M). (32) In a follow-up study, Priest et al. (2013) measured lung and liver retention and excretion of 3H at times up to 365 d after intratracheal instillation of tritiated pump oil into rats. To provide better dispersion in the lungs, the oil was mixed with an equal volume of saline solution to form an emulsion (median oil droplet diameter ∼2 µm). Within 1 d, approximately 17% ILD cleared to faeces, presumably by mucociliary clearance, and approximately 1% was excreted in urine. Subsequent lung retention was well fit by a single exponential function with a half-time of 223 d (with clearance mainly to faeces). That is much longer than would be expected for insoluble particles in rats (see, for example, ICRP, 2002a, Annex C), but the reason is not known; macrophage transport could be less effective for oil droplets than for particles, or clearance could have been impaired by the large mass administered (∼50 mg of oil). There was some rapid absorption, possibly, in part, from 3H in the aqueous phase. The first measurement of the liver was approximately 1% ILD, but it decreased rapidly up to 56 d, after which it increased. Absorption parameter values were not estimated here. The authors inferred that the behaviour indicated assignment to Type S, but that Type M could not be excluded.
(c) Tritium-contaminated glass
(33) Cool and Maillie (1983) followed loss of tritium into simulated lung fluid from fragments of tritium-filled glass microballoons used in laser fusion research for 150 d. The fraction of total tritium lost during the first 100 d ranged between 16% and 30% for different glass samples. Dissolution kinetics were reported as the fraction lost per day, which decreased from approximately 2% initially to approximately 0.04% at 100 d. Average parameter values calculated here were (approximately): fr = 0.2, sr = 0.1 d−1, and ss = 0.0002 d−1, consistent with assignment to Type M. Cool and Maillie (1984) followed the tissue distribution and excretion of tritium for 80 and 180 d, respectively, following intratracheal instillation into rats of fragments of tritium-labelled glass microballoons. There is insufficient information given for absorption parameter values to be estimated here. However, the authors reported that results obtained in vivo were in good agreement with the in-vitro data obtained from the same type of glass. A large percentage of the tritium present in the glass matrix at the start of the experiments remained with it. The main difference was that, generally, a greater proportion of the tritium was associated with the slower phase of tritium dissolution in vivo than in vitro. The uniform distribution of tritium activity found within the various soft tissues of the body was consistent with the hypothesis that tritium lost from the glass matrix is converted to HTO.
(d) Luminous paint
(34) Balonov et al. (1984, 1995) reported that, following intratracheal instillation of ‘Soviet luminous powder (PS-A)’ into rats, lung specific activity showed essentially no decrease within 5 months, and hence should be assigned to Publication 30 Class Y (ICRP, 1979). This indicates that such compounds should be assigned to Type M or S. (35) Results of 5 d in-vitro studies of the dissolution of samples of commercial luminous paint powder made from tritium-labelled polystyrene in bovine serum (Rudran, 1988a) were described as, on average, 12% dissolved on the first day and approximately 2% of remaining activity on subsequent days, i.e. fr of approximately 0.12, sr of > 1 d−1, and ss of approximately 0.02 d−1, consistent with assignment to Type M.
(e) Metal and carbon tritides
Tritiated lanthanum nickel aluminium alloy
(36) The results of a 72 d in-vitro study of the dissolution in serum simulant of a sample of tritiated metal (LaNi4.25Al0.75, known as LANA.75) powder particles taken from a process line were expressed as a two-component exponential retention function (Farfán et al., 2012). This gives fr = 0.995, sr = 1.177 d−1, and ss = 0.042 d−1, and assignment to Type F. The authors noted that dissolution was much faster than observed previously for other metal and carbon tritides. They also discussed the possibility that the slow component might have been due, at least in part, to retention and slow release of tritium from the apparatus; however, it was too small to affect the dose assessments. Specific values are not adopted here (Table 2.3) because only in-vitro data are available; instead, LaNi4.25Al0.75 tritide is assigned to Type F.
Titanium tritide
(37) Balonov et al. (1984, 1995) reported that, following inhalation by rats, titanium tritide (TiT) showed slow lung clearance, and hence should be assigned to Publication 30 Class Y (ICRP, 1979). This indicates that TiT should be assigned to Type M or S. (38) Measurements were made up to 4 months after intratracheal instillation of TiT [1 µm count median diameter (CMD); refer to Glossary from Part 1 (ICRP, 2015)] into rats, and simulation modelling was applied to obtain a time-dependent absorption function (fractional absorption rate) (Cheng et al., 1999). Fitting the HRTM dissolution model to the data gave parameter values: fr = 0.6, sr = 0.71 d−1, and ss = 2 × 10–4 d−1 with an upper bound on fA of 0.6 (Cheng, 2009) consistent with assignment to Type M. Results of a 30 d in-vitro study of the dissolution of the same powder in synthetic serum ultrafiltrate (SUF) (Cheng et al., 1997) were expressed as a two-component exponential retention function, giving fr = 0.24, sr = 0.71 d−1, and ss = 0.021 d−1. This dissolution rate is broadly similar to the absorption rate in vivo (initially lower, but higher after a few days), and also consistent with assignment to Type M. Dissolution in the same system of a sample of coarse dust (103 µm CMD) was much slower, but still consistent with assignment to Type M. The results indicated that loss of tritium was related to diffusion, and hence increases with the specific surface area of the particles. Although specific parameter values for titanium tritide based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, titanium tritide is assigned to Type M.
Zirconium tritide
(39) Measurements were made up to 6 months after intratracheal instillation of zirconium tritide (0.3 µm CMD) into rats, and simulation modelling was applied to obtain a fractional absorption rate (Zhou and Cheng, 2004). Fitting the HRTM dissolution model to the data gave parameter values: fr = 0.0995, sr = 0.058 d−1, and ss = 3.9 × 10–4 d−1 with an upper bound on fA of 0.1 (Zhou et al., 2010), consistent with assignment to Type M. Results of 200 d in-vitro studies of the dissolution in SUF of the same powder (Zhou and Cheng, 2004) were expressed as a two-component exponential retention function, with fr = 0.048, sr = 0.016 d−1, and ss = 1.8×10–3 d−1. This dissolution is somewhat faster than the absorption in vivo, but also consistent with assignment to Type M. Although specific parameter values for zirconium tritide based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, zirconium tritide is assigned to Type M.
Carbon tritide
(40) The results of a 110 d in-vitro study of the dissolution in SUF of carbon tritide (1 µm CMD) samples taken from a test fusion reactor were expressed as a fractional absorption rate (Cheng et al., 2002a). Fitting the HRTM dissolution model to the data gave parameter values: fr = 0.035, sr = 0.396 d−1, and ss = 3.72×10–4 d−1 (Cheng, 2009), consistent with assignment to Type S. (41) The results of a 14 d in-vitro study of the dissolution in serum simulant of ‘coarse’ and ‘fine’ tritium-loaded carbon particles taken from another test fusion reactor were expressed as two-component exponential retention functions (Hodgson et al., 2004). For ‘coarse’ particles, fr = 0.05, sr = 500 d−1, and ss = 6.3 × 10–3 d−1, giving assignment to Type M. For ‘fine’ particles, fr = 0.003, sr = 500 d−1, and ss = 3.6 × 10–4 d−1, giving assignment to Type S. Hodgson et al. (2006, 2007) measured dissolution in serum simulant of three samples from two batches of tritium-loaded carbon particles from the same reactor for 100 d. Retention of undissolved tritium was expressed as a three-component exponential function. (To take account of the three components in software that implements the HRTM with only two, dose coefficients were calculated by treating each sample as a mixture of two materials.) For one batch, results for two samples gave assignment to Type M and the third to Type S. For the other batch, results for all three samples gave assignment to Type S. (42) Specific values are not adopted here (Table 2.3) because in-vitro data alone are available. Instead, carbon tritide is assigned to Type S.
Hafnium tritide
(43) Measurements were made up to 6 months after intratracheal instillation of hafnium tritide (1 µm CMD) into rats, and simulation modelling was applied to obtain a fractional absorption rate (Zhou and Cheng, 2003). Fitting the HRTM dissolution model to the data gave parameter values: fr = 3.07 × 10–4, sr = 2.72 d−1, and ss = 1.22 × 10–5 d−1 with an upper bound on fA of 3.07 × 10–4 (Cheng, 2009), consistent with assignment to Type S. Results of 200 d in-vitro studies of the dissolution in SUF of similar powders (Inkret et al., 2001; Cheng et al., 2002b) were expressed as two-component exponential retention functions, giving (approximately) fr = 1×10–3, sr = 0.015 d−1, and ss = 2.5 × 10–6 d−1. This dissolution is broadly similar to the absorption in vivo (initially lower, but higher after a few days), and also consistent with assignment to Type S. Although specific parameter values for hafnium tritide based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, hafnium tritide is assigned to Type S.
2.2.1.3. Rapid dissolution rate for tritium
(44) The evidence of rapid uptake of tritiated gases from the lung indicates a rapid rate of absorption on the order of 100 d−1. A value of 100 d−1 is applied here to all Type F forms of hydrogen.
2.2.1.4. Extent of binding of tritium to the respiratory tract
(45) The evidence of rapid uptake of tritiated gases from the lung indicates that there is probably little binding of tritium. It is therefore assumed that the bound state can be neglected for tritium, i.e. fb = 0.0.
2.2.2. Ingestion
2.2.2.1. Tritiated water (HTO)
(46) Investigations in humans have shown that hydrogen in the form of deuterium oxide or tritiated water is absorbed from the gastrointestinal tract rapidly and virtually completely (Pinson and Langham, 1957; Etnier et al., 1984; Travis et al., 1984).
2.2.2.2. Organic compounds
(47) Studies using rodents indicate that absorption of the intact molecule is variable for many forms of biogenic tritiated organic compounds; according to the authors, it ranges from approximately 50% for a few specific compounds (Rochalska and Szot, 1977; Takeda, 1982, 1991) to almost 100% for most compounds, including [3H]-cortisol, [3H]-glucose, and [3H]-amino acids (Balonov et al., 1993; Taylor, 2008). (48) Similar experiments have shown that approximately 90% of ingested [3H]-thymidine is catabolised into [3H]-thymine in the small intestine, and that both compounds pass across the gut by simple diffusion (Lambert and Clifton, 1968). Balonov et al. (1993) showed that 10–20% of [3H]-thymidine and 60–100% of [3H]-deoxycytidine are absorbed from the gastrointestinal tract of rats in intact form, and the rest is catabolised to HTO and other metabolites. The absorbed intact fractions of [3H]-thymidine and [3H]-deoxycytidine are smaller in mice (Feinendegen et al., 1980) than in rats, and unknown in humans. (49) Although absorption of biogenic organic tritium compounds is likely to vary substantially, it is conservatively assumed here, as in Publications 30 (ICRP, 1979) and 56 (ICRP, 1989), that absorption is complete unless specific information is available to indicate otherwise, i.e. the default assumption for all organic tritium compounds is that fA = 1.
2.2.2.3. Insoluble compounds
(50) Insoluble compounds such as metal tritides and luminous compounds are not absorbed directly from the gastrointestinal tract. In-vitro experiments showed that these substances, when in contact with water, gradually release 0.5–5% of the activity, which passes into solution in the form of oxide and low molecular organic compounds (Balonov et al., 1984). This fraction may then be absorbed and cause a systemic burden. (51) After oral administration of a suspension containing titanium tritide (TiT) particles to rats, the HTO concentration in body water increased slightly during the 1–1.5 d of the residence of TiT in the gastrointestinal tract. Total absorption in these conditions was less than 0.1 (Balonov et al., 1984). (52) Following oral administration of [3H]-labelled luminous compounds to rats, less than 5% of the administered activity was absorbed as HTO after dissolution (Balonov et al., 1984). Measurements of absorption in cats showed that absorption of tritium from luminous paints depended on the plastic substrate involved, with values of 0.007 for polystyrene, approximately 0.03 for silicone rubber, and 0.8 for polyester (Wawerna, 1973; Hill and Johnson, 1993).
2.2.2.4. fA values for ingestion
(53) For both tritiated water and organic compounds, an fA of 1 is adopted in this report, although it is recognised that absorption may be substantially less than complete in the case of some organic compounds. For metal tritides and luminous paints, the available data indicate that an fA value of 0.1 is generally more appropriate.
2.2.3. Systemic distribution, retention, and excretion
2.2.3.1. Summary of the database
(a) Tritiated water
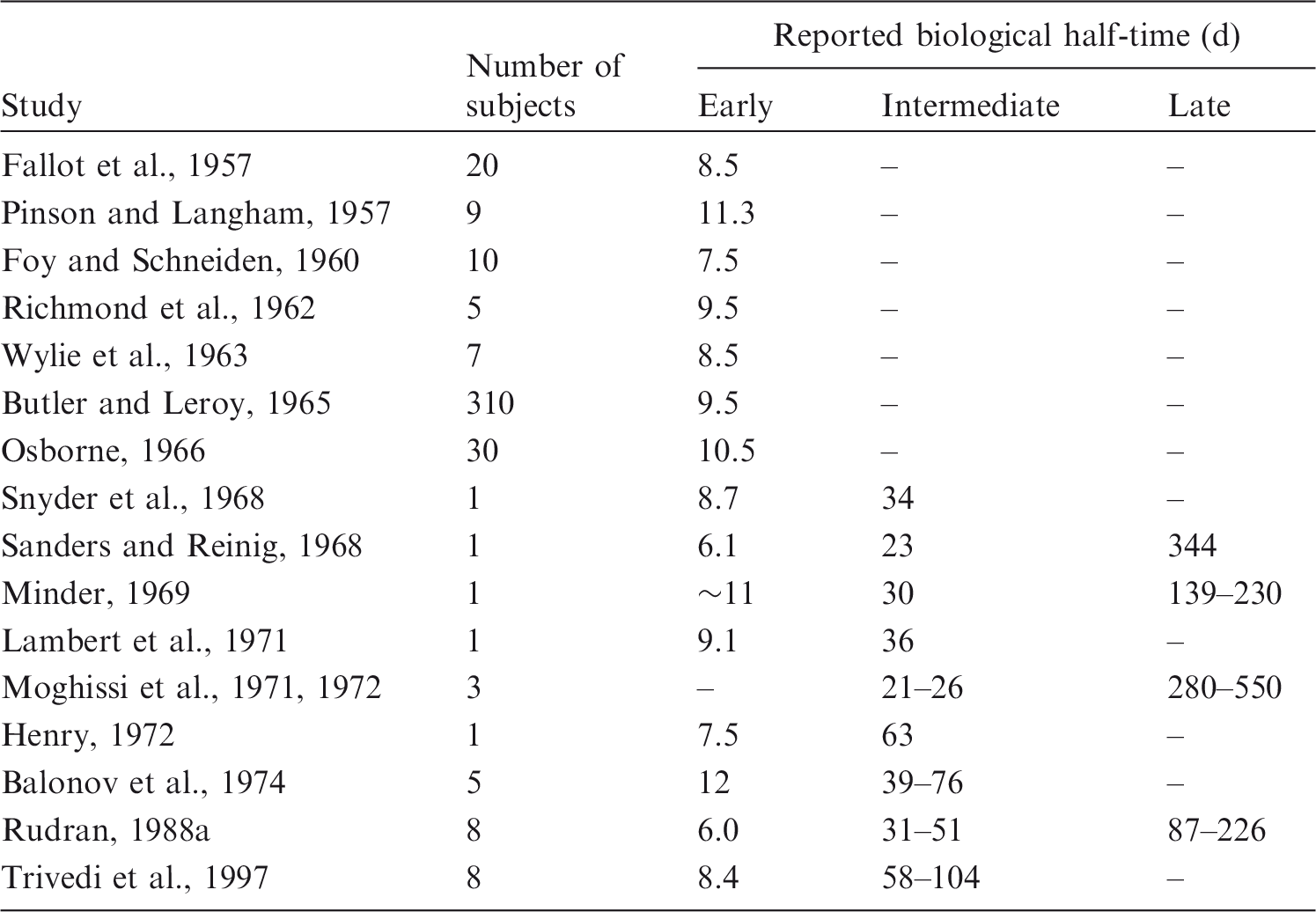
(54) Tritiated water (HTO) mixes rapidly with whole-body water after its entry into blood (Pinson and Langham, 1957; Moore, 1962; Balonov et al., 1974). In human subjects, the blood tritium concentration stabilised within approximately 1 h of intravenous injection of HTO (Moore, 1962; Balonov et al., 1974). Human studies using deuterium or HTO have confirmed that equilibration of HTO throughout the body water pool is essentially complete within 1 h of intake (Balonov et al., 1974; Davies et al., 2001; La Forgia and Withers, 2002). (55) A portion of tritium reaching blood as HTO becomes organically bound to carbon atoms due to biosynthesis in the body. The obtained OBT is generally non-exchangeable with hydrogen in body water, and has a lower rate of turnover than HTO. OBT refers here to carbon-bound tritium formed from HTO in living systems, including the human body, through natural biological processes. Some tritium atoms entering the body as HTO replace hydrogen bound with oxygen, sulphur, nitrogen, or phosphorus atoms in the tissues to form generally unstable bonds with those elements, and further tritium kinetics may be similar to HTO kinetics. These weakly bound forms of tritium are not considered here as OBT. The extent of organic binding of tritium reaching blood as HTO and the turnover time of OBT in a given tissue depend on the tissue’s metabolic activity (Smith, 1986; Taylor, 1989; Konig, 1990; Taylor et al., 1990). In general, the binding of tritium is greater, but the retention time of bound tritium is shorter, in metabolically active tissues such as liver and intestine than in skin, brain, and other tissues where metabolic activity is less pronounced (Smith, 1986). (56) Measurements on laboratory animals indicate that 1–5% of HTO entering blood becomes incorporated into organic components of tissues (Takeda and Kassida, 1979; Diabaté and Strack, 1993). On the basis of kinetic analysis of urinary excretion data for human subjects following acute intake of HTO (Sanders and Reinig, 1968; Snyder et al., 1968; Lambert et al., 1971; Balonov et al., 1974, 1984; Rudran, 1988b; Trivedi et al., 1997, 2000), it is estimated that 0.5–3% of the absorbed tritium typically binds to organic components of tissues. (57) Data from studies of laboratory animals and human subjects exposed to HTO indicate that whole-body retention can be described reasonably well as the sum of two or three exponential terms (Sanders and Reinig, 1968; NCRP, 1979; Taylor, 2003). The first two terms presumably represent HTO in body water and tritium incorporated into organic compounds within the tissues. A small, third component of retention identified in a few long-term studies may be associated with incorporation of tritium into structural tissues. This third retention component is less well characterised than the first two due to its small size, the scarcity of long-term observations of tritium retention in humans, and the potential influence of previous chronic intakes of HTO or tritiated luminous compounds. Data for human subjects indicate that the removal half-time of HTO in body water ranges from 4 to 18 d, with an average of approximately 10 d (Butler and Leroy, 1965). Estimated half-times for the second and third compartments are typically approximately 30–40 d and a few hundred days, respectively, but depend on the starting and ending times of the observation period and subjective distinctions between intermediate- and long-term components of retention. Estimated biological half-times of different components of tritium retention based on studies of human subjects exposed to HTO are summarised in Table 2.4. Reported biological half-times* for urinary excretion of tritium by humans exposed to tritiated water, tritium gas, or other inorganic forms of tritium. Values listed for groups of subjects are means except where ranges of values are indicated.
(b) Tritiated organic compounds
(58) Tritium taken into the body in the form of labelled biochemical substrates such as amino acids, tritiated glucose, or DNA precursors may be oxidised and enter the body water as HTO, or may be incorporated into the organic constituents of the body as OBT without first being converted to HTO. Soluble organic compounds of tritium entering the blood are incorporated into body tissues to an extent that depends on the specific chemical compound and the metabolic activity of the individual tissues. Tritium bound to carbon will normally be released through enzyme-mediated breakdown of the molecule in which the carbon atom is situated (Smith, 1986). The rate of such breakdown may be rapid for small molecules but slow for carbon-bound tritium incorporated into structural proteins such as collagen, or the phospholipids of some nerve cells. (59) Animal studies have demonstrated that much more OBT is present in tissues after intakes of tritiated biochemical substrates than after equal intakes of tritium as HTO (Mewissen et al., 1979; Takeda, 1982, 1991; Rodgers, 1992; Balonov et al., 1993). In rats fed HTO, tritiated amino acids, or tritiated DNA/RNA precursors for 22 d, the greatest concentrations of OBT were found after exposure to amino acids, with intermediate concentrations found after exposure to DNA/RNA precursors (Takeda, 1991). In mice administered HTO or tritium-labelled amino acids in diet for 56 d, the longer-term component of retention, attributable to OBT in tissues, accounted for approximately 50% of total body activity after administration of amino acids and approximately 15% after administration of HTO (Rodgers, 1992). In mice and rats administered tritiated glucose, various amino acids, and DNA precursors by intraperitoneal injection or ingestion, both initial and long-term retention of OBT were higher by a factor of 1.5–100 after administration of labelled biochemical substrates than after HTO intake (Balonov et al., 1993). (60) There is little information on the biokinetics of many of the tritiated organic compounds that may be encountered in the workplace. Available information indicates that tritium retention in the human or animal body after intake of 3H-labelled substances may vary greatly from one substance to another (Etnier et al., 1984; Rodgers, 1992; Richardson and Dunford, 2003a; Taylor, 2008). (61) On the basis of a review of the biokinetics of 11 xenobiotic tritiated organic compounds, Taylor (2008) estimated that the clearance half-time was less than 40 d in all cases. Some organic compounds may be incorporated directly into structural components and retained for much longer times.
(c) Elemental tritium
(62) Approximately 1–2% of inhaled tritium gas (HT) is dissolved in the blood and body fluids, and the rest is exhaled rapidly (Pinson and Langham, 1957; Peterman et al., 1985b). Experimental studies by Pinson and Langham (1957) showed that rats and humans slowly oxidise the retained HT to HTO. The rate of oxidation was approximately 50 times faster in the rat than in humans. Conversion from HT to HTO presumably results from microbial action in the large intestine, as mammalian tissues do not contain the hydrogenase enzyme necessary for the conversion of HT to HTO (Ichimasa et al., 1988). (63) Pinson and Langham (1957) found that equivalent rates of appearance of tritium in body fluids of humans following inhalation of HT and HTO occurred when the specific activity of HT in ambient air was approximately 15,000 times that of HTO. This indicates that approximately 0.007% of the inhaled HT was ultimately converted to HTO in vivo. Peterman et al. (1985a) repeated the experiments of Pinson and Langham (1957) with a larger group of human subjects, and obtained reasonably consistent results.
(d) Some other studied forms of tritium
(64) Results of in-vitro studies by Balanov et al. (1984), Cheng et al. (1997), Inkret et al. (2001), and Zhou and Cheng (2003) indicate that tritium is released from metal tritides into simulated lung fluids as HTO. (65) Eakins et al. (1975) studied the rate of urinary excretion of tritium in human volunteers whose skin had been exposed by contact with tritium-gas contaminated surfaces. Over the first several days, urinary tritium was mainly in the form of tritiated organic compounds, which were excreted in a biphasic pattern with half-times of approximately 0.2 d (range 0.1–0.3 d) and 1.7 d (range 1.1–1.9 d). The concentration of HTO in urine declined with a half-time of approximately 10 d. Excretion of tritium in organic form peaked approximately 24 h after exposure, at which time the concentration of tritium in organic form was more than 100 times greater than that of tritium as HTO. Similar results were observed for exposures to different areas of the skin, and from various contaminated metal and glass surfaces. (66) Trivedi (1995) studied the percutaneous absorption and systemic biokinetics of tritium-gas contaminated pump oil in male hairless rats. Skin-contact exposure with the pump oil resulted in uptake of tritiated organic compounds and HTO into blood. The systemic biokinetics indicated that absorbed tritium was mainly in organic form, most of which was transferred from the skin with a half-time of 1.7 d. A second, long-term component of retention of organic tritium with a removal half-time of 27.6 d accounted for < 3% of the tritium retained in the skin. HTO in the skin also showed two components of retention, with half-times of 3.7 and 18.1 d. A significant level of the organic form was excreted shortly after exposure. Elevated levels of tritium were found in the liver and kidneys. Overall, approximately 60% of the activity applied to skin was excreted in faeces, mostly in organic form, and 4% was excreted in urine. The remaining activity (∼36%) may have been removed gradually from the skin to the environment. The exposed skin was estimated to receive the highest dose of any tissue, primarily due to retention of the organic form of tritium at the point of contact with the contaminated pump oil. (67) Balonov (1983) and Balonov et al. (1984) studied absorption of tritium after ingestion, intratracheal instillation, and skin application of tritiated polystyrene-based luminous compounds to rats and volunteers. Following intake or skin application of tritiated luminous compounds, tritium is absorbed both as HTO and foreign organic low-molecular-weight compounds in comparable quantities. A substantial fraction of foreign tritium compounds is excreted quickly in urine and faeces, another fraction is retained for some days in liver and/or kidney of rats, and the rest catabolises to HTO.
2.2.3.2. Biokinetic model for systemic tritium
(a) Previous models
(68) Publication 56 (ICRP, 1989) recommended a two-component model to represent the behaviour of tritium that enters the human body as HTO. That model assumes that 97% of the tritium is eliminated with a biological half-time of 10 d, and 3% becomes organically bound and is eliminated with a biological half-time of 40 d. (69) The authors of Publication 56 (ICRP, 1989) interpreted the available data as indicating that 9–45% of ingested OBT is incorporated into organic constituents of tissues, and that, on average, approximately nine times more OBT is present in body tissues after intakes of OBT than after intakes of HTO. Publication 56 recommended a default model for unknown tritiated organic compounds in the environment, in which it is assumed that 50% of the OBT entering the systemic circulation enters into bonds with carbon and is cleared with the same half-time as carbon, assumed to be 40 d in Publication 56. The remaining 50% is assumed to be metabolised rapidly to HTO and removed from the body with a biological half-time of 10 d. (70) Taylor (2003) re-evaluated data on tritium excretion by human subjects exposed to HTO in an effort to develop a biokinetic model for HTO that could be used for protection planning and interpretation of bioassay data collected at early, intermediate, or late times after exposure. He proposed a three-component exponential model with half-times of 10 d (99%), 40 d (0.98%), and 350 d (0.02%). (71) Relatively sophisticated, physiologically based biokinetic models for dietary tritium have been proposed. Richardson and Dunford (2003a,b) designed a generic model framework for hydrogen, carbon, nitrogen, and oxygen, with the goal of predicting the biokinetics of each of these elements on the basis of the metabolic reactions of the principal nutrients: carbohydrates, fats, and proteins. Galeriu et al. (2009) and Galeriu and Melintescu (2010) proposed a biokinetic model for tritium based on organ-specific metabolic rates.
(b) Systemic models for tritium used in this publication
(72) It is not feasible to derive specific biokinetic models for systemic tritium for the many different physicochemical forms of tritium encountered in the workplace. Two different systemic models, referred to as the HTO systemic model and the OBT systemic model, are used to derive dose coefficients for two broad classes of tritium compounds with relatively fast and relatively slow removal from the body, respectively. The OBT systemic model is applied to intake of biogenic tritiated organic compounds. The HTO systemic model is applied to intake of all other forms of tritium, including foreign (non-biogenic) tritiated organic compounds.
HTO systemic model
(73) The HTO systemic model includes compartments representing blood, extravascular body water that exchanges rapidly with blood, and two components of retention of tritium converted to OBT in vivo. The model structure, which is broadly similar to a number of previously proposed structures for HTO (NCRP, 1979; Saito, 1992; Hill and Johnson, 1993), is shown in Fig. 2.1. Parameter values are given in Table 2.5. Excretion is from the blood compartment alone. The transfer coefficient from blood to excreta is set to yield an initial removal half-time from the body of 10 d. The transfer coefficients from OBT-1 and OBT-2 back to extravascular HTO correspond to half-times of 40 d and 1 y, respectively; the net retention half-times in these compartments are slightly longer than 40 d and 1 y due to recycling of activity. Specific excretion pathways are not shown in Fig. 2.1, but the following division is assumed on the basis of reference data for water balance (ICRP, 2002b): urine, 55%; faeces, 4%; exhalation, 12%; and loss through skin (sweat plus insensible loss), 29%. (74) Model predictions of the blood content of tritium as a function of time after intravenous injection of HTO are compared in Fig. 2.2 with estimates based on short- and long-term observations of Moore (1962), and short-term observations of Balonov et al. (1974) for human subjects exposed to HTO. The data of Moore (1962) were reported as concentrations of tritium in blood plasma. Derived estimates of tritium in whole blood are based on the assumptions that plasma water represents two-thirds of blood water, and red blood cell (RBC) water equilibrates with plasma water during the first few minutes after injection. The data of Balonov et al. (1974) were reported as relative concentrations over time in whole blood normalised to 1.0 at equilibrium, with equilibrium assumed to be reached within a few hours of injection. These data were converted to percentages of injected tritium by assuming that blood contains 10% of whole-body HTO at equilibrium, based on the estimate that blood water represents 10% of whole-body water. (75) Model predictions of urinary excretion of tritium as a function of time after acute uptake of tritium to blood are compared in Fig. 2.3 with data for five individual human subjects of five different long-term studies of whole-body retention following exposure to HTO (Sanders and Reining, 1968; Snyder et al., 1968; Balonov et al., 1974; Rudran, 1988a; Trivedi et al., 1997). The studies by Balonov et al., Rudran, and Trivedi et al. each involved several (five to eight) subjects, but in each case, the published paper only provided detailed data for one illustrative subject. The study by Balonov et al. involved ingestion of HTO as part of a controlled biokinetic study, and the other four studies involved accidental exposure to HTO in the workplace. In two of the cases of accidental exposure, an effort was made to accelerate the removal of tritium from the body at early times after intake, either by administration of an oral diuretic (Sanders and Reinig, days 3–35) or by increasing fluid intake (Trivedi et al., days 1–32). The observations and model predictions shown in Fig. 2.3 are normalised to a urine concentration of 1.0 on day 1. Structure of the tritiated water (HTO) systemic model. Transfer from blood to excreta (or excretion pathways) is divided as follows: 55% to urinary bladder contents; 4% to right colon contents; 12% exhaled with no retention in lungs; and 29% removed through the skin (sweat plus insensible loss) with no retention in skin. OBT, organically bound tritium; T½, half-life. Transfer coefficients (d−1) in the tritiated water (HTO) systemic model. OBT, organically bound tritium. For purposes of dose calculations, these compartments are assumed to be distributed uniformally in the body. 55% to urinary bladder contents, 4% to right colon contents, 12% exhaled, and 29% lost through skin. Observations and model predictions of blood content of tritium following intravenous injection of tritiated water. Observations and model predictions of urinary excretion of tritium as a function of time after acute intake of tritiated water by human subjects. Data and model predictions are normalised to a urine concentration of 1.0 on day 1.




OBT systemic model
(76) The model for systemic tritium applied to intake of biogenic tritiated organic compounds is a modification of the model for OBT applied in Publication 56 (ICRP, 1989) (Fig. 2.4). It is assumed here that 50% of tritium initially entering blood transfers immediately to the OBT-1 compartment, and 50% is converted immediately to HTO within the blood compartment. Tritium entering the OBT-1 or blood compartments subsequently follows the HTO model defined in Fig. 2.1. For application to individual organic tritium compounds, the division of absorbed activity between the OBT-1 and blood compartments can be modified as allowed by specific information. (77) The OBT model with the default initial division of activity between the OBT-1 (50%) and blood (50%) compartments predicts that OBT would represent approximately 65–70% of whole-body tritium in a worker who is chronically exposed to a biogenic tritiated organic compound. The model for HTO adopted in this publication predicts that OBT would represent approximately 5–6% of whole-body tritium in a worker who is chronically exposed to HTO. The OBT systemic model is not recommended for assessment of intake of tritiated DNA precursors (e.g. [3H]-thymidine, [3H]-deoxycytidine) because the concept of tissue dose may not be applicable to these forms of tritium. The organically bound tritium (OBT) model applied to tritium entering the systemic circulation after intake of a biogenic tritiated organic compound. Tritium entering the OBT-1 or blood compartments subsequently follows the tritiated water (HTO) model defined earlier. For application to individual organic tritium compounds, the division of absorbed activity between the OBT-1 and blood compartments can be modified as allowed by specific information. T½, half-life.

2.3. Individual monitoring
(78) Tritium intakes are generally monitored though measurement of the activity excreted in urine. The most common method of analysis is liquid scintillation counting (Table 2.6). (79) Currently, most laboratories do not perform faecal monitoring of tritium routinely, and therefore this method is not recommended here. However, faecal monitoring of workers exposed to particulate forms of tritium might be desirable. Atomic Energy of Canada Ltd (Trivedi et al., 1993) has published a method to measure OBT in faeces, with a detection limit of 5 Bq g–1. Monitoring techniques for 3H. The achievable value for tritium in urine, 10 Bq l−1, is possible but not always practical in routine monitoring of workers. In general, the background tritium excretion in urine is higher than 10 Bq l−1.

2.4. Dosimetric data for tritium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 3H compounds.

AMAD, activity median aerodynamic diameter; OBT, organically bound tritium.
Dose per activity content of 3H in daily excretion of urine (Sv Bq−1); 5µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
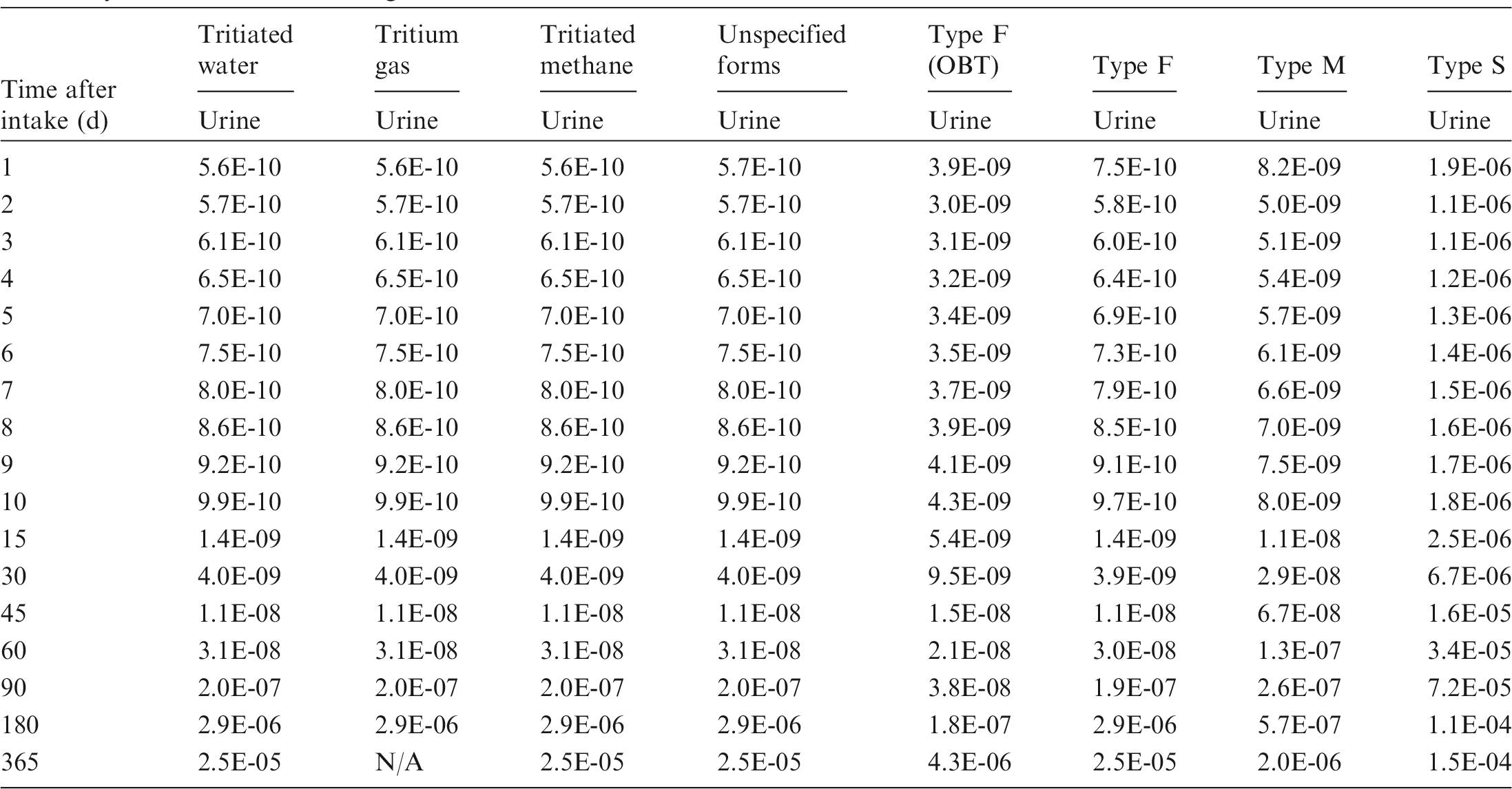
OBT, organically bound tritium.

Daily urinary excretion of 3H following inhalation of 1 Bq tritiated water.

Daily urinary excretion of 3H following inhalation of 1 Bq tritium gas.
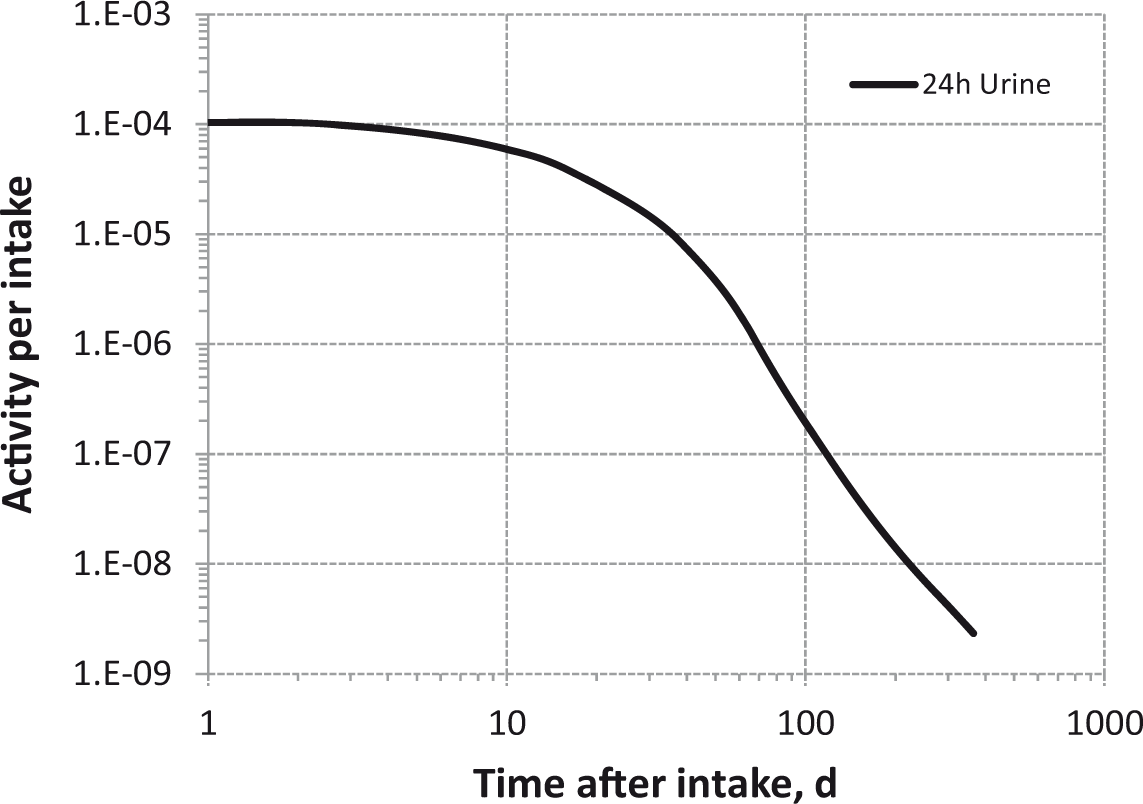
Daily urinary excretion of 3H following inhalation of 1 Bq tritiated methane.

Daily urinary excretion of 3H following inhalation of 1 Bq unspecified gas or vapour forms.

Daily urinary excretion of 3H following inhalation of 1 Bq Type F (biogenic organic compounds).
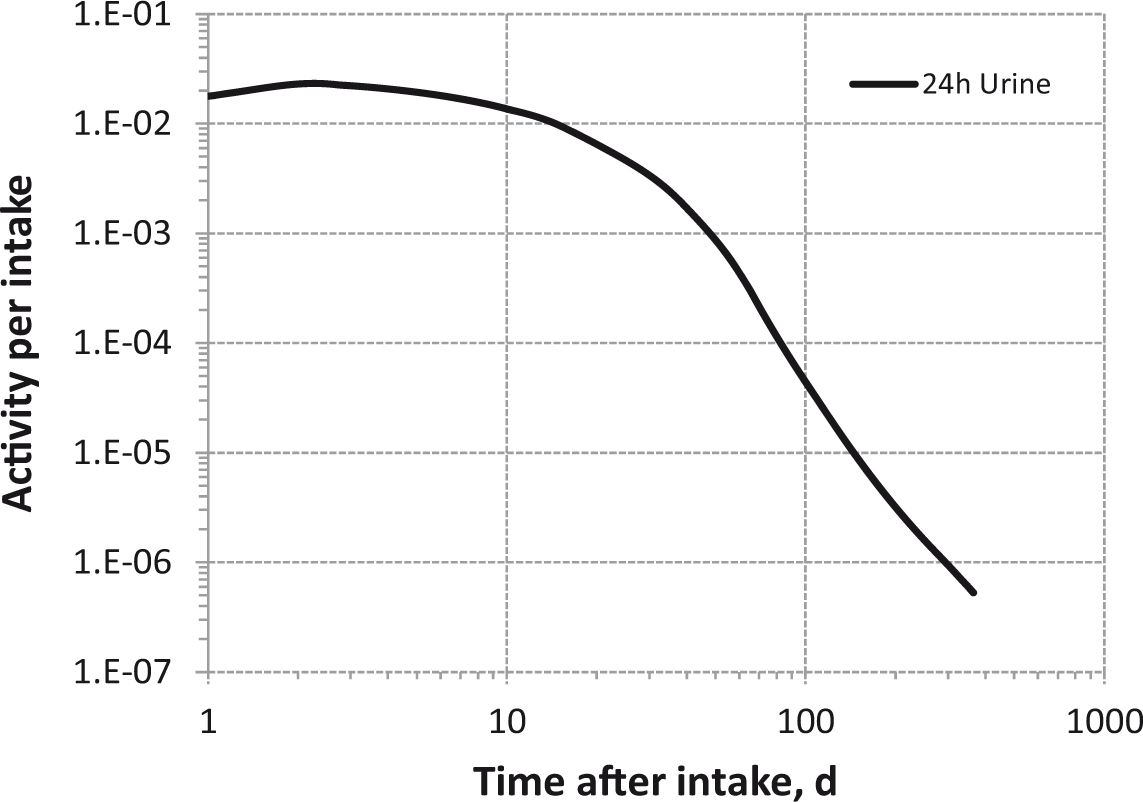
Daily urinary excretion of 3H following inhalation of 1 Bq Type F (LaNi4.25Al0.75 tritide).
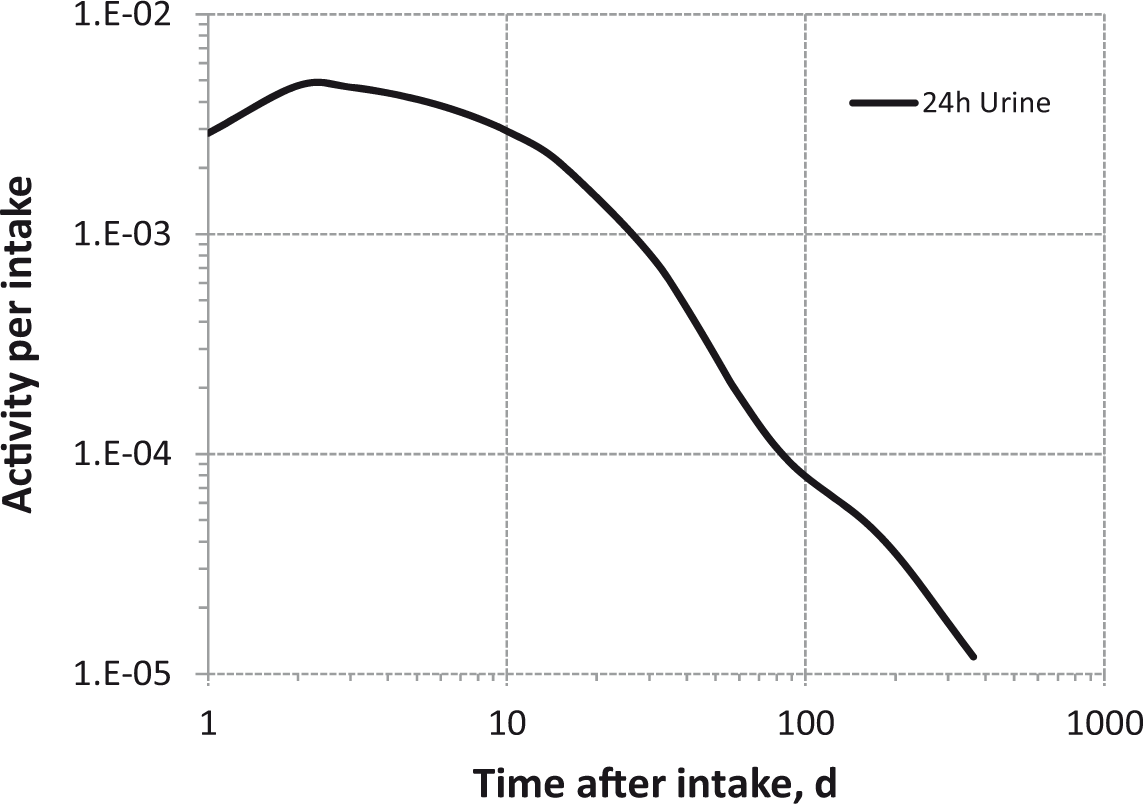
Daily urinary excretion of 3H following inhalation of 1 Bq Type M.

Daily urinary excretion of 3H following inhalation of 1 Bq Type S.
2.5. References
3. CARBON (Z = 6)
3.1. Chemical forms in the workplace
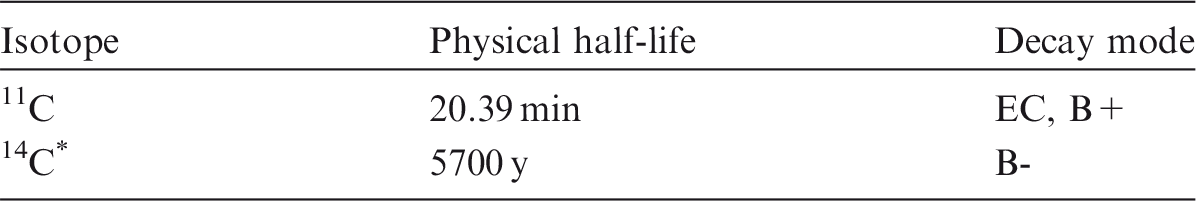
(80) Carbon is a non-metal that occurs mainly in oxidation states II and IV. It may be encountered in industry in a variety of chemical forms, including carbon monoxide, carbon dioxide, and methane, as well as in a wide range of organic carbon compounds and particles containing 14C. (81) Only two isotopes of carbon are of importance for radiological protection, 11C and 14C. Table 3.1 shows the isotopes of carbon addressed in this publication. Due to its short half-life, and the penetrating 511 keV annihilation radiation it emits, external irradiation from 11C may well be a greater hazard than internal exposure. Isotopes of carbon addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. EC, electron-capture decay; B+, beta-plus decay; B-, beta-minus decay
3.2. Routes of intake
(82) It is not feasible to provide biokinetic models, dose coefficients, and bioassay functions for the large number of carbon compounds with potentially distinct biokinetic behaviour. Hence, systemic biokinetic models and dosimetric information are only given for selected forms. It is the responsibility of employers to assess doses to ensure appropriate protection for forms for which dose coefficients are not provided.
3.2.1. Inhalation
(83) Some information on absorption from the respiratory tract is available for inhaled gases of carbon in man and in experimental animals. Some information is also available on the behaviour of 14C-labelled compounds and particles, mainly in rats, and on forms of carbon labelled with other radionuclides. (84) Absorption parameter values and types, and associated fA values for gas and vapour forms of carbon are given in Table 3.2 and for particulate forms in Table 3.3. (85) Exposures to both gas/vapour forms and particulate forms of carbon are common, and it is therefore recommended in the OIR series that 50% particulate and 50% gas/vapour should be assumed in the absence of information (ICRP, 2002a). Deposition and absorption for gas and vapour forms of carbon.* ET1, anterior nasal passage; ET2, posterior nasal passage, pharynx, and larynx; BB, bronchial; bb, bronchiolar; AI, alveolar-interstitial. For carbon in unspecified gas or vapour form (including unspecified organic vapours), the default option for gases and vapours is recommended: 100% total deposition in the respiratory tract; default distribution between regions¶ and Type F absorption. Percentage deposited refers to how much of the material in the inhaled air remains in the body after exhalation. Almost all inhaled gas molecules contact airway surfaces, but usually return to the air unless they dissolve in, or react with, the surface lining. In the case of methane, a small fraction is absorbed into body fluids, and of that, a fraction is metabolised and the rest is subsequently exhaled. CO, systemic model for carbon monoxide; CO2, systemic model for carbon dioxide/bicarbonate; C, generic systemic model for other 14C compounds (Section 3.2.3.2). As instantaneous absorption to blood is assumed, calculations can be performed assuming direct injection into blood, and the regional deposition does not need to be considered. However, for completeness, the default distribution is assumed. Default distribution between regions (20% ET2, 10% BB, 20% bb, and 50% AI). Not applicable for Type V absorption because all activity deposited in the respiratory tract is absorbed instantaneously. Absorption parameter values for inhaled particulate forms of carbon and ingested carbon.
*
Following uptake into body fluids, the generic systemic model for carbon is used (Section 3.2.3.2), with the exception of barium carbonate, for which the carbon dioxide/bicarbonate systemic model (Section 3.2.3.2) is applied to the absorbed carbon. It is assumed that the bound state can be neglected for carbon, i.e. fb=0. The value of sr for Type F forms of carbon (100 d−1) is element-specific. The values for Types M and S (3 d−1) are the general default values. See text for summary of information on which parameter values are based, and on ranges of parameter values observed for individual materials. For barium carbonate, Type F default parameter values are used for absorption from the respiratory and alimentary tracts, but a specific systemic model, carbon dioxide/bicarbonate, is used for absorbed carbon. Materials (e.g. elemental carbon) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of carbon (1.0). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (1) for ingestion of the radionuclide.


3.2.1.1. Gases and vapours
(a) Carbon monoxide (CO)
(86) Carbon monoxide at high concentration is a potent asphyxiant; for that reason, its human respiratory physiology has been studied extensively (Lipsett et al., 1994). Carbon monoxide diffuses readily across the membranes of the gas exchange [alveolar-interstitial (AI)] region (Crapo et al., 1982). Although carbon monoxide only has low solubility in biological fluids, once absorbed into the pulmonary circulation, it binds avidly to haemoglobin molecules within RBCs. Peterson and Stewart (1970) estimated the biological half-life of carbon monoxide in the blood to be between 150 and 200 min, and these values, together with the haemoglobin content of the blood of a reference worker (ICRP, 2002b), can be used to estimate that 0.4 of the inhaled carbon monoxide becomes bound to haemoglobin (ICRP, 1981). On that basis, for carbon monoxide, it is assumed that there is effective deposition of 40% of the inhaled activity in the respiratory tract with instantaneous (Type V) absorption (Table 3.2). It is assumed that the 14C-carboxyhaemoglobin formed releases 14C to the environment via the lungs with a biological half-time of 200 min (Section 3.2.3.2).
(b) Carbon dioxide (CO2)
(87) Release to the environment of blood-borne carbon dioxide resulting from tissue carbon metabolism is a central function of the respiratory system, and the transport processes have been documented in detail (Guyton and Hall, 2000). Due to the very high solubility of carbon dioxide and the associated bicarbonate ion in tissue fluids, carbon dioxide is transferred 20 times more rapidly than oxygen across the alveolar membrane (Guyton and Hall, 2000). Thus, despite the net flow of carbon dioxide into the alveolar space, inhaled radioactive CO2 equilibrates rapidly with blood-borne carbon dioxide/bicarbonate, and is absorbed quantitatively into the circulation. On that basis, for carbon dioxide, it is assumed here that there is 100% deposition in the respiratory tract with instantaneous (Type V) absorption (Table 3.2). The carbon dioxide/bicarbonate systemic model (Section 3.2.3.2) is applied to the absorbed material.
(c) Methane (CH4)
(88) The dosimetric implications of inhaling methane gas were examined by Phipps et al. (1990). They made the conservative assumption that 1% of methane was metabolised, based on observations by Dougherty et al. (1967) which indicated that approximately 0.3% of methane infused into sheep was converted to carbon dioxide. Carlisle et al. (2005) and Didychuk et al. (2014) investigated the extent of oxidation and organic fixation of 3H and 14C following inhalation of a mixture of 3H- and 14C-labelled methane by rats. In a pilot study, Carlisle et al. (2005) measured retention of activity in skin, liver, brain, and carcass 1 and 24 h after a 4 h exposure. They estimated that uptake was approximately 0.1% of intake. Most (82–95%) of the retained 14C was organically bound. In a more comprehensive study using the same methods, Didychuk et al. (2014) followed retention in these tissues up to 14 d after exposure. They estimated that uptake was approximately 0.3% of intake for both 3H and 14C. For methane, it is therefore assumed here that there is 0.3% deposition in the respiratory tract with instantaneous (Type V) absorption (Table 3.2). It is also assumed here that the carbon in the absorbed methane follows the systemic model for methane (Section 3.2.3.2).
(d) Industrial organic chemicals
(89) As part of a programme to study the disposition of selected industrial organic chemicals thought to pose an inhalation health risk to humans, biokinetic studies were conducted on several which might be inhaled in vapour form, including benzene, dichloropropene, methyl bromide, butadiene, isoprene, butoxyethanol, and isobutene. Brief summaries of relevant information follow, but no systemic model, dose coefficients, or bioassay functions are given here for these compounds. Except where noted otherwise, these studies followed retention, metabolism, and excretion for approximately 3 d after a 6 h inhalation exposure of rats to a vapour of the 14C-labelled compound.
Benzene (C6H6)
(90) Krins et al. (2003) conducted a study of the distribution, retention, and excretion of 14C-labelled benzene based on existing pharmacokinetic models. They reported that in humans exposed to 55 ppm for 4 h, approximately 30% of inhaled benzene is absorbed into blood (Nomiyama and Nomiyama, 1974a,b). Studies on rats, however, showed that retention during exposure is highly dependent on the concentration of benzene in the inhaled air (Sabourin et al., 1987).
Dichloropropene (DCP)
(91) It was estimated that 38% of inhaled dichloropropene was absorbed (Bond et al., 1985a; Dutcher et al., 1985). The results indicated that the absorbed dichloropropene is metabolised rapidly in tissues, and the metabolites are excreted.
Methyl bromide
(92) It was estimated that 48% of inhaled methyl bromide was absorbed at the lower concentrations used, but the fraction decreased to 27% at the highest concentration (Bond et al., 1985b; Medinsky et al., 1985). The results indicated that the absorbed methyl bromide is metabolised rapidly in tissues (> 90% within 1 h) and the metabolites are excreted; approximately 20% of the amount in tissues immediately after exposure was retained at 65 h.
1,3-Butadiene
(93) Interspecies differences were investigated. Approximately 20% of inhaled butadiene was absorbed (and retained at the end of exposure) in rats and mice at the lowest concentrations used, with the fraction decreasing to 2–4% at the highest concentrations (Bond et al., 1986a). Bond et al. (1987) followed the tissue distribution of 14C for 13 d after 3.4 h inhalation exposures of rats and mice. In both species, approximately 90% of 14C present in the lungs at the end of exposure cleared with a half-time of several hours, and the rest cleared with a half-time of approximately 1 week. In monkeys, the fraction absorbed and excreted within 4 d was lower, at approximately 3%, than in rats and mice exposed to the same concentration (Dahl et al., 1991).
Isoprene (2-methyl-1,3-butadiene)
(94) In rats, approximately 20% of inhaled isoprene was absorbed (and retained at the end of exposure) at the lowest concentration used, with the fraction decreasing to approximately 4% at the highest concentration. Mice showed similar absorption but less change with concentration (Dahl et al., 1987; Bond et al., 1991).
Butoxyethanol
(95) As part of a wider study of the biokinetics and metabolism of glycol ethers administered by different routes, Sabourin et al. (1992) followed the retention and excretion of 14C for 66 h after 6 h inhalation exposures of rats to [14C]butoxyethanol. It was estimated that approximately 20% of inhaled butoxyethanol was absorbed.
Isobutene (2-methyl-1-propene)
(96) Approximately 8% of inhaled isobutene was absorbed (and retained at the end of exposure) at the lowest concentrations used, with the fraction decreasing to approximately 2% at the highest concentration (Henderson et al., 1993).
(e) Other volatile organic forms
(97) The volatility and solubility in body fluids of organic compounds have wide ranges. Smith et al. (1983) reported a wide range of urinary excretion patterns in workers following inhalation of a variety of 14C-labelled organic compounds. However, information was not reported that would enable estimation of the fraction of inhaled material that was deposited in the respiratory tract, nor the rate at which it was absorbed. In the absence of specific information, the default option for gases and vapours is taken. As for tritium (Section 2.2.1.1.), for carbon (gas or vapour) in unspecified organic forms, it is assumed here that there is 100% deposition in the respiratory tract (with default regional distribution, Table 3.2) and Type F absorption.
3.2.1.2. Particulate materials (liquid and solid)
(a) 14C-labelled compounds
(98) Some information is available for 14C-labelled compounds administered to rats. For the 14C-labelled carbon compounds considered in the following sections, the systemic behaviour is specific to each compound. In these cases, no systemic model, dose coefficients, or bioassay functions are given here.
DTPA (diethylenetriaminepentaacetic acid)
(99) Absorption of DTPA from the respiratory tract has been studied in detail, mainly because of the use of DTPA as a decorporation agent for treating intakes of actinides, and interest in its administration by inhalation. Crawley and Haines (1979b) reported rapid lung clearance of 14C following pulmonary instillation of 14C-DTPA into rats, with < 1% ILD retained in the lungs at 1 d, and 0.03% ILD retained at 7 d. Dudley et al. (1980a) determined absorption of 111In-DTPA from the nasopharyngeal, tracheobronchial, and pulmonary regions of beagle dogs, to be 16%, 48%, and 90%, respectively, following instillation. Nasopharyngeal absorption was slightly higher following nasal inhalation (23%) than following nasal instillation (16%). In rats, Dudley et al. (1980b) found nasopharyngeal absorption to be much higher (68%) following nasal inhalation than following instillation (19%). In complementary experiments, Dudley et al. (1980a,b) found absorption from the alimentary tract to be approximately 8% in dogs and 4% in rats. Stather et al. (1983) followed the biokinetics of 14C for 1 week after inhalation of 14C-labelled DTPA by two healthy volunteers. Studies were carried out on the same subjects following intravenous injection, and in one case by ingestion (which indicated that approximately 3% was absorbed from the alimentary tract). Modelling by the authors gave an estimated rate of absorption from lungs to blood of approximately 13 d−1 (fr∼1), giving assignment to Type F. A similar absorption rate (∼10 d−1) has been obtained with 99mTc-labelled DTPA, which has been used extensively to study pulmonary epithelial permeability in man (see technetium inhalation section 15.2.1.).
Potassium cyanide
(100) 14C-labelled potassium cyanide (K14CN) is an important precursor in the synthesis of organic compounds. Crawley and Goddard (1977) studied its behaviour following administration to rats by intravenous injection, pulmonary and gastric intubation, and skin absorption. The biokinetics following pulmonary intubation was very similar to that following intravenous injection, showing that K14CN was absorbed completely and rapidly from the lungs (fr∼1.0, sr > 100 d−1), giving assignment to Type F. Absorption following gastric intubation was somewhat slower.
Methanol
(101) Crawley (1977) reported that the behaviour of 14C following administration of 14C-labelled methanol (14CH3OH) to rats by pulmonary intubation was very similar to that following intravenous injection. Details were only given for the latter, but indicated that 14C-methanol was absorbed completely and rapidly from the lungs (fr∼1.0, sr > 100 d−1), giving assignment to Type F.
Sodium acetate
(102) Crawley and Haines (1978) reported that the behaviour of 14C following administration of 14C-labelled sodium (2-14C) acetate to rats by pulmonary intubation was very similar to that following intravenous injection, but few details were given. By 1 d, most tissue levels were below 1% of the injected activity, indicating assignment to Type F.
Nitrobenzene
(103) Crawley and Haines (1979a) reported that following pulmonary intubation of 14C-labelled nitrobenzene into rats, lung clearance was very rapid. Retention could be described by a three-component exponential function with half-lives of 2.5 min (99%), 0.75 d (0.7%), and 5 d (0.3%), giving fr of approximately 0.99 and sr of approximately 400 d−1, and assignment to Type F.
14C labelled drugs
(104) Brown and Schanker (1983) measured the absorption rate of a range of 14C-labelled drugs for up to 1 h after inhalation by rats. For lipid-insoluble compounds, the half-time (range 1.4–35 min) tended to increase with molecular mass [range 60–300 daltons (Da)]. Lipid-soluble compounds were absorbed more rapidly (range 0.25–6 min), with less clear dependence on molecular mass (range 80–700 Da).
Azodicarbonamide
(105) Studies were also conducted with azodicarbonamide (ADA). Mewhinney et al. (1987) followed the kinetics of 14C for 102 d after inhalation of 14C-ADA by rats. In complementary experiments, 30% of administered ADA was absorbed following gavage and 90% following intratracheal instillation. The lungs contained approximately 0.5% ILD at 3 d after intratracheal instillation, and there was similar rapid lung clearance after inhalation. Results suggested that ADA was converted rapidly to biurea, most of which was eliminated rapidly in urine.
(b) Atmospheric pollutants
(106) Bond et al. (1986a,b) summarised studies of the biokinetics, following inhalation by rats, of 14C- or 3H-labelled chemicals selected as representative of different important chemical classes found in atmospheric pollutants: benzo[a]pyrene, aminoanthracene, nitropyrene, and phenanthridone. The chemicals were inhaled in pure form and, in some cases, associated with carbonaceous (diesel exhaust), organic (coal tar), or inorganic (gallium oxide) particles. Lung retention and excretion of the labels were followed for up to 26 d after inhalation. For all four compounds, in pure form, > 99% cleared from the lungs with a half-time < 1 d. Association with particles increased lung retention in some cases but not others. For benzo[a]pyrene associated with coal tar, a similar fraction (> 99%) cleared rapidly, with gallium oxide slightly less (98%), and diesel soot only 50%. For amino-anthracene associated with coal tar, rapid clearance was less (92%). For nitropyrene associated with gallium oxide, > 99% cleared rapidly, and 92% for diesel soot. Bond et al. (1985c) followed lung retention of 14C for 4 d after instillation of 14C-labelled anthracene, benz[a]athracene, 1-nitropyrene, 6-nitrobenzo[a]pyrene, and dibenzo[c,g]carbazole into the lungs of rats. They found that the retention half-time of the small fraction that was retained beyond 2 d increased with lipophilicity (as measured by the octanol:water partition coefficient) over the range 26–63 h.
(c) Low molecular mass organic compounds
(107) Henderson et al. (1988) reported that a wide range of inhaled organic compounds with molecular mass < 300 Da, including those studied by Bond et al. (1985c, 1986a,b), are cleared rapidly (t½ < 12 h) from the lungs of rats. They determined lung retention of a series of dyes (easily traced without radiolabels) of varying molecular mass and lipophilicity (which increases with molecular mass) up to 24 h after instillation into rat lungs. For organic-soluble compounds, the fraction of the ILD retained in the lungs at 24 h increased from approximately 3% for molecular mass of 250 Da to approximately 90% for 400 Da. However, retention of a compound [1,5-di(2-sulfo-p-toluidino) anthraquinone] of higher molecular mass (576 Da) but containing a polar functional group was only 21%. The authors concluded that both molecular mass and lipophilicity are important in determining lung retention.
(d) Barium carbonate
(108) Crawley and Haines (1979a) followed retention and excretion of 14C following pulmonary intubation of a suspension of barium 14C-labelled carbonate into rats. Lung retention decreased rapidly from 70% ILD at 6 h to 0.2% at 8 d, indicating assignment to Type F. Kramer et al. (1996) measured lung retention of 14C for 550 d after accidental inhalation of barium 14C-labelled carbonate by a worker. Most of the activity remaining in the lung at 2 d after the presumed intake (the first in-vivo measurement) cleared rapidly, with an effective half-time of 0.77 d, also indicating assignment to Type F. The carbon dioxide/bicarbonate systemic model (Section 3.2.3.2) is applied to the absorbed carbon. Although in-vivo data are limited, specific parameter values for barium carbonate are adopted in Table 3.3. It is assigned Type F parameter values for absorption from the respiratory and alimentary tracts, but presented separately to emphasise the difference in systemic model from that used by default for particulate forms of carbon. Thus, it provides dose coefficients and bioassay functions which can be used for other Type F particulate forms of carbon for which the carbon dioxide/bicarbonate systemic model is found to be appropriate.
(e) 14C-labelled particles
(109) Some information is also available from experimental studies on 14C-labelled particles, for which carbon released in the lungs would reasonably be expected to follow the generic systemic model for carbon (Section 3.2.3.2).
Elemental carbon
(110) Johnson (1989) followed the biokinetics of 14C for 146 d after administration to rats by intratracheal instillation of 14C-bearing material obtained from air filters during re-tubing of a CANDU reactor (Greening, 1989). No 14C above background was detected in urine or liver, indicating negligible dissolution in the lungs (or alimentary tract). After the first few days, lung clearance was very slow, with more than 70% ILD retained at 146 d, giving assignment to Type S. Oberdörster et al. (2002) reported significant translocation of particles to the liver following inhalation by rats of ultrafine (median diameter 22 nm) 13C-carbon particles. However, far less translocation to liver was observed by this group in a similar experiment using 192Ir-labelled carbon particles (Kreyling et al., 2009).
Diesel exhaust particles
(111) Lee et al. (1983) followed the biokinetics of 14C for 365 d after inhalation of 14C-labelled diesel exhaust particles by rats and guinea pigs. Lung retention at 180 d was approximately 15% ILD in rats and 80% ILD in guinea pigs, with no 14C detected in other tissues after the first day, indicating Type S behaviour. Similar lung retention in rats was observed in other studies (Chan et al., 1981; Lee et al., 1987).
(f) Carbon particles labelled with isotopes of other elements
(112) Carbon particles may also contain other elements, which may or may not be chemically bound to the particle matrix. For such particles, some information may be available from studies with particles labelled with a radioisotope of one of the other elements. For details, refer to the section dealing with the labelling radioelement.
Carbon ‘tritide’ (tritium-loaded carbon particles) (Section 2.2.1.2)
(113) The results of in-vitro dissolution tests are consistent with assignment to Type S.
Technetium-labelled carbon particles (Section 15.2.1)
(114) The results of human inhalation studies suggest that it is more likely to be Type M or S than Type F.
3.2.1.3. Rapid dissolution rate for carbon
(115) Very rapid uptake of carbon (100 d−1 or more) has been observed for several chemical forms. A value of 100 d−1 is applied here to all Type F forms of carbon.
3.2.1.4. Extent of binding of carbon to the respiratory tract
(116) The evidence of rapid uptake of carbon gases and several solid and liquid forms from the lung indicates that that there is probably little binding of carbon. It is therefore assumed that the bound state can be neglected for carbon, i.e. fb = 0.0.
3.2.2. Ingestion
(117) The uptake of carbon from the gastrointestinal tract is highly dependent on the form in which it is ingested. Absorption is almost complete for carbon administered as [14C]-labelled inorganic compounds, such as potassium cyanide (Crawley and Goddard, 1977), or [14C]-labelled organic compounds, such as methyl methacrylate (Bratt and Hathway, 1977). Absorption may be much lower for some other organic or inorganic compounds, such as polydiethylstilboesterol, octanoic acid, or hydrolysed polyacrylonitrile grafted cellulose (Lai et al., 1978). (118) Publication 30 (ICRP, 1981) recommended that, in the absence of compound-specific information, organic compounds labelled with radioactive isotopes of carbon should be assumed to be absorbed completely from the gastrointestinal tract, and this recommendation is retained here for all chemical forms (i.e. fA = 1).
3.2.3. Systemic distribution, retention, and excretion
3.2.3.1. Summary of the database
(119) The biokinetics of systemic radiocarbon depends on the carbon compound taken into the body, and presumably the location of the radioactive atom within the molecule (Taylor, 2004). Internally deposited 14C-labelled compounds have shown residence times varying from a few hours to several months in human volunteers (Stather et al., 1981; Stenström et al., 1996; Taylor, 2004). The distribution of radiocarbon in the body, and the fractions of ingested or inhaled activity lost by exhalation, urinary excretion, and faecal excretion also depend on the nature of the carbon compound taken into the body. (120) Variation in the biokinetics of carbon compounds is illustrated in Table 3.4, which is based on a review of the literature and a biokinetic and dosimetric analysis of the collected data (Taylor, 2004, 2007). The relative dose estimates represent the effective dose coefficient derived from the compound-specific information, divided by the effective dose coefficient based on a generic biokinetic model for carbon introduced in Publication 30 (ICRP, 1981). That model assumes that internally deposited carbon is distributed uniformly in the body and removed with a half-time of 40 d (ICRP, 1981). The 7 d retention values and relative dose estimates given in Table 3.4 are rough estimates in some cases, and the effective dose estimates are based on tissue weighting factors that have since been replaced (ICRP, 2008). Nevertheless, the data demonstrate the large differences in the biokinetics of different carbon compounds in the body, and, as a result, a wide variation in radiation dose per intake of carbon compounds for a given mode of intake. Retention of 14C in the human body at 7 d, and relative effective dose estimates for intake of various [14C]-labelled compounds, as estimated by Taylor (2004, 2007) on the basis of a review of biokinetic models and data for carbon. IV, intravenous. Multiple of effective dose based on the International Commission on Radiological Protection’s generic model for carbon introduced in Publication 30 (ICRP, 1981). Estimates based on data for rats.

3.2.3.2. Biokinetic models for systemic carbon
(121) In this publication, compound-specific biokinetic models are applied to carbon that reaches the systemic circulation as carbon monoxide, carbon dioxide, bicarbonate, or methane. A common model is applied to carbon dioxide and bicarbonate. A generic systemic model for carbon is applied to unspecified forms of carbon. For example, the generic model is used to develop dose coefficients for inhalation of particulate forms of carbon described as Type F, Type M, or Type S material.
(a) Inhaled carbon monoxide
(122) Inhaled carbon monoxide diffuses readily across the membranes of the AI region of the lung and enters the pulmonary blood, where it is bound to haemoglobin (ICRP, 1987). It is released from haemoglobin and removed from the body in expired air over a period of hours. (123) The model for inhaled carbon monoxide used in this publication is the model applied in Publication 30 (ICRP, 1981) and Publication 71 (ICRP, 1995). It is assumed that 40% of inhaled carbon monoxide is absorbed instantly into blood and bound to haemoglobin, and 60% is exhaled instantly. Carbon monoxide is assumed to be lost from blood to the environment via the lungs with a biological half-time of 200 min (Glass et al., 1968; Peterson and Stewart, 1970).
(b) Inhaled carbon dioxide
(124) Inhaled carbon dioxide is transferred rapidly across the alveolar membrane into blood (Guyton and Hall, 2000). Carbon dioxide is also formed in the body during the metabolism of organic substances. As most of the absorbed or internally produced carbon dioxide is converted to bicarbonate after entering the blood (Guyton and Hall, 2000), data from metabolic studies involving intravenous injection of [14C]bicarbonate provide information on the systemic biokinetics of carbon inhaled as carbon dioxide. (125) The model for inhaled carbon dioxide introduced in Publication 30 (ICRP, 1981) and applied in Publication 68 (ICRP, 1994) and Publication 71 (ICRP, 1995) depicts whole-body retention of carbon as the sum of three exponential terms:
(126) The model applied in this publication to carbon inhaled as carbon dioxide, and, more generally, to carbon reaching the blood as carbon dioxide or bicarbonate, is a modification of a model proposed by Leggett (2004). The structure of the modified model is shown in Fig. 3.1. Parameter values are listed in Table 3.5. The modifications to the original model were made to simplify implementation of the model by reducing the total numbers of compartments and pathways, and improve predictions of the long-term urinary excretion rate by including additional phases of transfer from soft tissues to the urinary excretion pathway. The modified model adds a blood compartment (Blood 2 in Fig. 3.1) and some paths of movement of carbon to the original model, but simplifies the original model overall by eliminating compartments and pathways depicting rapid exchange of activity between blood and peripheral compartments. (127) In the present model, absorbed carbon is assigned to Blood 1. Activity leaves Blood 1 at a rate of 100 d−1 (t½ = 10 min), with 60% of the outflow assigned to ST0, 1.8% to ST1, 0.3% to ST2, 0.44% to ST3, 0.15% to bone surface, 0.01% to bone volume, 36.2% to excreta through exhalation, 0.3% to excreta via skin, 0.65% to bladder contents, and 0.15% to right colon contents. Removal half-times from ST0, ST1, ST2, and ST3 are 20 min, 0.5 d, 3 d, and 40 d, respectively. It is assumed that 4% of outflow from ST1, ST2, and ST3 enters Blood 2, and all other outflow from the four soft tissue compartments returns to Blood 1. Activity transfers from Blood 2 to the urinary bladder contents at a rate of 1000 d−1 (t½ = 1 min). Based on estimates of the relative masses of trabecular and cortical bone replaced per unit time in an adult human, 60% of carbon entering bone is assigned to trabecular bone, and 40% is assigned to cortical bone. The trabecular and cortical bone surface compartments are assumed to lose carbon to Blood 1 with a half-time of 40 d. The bone volume compartments are assumed to lose carbon to Blood 1 at the rate of bone turnover, which differs for trabecular and cortical bone. (128) Total-body retention of carbon following acute input of carbon dioxide or bicarbonate into blood based on the present model agrees closely with predictions based on the original model (Leggett, 2004). Also, in agreement with the original model, the present model predicts that exhalation, urinary excretion, faecal excretion, and loss through skin accounts for 96.8%, 2%, 0.4%, and 0.8%, respectively, of the total loss of activity from the body over an extended period. The present model predicts slower accumulation of activity in bone than the original model, but the two models predict similar levels of activity in bone beyond a few days after acute input of activity to blood. For example, the present model predicts that bone contains 0.41% of intake at 1 d, 0.36% at 10 d, and 0.098% at 100 d after intake, compared with predictions of 0.89% at 1 d, 0.38% at 10 d, and 0.096% at 100 d based on the original model. In view of the uncertainty in the early distribution of radiocarbon in bone, a relatively long residence time of carbon on bone surface (40 d) is assigned in the original model as a dosimetrically cautious measure. Transfer coefficients for the systemic model used in this publication for radiocarbon assumed to reach blood as carbon dioxide or bicarbonate. ST, soft tissue. *36.2 d−1 in expired air and 0.3 d−1 via skin. Structure of the systemic model used in this publication for carbon taken into the body as carbon dioxide or bicarbonate [modification of a model by Leggett (2004)]. ST, soft tissue.
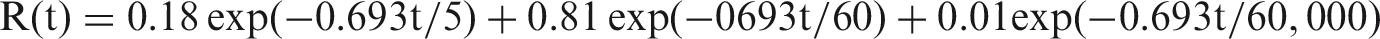

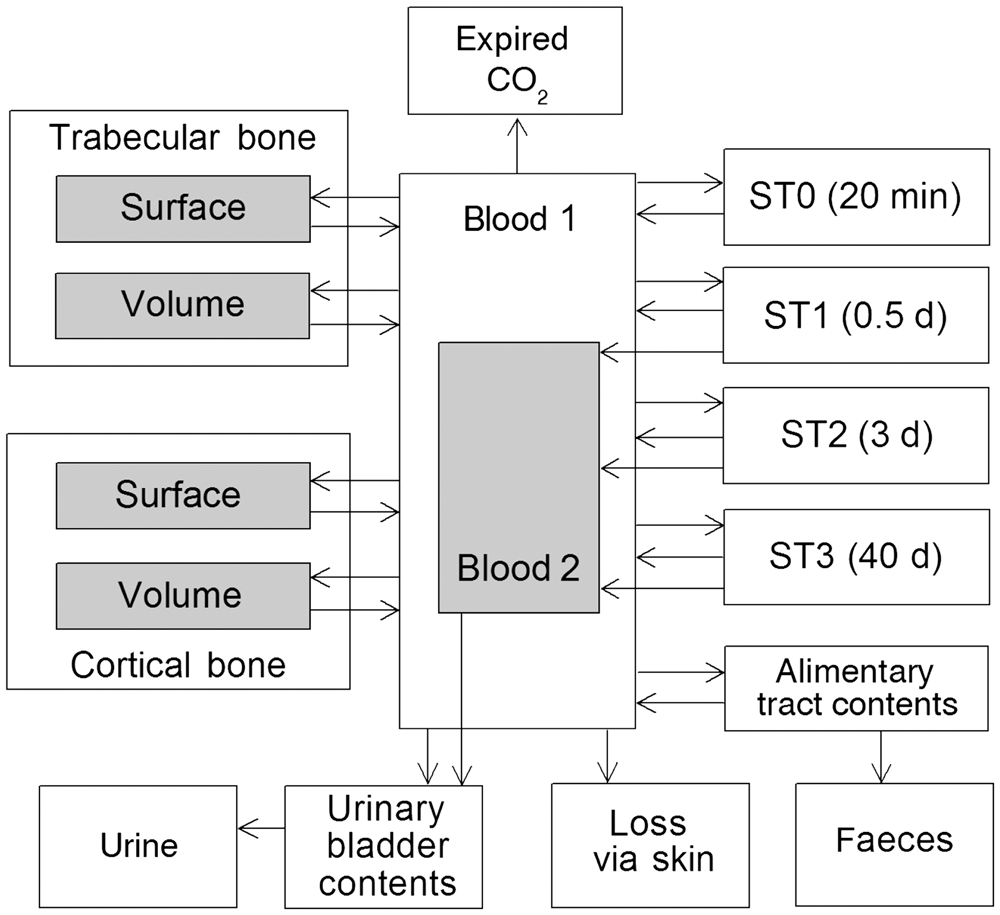
(c) Generic model for systemic carbon
(129) In Publication 30 (ICRP, 1981) and Publication 68 (ICRP, 1994), a generic biokinetic model is applied to 14C-labelled compounds for which specific biokinetic data are not available. It is assumed in that model that inhaled or ingested 14C-labelled compounds are distributed instantly and uniformly throughout all organs and tissues of the body, where they are retained with a biological half-time of 40 d. The half-time of 40 d is based on balance considerations, assuming daily carbon intake of 0.3 kg and a carbon pool of mass 16 kg in Reference Man (ICRP, 1975):
(130) For general radiological protection purposes, a generic biokinetic model proposed by Manger (2011) (but with rounded parameter values) is applied in this publication to radiocarbon absorbed into blood following intake in forms other than carbon monoxide, carbon dioxide, bicarbonate, or methane. The model is less conservative than the generic model introduced in Publication 30 (ICRP, 1981), but still accounts for the possibility that a dosimetrically significant portion of absorbed radiocarbon may be retained in the body for an extended period. Based on its design and on comparison of dose estimates with biokinetic models for a number of specific forms of carbon, the generic model adopted for use in this publication seems more likely to overestimate than underestimate dose per intake of 14C in the workplace. (131) The generic model structure and its connections to the respiratory and alimentary tract models and urinary bladder are shown in Fig. 3.2. Baseline transfer coefficients for systemic pathways are listed in Table 3.6. (132) The generic model is based on consideration of retention times and rates of loss along specific excretion pathways identified in published studies of 14C-labelled carbon compounds. The model was designed with the goal of providing cautious but not unnecessarily conservative estimates of dose per intake of unknown forms of radiocarbon, as judged from published biokinetic data for carbon compounds. It was also considered that the model should be adaptable to case-specific information, such as measurement of the rates of urinary excretion and exhalation of activity following exposure to a carbon compound in the workplace. (133) The excretion pathways addressed in the model are urinary and faecal excretion and exhalation. Three systemic compartments are used to represent blood, relatively short-term retention in systemic tissues, and relatively long-term retention in systemic tissues. The short-term compartment represents losses with a half-time of a few days, which typically accounts for most of the loss of the label from the body as indicated by published studies of different carbon compounds. The long-term compartment depicts the longer removal half-times depicted in several models for specific carbon compounds. This long-term retention is generally associated with adipose tissue in these models. The carbon dioxide and bicarbonate model defined in Fig. 3.1 and Table 3.5 is included as a submodel that describes the fate of labelled carbon dioxide produced in systemic tissues by metabolism of the initial form of carbon that reaches blood. Carbon dioxide produced in systemic tissues is assumed to move instantly to Blood 1 in the carbon dioxide model (Fig. 3.1). (134) For the case of ingested radiocarbon, activity moves through the alimentary tract as described in the Human Alimentary Tract Model, and is nearly completely (99%) absorbed to blood from the small intestine contents. The transfer rate from blood to all destinations combined is 3 d−1. It is assumed that 50% of the outflow from blood goes to the urinary bladder contents, 40% to the short-term systemic compartment, and 10% to the long-term systemic compartment. The removal half-time from the short-term systemic tissue compartment is 3 d. Outflow from this compartment is divided as follows: 40% returns to blood, 30% is secreted into the right colon contents and is subsequently excreted in faeces, and 30% moves to Blood 1 in the carbon dioxide model (Fig. 3.1). Carbon entering the long-term retention compartment is assumed to be metabolised slowly to carbon dioxide, which moves to Blood 1 in the carbon dioxide model with a half-time of 70 d. (135) For the case of inhaled radiocarbon, activity enters the HRTM and is absorbed to blood or transported to the alimentary tract over time. Activity moving from the HRTM to blood or to the alimentary tract is treated as described above for the ingestion case. (136) The baseline transfer coefficients for the systemic pathways (Table 3.6) were determined by fitting average excretion rates determined in studies involving administration of different carbon compounds. Average fractional excretion along the major excretion pathways was estimated as 0.59 for urine (range 0.01–1.00), 0.24 for exhalation (range 0–0.95), and 0.17 for faeces (range 0–0.99) (Baker et al., 1954; Fukushima et al., 1954; Hellman et al., 1954, 1955; Berlin and Tolbert, 1955; Migeon et al., 1956; Sandberg and Slaunwhite, 1957; Fine et al., 1962; Crawley, 1977; Crawley and Haines, 1978; Stather et al., 1981; ICRP, 1987, 1998; Thierens et al., 1994; Eriksson et al., 1998; Webber et al., 2003). Up to three phases of urinary excretion were determined in different studies, depending, in part, on the length of the observation period (Fukushima et al., 1954; Berlin and Tolbert, 1955; Hellman et al., 1955; Migeon et al., 1956; Sandberg and Slaunwhite, 1957; Crawley, 1977; ICRP, 1987; Kramer et al., 1996; Eriksson et al., 1998; Webber et al., 2003). The average half-time was 0.43 d (range 0.07–1.0 d) for the fastest phase, 3.3 d (range 0.29–7.0 d) for the intermediate phase, and 70 d for the slowest phase (range 33–620 d). The fast phase typically represented 85% or more, and the intermediate phase represented approximately 5% of total urinary excretion. In the generic model, the fast phase of loss is represented mainly by transfer from blood to the urinary bladder contents, and removal half-times and pathways from the two systemic tissue compartments are used to account for the intermediate and long-term phases of loss inferred from the published data. (137) Cumulative activity of intravenously injected 14C in the body based on the generic model was compared with predictions of published biokinetic models for benzene, glycocholic acid, triolein, urea, inulin, and xylose. These compounds represent some of the longest and some of the shortest retention times that have been determined for carbon compounds. Comparison was also made with the generic systemic model for carbon used in Publication 71 (ICRP, 1995). Results of the comparison are shown in Table 3.7. Transfer coefficients for the generic model for systemic carbon.
*
In addition to transfer coefficients in the CO2 model (Table 3.5). Initially enters Blood 1 in the CO2 model. Comparison of cumulative activity in the body as predicted by the generic model and by existing models for various specific carbon compounds following intravenous injection of 14C. Assumes uniform distribution in body and biological half-time of 40 d. Generic structure for radiocarbon-labelled substances. HRTM, Human Respiratory Tract Model; HATM, Human Alimentary Tract Model.



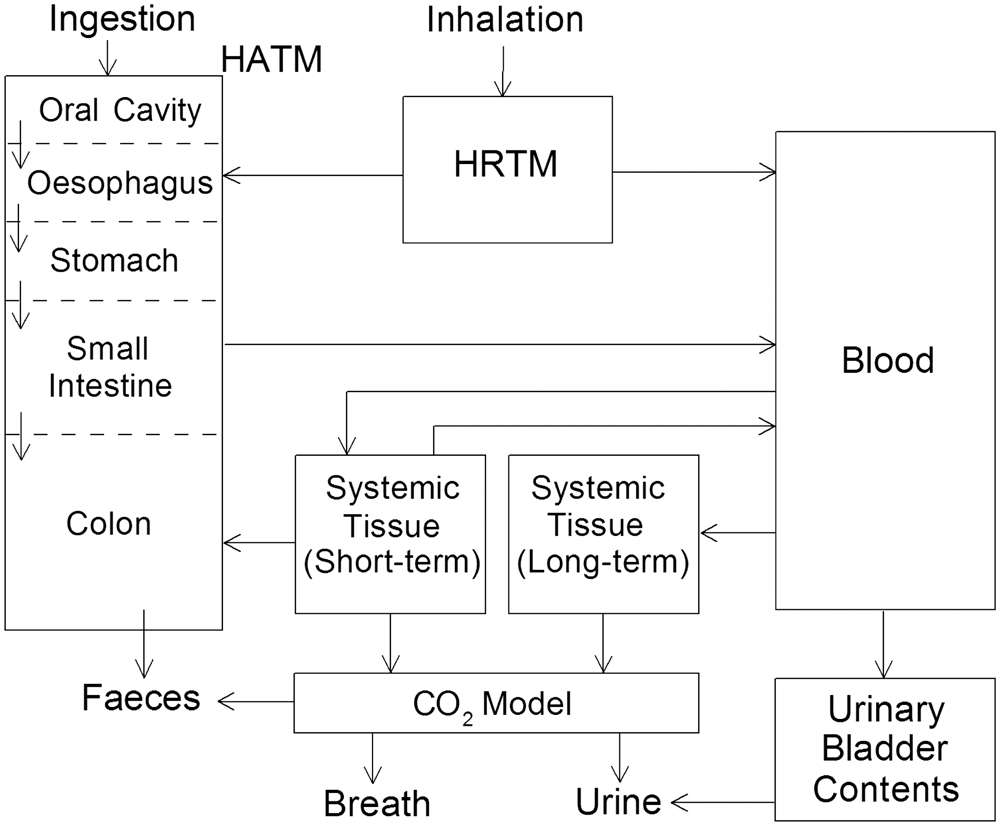
(d) Inhaled methane
(138) Oxidation of a small portion of methane that reaches the systemic circulation was demonstrated in sheep by trapping 14CO2 following arterial infusion of 14C-labelled methane. Most of the infused methane was exhaled unchanged (Dougherty et al., 1967). In rats exposed to 14C-labelled methane mixed with breathing air, an estimated 72% of the label that reached blood was exhaled in the first hour (Carlisle et al., 2005; Didychuk et al., 2014). Measurements over 14 d post inhalation indicated that most of the retained 14C was organically bound. The preponderance of organically bound label was removed from the body with a half-time of approximately 1.3 d, but a few percent was removed with a half-time of 7 d or longer. (139) The model for systemic methane used in this publication is a modification of the generic model for carbon described above. The generic model structure (Fig. 3.2) is modified by addition of a blood compartment that receives methane absorbed from the respiratory tract. It is assumed that the carbon label leaves this blood compartment with a half-time of 10 min (removal rate of 100 d−1), with 70% removed in expired air and 30% entering the short-term tissue compartment of the generic model structure. The parameter values of the generic model for carbon are modified for application to methane to depict faster removal from the short-term tissue compartment, lower transfer from the short-term tissue compartment to the carbon dioxide model, and lower transfer from blood to the long-term tissue compartment than predicted by the transfer coefficients listed in Table 3.6. It is assumed for application to methane that carbon is removed from the short-term compartment in Fig. 3.2 with a half-time of 1 d, with 60% going to the blood compartment of the generic model structure, 30% going to the right colon contents, and 10% entering the blood compartment of the carbon dioxide model. It is assumed that carbon leaves the blood compartment of the generic model structure at a rate of 3 d−1, with 3% going to the long-term tissue compartment, 47% to the short-term retention compartment, and 50% to the urinary bladder contents. As in the generic model, carbon transfers from the long-term tissue compartment to the blood compartment of the carbon dioxide model with a half-time of 70 d. The rate of production of carbon dioxide from absorbed methane based on these modified parameter values is reasonably consistent with the rate observed in sheep in the study by Dougherty et al. (1967). The model predicts whole-body retention of carbon that is moderately higher than observed in rats over the first 14 d after inhalation of labelled methane (Didychuk et al., 2014).
3.3. Individual monitoring
(140) 14C intake is generally monitored though measurement of the activity excreted in urine. The most common method of analysis is liquid scintillation counting (Table 3.8). Measurement of activity in exhaled breath may be used for 14C-labelled organic material metabolised to carbon dioxide, but there is no information on detection limits or routine use of the technique. Monitoring techniques for 14C.

3.4. Dosimetric data for carbon
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 14C compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 14C in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Daily urinary excretion of 14C following inhalation of 1 Bq carbon dioxide.
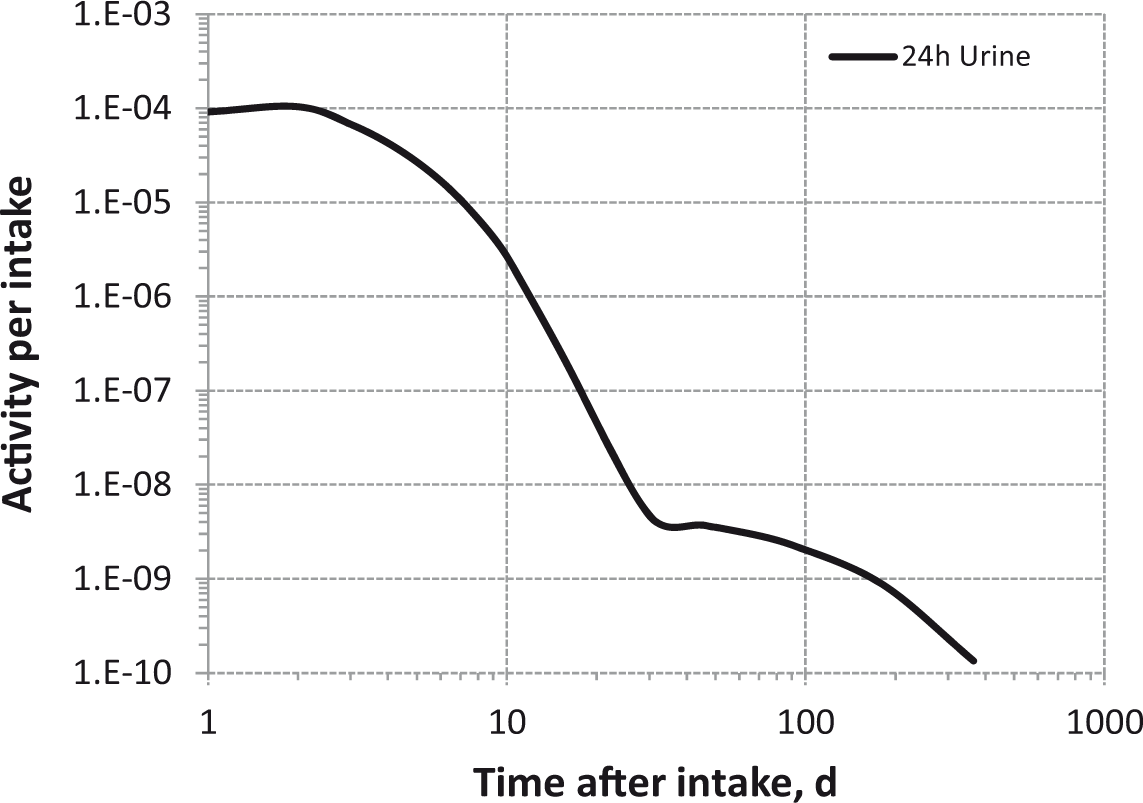
Daily urinary excretion of 14C following inhalation of 1 Bq methane.
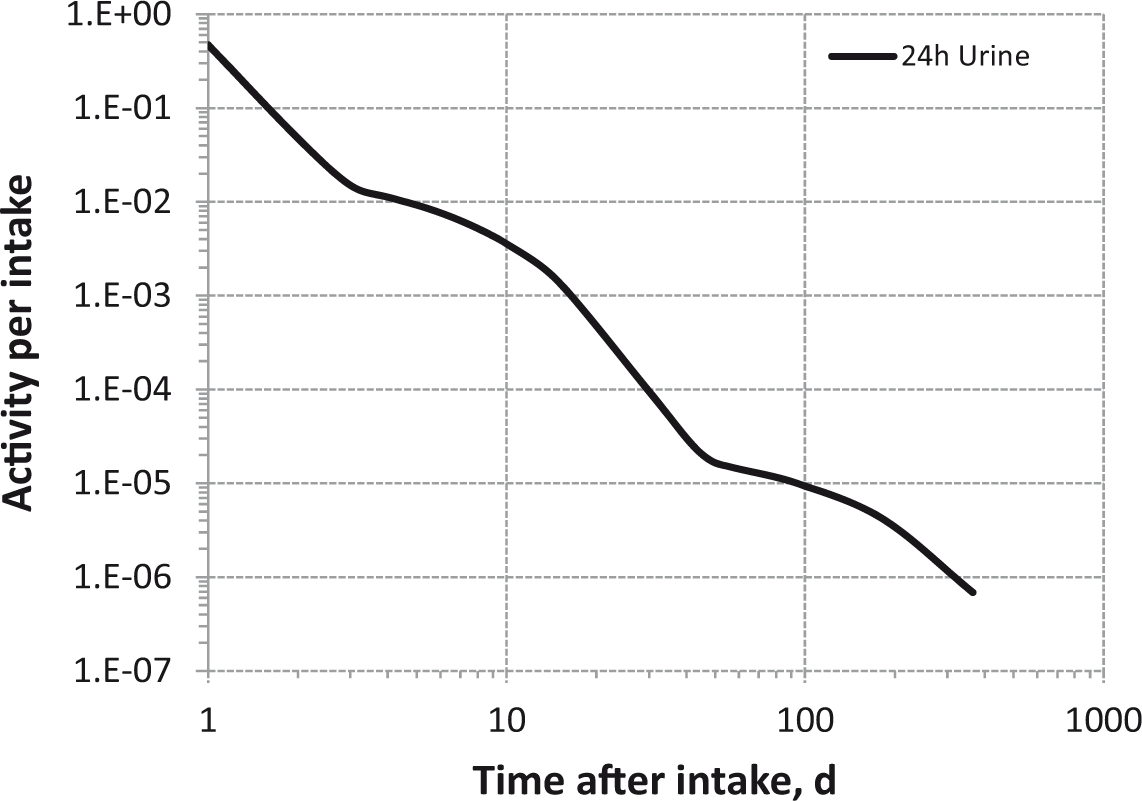
Daily urinary excretion of 14C following inhalation of 1 Bq unspecified organic compounds.

Daily urinary excretion of 14C following inhalation of 1 Bq Type F (barium carbonate).

Daily urinary excretion of 14C following inhalation of 1 Bq Type F.

Daily urinary excretion of 14C following inhalation of 1 Bq Type M.
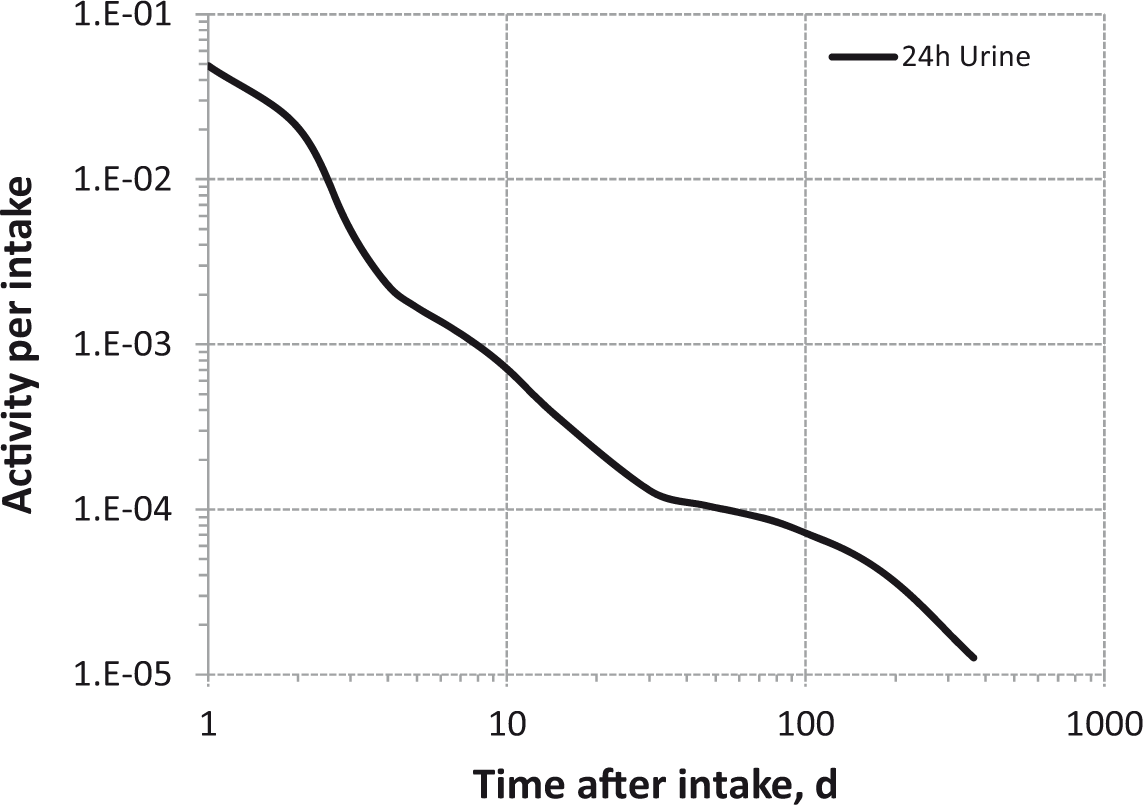
Daily urinary excretion of 14C following inhalation of 1 Bq Type S.
3.5. References
4. PHOSPHORUS (Z = 15)
4.1. Chemical forms in the workplace
(141) Phosphorus is a non-metal that occurs in numerous oxidation states, with V being the most common. It is able to react chemically with many other elements to form organic and inorganic compounds. The most common phosphorus compounds in solution are phosphates, which occur in different forms depending on the pH (e.g. HPO42−, PO43−). Phosphorus may be encountered in industry in a variety of chemical forms, including the oxide, hydride, halide, phosphate, and phosphide, and also organophosphorus and organophosphate. (142) 32P and 33P are used routinely to produce radiolabelled compounds. Table 4.1 shows the isotopes of phosphorus addressed in this publication. Isotopes of phosphorus addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B-, Beta-minus decay

4.2. Routes of intake
4.2.1. Inhalation
(143) Information on absorption from the respiratory tract is available from a few experimental studies on the behaviour of inhaled phosphorus. However, most of it relates to phosphates for which the cation, rather than the phosphorus itself, was radiolabelled. (144) Absorption parameter values and types, and associated fA values for particulate forms of phosphorus are given in Table 4.2. Absorption parameter values for inhaled and ingested phosphorus. It is assumed that the bound state can be neglected for phosphorus, i.e. fb=0. The values of sr for Type F, M, and S forms of phosphorus (1 d−1) are element-specific. Materials (e.g. sodium phosphate) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied; i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of phosphorus (0.8). These calculated values are not rounded for purposes of consistency. Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.8) for ingestion of the radionuclide.

4.2.1.1. Particulate materials
(a) Sodium phosphate
(145) Schiessle (1956, 1957) followed retention of 32P in guinea pigs for 28 d after inhalation of Na3(32PO4). Most of the ILD was absorbed over this period, but not very rapidly; there was little transfer to blood at the end of the 25 min exposure, approximately 40% ILD remained after 1 d, and 9% ILD remained after 28 d. (The author noted that there was greater uptake to bone compared with liver than after intravenous injection of 32P.) Specific parameter values were estimated here (i.e. by the Task Group) to be: fr = 0.8, sr = 1 d−1 (t½∼17 h), and ss = 0.02 d−1 (t½∼3 d), consistent with assignment to Type F. Although specific parameter values for sodium phosphate based on in-vivo data are available, they are not adopted here because inhalation exposure to it is so unlikely. Instead, sodium phosphate is assigned to Type F. However, the data are used as the basis of the default rapid dissolution rate for phosphorus.
(b) Phosphates labelled with isotopes of other elements
(146) For details relating to zinc and yttrium, refer to the sections dealing with the labelling radioelement. Details are given here for stannic phosphate because inhalation of tin has not yet been covered elsewhere in the OIR report series.
Zinc phosphate [Zn3(PO4)2] (see Section 9.2.1)
(147) The results of a study of inhalation of 65Zn3(PO4)2 by dogs were consistent with assignment to Type M.
Yttrium phosphate (YPO4) (see Section 11.2.1)
(148) The results of a study of inhalation of 91YPO4 by dogs were consistent with assignment to Type M. The authors (Newton et al., 1971) noted that following both inhalation and gavage of 91YPO4, the ratio of deposition in the skeleton to that in the liver was lower than following inhalation of other forms of 91Y.
Stannic phosphate
(149) Morrow et al. (1968) followed lung clearance of 113Sn for 7 d after inhalation of 113Sn3(PO4)2 by dogs and rats, but few details were given. Lung retention in dogs was described by a two-component exponential function with half-times of 2 d (28%, clearance rate 0.35 d−1) and 59 d (clearance rate 0.012 d−1), giving predicted lung retention at 30 d and 180 d of 50% and 8% ILD, respectively, and indicating Type M behaviour.
4.2.1.2. Rapid dissolution rate for phosphorus
(150) The value of sr estimated for sodium phosphate above, 1 d−1, is applied here to all Type F forms of phosphorus. As it is lower than the general default value of 3 d−1 for Type M and S materials, it is also applied to Type M and S forms of phosphorus.
4.2.1.3. Extent of binding of phosphorus to the respiratory tract
(151) Evidence from the sodium phosphate study outlined above suggests that there is probably little binding of phosphorus. It is therefore assumed that the bound state can be neglected for phosphorus, i.e. fb = 0.0.
4.2.2. Ingestion
(152) Intake of phosphorus is mainly through the diet in the form of inorganic phosphate and phosphorus-containing biomolecules such as nucleic acids and phospholipids. According to Eakins et al. (1966), fractional absorption of 32P from the gastrointestinal tract is approximately 0.75 when it is ingested as phosphate under normal dietary conditions, and is above 0.9 while fasting. The Food and Nutrition Board of the US Institute of Medicine reports absorption values ranging from 0.55 to 0.70 in adults and from 0.65 to 0.90 in infants and children (FNB, 1997). (153) Animal studies have shown that maximal absorption of phosphate occurs in the ileum for mice, and in the duodenum and the jejunum for rats (Radanovic et al., 2005; Stauber et al., 2005; Marks et al., 2006). Absorption of phosphorus can be reduced by the simultaneous administration of unusually high levels of calcium (FNB, 1997). According to recent findings, the intestinal transport process of inorganic phosphate is known to occur by both a sodium-independent, non-saturable process and via an active process mediated by sodium-phosphate cotransporters (Katai et al., 1999a). Studies by Katai et al. (1999b) and Kirchner et al. (2008) with rats showed that transporter-mediated absorption of inorganic phosphate is inhibited by nicotinamide and fructose, respectively. Intestinal sodium-dependent phosphate absorption was reduced significantly (reduction between 35% and 60%) in mice and rats with simulated inflammable bowel diseases (Chen et al., 2009). (154) In Publication 30 (ICRP, 1979), the recommended absorption value was 0.8 for all compounds of the element. This value is used here; that is, fA = 0.8 for all compounds.
4.2.3. Systemic distribution, retention, and excretion
4.2.3.1. Summary of the database
(155) Phosphorus represents approximately 1% of the weight of the human body. In adults, approximately 85% of the phosphorus is in bone, 9% is in muscle, and 6% is in the remaining tissues and fluids. Most of the phosphorus in blood is contained in the RBCs (Eakins et al., 1966; ICRP, 1975; Parfitt and Kleerekoper, 1980). (156) Intravenously administered radiophosphorus is distributed throughout the extracellular fluids within a few minutes. Kinetic analysis indicates that the rapidly exchangeable pool is larger than the extracellular pool, and thus presumably includes a portion of the intracellular phosphorus. Radiophosphate is incorporated rapidly into organic compounds in the body. The tissue turnover rate of phosphate, as measured by exchange of the tracer, depends on the rate of glycolysis of the tissue and is relatively low in resting muscle, intermediate in liver, and high in RBCs (Parfitt and Kleerekoper, 1980). (157) Following intravenous administration of 32P to healthy human subjects, the activity concentration in RBCs exceeded that in plasma by approximately a factor of three after 2 h, and by an order of magnitude after 6 h (Erf and Lawrence, 1941). The RBC concentration apparently peaked during the first few hours, and then declined roughly in parallel with the plasma disappearance curve (Fig. 4.1). (158) The rate of biological removal of 32P from the body varied widely in human subjects following intravenous injection of Na2H32PO4 (Erf et al., 1941; Hevesy, 1948; Weijer et al., 1962; Eakins et al., 1966). On average, approximately one-quarter of the administered amount was excreted in urine and faeces during the first 6 d, with urinary excretion generally representing more than 90% of total excretion. Average daily urinary excretion of activity as measured in four human injection studies is summarised in Fig. 4.2. (159) 32P was measured in samples of tissue collected at autopsy 2, 11, 18, and 19 d after the end of its therapeutic administration to four patients (A–D, respectively) terminally ill with leukaemia (Erf, 1941; Erf and Friedlander, 1941). The activity was administered either orally or intravenously as sodium phosphate over periods of approximately 73, 5, 65, and 1 d to Patients A–D, respectively. The 32P concentration was consistently elevated in liver. Concentrations in bone, spleen, kidney, and lymph nodes approached or slightly exceeded values for liver in some subjects. Relatively low concentrations were observed in brain, lung, and skeletal muscle, but skeletal muscle contained a substantial portion of total body activity due to its relatively large mass. Detailed autopsy measurements tabulated for Patient D also indicated relatively low concentrations (typically 10–50% of that in liver) in the gallbladder, urinary bladder, stomach, intestines, fat, bone marrow, pancreas, prostate, skin, thyroid, and other tissues. The investigators concluded that the relative concentrations of 32P in tissues did not depend on the type of leukaemia, age of the patient, mass and route of administered activity, or mass of the sodium phosphate carrier. On the basis of tissue masses for a reference adult male (ICRP, 2002), it is estimated that bone, liver, kidneys, muscle, and other soft tissue contained approximately 12–20%, 6–7%, 0.8–1.3%, 46–54%, and 19–24%, respectively, of whole-body 32P in these subjects. (160) The behaviour of phosphorus in bone resembles that of calcium. Rapid uptake of both elements occurs on all bone surfaces, with considerable variability in the uptake rate between different bones and different surfaces of the same bone. Within a period of hours or days, radioisotopes of phosphorus or calcium diffuse throughout bone volume. Both elements can penetrate into the interior of bone crystal. The exchangeable and non-exchangeable fractions of the total bone mineral are approximately the same for phosphorus and calcium (Neuman and Neuman, 1958; Parfitt and Kleerekoper, 1980). (161) As is the case for calcium, uptake of phosphorus is considerably greater in forming or growing bone than in mature bone. Phosphorus and calcium both show high concentrations in forming osteons (Parfitt and Kleerekoper, 1980). In rats injected intraperitoneally with 32P, skeletal uptake decreased with increasing age at injection, from approximately 90% of the injected amount at 15 d of age to approximately 17% at 170 d of age (Bonner, 1948). (162) Stather (1974) compared the distribution and retention of 32P and the alkaline earths 45Ca, 85Sr, and 133Ba in the mouse skeleton. At 24 h after intraperitoneal injection into 8-week-old mice, the distribution of the four radionuclides was virtually the same throughout the skeleton, but skeletal content as a percentage of injected activity differed from one radionuclide to another: 32P, 21.6%; 45Ca, 61.5%; 85Sr, 37.3%; and 133Ba, 48.8%. The skeletal burden represented approximately 37% of whole-body 32P compared with approximately 90% of whole-body 85Sr. (163) Bauer and Carlsson (1955) compared the uptake of 32P and 45Ca by bone (tibial shaft) and incisors in adult rats over the first 5 d after simultaneous subcutaneous injection of these radionuclides. The percentage of the administered 45Ca found in bone was consistently approximately 2.3 times the percentage of administered 32P in the same bone samples at corresponding times after administration. The ratio of uptake of 45Ca and 32P was approximately the same for incisors as for bone. (164) During the first 2 d after administration of 32P as phosphate to rats by various routes, most of the absorbed activity was found in the muscle and bone (Cohn and Greenberg, 1938). Among the viscera, liver showed the highest activity concentration, followed by the stomach plus small intestine. (165) Following intraperitoneal administration of 32P to rats, the skeleton, muscle, and liver contained approximately 45%, 19%, and 9.2% (corrected for radioactive decay) of the administered amount, respectively, at 1 d; 42%, 11%, and 1.3%, respectively, at 7 d; and 34%, 16%, and 1.8%, respectively, at 14 d (Friedell and Storaasli, 1949). Total excretion amounted to 46% of the administered amount over the first 2 weeks. (166) At 23 h–7 d after oral, intraperitoneal, or subcutaneous administration of 32P to rats, the skeleton, muscle, liver, and kidneys contained 31–58%, 34–55%, 4.7–12%, and 1.0–2.6%, respectively, of the retained activity (Ely, 1940). (167) Following oral administration of biologically incorporated 32P to rats, the peak contents of skeleton and liver were 13.5% and 3.6%, respectively, of the ingested amount. From 1 to 20 d, the liver content declined with a biological half-time of approximately 5 d (Kawin and Palmer, 1958). (168) Following intravenous injection of 32P into swine of different ages, highest concentrations in non-skeletal tissues at early times were found in bile, liver, kidneys, and thymus (Smith et al., 1951, 1952). Following intravenous injection of 32P into sheep of different ages, highest concentrations in non-skeletal tissues at early times were found in bile, thymus, liver, tongue, and kidneys (Smith et al., 1952). Distribution of activity in blood following intravenous administration of 32P to healthy human subjects. Data points from Erf and Lawrence (1941). Curves generated by the systemic model used in this publication. Daily urinary excretion of phosphorus following intravenous injection into human subjects [data summarised by Eakins et al. (1966)]. The curve shows predictions of the systemic biokinetic model for phosphorus used in this publication.
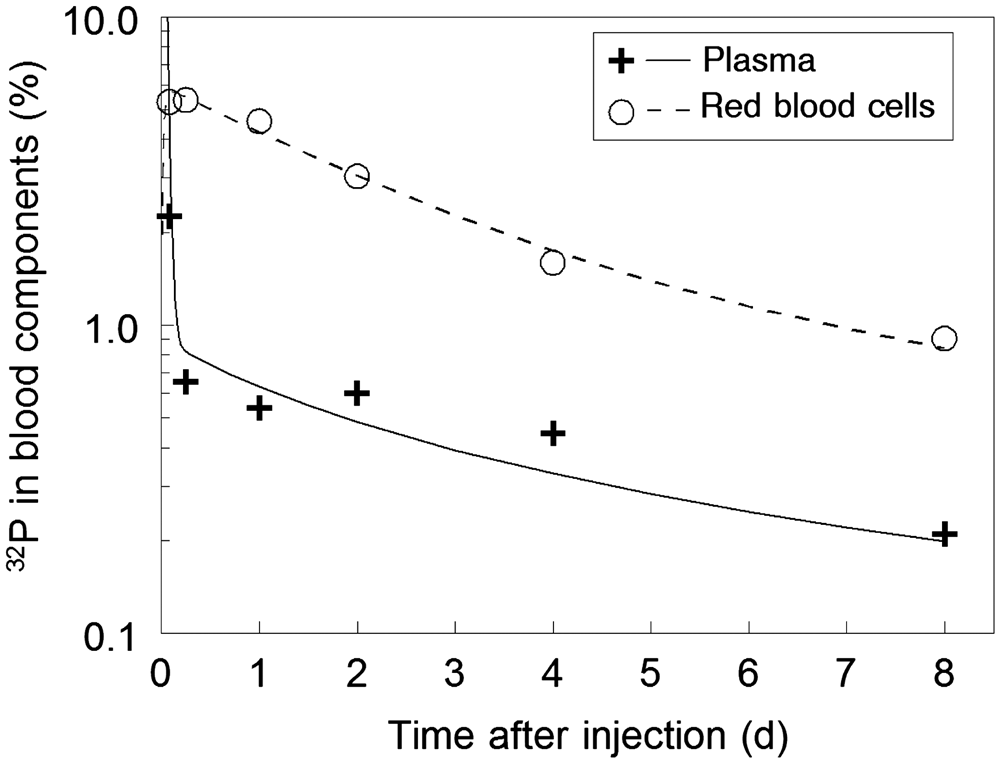
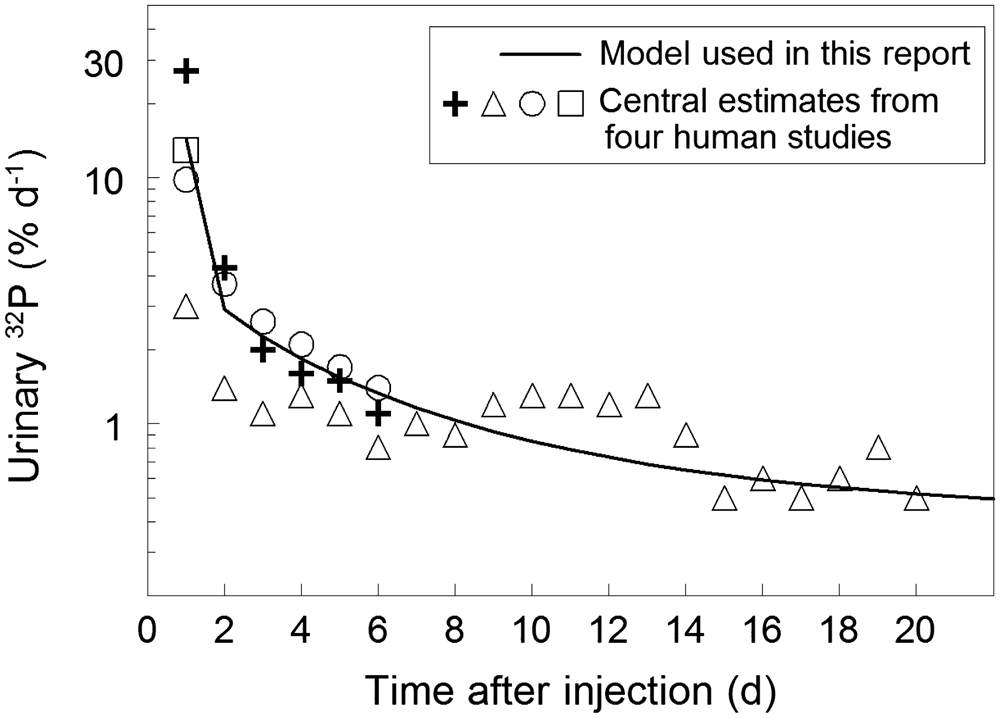
4.2.3.2. Biokinetic model for systemic phosphorus
(169) Dyson (1966) proposed the compartment model for phosphorus shown in Fig. 4.3 for use in radiological protection. The flow rates are given in terms of the movement of stable phosphorus at equilibrium. It is assumed that 1 g of phosphorus is absorbed daily from dietary phosphorus. Absorbed phosphorus is removed from plasma with a half-time of 0.5 d. Outflow from plasma is divided as follows: 15% is lost in excreta, 30% deposits in bone, 15% enters cell fluids, and 40% enters other soft tissue components. Phosphorus is returned to plasma from cell fluids and other soft tissue components with half-times of 2 d and 19 d, respectively. The removal half-time from bone to plasma is not specified, but is assumed to be long compared with the radiological half-lives of radioisotopes of phosphorus. (170) The biokinetic model for systemic phosphorus used in Publication 30 (ICRP, 1979) and Publication 68 (ICRP, 1994) is based on the model proposed by Dyson (1966). As implemented in Publication 68 (ICRP, 1994), activity leaves blood with a half-time of 0.5 d and is distributed as follows: 15% goes to excretion pathways; 30% goes to mineral bone; and 55% is distributed uniformly in remaining tissues. These remaining tissues are divided into two compartments, one receiving 15% of activity leaving blood and having a removal half-time of 2 d, and the other receiving 40% and having a half-time of 19 d. Activity is retained permanently in bone. A urinary:faecal excretion ratio of 9:1 is assigned. A phosphorus isotope entering bone is assigned to bone surface if the half-life is ≤15 d, and otherwise is assigned to bone volume. (171) The structure of the biokinetic model for systemic phosphorus used in this publication is shown in Fig. 4.4. This is a variation of the generic model structure for bone-volume-seeking radionuclides used in the OIR series. (172) Parameter values are listed in Table 4.3. These values are set for consistency with the observed behaviour of radiophosphorus in human subjects and laboratory animals, and the distribution of stable phosphorus in adult humans including: the blood kinetics (Fig. 4.1), urinary excretion rate (Fig. 4.2), and cumulative faecal excretion of 32P in healthy human subjects during the early days or weeks after its intravenous administration; the distribution of 32P in leukaemic human subjects during the early days or weeks after intravenous or oral administration (Erf, 1941; Erf and Friedlander, 1941); the steady-state whole-body content and distribution of phosphorus in adult human subjects as determined mainly from autopsy measurements (ICRP, 1975; Iyengar et al., 1978); and the time-dependent distribution of 32P in laboratory animals following administration by various routes (e.g. Cohn and Greenberg, 1938; Ely, 1940; Friedell and Storaasli, 1949; Smith et al., 1951, 1952; Kawin and Palmer, 1958). (173) Phosphorus is assumed to leave blood plasma at a rate of 40 d−1, corresponding to a removal half-time of 25 min. The outflow from plasma is divided as follows: 6% to RBCs; 11% to the urinary bladder contents; 0.5% to the right colon contents; 20% to bone surface; 10% to Liver 1; 1% to a kidney compartment called ‘urinary path’; 0.35% to a kidney compartment called ‘other kidney tissue’; and the remaining 51.15% to other soft tissue. Phosphorus is removed from Liver 1 with a half-time of 0.5 d, with 25% going to Liver 2 and 75% returning to plasma. Phosphorus is removed from RBCs to plasma with a half-time of 1 d, removed from Liver 2 and other kidney tissue to plasma with a half-time of 20 d, and removed from urinary path to urinary bladder contents with a half-time of 1 d. Other soft tissue is divided into three compartments – ST0, ST1, and ST2 – representing fast, intermediate, and slow turnover, respectively. These compartments receive 25.45%, 25.45%, and 0.25% of outflow from plasma, respectively, and return activity to plasma with half-times of 2 d, 20 d, and 5 y, respectively. The biokinetics of phosphorus in the skeleton is assumed to be identical to that of calcium, including the division of deposited activity between cortical and trabecular bone surfaces. Fractions 0.445 and 0.555 of the deposited amount (8.9% and 11.1% of the amount reaching blood) are assigned to cortical and trabecular surfaces, respectively. The transfer coefficients describing translocation of phosphorus within the skeleton and return from skeletal compartments to blood plasma are taken from the ICRP’s systemic model for calcium (ICRP, 1995). Transfer coefficients (d−1) in the biokinetic model for systemic phosphorus. ST, soft tissue; RBC, red blood cells; Exch, exchangeable; Nonexch, non-exchangeable. ST0, ST1, and ST2 represent other soft tissues with fast, intermediate, and slow turnover, respectively. Compartmental model of the biokinetics of systemic phosphorus proposed by Dyson (1966). The flow rates are given in terms of daily transfers of stable phosphorus at equilibrium. T½, half-life. Structure of the model for systemic phosphorus used in this publication. Exch, exchangeable; Nonexch, non-exchangeable; RBC, red blood cells; ST, soft tissue.

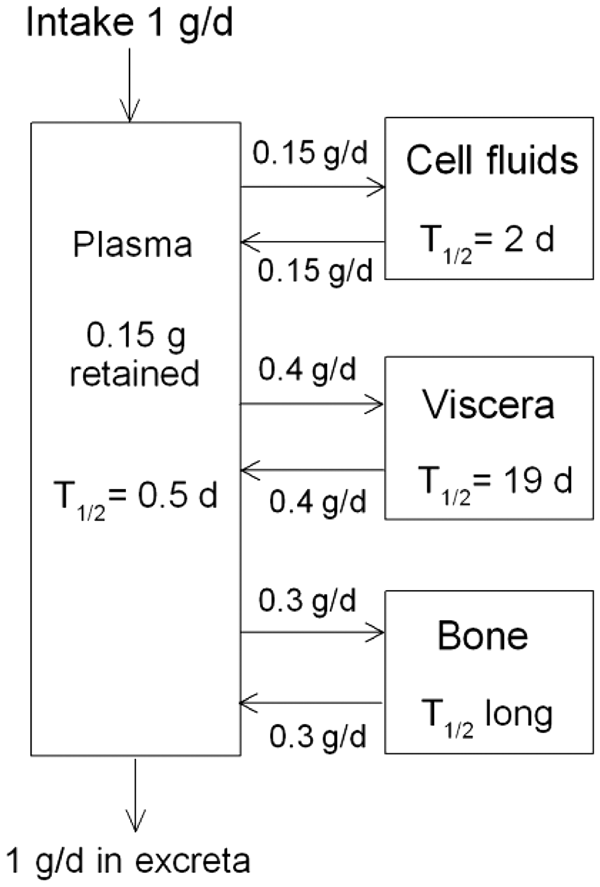

4.3. Individual monitoring
(174) 32P is a pure beta emitter. Monitoring of individuals is done through urine bioassay techniques, typically liquid scintillation (Table 4.4). Monitoring techniques for 32P.

4.4. Dosimetric data for phosphorus
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 32P compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 32P in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.

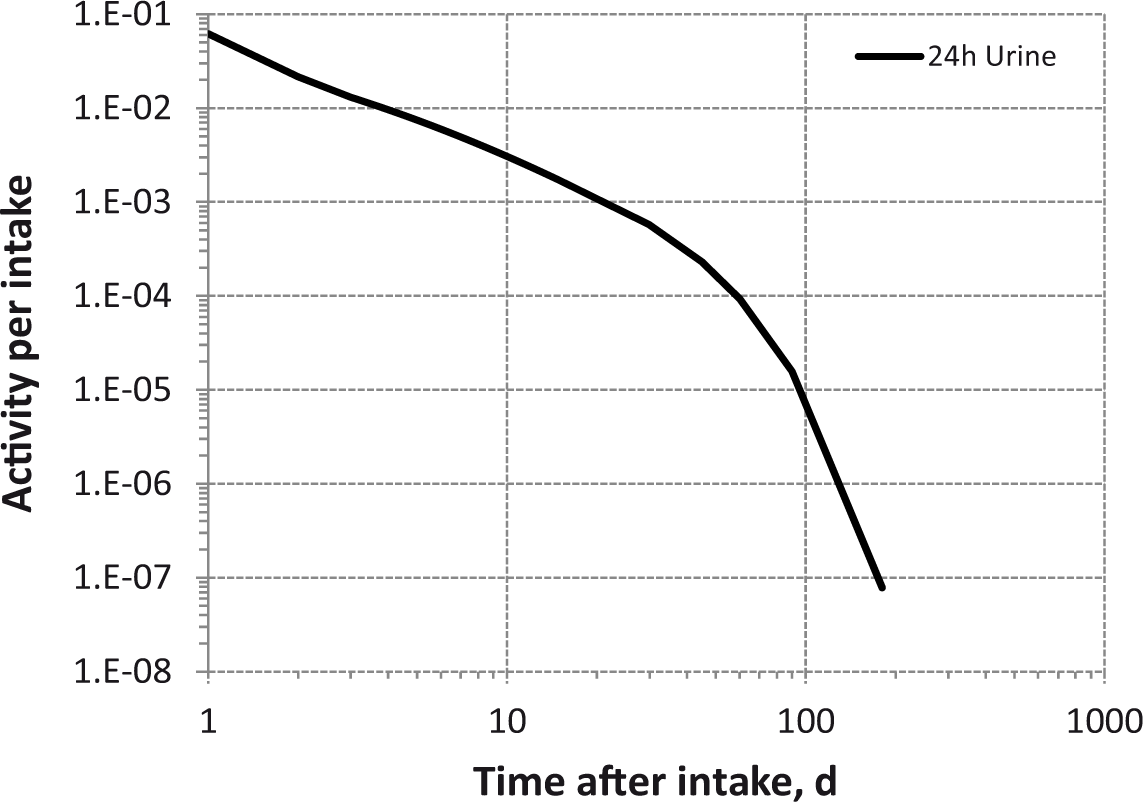
Daily urinary excretion of 32P following inhalation of 1 Bq Type F.
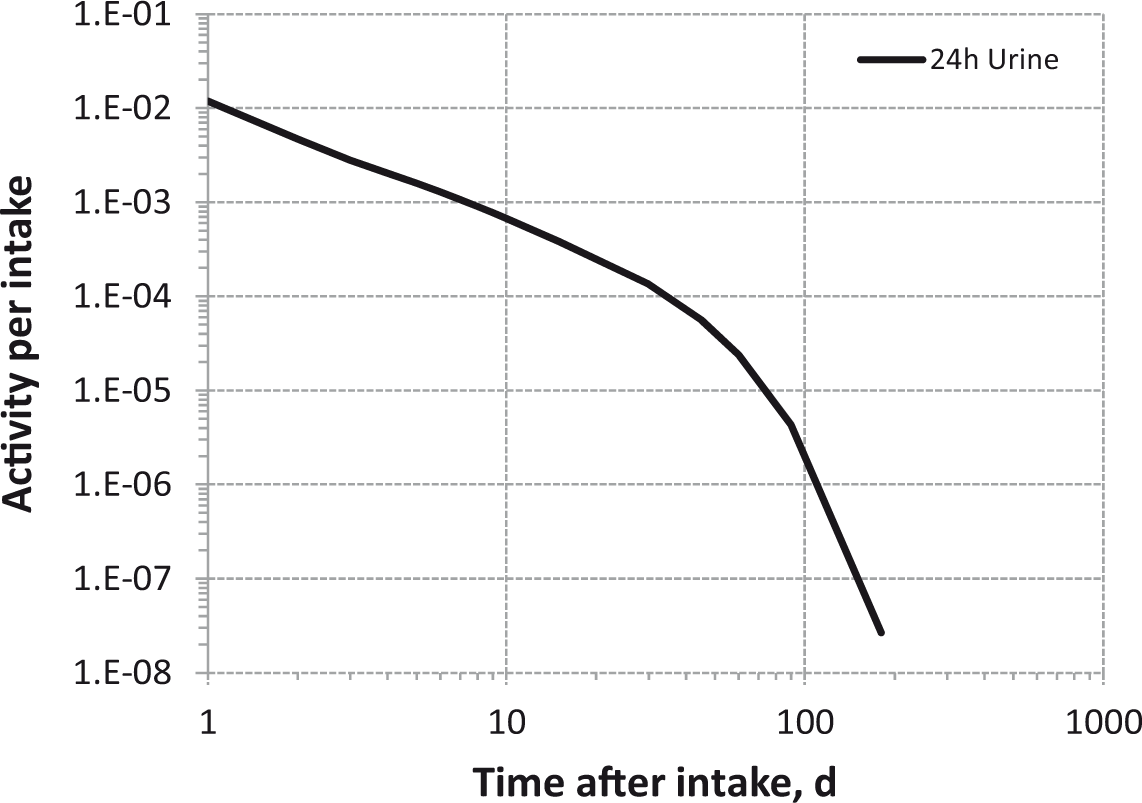
Daily urinary excretion of 32P following inhalation of 1 Bq Type M.

Daily urinary excretion of 32P following inhalation of 1 Bq Type S .
4.5. References
5. SULPHUR (Z = 16)
5.1. Chemical forms in the workplace
(175) Sulphur is a non-metal that occurs mainly in oxidation states -I, -II, II, IV, and VI. It is able to react chemically with many other elements, forming organic and inorganic compounds. The most common sulphur compound in solution is sulphate (SO42−). 35S is the only isotope of radiological significance that may be encountered in the workplace. Table 5.1 shows the isotopes of sulphur addressed in this publication. Sulphur may occur in industry in a number of different chemical forms, including the gases hydrogen sulphide (H2S), sulphur dioxide (SO2), and sulphur trioxide (SO3); fluids or their vapours such as carbon disulphide (CS2); and solid compounds such as barium sulphate (BaSO4). In research laboratories, it can be present in a wide variety of compounds. Isotopes of sulphur addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B-, Beta-minus decay

5.2. Routes of intake
5.2.1. Inhalation
(176) Some information on absorption from the respiratory tract is available for inhaled gases of sulphur in man and in experimental animals. Most of the information available on inhaled particulate forms of sulphur relates to sulphates. (177) Absorption parameter values and types, and associated fA values for gas and vapour forms of sulphur are given in Table 5.2 and for particulate forms in Table 5.3. (178) Exposures to both gas/vapour forms and particulate forms of sulphur are common, and it is therefore recommended in the OIR series that 50% particulate and 50% gas/vapour should be assumed in the absence of site-specific information (ICRP, 2002). Deposition and absorption for gas and vapour forms of sulphur.
*
ET1, anterior nasal passage; ET2, posterior nasal passage, pharynx, and larynx; BB, bronchial; bb, bronchiolar; AI, alveolar-interstitial. For sulphur in unspecified gas or vapour form, the default option for gases and vapours is recommended: 100% total deposition in the respiratory tract; default distribution between regions;§ and Type F absorption. Percentage deposited refers to how much of the material in the inhaled air remains in the body after exhalation. Almost all inhaled gas molecules contact airway surfaces, but usually return to the air unless they dissolve in, or react with, the surface lining. For the forms of sulphur considered here, it is assumed that most, if not all, of the inhaled sulphur is absorbed into body fluids. Systemic model for inorganic sulphur, Section 5.2.3.2; systemic model for organic sulphur, Section 5.2.3.2. Default distribution between regions (20% ET2, 10% BB, 20% bb, and 50% AI). Absorption parameter values for inhaled particulate forms of sulphur and ingested sulphur.
*
Following uptake into body fluids, the systemic model for inorganic sulphur is used (see Section 5.2.3.2). It is assumed that the bound state can be neglected for sulphur, i.e. fb=0. The values of sr for Type F, M, and S forms of sulphur (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. caesium sulphate) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of sulphur (1.0). In the case of thorium sulphate, the thorium is assigned to Type M and the sulphur to Type F. Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the highest value for ingestion of the radionuclide (fA =1).


5.2.1.1. Gases and vapours
(a) Sulphur dioxide (SO2)
(179) In two human studies (Speizer and Frank, 1966; Andersen et al., 1974), approximately 85% of inhaled SO2 was deposited, all in the ET airways. In dogs, more than 95% of the inhaled gas was deposited in the ET airways during nose breathing and 50–90% during mouth breathing (Frank et al., 1967, 1969). A further study with dogs, in which the trachea was perfused with SO2, gave 90% deposition in the trachea (Balchum et al., 1960). Studies exposing rabbits to different concentrations of SO2 gave 80% respiratory tract deposition at low concentrations (0.05 ppm), 98% at high concentrations (700 ppm) (Strandberg, 1964), and more than 90% upper airway deposition at concentrations between 100 and 300 ppm (Dalhamn and Strandberg, 1961). Absorption to blood of SO2 deposited in the respiratory tract of dogs was consistent with assignment to Type F (Balchum et al., 1960; Frank et al., 1967). For SO2, it is assumed here that there is 100% deposition in the respiratory tract (with default regional distribution, Table 5.2) and Type F absorption.
(b) Carbon disulphide (CS2)
(180) Studies have been performed with CS2 in mice, rats, dogs, and man (McKee et al., 1943; Teisinger and Souček, 1949; McKenna and DiStefano, 1977; Bergman et al., 1984). In all cases, CS2 was taken up by the respiratory tract and absorbed into the blood. However, there is no information on the fraction of inhaled vapour deposited, or on the site of deposition. McKenna and DiStefano (1977) observed that CS2 uptake into blood was characterised by a single exponential with a half-life of 19.3 min, consistent with assignment to Type F. For CS2, it is assumed here by default that there is 100% deposition in the respiratory tract (with default regional distribution, Table 5.2) and Type F absorption.
(c) Hydrogen sulphide (H2S)
(181) Patty (1963) reported that H2S is absorbed through the lung and that H2S does not appear in exhaled breath, indicating that a large fraction is absorbed. In the absence of any real quantitative data on the fraction of H2S absorbed, the default option for gases and vapours is taken. For H2S, it is assumed here that there is 100% deposition in the respiratory tract (with default regional distribution, Table 5.2) and Type F absorption.
(d) Carbonyl sulphide (COS)
(182) Little has been published on the uptake of COS. Patty (1963) noted that COS decomposes in water to H2S and carbon dioxide. On this basis, it is assumed that the uptake of COS is the same as that of H2S; in the absence of specific information, the default option for gases and vapours is taken. For COS, it is assumed here that there is 100% deposition in the respiratory tract (with default regional distribution, Table 5.2) and Type F absorption.
5.2.1.2. Particulate materials
(183) No detailed information is available on the rate of absorption of sulphur following respiratory tract deposition of particulate compounds other than sulphates (see below). However, a few cases of accidental exposure of humans to 35S compounds have been reported.
(a) Elemental sulphur
(184) A worker was contaminated internally and externally following the explosion of a glass vial containing elemental 35S dissolved in benzene (Maass et al., 1963). Similar amounts of 35S were excreted in urine and faeces during the first few days, and levels in plasma and urine fell rapidly, suggesting rapid absorption from the lungs, and hence Type F behaviour.
(b) Methionine
(185) Smith et al. (1983) reported urinary excretion functions measured in three workers following separate accidental intakes of 35S-labelled L-methionine, probably by inhalation. Information was not reported that would enable estimation of the rate at which the deposited material was absorbed from the respiratory tract. However, in two cases, urinary excretion was fit by a single exponential with a half-time of approximately 0.6 d, suggesting Type F behaviour.
(c) Other compounds
(186) Two workers were contaminated with 35S while segregating waste of unknown chemical composition formed by irradiating KCl targets (Spate et al., 1985). Urine monitoring in both subjects indicated that approximately 90% cleared with a half-time of approximately 6 h, and the rest cleared with a half-time of approximately 6 d. From this, it was inferred that the activity dissolved rapidly in the lungs, indicating Type F behaviour.
(d) Sulphates
(187) For details of experiments, see the element section for the relevant cation. Those in OIR Part 2 and Part 3 are listed below. However, in the studies of the biokinetics of inhaled (or instilled) sulphates, only the cation was radiolabelled, and therefore caution must be used in drawing inferences about the behaviour of the anion. For sulphates that are insoluble in both aqueous media and in vivo (e.g. barium sulphate), it is reasonable to assume that the compound will dissociate slowly, and the behaviour of the sulphate moiety will be broadly similar to that of the metal. However, other sulphates, such as those of caesium, nickel, and thorium, are very soluble in aqueous media, and would be expected to dissociate into the respective metal and sulphate ions in vivo, each of which will follow its specific biokinetic pattern. In particular, following deposition in the lungs of thorium sulphate, like other water-soluble forms of thorium, most of the thorium is retained in particulate form and so is assigned to Type M. However, it is reasonable to assume that the sulphur would be absorbed rapidly (Type F). It should also be noted that solubility in water is not a reliable guide to solubility in vivo. When 90SrSO4, which is insoluble in water, was inhaled by mice, the 90Sr was absorbed rapidly.
Barium sulphate (see OIR Part 3)
(188) Studies of respiratory tract clearance in several species indicate a wide range of absorption rates, and BaSO4 is assigned to Type M.
Caesium sulphate (see OIR Part 3)
(189) Measurements following accidental human inhalation indicate Type F behaviour.
Radium sulphate (see OIR Part 3)
(190) Measurements following accidental human inhalation were difficult to interpret, and no assignment was made.
Strontium sulphate (see Section 10.2.1.)
(191) Measurements following inhalation by mice and dogs indicate Type F behaviour.
Thorium sulphate (see OIR Part 3)
(192) Measurements following intratracheal instillation into rats indicate Type M behaviour.
5.2.1.3. Rapid dissolution rate for sulphur
(193) No reliable estimates have been made of the rapid dissolution rate of sulphur in particulate form. The general default value of 30 d−1 is therefore applied to all Type F forms of sulphur.
5.2.1.4. Extent of binding of sulphur to the respiratory tract
(194) The evidence of rapid uptake of sulphur gases from the lung indicates that there is probably little binding of sulphur. It is therefore assumed that the bound state can be neglected for sulphur, i.e. fb = 0.0.
5.2.2. Ingestion
(195) Bauer (1976) showed that sulphur ingested as radioactive sulphate (35S) by eight volunteers was absorbed completely in tracer amounts. Volwiler et al. (1955) reported that the fractional absorption of sulphur given as organic compounds to adult men was greater than 0.6. Schulz (1984) reported that orally administered thiosulphate (S2O32−) in humans was not absorbed from the gastrointestinal tract, but thiocyanate (CNS−) was absorbed completely. (196) Results obtained by Dziewiatkowski (1949) for the excretion of 35S in rats after oral administration as the sulphate or sulphide indicated that absorption was 0.9 or greater. Minski and Vennart (1971) measured the absorption of [35S]-methionine in rats and obtained a mean value of approximately 0.9. Elemental sulphur was found to be absorbed less well, with values in rats of approximately 0.1 (Dziewiatkowski, 1962). (197) Publication 30 (ICRP, 1980) recommended absorption values of 0.8 for inorganic forms of sulphur and 0.1 for elemental sulphur. In Publication 67 (ICRP, 1993), a value of 1 was adopted for dietary intakes. In this publication, recommended fA values are 0.1 for elemental sulphur and thiosulphate, and 1 for all other forms.
5.2.3. Systemic distribution, retention, and excretion
5.2.3.1. Summary of the database
(a) Inorganic sulphur
(198) Andrews et al. (1960) measured the rate of disappearance of 35S from blood following its intravenous administration as sulphate (H2SO4) to an adult male subject with chondrosarcoma. The measurements indicated two components of biological removal from blood with half-times of 0.35 d (94%) and 5.6 d (6%). (199) Schulz (1984) showed that after intravenous injection of thiosulphate into humans, the compound left plasma with a half-time of approximately 15 min. Most of the thiosulphate was oxidised to sulphate or incorporated into endogenous sulphur compounds. A small proportion was excreted in urine. Following oral administration of thiocyanate to human subjects, sulphur was almost completely absorbed into the blood and cleared from the serum with a half-time of approximately 3 d. Elimination was mainly renal (Schulz, 1984). (200) Following intravenous injection of dilute H235SO4 into 15 normal human subjects, an average of 4.5% (range 1.3–8.8%) of the administered activity was excreted in urine within 18 min, and approximately half was excreted within 4–9 h (Walser et al., 1953). In a similar study involving dogs, an average of 3.6% (range 1–6%) of the administered activity was excreted within 25–30 min after injection. Following prior water loading by stomach tube in another group of dogs, mean urinary excretion in the first 25–30 min increased to 5.6% (range 3.7–8.2%). (201) In a study involving intravenous administration of 35S to 21 patients with chondrosarcoma or chordoma, an estimated 70–90% of administered activity was excreted in the urine in the first 3 d (Woodard et al., 1976). Studies of the blood kinetics in six of these patients indicated a major component with a removal half-time of 0.4–0.7 d. Measurements of activity in tissues obtained from biopsies or autopsies indicated high initial uptake in red bone marrow and epiphyseal cartilage. Uptake in other types of cartilage and in samples of skin, fibrous tissue, and muscle was relatively low, but subsequent loss from these tissues was slow. (202) In studies of the behaviour of intravenously injected inorganic 35S in human subjects and laboratory animals, it was found that a significant portion of the 35S accumulated in the cartilage and bone (Denko and Priest, 1957; Buck and Heagy, 1958; Gottschalk et al., 1959). Activity depositing in these tissues was removed with a biological half-time of several days. (203) Minski and Vennart (1971) studied the biokinetics of 35S in 76 rats following its intravenous administration as the inorganic form Na235SO4 or the organic form 35S-L-methionine. Following administration of inorganic 35S, the cartilage and marrow had the greatest integrated activity per unit mass, and the soft tissue had the lowest integrated activity. 35S was eliminated from the body at a faster rate when administered as sodium sulphate than when administered as methionine. The authors determined the retained fraction of administered activity in several tissues, and presented results as tissue-specific retention functions. (204) Studies in rats showed that after intravenous injection of 99mTc35S-sulphur colloid, the rates of clearance of 99mTc and 35S from blood and their accumulation in liver and bone were markedly different. The colloid particles were apparently broken down in vivo with the release of sulphur (Frier et al., 1981).
(b) Gaseous inorganic compounds of sulphur
Hydrogen sulphide (H2S)
(205) H2S entering blood is oxidised rapidly to pharmacologically inert compounds such as thiosulphate and sulphate, and excreted in urine (Patty, 1963; Vennart and Ash, 1976).
Carbon disulphide (CS2)
(206) CS2 is insoluble in water. Results of several studies (McKee et al., 1943; Teisinger and Souček, 1949; McKenna and DiStefano, 1977; Bergman et al., 1984) indicate that CS2 is taken up by fat, reaching equilibrium in humans after 1–2 h under continuous exposure. Some activity from the fat reserves is then metabolised and ultimately excreted in urine. McKee et al. (1943) showed that 85–90% of CS2 in the body is metabolised, and the remaining non-metabolised CS2 is eliminated unchanged, mostly in the breath. There is extensive metabolic incorporation of S released from CS2 during biotransformation. Bergman et al. (1984) showed that, after initial concentration in liver and kidneys, 35S labelled metabolites are eliminated rapidly from the body, probably in inorganic form.
Carbonyl sulphide (COS)
(207) COS decomposes in water to form H2S and carbon dioxide. The 35S moiety of COS is assumed to behave like H2S when in the bloodstream. The toxic effects of COS after inhalation appear to result from the toxicity of the H2S produced, supporting the assumption that the 35S label can be treated as though it were H2S (Vennart and Ash, 1976).
Sulphur dioxide (SO2)
(208) SO2 entering the blood is expected to dissolve, and produce sulphite and sulphate ions.
(c) Organic compounds of sulphur
(209) Minski and Vennart (1971) studied the distribution and retention of 35S in rats following intravenous administration of the organic form 35S-L-methione and the inorganic form Na235SO4. 35S administered in organic form was removed from blood more slowly and distributed in tissues more uniformly than 35S administered in inorganic form. Blood disappearance of 35S administered in organic form was described as the sum of three exponential terms: 34% was removed with a half-time of 0.16 d, 14% was removed with a half-time of 4.1 d, and 52% was removed with a half-time of 60.5 d. The cumulative activity in the whole body was an order of magnitude higher for 35S administered as methionine than for 35S administered as sodium sulphate. The cartilage and intestines showed the highest cumulative activity per unit mass of tissue following injection of inorganic 35S, but relatively low cumulative activity per unit mass compared with several other tissues following its injection as organic 35S. The half-time in blood following administration of the organic form was 40 times larger than that following administration of the inorganic form. (210) Taking account of these data in conjunction with the dietary intake and whole-body content of sulphur in adult humans (ICRP, 1975), Vennart and Ash (1976) proposed that organic sulphur ingested in food should be assumed to be absorbed completely from the gastrointestinal tract, distributed uniformly throughout the body tissues, and eliminated with a single biological half-time of 140 d. These assumptions form the basis for the systemic model for organic sulphur adopted in Publication 30 (ICRP, 1980) and also applied in Publication 67 (ICRP, 1993). In Publication 67, a urinary:faecal excretion ratio of 9:1 was assigned.
5.2.3.2. Biokinetic model for systemic sulphur
(a) Inorganic sulphur
(211) Data for human subjects indicate that following entry of inorganic forms of sulphur into blood, there is a rapid phase of excretion with a half-time of approximately 0.3 d, followed by a slower phase of elimination with a half-time of at least 7 d and possibly as much as 80 d (ICRP, 1980, 1993). Studies of dietary sulphur suggest that two components of retention with half-times of this order are insufficient to explain the whole-body content of 140 g of sulphur given for Reference Man (ICRP, 1975), and that at least one longer-term component of retention must be present. (212) The biokinetic model for inorganic sulphur used in Publication 67 (ICRP, 1993) assumes a removal half-time from blood of 0.25 d. The fraction 0.8 is assumed to be excreted promptly, and fractions 0.15 and 0.05 are assumed to be distributed uniformly throughout the body and removed with biological half-times of 20 and 2000 d, respectively. (213) The structure of the systemic model for inorganic sulphur used in this publication is shown in Fig. 5.1. Transfer coefficients are listed in Table 5.4. Sulphur is assumed to be removed from blood at a rate of 2.5 d−1. Deposition fractions in tissue compartments and excretion pathways are based on data from human studies by Woodard et al. (1976), Andrews et al. (1960), Gottschalk et al. (1959), Maass et al. (1963), and Denko and Priest (1957), and rat studies by Dziewiatkowski (1945, 1949, 1953), Denko and Priest (1957), Minski and Vennart (1971), and Singher and Marinelli (1945). The assumed distribution of activity leaving blood is as follows: 72% goes to urinary bladder contents, 10% goes to cartilage, 8% goes to right colon contents, 7% goes to other, and 3% goes to red marrow. The retention half-times in compartments were set for reasonable consistency with data for human subjects or rats summarised earlier. Structure of the systemic biokinetic model for inorganic sulphur used in this publication. Transfer coefficients for inorganic sulphur in adults.


(b) Organic compounds of sulphur
(214) The structure of the systemic model for organic sulphur used in this publication is presented in Fig. 5.2. Transfer coefficients are listed in Table 5.5. The model is based largely on data for rats (Minski and Vennart, 1971), but depicts a longer residence time of sulphur in tissues than indicated by that data on the basis of balance considerations for stable sulphur in human diet and the whole body. (215) Model predictions of the clearance of intravenously injected organic sulphur from blood are consistent with the clearance pattern determined for rats following intravenous administration of 35S-L-methione (Minski and Vennart, 1971). Structure of the systemic biokinetic model for organic sulphur used in this publication. Transfer coefficients for organic sulphur in adult humans. SI, small intestine.


Applicability of the 35S-L-methionine model
(216) For general radiological protection purposes, this modified biokinetic model for 35S-L-methionine could be applied with caution to other organic forms of sulphur in the absence of other compound-specific data. However, this model should not be used for the interpretation of bioassay data.
5.2.3.3. Treatment of radioactive progeny
(217) The only radioactive progeny of a sulphur isotope addressed in this publication is 38Cl (t½ = 37.24 min), produced by decay of 38S. It is assumed for dosimetric purposes that 38Cl decays at its site of production in the body.
5.3. Individual monitoring
(218) 35S intake is generally monitored though measurement of the activity excreted in urine. The most common method of analysis is liquid scintillation counting (Table 5.6). Monitoring techniques for 35S. Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 35S compounds. AMAD, activity median aerodynamic diameter. Dose per activity content of 35S in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work. Sulphur dioxide, carbon disulphide, hydrogen sulphide, carbonyl sulphide, and other unspecified inorganic gases and vapours.


5.4. Dosimetric data for sulphur
Daily urinary excretion of 35S following inhalation of 1 Bq sulphur dioxide, carbon disulphide, hydrogen sulphide, carbonyl sulphide, and other unspecified inorganic gases and vapours. Daily urinary excretion of 35S following inhalation of 1 Bq other organic gases or vapours. Daily urinary excretion of 35S following inhalation of 1 Bq Type F. Daily urinary excretion of 35S following inhalation of 1 Bq Type M. Daily urinary excretion of 35S following inhalation of 1 Bq Type S.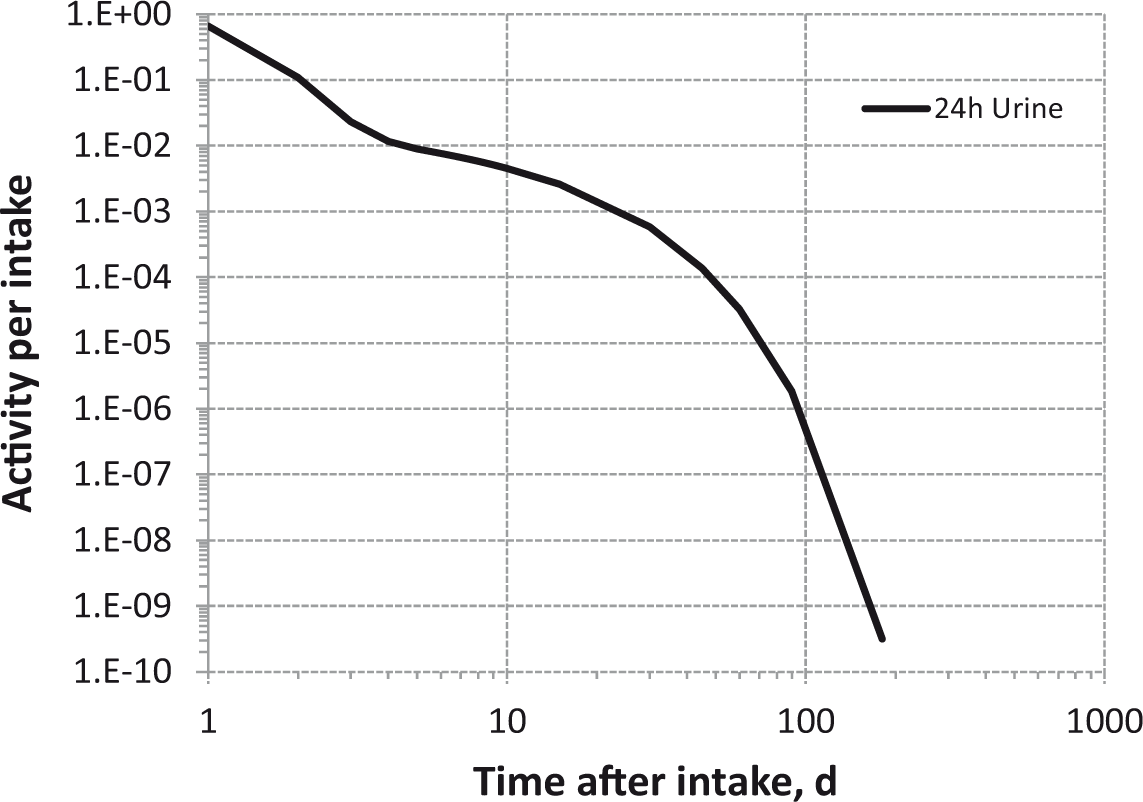



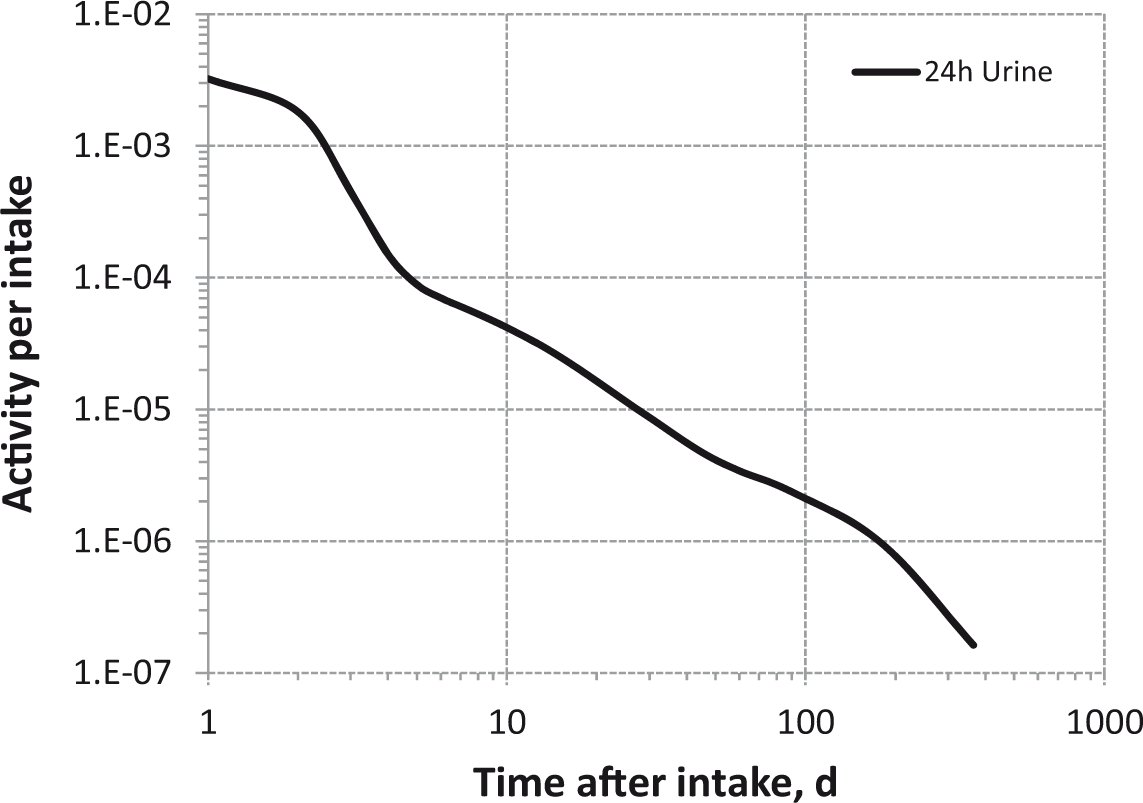
5.5. References
6. CALCIUM (Z = 20)
6.1. Chemical forms in the workplace
(219) Calcium is an alkaline earth element that mainly occurs in oxidation state II. It is an essential element for life. Chemical forms encountered in industry include oxides, phosphates, nitrates, sulphides, chlorides, carbonates, and fluorides. 45Ca and 47Ca are occasionally used in research and in medicine. Table 6.1 shows the isotopes of calcium addressed in this publication. Isotopes of calcium addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B-, Beta-minus decay; EC, electron-capture decay

6.2. Routes of intake
6.2.1. Inhalation
(220) No information was found on the behaviour of inhaled calcium in man. Information on absorption from the respiratory tract is available from experimental studies of calcium chloride. (221) Absorption parameter values and types, and associated fA values for particulate forms of calcium are given in Table 6.2. Absorption parameter values for inhaled and ingested calcium. It is assumed that the bound state can be neglected for calcium, i.e. fb=0. The value of sr for Type F forms of calcium (70 d−1) is element-specific. The values for Types M and S (3 d−1) are the general default values. Materials (e.g. calcium chloride) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of calcium (0.4). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract.

6.2.1.1. Particulate materials
(a) Calcium chloride
(222) Schiessle et al. (1964) followed the retention of 45Ca in the lungs of guinea pigs for 28 d after inhalation of CaCl2. Most of the ILD was absorbed very rapidly; at 1 d, less than 1% of the ILD remained, consistent with assignment to Type F. Specific parameter values were estimated here (i.e. by the Task Group) to be: fr = 0.996, sr = 70 d−1 (t½∼14 min), and ss = 0.07 d−1 (t½∼10 d), consistent with assignment to Type F. Although specific parameter values for calcium chloride based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, calcium chloride is assigned to Type F. However, the data are used as the basis for the default rapid dissolution rate for calcium. Hence, specific parameter values for calcium chloride would be the same as default Type F calcium parameter values.
6.2.1.2. Rapid dissolution rate for calcium
(223) The value of sr estimated for CaCl2 above, 70 d−1, is applied here to all Type F forms of calcium.
6.2.13. Extent of binding of calcium to the respiratory tract
(224) Evidence from the calcium chloride study outlined above suggests that there is probably little binding of calcium. It is therefore assumed that the bound state can be neglected for calcium, i.e. fb = 0.0.
6.2.2. Ingestion
(225) Calcium is the first member of the alkaline earth metal series, and it may exist under physiological conditions partly as a divalent cation and partly as complexes with proteins and other ligands. However, unlike strontium, barium, and radium, the other alkaline earth elements, calcium is an essential element and physiological mechanisms facilitate its intestinal absorption. (226) Calcium absorption has been measured in numerous volunteer studies, and the reported mean absorption values were in the range 0.2–0.5 in most cases (Samachson, 1963; DeGrazia and Rich, 1964; Lutwak, 1969; Mautalen et al., 1969; Jovanovic, 1978; Cochet et al., 1983; Marchandise et al., 1986; Spencer et al., 1987; Harvey et al., 1988; Heaney et al., 1989, 1999). Greater mean values of 0.6 (Sambrook et al., 1985) and 0.7 (Rumenapf and Schwille, 1987) have also been reported for normal volunteers. These differences may probably be explained by the large interindividual differences in calcium absorption observed in healthy subjects, with individual values ranging from 0.3 to 0.6 (Barger-Lux and Heaney, 1995) or even from 0.4 to 0.9 (Isaksson et al., 2000). Indeed, calcium absorption depends first on the intraluminal concentration of ionised calcium (Schachter et al., 1960), which can be reduced locally by the presence of calcium binding agents such as EDTA or citrate ions (Rumenapf and Schwille, 1987). Additional variability may be associated with morphological factors as calcium absorption is positively correlated with body size (Davies et al., 2000; Barger-Lux and Heaney, 2005) and many nutritional factors. It is known that fractional calcium absorption is increased by high intakes of vitamin D, by a high protein or carbohydrate diet, by calcium deficiency or low calcium intake, and by pregnancy or lactation (Allen, 1982; Spencer et al., 1987; Heaney et al., 1989; Cashman and Flynn, 1996; Griffin et al., 2002; Kerstetter et al., 2005; Holloway et al., 2007). On the other hand, caffeine intake or oral supplementation with magnesium decreased calcium absorption in humans (Barger-Lux and Heaney, 1995; De Swart et al., 1998; Heaney, 2002). (227) Calcium absorption is known to occur mainly from the small intestine (ICRP, 2006). However, a few percent of calcium may also be absorbed from other sites, such as the colon, which, at 26 h after ingestion, can absorb as much as 4% of calcium provided to healthy perimenopausal women (Barger-Lux et al., 1989). (228) In Publication 30 (ICRP, 1980) and Publication 71 (ICRP, 1995), an absorption value of 0.3 was recommended. As absorption appears to be generally greater than 0.3 in normal subjects, an fA value of 0.4 for all chemical forms is adopted here.
6.2.3. Systemic distribution, retention, and excretion
6.2.3.1. Summary of the database
(229) The biokinetics of calcium in the human body has been investigated extensively in physiological and clinical studies, and in radiobiological studies comparing the behaviour of isotopes of the alkaline earth elements. Reviews and bibliographies can be found in Publication 20 (ICRP, 1973), Publication 71 (ICRP, 1995), and an article by Leggett (1992). The primary datasets underlying specific parameter values in the model for systemic calcium used in this publication are summarised below.
6.2.3.2. Biokinetic model for systemic calcium
(230) The structure of the model for systemic calcium is shown in Fig. 6.1. All soft tissues including the liver and kidneys are included in the three ‘other’ (soft tissue) compartments – ST0, ST1, and ST2 – corresponding to rapid, intermediate, and slow exchange of activity with blood, respectively. These soft tissue compartments are defined on a kinetic basis rather than an anatomical or physiological basis, but ST0 may correspond roughly to interstitial fluids plus some rapidly exchangeable cellular calcium (Heaney, 1964; Harrison et al., 1967; Hart and Spencer, 1976); ST1 may be a composite of several pools with slower exchange rates, including mitochondrial calcium, cartilage calcium, and exchangeable dystrophic calcium (e.g. arterial plaque and calcified nodes) (Heaney, 1964; Borle, 1981); and ST2 may be associated with relatively non-exchangeable dystrophic calcium that gradually accumulates in the human body (Heaney, 1964). (231) Blood is treated as a uniformly mixed pool that exchanges calcium with soft tissues (ST0, ST1, ST2) and bone surface compartments. Calcium is assumed to be lost from the body by urinary or faecal excretion alone. Activity going to urine is first transferred from blood to urinary bladder contents, and activity going to faeces is first transferred from blood to the right colon contents. Structure of the model for systemic calcium. ST, soft tissue; exch, exchangeable; nonexch, non-exchangeable.

6.2.3.3. Parameter values
(232) The parameter values applied to systemic calcium in this publication are the same as those applied in Publication 71 (ICRP, 1995). These values are listed in Table 6.3. The selection of each parameter value is described briefly in the following, and explained in more detail by Leggett (1992). (233) Kinetic analysis of plasma disappearance curves for normal subjects intravenously injected with radioisotopes of the alkaline earth elements indicates that these elements initially leave blood plasma at a rate of several hundred plasma volumes per day, and equilibrate rapidly with an extravascular pool (presumably consisting largely of interstitial fluids) roughly three times the size of the plasma pool (Heaney, 1964; Harrison et al., 1967; Hart and Spencer, 1976). The present model does not depict the rapid exchange of calcium between blood and this extravascular pool. However, the model includes a soft tissue compartment (ST0) that receives more than half of the activity leaving blood, returns activity to blood with a half-time of a few hours, and contains three times as much activity as blood at times more than a few hours after input of calcium to blood. This compartment is used to account for relatively high concentrations of calcium tracers observed in soft tissues during the first few hours after injection, and to help maintain the proper amount of material in blood. A total transfer rate from blood of 15 d−1 (i.e. a removal half-time of ln(2)/15 d = 0.04621 d) yields reasonable fits to blood disappearance curves for calcium or strontium tracers at times beyond 1–2 h after injection into human subjects (Barnes et al., 1961; Heaney, 1964; Heaney et al., 1964; Harrison et al., 1967; Neer et al., 1967; ICRP, 1973; Newton et al., 1990). (234) It is assumed that 58% of calcium leaving blood moves to the rapid-turnover soft tissue compartment ST0; this is the balance of outflow from blood after deposition percentages in other compartments are assigned. The corresponding transfer rate from blood to ST0 is 0.58 × 15 d−1 = 8.7 d−1. Based on the assumed relative amounts of calcium in ST0 and blood, the transfer rate from ST0 to blood is set at one-third of the transfer rate from blood to ST0, or 2.9 d−1. (235) Readily exchangeable calcium in soft tissues, meaning calcium that is turned over to a substantial extent in a period of hours or days, is represented in this model as the sum of calcium in ST0 and ST1. The amount of readily exchangeable stable calcium in soft tissues is approximately 0.35% of whole-body calcium in a middle-aged adult human (Heaney, 1964; Borle, 1981). As blood contains approximately 0.03% of whole-body calcium in the adult (ICRP, 1975), the three-fold larger compartment ST0 is estimated to contain 0.09% and ST1 is estimated to contain approximately 0.26% (0.35–0.09%) of whole-body calcium during chronic intake. Parameter values for ST1 are set to reproduce these steady-state conditions while approximating soft tissue retention data for terminally ill human subjects intravenously injected with 45Ca at times up to 124 d before death (Schulert et al., 1959). This is accomplished by assigning a deposition fraction of 0.1 to ST1 and a removal half-time to blood of 4 d. The derived transfer rate from blood to ST1 is 0.1 × 15 d−1 = 1.5 d−1, and the transfer rate from ST1 to blood is ln(2)/4 d = 0.1733 d−1. (236) Parameter values for ST2 are set for consistency with estimates of the accumulation of relatively non-exchangeable calcium in adult humans (Heaney, 1964), an estimate of the fraction of whole-body calcium in soft tissues under conditions of chronic exposure (Schlenker et al., 1982), and the observed retention of 45Ca in human soft tissues at 3 months after injection (Schulert et al., 1959). Reasonable agreement with these three values is achieved by assuming that ST2 receives 0.005% of outflow from blood and that the removal half-time from ST2 to blood is 5 y. The resulting transfer rate from blood to ST2 is 0.00005 × 15 d−1 = 0.00075 d−1, and the transfer rate from ST1 to blood is ln(2)/(5 × 365 d) = 0.00038 d−1. (237) Data for laboratory animals indicate that fractional deposition on bone surfaces is similar for calcium, strontium, barium, and radium. This is inferred from the similar skeletal contents of these elements in the first few hours after injection (Bligh and Taylor, 1963; Kshirsagar et al., 1966; Domanski et al., 1969, 1980). Use of a common bone-surface deposition fraction for all four elements is consistent with autoradiographic measurements of surface activity in bone samples taken at autopsy from subjects injected with radiocalcium at 0.6 d or longer before death (Riggs et al., 1971; ICRP, 1973); measurements of radiocalcium and radiostrontium in bone samples from subjects injected 3 h or longer before death (Schulert et al., 1959); and external measurements of the build-up of radiocalcium (Anderson et al., 1970; Heard and Chamberlain, 1984) and radiobarium (Korsunskii et al., 1981) after intravenous injection. Based on these data, 25% of calcium, strontium, barium, or radium leaving blood is assigned to the two bone surface compartments combined. The transfer rate from blood to cortical and trabecular surface compartments combined is 0.25 × 15 d−1 = 3.75 d−1. (238) The initial distribution between different bones of the skeleton and between the two bone types (cortical and trabecular) appears to be similar for calcium, strontium, barium, and radium (Ellsasser et al., 1969; Wood et al., 1970; Liniecki, 1971; Stather, 1974; Lloyd et al., 1976). Relative deposition of alkaline earth elements on cortical and trabecular bone surfaces is based on the estimated calcium turnover rate of each bone type. This approach agrees with measurements on laboratory animals (Kshirsagar et al., 1966; Norrdin and Arnold, 1980). As an average over adult ages, deposition on trabecular bone surface is estimated to be 1.25 times the deposition on cortical bone surface (Leggett et al., 1982). The transfer rate from blood to trabecular surface is (1.25/2.25) × 3.75 d−1 = 2.08 d−1, and the transfer rate from blood to cortical surface is (3.75 - 2.08) d−1 = 1.67 d−1. (239) The removal half-time of calcium from the bone surface compartments to all destinations (blood and the exchangeable bone volume compartments) is estimated as 1 d. This is based on autoradiographic measurements of surface activity in human and canine bone samples taken at times ranging from a few hours to a few days after intravenous injection of 45Ca (Riggs et al., 1971; Groer et al., 1972; Groer and Marshall, 1973; ICRP, 1973). (240) Parameter values for the exchangeable bone volume compartments are estimated from whole-body measurements using data for times after bone surfaces and soft tissues have largely cleared of activity, but before loss from bone resorption becomes an important consideration. Based on whole-body retention curves for human subjects injected with radioisotopes of calcium, strontium, barium, or radium (Bishop et al., 1960; Spencer et al., 1960; Heaney et al., 1964; Harrison et al., 1967; Maletskos et al., 1969; Phang et al., 1969; Carr et al., 1973; Likhtarev et al., 1975; Malluche et al., 1978; Henrichs et al., 1984; Newton et al., 1990, 1991), the fraction of activity that moves from a bone surface compartment back to blood is assumed to be the same for all four elements. Specifically, five-sixths of activity leaving a bone surface compartment is assumed to return to blood, and one-sixth is assumed to transfer to the corresponding exchangeable bone volume compartment. The transfer rate from a bone surface compartment to the corresponding exchangeable bone volume compartment is (1/6) × ln(2)/1 d = 0.116 d−1, and the transfer rate from a bone surface compartment to blood is (5/6) × ln(2)/1 d = 0.578 d−1. (241) Element-specific removal half-times from the exchangeable bone volume compartments are based, in part, on fits to the intermediate-term retention data indicated above. However, it is also considered that the assigned half-times should increase roughly in proportion to the likelihood of entering non-exchangeable sites in bone mineral, as suggested by data from in-vitro experiments with hydroxyapatite crystals and whole-body retention patterns for alkaline earth elements in human subjects. A removal half-time of 100 d is assigned to calcium, compared with values of 80 d for strontium, 50 d for barium, and 30 d for radium (Leggett, 1992). As the data do not allow the derivation of removal half-times as a function of bone type, the same half-time is applied to cortical and trabecular exchangeable bone volume compartments. (242) Discrimination between alkaline earth elements by bone is accounted for by fractional transfer of activity from exchangeable to non-exchangeable bone volume. It is assumed, in effect, that calcium, strontium, barium, and radium are all equally likely to become temporarily incorporated in bone mineral after injection into blood, but that the likelihood of reaching a non-exchangeable site in bone crystal decreases in the order calcium > strontium > barium > radium. Fractional transfers of calcium, strontium, barium, and radium from exchangeable to non-exchangeable bone volume are set at 0.6, 0.5, 0.3, and 0.2, respectively, for consistency with whole-body and skeletal retention data on these elements (Bishop et al., 1960; Spencer et al., 1960; Heaney et al., 1964; Harrison et al., 1967; Maletskos et al., 1969; Phang et al., 1969; Carr et al., 1973; Likhtarev et al., 1975; Malluche et al., 1978; Henrichs et al., 1984; Newton et al., 1990, 1991), as well as results of in-vitro measurements on hydroxyapatite crystals (Neuman, 1964; Stark, 1968). The derived transfer rate from an exchangeable trabecular or cortical bone volume compartment to the corresponding non-exchangeable bone volume compartment is 0.6 × ln(2)/100 d = 0.004159 d−1, and the transfer rate to the corresponding bone surface compartment is 0.4 × ln(2)/100 d = 0.002773 d−1. (243) Biological removal from the non-exchangeable bone volume compartments of cortical and trabecular bone is assumed to result from bone turnover. The average bone turnover rates during adulthood are estimated to be 3% y−1 and 18% y−1 for cortical and trabecular bone, respectively (ICRP, 2002). The corresponding transfer rates from the non-exchangeable bone volume compartments of cortical and trabecular bone to blood are 0.0000821 d−1 and 0.000493 d−1, respectively. Clearance of calcium from blood to urine and faeces has been studied in human subjects, many of them healthy (Bishop et al., 1960; Spencer et al., 1960; Barnes et al., 1961; Cohn et al., 1963; Heaney et al., 1964; Samachson, 1966; Phang et al., 1969; Carr et al., 1973; Newton et al., 1990). Based on the results of these studies, it is assumed that 4% of calcium leaving blood is transferred to urinary bladder contents and subsequently to urine, and 3% is transferred to right colon contents and subsequently to faeces. Therefore, the transfer rate from blood to urinary bladder contents is 0.04 × 15 d−1 = 0.6 d−1 and the transfer rate from blood to right colon contents is 0.03 × 15 d−1 = 0.45 d−1. Transfer coefficients for systemic calcium. ST, soft tissue; Exch, exchangeable; Nonexch, non-exchangeable. ST0, ST1, and ST2 are compartments within ‘other’ with fast, intermediate, and slow turnover, respectively.
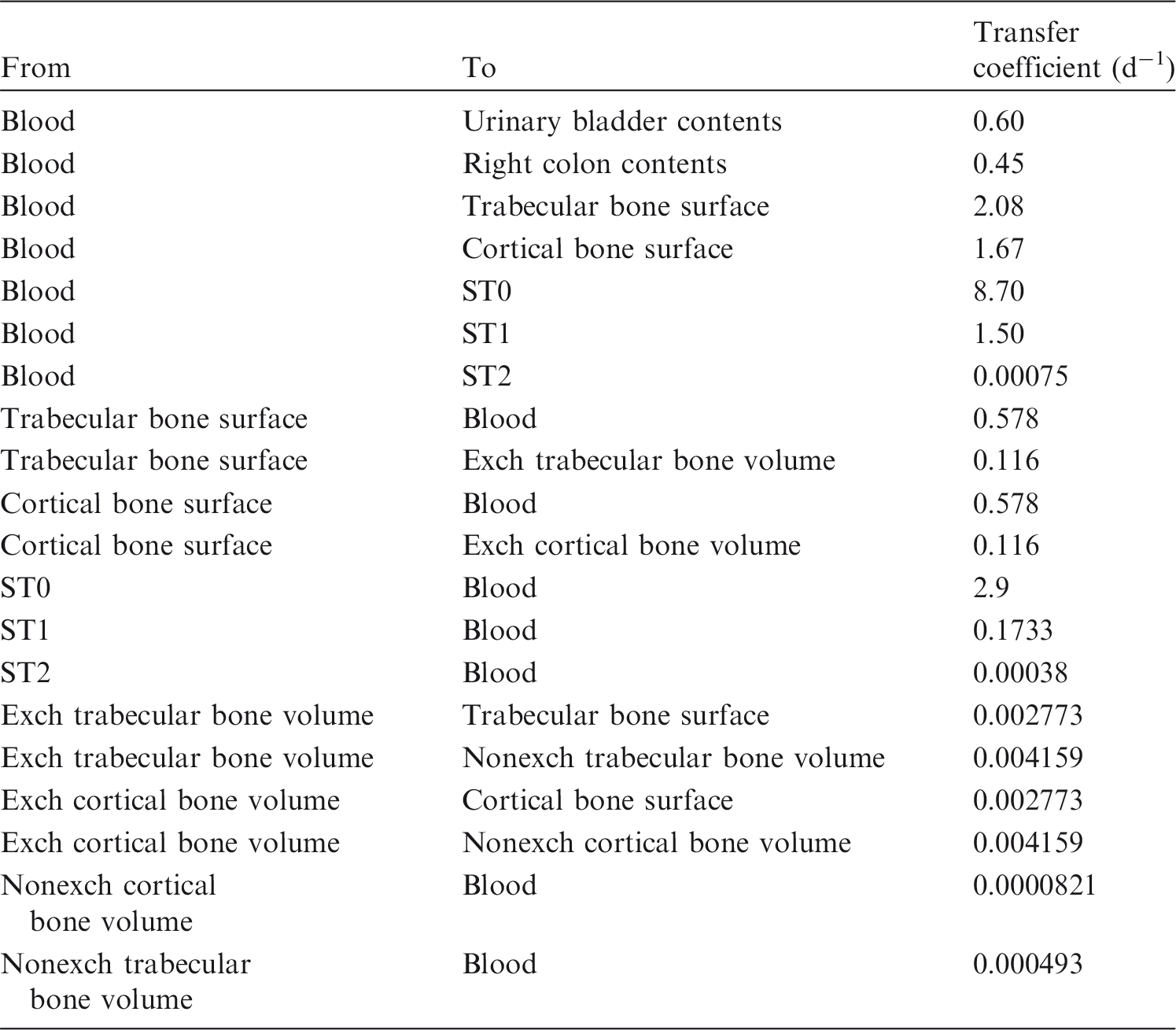
6.2.3.4. Treatment of radioactive progeny
(a) Experimental data
(244) The only calcium isotope addressed in this publication that decays to another radionuclide is 47Ca (t½ = 4.54 d), which decays to 47Sc (t½ = 3.35 d). The biological behaviour of 47Sc produced in vivo by decay of 47Ca has been investigated in rats (Taylor, 1966) and mice (Freed et al., 1975). (245) After intravenous administration of a mixture of 47Ca and 47Sc to rats, 47Sc accumulated primarily in liver, spleen, kidneys, and bone (Taylor, 1966). There was evidence that 47Sc also translocated to the liver and spleen after its production by decay of 47Ca at other sites in the body. Most of the 47Sc produced in vivo by decay of 47Ca arose in bone due to the high uptake and retention of 47Ca by bone. Nearly all of the 47Sc produced in bone was retained in bone for several days. (246) In mice, redistribution of 47Sc produced in the body following intravenous administration of 47Ca accounted for a large part of 47Sc found in soft tissues and blood (Freed et al., 1975). At times greater than 2 d after injection, 47Sc was contained largely in bone. It appeared that 47Sc escaped to some extent from its site of production in bone during the early hours after administration of 47Ca, but no preferential loss of 47Sc from bone was observed thereafter. At 1–11 d after injection, loss of 47Sc from bone was slower than that of 47Ca. After 11 d, the rate of loss of 47Sc from bone approached that of the parent, suggesting removal of both radionuclides by the process of bone resorption.
(b) General assumptions
(247) It is assumed in this publication that 47Sc produced by decay of 47Ca in soft tissues is removed to blood with a biological half-time of 3 d, and then follows the characteristic model for scandium, i.e. behaves as if injected into blood as a parent radionuclide. The removal half-time of 3 d is the shortest removal half-time of scandium from tissues in the characteristic model for scandium used here. 47Sc produced in a bone compartment of the calcium model is assumed to follow the characteristic model for scandium described below.
(c) Characteristic model for systemic scandium
(248) The structure of the characteristic model for scandium is a modification of the generic model structure applied in the OIR series to bone-surface-seeking radionuclides. Scandium is treated as a bone-surface seeker based on analogy with its chemical analogue yttrium. The spleen is added to the generic model structure because human and animal data indicate that it is an important repository for scandium. The generic structure is also modified with regard to routes of transfer to and from the marrow compartments, based on indications from animal studies of relatively high transfer of scandium from plasma to marrow (Rosoff et al., 1963, 1965; Hara and Freed, 1973; Byrd et al., 1975; Lachine et al., 1976). (249) Transfer coefficients in the characteristic model for scandium are set for consistency with the following data: (1) for human subjects, measurements of blood clearance over 3 d, urinary and faecal excretion rates over 15 d, whole-body retention over 1.5 y, and activity concentrations in autopsy tissues of subjects dying 5–7 months after injection (Rosoff et al., 1965); and (2) measurements of the time-dependent systemic distribution of activity in rats, mice, and rabbits (Durbin, 1960; Rosoff et al., 1963; Basse-Cathalinat et al., 1968; Hara and Freed, 1973; Byrd et al., 1975; Lachine et al., 1976). (250) Blood is divided into two compartments: Blood 1 and Blood 2. These represent two components of retention as indicated by data for intravenously injected 46Sc NTA in human subjects (Rosoff et al., 1965). Blood 1 is a central compartment that exchanges activity with Blood 2 and several tissue compartments. 47Sc migrating to blood from sites of production is assigned to Blood 1. Blood 2 represents scandium that is firmly bound to plasma proteins. (251) The total outflow rate from Blood 1 is 3 d−1. Blood 2 receives 15% of outflow from Blood 1, and loses scandium back to Blood 1 with a half-time of 1.5 d. This half-time is taken from the model applied in the OIR series to the chemically similar element yttrium. (252) The liver is divided into two compartments: Liver 1 and Liver 2. Liver 1 receives 20% of outflow from Blood 1. Activity is removed from Liver 1 with a half-time of 3 d, with 50% moving to Blood 1, 25% to Liver 2, and 25% to the small intestine contents (representing biliary secretion). Faecal excretion of scandium is assumed to arise solely from transfer of scandium from Liver 1 to the small intestine contents based on data of Rosoff et al. (1965) for a human subject. Almost all of the scandium secreted into the small intestine is lost in faeces because of the low rate of absorption of scandium from the small intestine to blood. Activity transfers from Liver 2 to Blood 1 with a half-time of 100 d. (253) The kidneys are represented as a single compartment that exchanges activity with Blood 1. This compartment receives 3% of outflow from Blood 1, and loses scandium to Blood 1 with a half-time of 20 d. Urinary excretion of scandium is represented as a direct transfer from Blood 1 to urinary bladder contents, without intermediate retention in the kidneys. Urinary bladder contents receives 1.8% of outflow from Blood 1. (254) Trabecular and cortical marrow each receive 5% of outflow from Blood 1. Activity is removed from the marrow compartments to Blood 1 with a half-time of 100 d. (255) The spleen receives 2% of outflow from Blood 1. The removal half-time from spleen to Blood 1 is 1 y. (256) Other soft tissues are divided into two compartments representing relatively fast (t½ = 3 d) and relatively slow (t½ = 100 d) return of scandium to Blood 1. These compartments receive 20% and 18.2% of outflow from Blood 1, respectively. The deposition fraction in the latter compartment is the balance of outflow from Blood 1 after all other deposition fractions in the model were assigned. (257) Bone surface receives 10% of outflow from Blood 1. The deposition on bone surface is divided equally between trabecular and cortical surfaces. The fate of scandium deposited on bone surfaces is described by the generic model for bone-surface seekers, except that scandium removed biologically from bone is assumed to return to blood rather than channelled through bone marrow. Thus, scandium is removed from cortical or trabecular bone surfaces at a rate proportional to (1.5 times) the turnover rate of that bone type. The assumed bone turnover rates are 3% y−1 for cortical bone and 18% y−1 for trabecular bone. One-third of activity removed from bone surfaces is buried in bone volume, and two-thirds transfers to Blood 1. Activity is removed from cortical or trabecular bone volume to Blood 1 at the rate of turnover of that bone type.
6.3. Individual monitoring
(258) 45Ca is a beta emitter. 45Ca intake is generally monitored though measurement of the activity excreted in urine. The most common method of analysis is liquid scintillation counting (Table 6.4). Monitoring techniques for 45Ca.

6.4. Dosimetric data for calcium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 45Ca compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 45Ca in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.

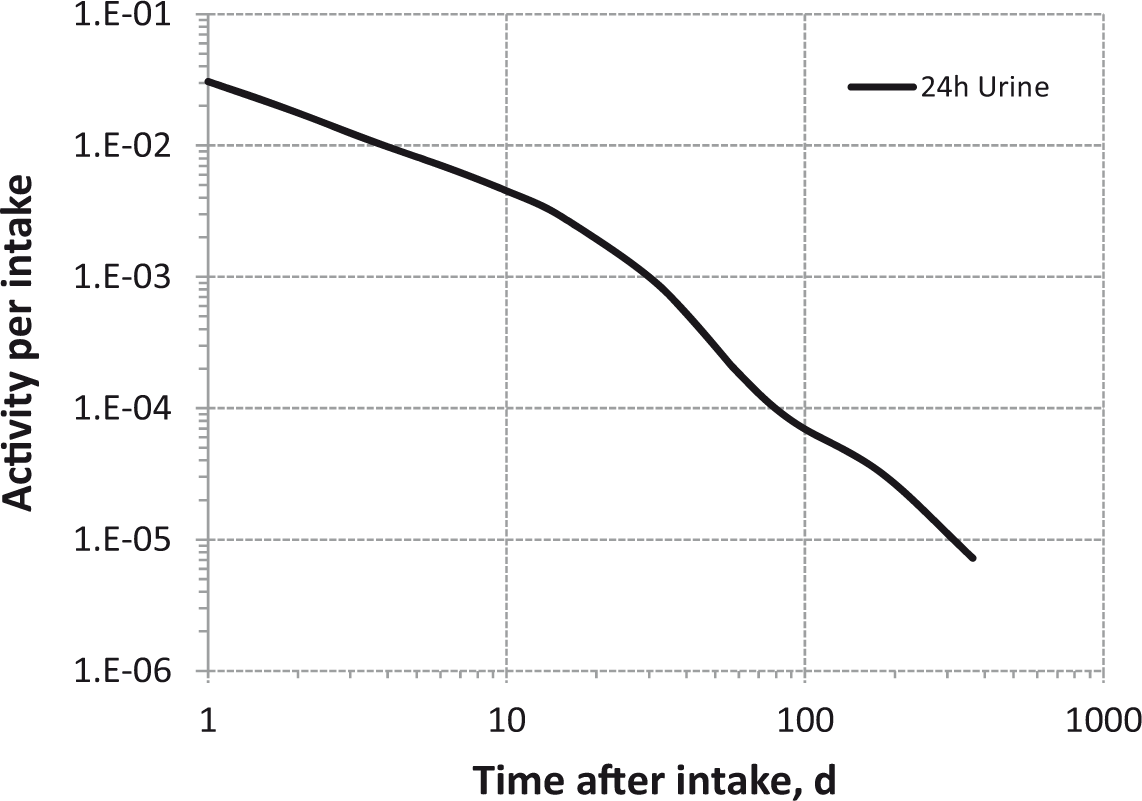
Daily urinary excretion of 45Ca following inhalation of 1 Bq Type F.
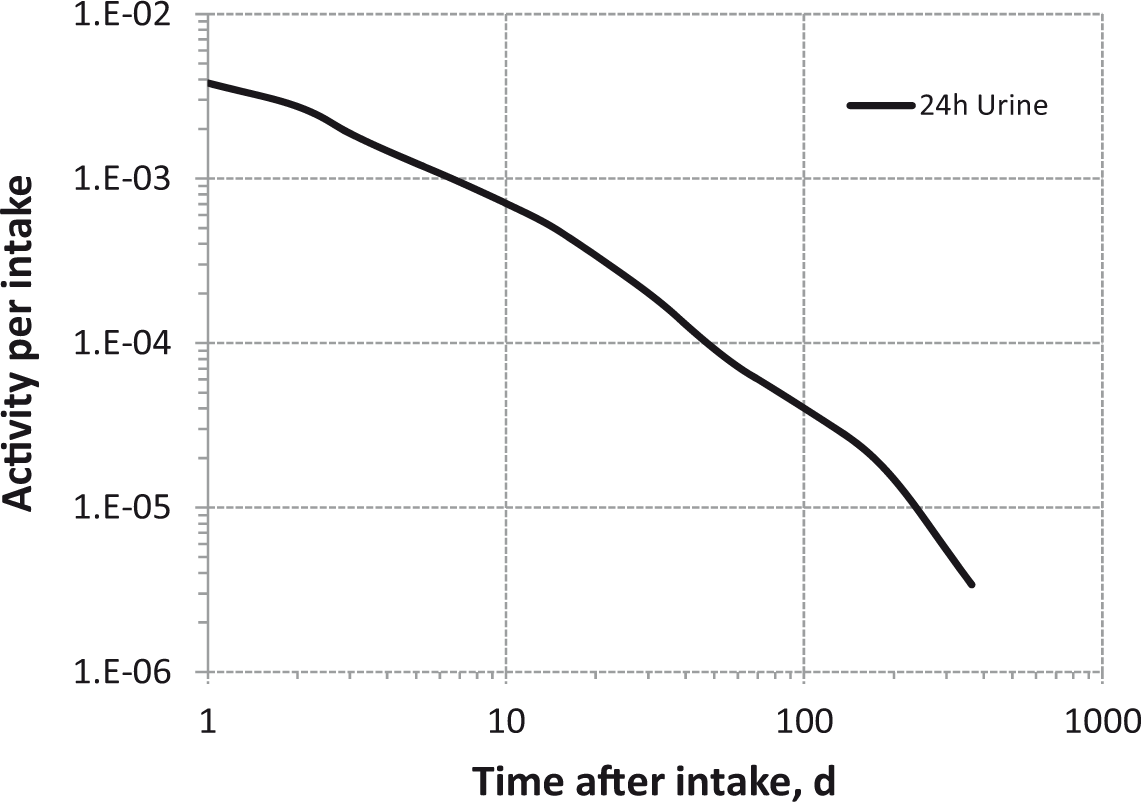
Daily urinary excretion of 45Ca following inhalation of 1 Bq Type M.

Daily urinary excretion of 45Ca following inhalation of 1 Bq Type S.
6.5. References
7. IRON (Z = 26)
7.1. Chemical forms in the workplace
(259) Iron is a transition metal, occurring mainly in oxidation states II and III. It is a vital constituent of plant and animal life, and is the key component of haemoglobin. Iron is used in industry in a variety of chemical forms, including oxides (FeO, Fe2O3, Fe3O4), chlorides, fluorides, and bromides. (260) The main radioactive isotope is 59Fe, which is used as ferrous citrate, chloride, or sulphate for diagnostic applications in hospitals. In the nuclear industry, 59Fe is an important neutron-activated corrosion product. It is likely to be present as oxides in different parts of the primary circuits of water-cooled reactors (Collier et al., 1994). Table 7.1 shows the isotopes of iron addressed in this publication. Isotopes of iron addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay.

7.2. Routes of intake
7.2.1. Inhalation
(261) Extensive information was found on the behaviour of iron inhaled in oxide form in both animals and in man because it has been used as a test material to study lung clearance. Some information on absorption from the respiratory tract was also found on other forms, such as the chloride. (262) Absorption parameter values and types, and associated fA values for particulate forms of iron are given in Table 7.2. Absorption parameter values for inhaled and ingested iron. It is assumed that the bound state can be neglected for iron, i.e. fb=0.0. The value of sr for Type F forms of iron (100 d−1) is element-specific. The values for Types M and S (3 d−1) are the general default values. Materials (e.g. ferric chloride) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied; i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of iron (0.1). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.1) for ingestion of the radionuclide.
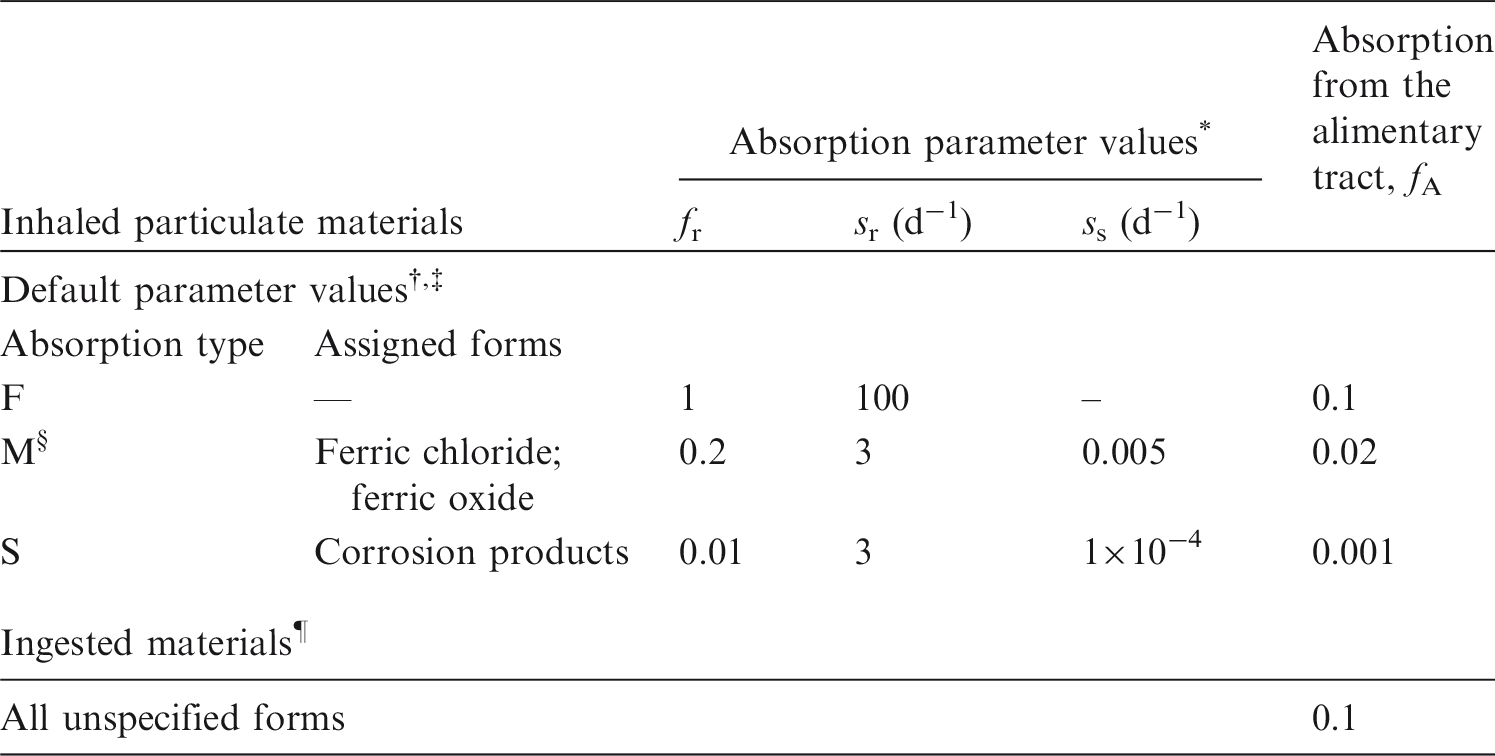
7.2.1.1. Particulate materials
(a) Iron chloride (FeCl3)
(263) Morrow et al. (1968) followed lung retention of 59Fe for 7 d after inhalation of 59FeCl3 by dogs and rats, but few details are given. Lung retention in dogs was represented by a two-component exponential function with half-times of 1.9 d (17%, clearance rate 0.36 d−1) and 85 d (clearance rate 0.0081 d−1), giving predicted lung retention at 30 d and 180 d of 65% and 19% ILD, and indicating Type M behaviour.
(b) Iron oxide (Fe2O3)
(264) Radiolabelled Fe2O3 has been used as a test material in many studies of the respiratory tract deposition and clearance of inhaled particles, including several human studies of lung retention lasting for 2–8 months [see review in Publication 66, Annex E, Table E.19 (ICRP, 1994)]. Over this period, retention could be represented adequately by a single exponential function, with a half-time of approximately 60–600 d, but less than 200 d in most cases, indicating Type M behaviour. The results are difficult to interpret as the retention followed was that of the label, which varied; in some cases, the label was 51Cr (Albert et al., 1967; Morrow et al., 1967a,b; Waite and Ramsden, 1971a; Ramsden and Waite, 1972), and in one case, the label was 237Pu (Waite and Ramsden, 1971b; Ramsden and Waite, 1972). As observed in Publication 30 (ICRP, 1980), this raises questions about the contributions to retention made by the iron oxide particle matrix itself, and by the chemical form of the label. However, after careful correction for leaching of the label, Ramsden and Waite (1972) estimated a retention half-time for the iron oxide matrix of approximately 270 d. (265) Some studies used material labelled with 59Fe itself. Results following inhalation of 59Fe2O3 by rats and dogs showed that lung retention could be fit by a single exponential with a rate of 0.01 d−1 (t½∼70 d) (Gibb and Morrow, 1962; Morrow et al., 1964, 1968). Calculations carried out here (i.e. by the Task Group) indicate that lung retention at 30 d and 180 d would be 71% and approximately 13% ILD. Similar experiments performed on rats showed similar results with a clearance rate of 0.011–0.013 d−1 (Muhle and Bellman, 1986). Other studies where 59Fe-labelled iron oxide particles were inhaled periodically by rats showed that lung retention followed a single exponential function with a rate from 0.008 to 0.011 d−1, depending on the age of the animals (Bellmann et al., 1991). (266) Studies on the retention of instilled iron oxide particles in human alveolar macrophages indicated that particles were cleared from the lungs with a rapid-phase clearance rate of 1.4 d−1 and a long-term clearance rate of approximately 0.006 d−1 (Lay et al., 1998). All these results indicate Type M behaviour.
(c) Magnetite (Fe3O4)
(267) Ferromagnetic iron oxide particles, Fe3O4, have also been used as a test material in studies of the lung retention of inhaled particles, measured using magneto-pneumography, i.e. measurement of the remanent magnetic field from particles within the chest, after application of a strong magnetic field. The results of measurements made in groups of volunteers for up to approximately 1 y after inhalation (Cohen et al., 1979; Freedman et al., 1988; Möller, 1991; Stahlhofen and Möller, 1991; Möller et al., 2001, 2004, 2006) are consistent with assignment to Type M. In particular, Möller et al. (2001) measured long-term retention of ferromagnetic iron oxide particles in healthy and diseased subjects. In healthy non-smokers, on average less than 10% ILD cleared from the lungs rapidly (within 2 d). This fraction was somewhat greater (10–20%) in smokers and patients with sarcoidosis, and considerably greater in patients with idiopathic pulmonary fibrosis (∼30% ILD) and chronic obstructive bronchitis (∼50% ILD). The mean (± standard deviation) half-time of the slow phase of lung clearance varied between groups as follows: young (20–39 y) healthy non-smokers, 124 ± 66 d; young cigarette smokers, 220 ± 74 d; older (40–65 y) healthy non-smokers, 162 ± 120 d; older smokers, 459 ± 334 d; patients with sarcoidosis, 275 ± 109 d; patients with idiopathic pulmonary fibrosis, 756 ± 345 d; and patients with chronic obstructive bronchitis (mostly ex-smokers), 240 ± 74 d. As lung clearance in healthy subjects was faster than measured in healthy human volunteers with inert particles like Teflon (Philipson et al., 1996), it was concluded that lung clearance was determined by particle dissolution in alveolar macrophages, which was impaired by cigarette smoking and the diseases investigated.
(d) Contaminated dusts (‘residues’) formed at nuclear power plants
(268) The biokinetics of 59Fe was followed for 84 d after intratracheal instillation into rats of a suspension of corrosion product ‘crud' particles (oxide-bearing debris, 5% 59Fe activity) from the primary containment of a water-cooled reactor (Collier et al., 1994). Few details are given, but it was assessed here that the results indicate Type S behaviour of the 59Fe present.
(e) Welding fumes
(269) Kalliomäki et al. (1978, 1983a, 1985) used magneto-pneumography to measure the lung contents of magnetic dusts in groups of welders with similar exposures. A single exponential model was applied to lung retention. Repeated measurements over a 6 y period on welders who worked with mild steel gave a clearance constant of 0.2 y−1 (t½∼3.5 y). Results of a cross-sectional study on stainless steel welders gave a half-life of 8.5 y. Both indicate Type S behaviour for at least some of the material. (270) To simulate occupational exposure, rats inhaled fumes from manual metal arc (MMA) or metal inert gas (MIG) welding of stainless steel for 1 h per working day for 4 weeks (Kalliomäki et al., 1983b,c). Lung contents of iron, chromium, manganese, and nickel were measured by neutron activation analysis for 106 d after the end of exposure. Retention of exogenous iron (i.e. that derived from the welding fumes) was also followed by magneto-pneumography. For the MMA welding fumes, results indicate Type M behaviour for all elements measured, except iron measured by neutron activation analysis (Type S). Clearance was slower following inhalation of MIG welding fumes, indicating Type S for all elements studied, except iron measured by magneto-pneumography (Type M). (271) Kalliomäki et al. (1986a,b, 1987) followed lung retention of 59Fe, 51Cr, and 58Co (as indicators of iron, chromium, and nickel, respectively) in rats for 106 d after intratracheal instillation of neutron-activated fumes from MMA or MIG welding of stainless steel or mild steel (59Fe only). Results indicate Type S behaviour for the 59Fe present in all fumes studied except MMA (mild steel) (Type M), Type S behaviour for the chromium and nickel present in MIG (stainless steel) fumes, and Type M for these elements in MMA (stainless steel) fumes.
(f) Other compounds
(272) Measurements following inhalation of neutron-activated fly ash by hamsters indicate Type M behaviour for the 59Fe present (Wehner and Wilkerson, 1981). Measurements following inhalation of neutron-activated volcanic ash by rats indicate Type M or S behaviour for the 59Fe present (Wehner et al., 1984).
7.2.1.2. Rapid dissolution rate for iron
(273) Little experimental information is available except for iron oxide, which is relatively insoluble. Although there is some experimental information for ferric chloride, which is probably absorbed more rapidly, it is insufficient to estimate the rapid dissolution rate. There is therefore no justification for choosing a rate different from the general default value of 30 d−1, which is applied here to all Type F forms of iron.
7.2.1.3. Extent of binding of iron to the respiratory tract
(274) The only experimental information for iron administered in solution relates to ferric chloride. This indicates Type M behaviour, suggesting that there could be significant binding of iron. However, there is insufficient information to estimate the extent of any bound state. Although it is not clear that the bound state for iron is negligible, it is assumed by default that fb = 0.
7.2.2. Ingestion
(275) The gastrointestinal absorption of iron has been studied extensively due to its important role in nutrition. (276) Freiman et al. (1963) reported a mean absorption value of 0.7 for a group of 16 volunteers aged between 27 and 60 y. Brozovic (1975) reviewed data from radioactive iron uptake studies involving a total of 990 normal human volunteers, and concluded that absorption values of 0.05–0.1 are usual. However, individual studies produced mean figures as high as 0.4 for men and 0.6 for women. Some of the variation may be caused by differences in the techniques used to measure absorption, but much of it is caused by dietary and physiological factors as reviewed by Brozovic (1975), Underwood (1977), Morris (1983), Lynch (1984), Cook et al. (1991), Whiting (1995), and Teucher et al. (2004). Human milk and organic acids (e.g. ascorbic, lactic, citric) are enhancers of iron absorption, while dietary fibres (e.g. pectins, cellulose), tannates in tea, polyphenols in coffee, and even calcium supplements in diet are potent inhibitors. Similarly, lowered iron status of the individual results in increased iron uptake, as shown by menstruating women and individuals with anaemia. Uptake is also increased during pregnancy. These latter points, associated with hormonal differences, result in higher iron absorption in females compared with males (Brozovic, 1975; Woodhead et al., 1991; Fletcher et al., 1994). (277) In some circumstances, iron is known to be retained in the wall of the small intestine. Study of whole-body retention of 59Fe in human volunteers after oral administration provided evidence of temporary retention of approximately 20% of the ingested 59Fe (Werner et al., 1987; ICRP, 2006). It was suggested that this iron was incorporated by macrophages lying under the epithelial layer and then transferred to goblet cells before being excreted back in the lumen of the intestine. All of these data are consistent with a half-time of intestinal retention of approximately 3 d (ICRP, 2006). (278) This iron retention in the intestine wall seems to be dependent on the iron status, and forms part of the mechanism operating to regulate iron absorption (Werner et al., 1987). (279) In Publication 30 (ICRP, 1980) and Publication 69 (ICRP, 1995), an absorption value of 0.1 was recommended for both males and females. An fA value of 0.1 is applied in this publication to all forms of ingested iron.
7.2.3. Systemic distribution, retention, and excretion
7.2.3.1. Overview of normal iron metabolism
(280) The biokinetics of iron has been investigated extensively in healthy human subjects as well as patients with iron deficiency or overload. The following overview of the physiological functions and normal biokinetics of iron in the human body is based mainly on the authoritative treatise by Bothwell et al. (1979) [see also Saito et al. (1964), Green et al. (1968), Munro and Linder (1978), Trubowitz and Davis (1982), and Barton and Edwards (2000)]. (281) The mass of iron in the human body is typically approximately 3.5–4.0 g in adult males and 2.0–2.5 g in adult females. The small mass of iron in the body does not reflect its important role in many physiological functions. This small mass is usually sufficient to maintain the normal physiological functions of iron because systemic iron has low rates of entry into the urinary bladder, gastrointestinal contents, and other excretion pathways, and is reused repeatedly by the body. (282) The body’s iron content may be divided into two categories: essential (functional) iron and storage iron. (283) Essential iron is the portion of the body’s iron representing integral components of molecules that fulfil well-defined physiological functions. For example, iron is an essential component of the oxygen-carrying proteins haemoglobin and myoglobin, and of numerous haem and non-haem enzymes involved in metabolic processes. The adult human body typically contains 30–40 mg of essential iron per kilogram of body mass. Approximately 80–85% of this is found in haemoglobin within the RBCs, and approximately 10–12% is found in myoglobin within muscle and other tissues. The remainder is distributed throughout the body tissues as haem enzymes (2–3% of body iron) and non-haem enzymes (3–4% of body iron). Essential iron typically represents approximately two-thirds of all iron in the body of adult males, and four-fifths or more of all iron in the body of premenopausal adult females. (284) Storage iron is an iron reserve in the body that assures an adequate supply of iron for normal physiological processes during periods of unusually low intake or rapid loss. It is stored as ferritin and haemosiderin, which hold iron in a relatively non-reactive form. Storage iron is located mainly in two tissues: the reticuloendothelial (RE) system and hepatic parenchyma. In most situations where body iron is increased, storage iron accumulates in both parenchymal and RE cells. The only condition in which selective parenchymal overload occurs is idiopathic haemochromatosis, in which there appears to be an associated defect in the way in which RE cells handle iron, with the result that RE stores are disproportionately small. (285) Typical iron requirements in males (i.e. uptake to blood from diet) are approximately 1.2 mg d−1, or 6% of a typical daily intake of 20 mg by an adult male. Iron balance is favourable in the adult male, as reflected by the rarity of nutritional iron deficiency in males. By 30 y of age, there is usually a reserve store of iron on the order of 1 g in males. (286) Iron balance is less favourable in the adult premenopausal female due to loss of circulating iron via menstruation. The amount of dietary iron required to replace this loss varies greatly, but the median value is probably approximately 0.4–0.5 mg d−1. The total daily requirement in the female is typically approximately 1.4 mg, but variation is great. Whole-body iron in the adult female is typically approximately 38 (34–42) mg kg−1. This corresponds to approximately 2300 mg of whole-body iron in a 60 kg female. Essential iron in the adult female is approximately 33 mg kg−1. This concentration is 10–20% lower than that in the male, reflecting differences in red cell mass and a larger amount of myoglobin in muscle in the male. The mean hepatic non-haem iron concentration is estimated as 0.1 mg g−1 liver in women, compared with approximately 0.27 mg g−1 liver in men. The average marrow storage of iron has been estimated as approximately 300 mg in adult males and 100 mg in adult females. (287) Iron is distributed within the body by blood plasma. Nearly all plasma iron is bound to the transport protein transferrin. The removal half-time of transferrin iron from plasma to tissues is approximately 90 min. Most of the transferrin-bound iron leaving plasma enters a circuit starting in the erythroid marrow. A portion enters the extravascular spaces and returns to plasma mainly via the lymphatics. The rest is delivered to the parenchymal tissues, mainly the liver. (288) The erythroid marrow takes up transferrin iron from plasma for incorporation into haemoglobin. Most of this iron appears in circulating RBCs in the next few days and remains there for the life of the cells. The life span of RBCs is typically approximately 4 months. The portion that does not appear in circulating RBCs consists of defective cells or extruded components of developing cells. This portion, called the wastage iron of erythropoiesis, typically represents 20–30% of iron that enters the erythroid marrow. This portion is collected by the body’s RE system, degraded, and returned to plasma.
7.2.3.2. Biokinetic model for systemic iron
(289) The structure of the systemic model for iron used in this publication is shown in Fig. 7.1. Baseline transfer coefficients are listed in Table 7.3. The model structure and parameter values have been modified slightly from a model developed to compare the normal biokinetics of iron with its biokinetics in individuals with haemochromatosis (Leggett et al., 2000). The parameter values were based on data for adult males. (290) Parameter values describing the fate of iron in the first few weeks after entry into blood plasma were based on results of radioiron studies on reasonably healthy male subjects. After the parameter values governing the early kinetics of iron had been set, values controlling long-term retention and excretion were set for consistency with estimated contents of various iron pools in a male of age 50 y, estimated daily losses of iron along various excretion pathways, and the assumption that 0.9 mg of iron is absorbed each day from food. The normal 50-year-old male is assumed to have a whole-body iron content of approximately 3.9 g, and this is assumed to be divided among major iron pools as follows: erythrocytes, 2300 mg; liver hepatocytes, 400 mg; liver RE cells, 50 mg, RE cells of bone marrow, 320 mg; spleen (mainly RE cells), 80 mg; other RE cells, 300 mg; erythroid marrow, 80 mg; plasma transferrin, 2.9 mg; remaining plasma, 0.4 mg; and remainder of the body (including several of the compartments shown in Fig. 7.1), approximately 400 mg (Bothwell et al., 1979). The precise whole-body and compartmental contents calculated for age 50 y depend to some extent on the age at which the calculation is started and the assumed compartmental contents at that age. The compartment contents given above for a 50-year-old male are based on a starting age of 15 y, with the initial iron content of a given storage pool being 30% of the value indicated above for age 50 y and the initial iron content of any other pool being 80% of the value indicated above for age 50 y. (291) Iron absorbed from the gastrointestinal or respiratory tract or returning to plasma after degradation of RBCs or wastage iron by the RE system enters a compartment in blood plasma called ‘other plasma’, which represents plasma iron not bound to transferrin. Most of the iron in other plasma transfers to plasma transferrin, but small amounts transfer into urinary bladder contents and right colon contents. Iron is removed from plasma transferrin with a half-time of 90 min, with approximately 85% moving to erythroid marrow (marrow synthesis), 5% to the hepatic parenchyma (Liver 1), and 10% to Other 1 (extravascular transferrin), representing relatively rapidly exchanging extravascular spaces. (292) Iron is removed from marrow synthesis with a half-time of 2 d, with 70% transferring to RBCs and the remaining 30%, representing ineffective erythropoiesis, transferring to a marrow RE compartment called ‘marrow transit’. The removal of aging erythrocytes from the circulation is depicted as a transfer from RBCs to marrow transit, representing phagocytosis by RE cells, plus a smaller transfer (approximately 10% of the total) from RBCs to other plasma, representing intravascular breakage of RBCs and release of the haemoglobin into the plasma. Most of the iron entering marrow transit is returned to other plasma with a half-time of 12 h. To account for relatively long-term storage of iron throughout the RE system, a small fraction of iron leaving marrow transit is distributed to the RE storage compartments in marrow, liver, spleen, and other tissues called, respectively, ‘marrow storage’, ‘Liver 2’, ‘spleen’, and ‘Other 3’. Iron is removed from these storage sites to marrow transit (and, therefore, largely to other plasma) over a period of months. The use of marrow transit as a central compartment within the RE system is a simplification of the real events, in that destruction of RBCs (including red cell precursors) does not actually occur entirely in the marrow, and iron entering or leaving RE cells in the liver, spleen, and other extraskeletal sites is not actually channelled through the marrow. (293) In addition to the RE system, an important storage site for iron is the hepatic parenchyma, represented in this model (for normal iron kinetics) by Liver 1. This compartment receives 5% of the outflow from plasma transferrin. Iron entering Liver 1 is returned over a period of months to plasma transferrin, except for a small amount representing biliary secretion that transfers to the small intestine contents. (294) It is assumed that most (80%) of the iron that transfers from plasma transferrin to extravascular transferrin returns to plasma over the next day or two, but a portion (20%) is taken up by Other 2, representing functional or storage iron not accounted for by explicitly identified tissues and fluids. Other 2 is also used to account for losses of iron due to exfoliation of skin, sweating, and losses in urine associated with exfoliation of kidney cells. Iron in Other 2 that is not lost in excreta returns over a period of months to Other 1 (extravascular transferrin). (295) In addition to the excretion pathways indicated above, iron is lost from the body in erythrocytes that enter the gut or urinary bladder. According to the model, approximately two-thirds of iron losses are in faeces and the remainder is in skin, sweat, and urine in normal adult males. Transfer coefficients for systemic iron.* RBC, red blood cells; RE, reticuloendothelial. *The transfer from RBCs to ‘other excreta’ shown in Fig. 7.1 is included to address loss of iron in RBCs during menstruation when applicable. The transfer coefficient for the premenopausal adult female is 0.0003 d−1. Structure of the biokinetic model for systemic iron used in this publication. RE, reticuloendothelial; RBC, red blood cells; SI, small intestine.


7.2.3.3. Treatment of radioactive progeny
(296) Two isotopes of iron addressed in this publication have radioactive progeny that contribute significantly to dose coefficients for the parent radionuclide: 52Fe, with chain members 52mMn (t½ = 21.1 min) and 52Mn (5.59 d); and 60Fe, with chain members 60mCo (10.5 min) and 60Co (5.27 y). The models for manganese and cobalt produced in vivo are modifications of the models applied in the OIR series to these two elements as parent radionuclides. The model for internally deposited cobalt is described in the section on cobalt in this publication (Section 8.2.3.). The model for internally deposited manganese, which will appear in a later part of this series, is available in the open literature (Leggett, 2011). Both models were amended by the addition of compartments representing the spleen and red marrow, which are represented explicitly in the systemic model for iron. Modifications of the cobalt model were based on biokinetic data for this element developed by Comar et al. (1946), Comar and Davis (1947), Barnaby et al. (1968), Smith et al. (1971), Hollins and McCullough (1971), Thomas et al. (1976), Kreyling et al. (1986), and André et al. (1989). Modifications of the manganese model were based on results of biokinetic or tissue distribution studies of this element by Fore and Morton (1952), Koshida et al. (1963), Tipton and Cook (1963), Furchner et al. (1966), and Dastur et al. (1971). (297) The compartment in the iron model called ‘other plasma’ is identified with the plasma compartment in the manganese model. Manganese produced in tissue compartments in the model for iron is assumed to transfer to plasma with the following half-times: 1 min for the blood compartment of the iron model that is not included in the manganese model (plasma transferrin), 83.2 d for RBCs (based on a mean lifetime of 120 d for RBCs), and 2 d for all other iron compartments (the removal half-time to blood for most compartments in the characteristic model for manganese). Manganese is assumed to leave plasma at a rate of 1000 d−1, with 30% going to liver, 5% going to kidneys, 5% going to pancreas, 1% going to right colon contents, 0.2% going to urinary bladder contents, 0.5% going to bone surface, 0.02% going to RBCs, 0.1% going to brain, 0.3% going to spleen, 0.1% going to red marrow, and the remaining 57.78% going to other soft tissue. The liver is divided into two compartments: Liver 1 and Liver 2. Manganese depositing in the liver is assigned to Liver 1. Manganese is removed from Liver 1 with a half-time of 1 d, with 20% of outflow going to small intestine contents via biliary secretion and 80% entering Liver 2. Activity transfers from Liver 2 to plasma with a half-time of 2 d. Activity is removed from pancreas with a half-time of 1 d, with outflow equally divided between plasma and small intestine contents. The transfer from pancreas to small intestine contents represents secretion in pancreatic juice. Activity transfers from kidneys to plasma with a half-time of 2 d, and from brain to plasma with a half-time of 150 d. The removal half-time from RBCs is 83.2 d, as assumed for manganese produced by decay of iron in RBCs. Activity depositing on bone surfaces is divided equally between cortical and trabecular surfaces, and leaves bone surfaces with a half-time of 40 d, with 99% returning to plasma and 1% entering the corresponding bone volume compartment. Activity is removed from cortical or trabecular volume at the reference turnover rate for the specific bone type in adults as given in Publication 89 (ICRP, 2002). Other soft tissue is divided into ST0, ST1, and ST2 representing fast, intermediate, and slow turnover of manganese. ST1 receives 14.6% of activity leaving plasma, ST2 receives 4%, and ST0 receives 39.18% (the amount remaining after all other deposition fractions in the model were assigned). Activity is returned from ST0, ST1, and ST2 to plasma with half-times of 30 min, 2 d, and 40 d, respectively. (298) Cobalt produced in tissue compartments in the model for iron is assumed to be transferred to the central blood compartment in the cobalt model (identified with other plasma in the iron model) with the following half-times: 1 min for RBCs and plasma transferrin, 2 d for compartments of the liver, 30 d for spleen and compartments of red marrow, and 7 d for other compartments. The subsequent biokinetics of cobalt entering or produced in the central blood compartment is described by the systemic model for internally deposited cobalt (see the section on cobalt in this publication (Section 8.2.3.)), with the following modifications for application to cobalt as a progeny of iron. The spleen and red marrow are each added to the model as individual compartments that exchange cobalt with the central blood compartment. These compartments are assumed to receive 0.5% and 1% of outflow from the central blood compartment, respectively. Depositions in the compartments of other soft tissues with relatively fast and intermediate turnover rates are reduced from 9% and 5%, respectively, in the original model to 8% and 4.5%, respectively. Cobalt is removed from the spleen and red marrow to the central blood compartment with a half-time of 30 d.
7.2.3.4. Differences between sexes
(299) The premenopausal adult female typically absorbs a greater portion of dietary iron and has faster turnover of body iron than the adult male due to higher iron requirements. The mass of whole-body iron is typically 50–100% greater in the adult male due to the combination of a larger body mass and a substantially larger mass of storage iron than the adult female. Despite the higher fractional uptake of iron from diet by females, the mass of storage iron in the premenopausal adult female is typically only approximately one-quarter of that in the adult male due to lower dietary intake of iron by females and substantial losses of iron via menstruation (Bothwell et al., 1979).
7.3. Individual monitoring
(300) 59Fe is a high-energy gamma emitter. Monitoring of 59Fe is generally accomplished through whole-body measurement. Urine bioassay monitoring is also used in monitoring for 59Fe (Table 7.4). Monitoring techniques for 59Fe.

7.4. Dosimetric data for iron
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 59Fe compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 59Fe in total body and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
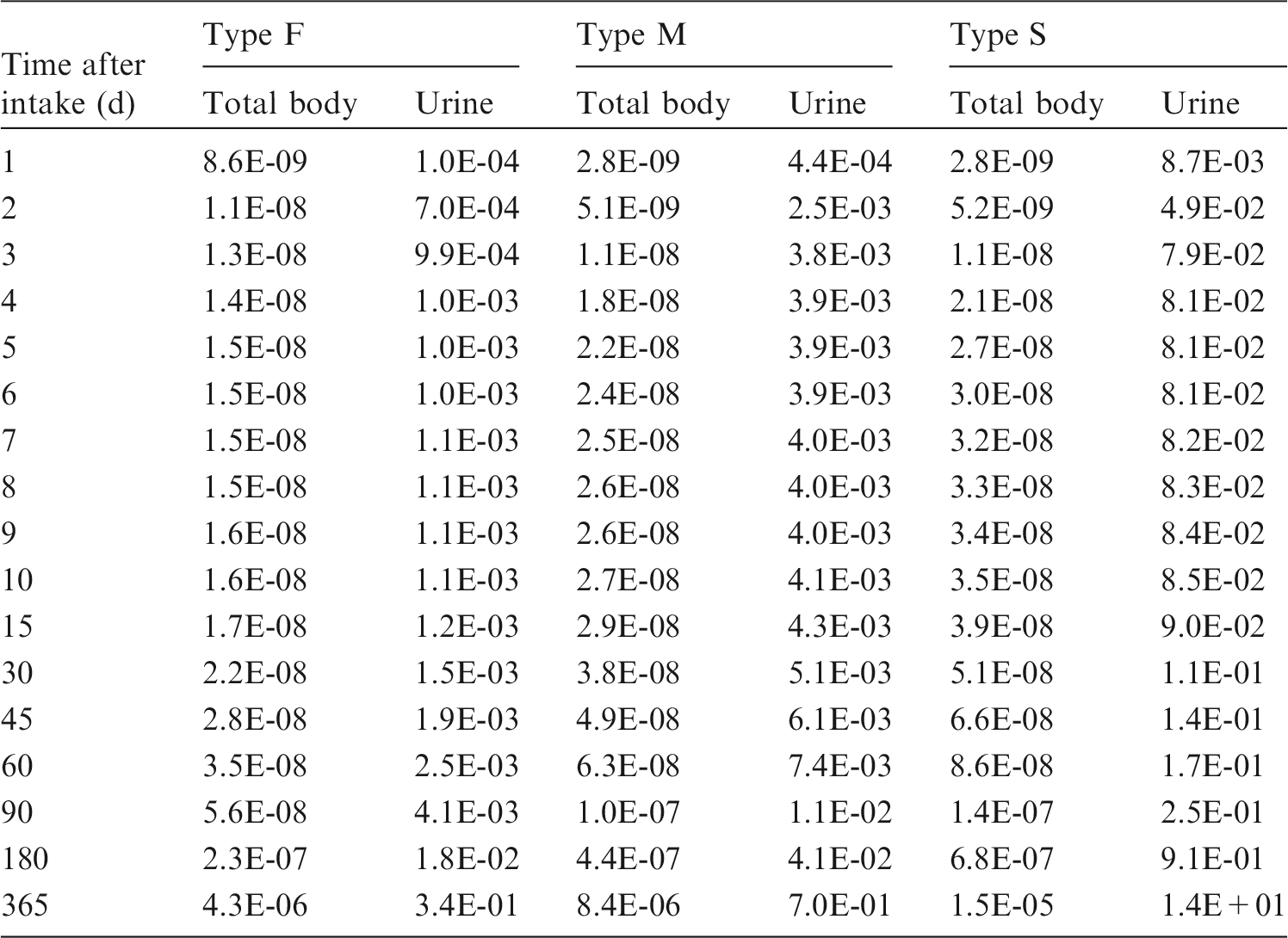

Total body content and daily urinary excretion of 59Fe following inhalation of 1 Bq Type F.

Total body content and daily urinary excretion of 59Fe following inhalation of 1 Bq Type M.
Total body content and daily urinary excretion of 59Fe following inhalation of 1 Bq Type S.
7.5. References
8. COBALT (Z = 27)
8.1. Chemical forms in the workplace
(301) Cobalt is a transition metal that occurs mainly in oxidation states II and III. Cobalt may be encountered in industry in a variety of chemical forms, including metal dusts, oxides (CoO, Co3O4), and soluble salts such as nitrates and chlorides. (302) 60Co is an important activation product produced in nuclear power plants, and could also be present in fragments of irradiated fuel. (303) Significant quantities of 57Co and 60Co are used as sealed sources in medicine (nuclear medicine, radiotherapy) and in the food industry for sterilisation. Table 8.1 shows the isotopes of cobalt addressed in this publication. Isotopes of cobalt addressed in this publication. Dose coefficients and bioassay data for these radionuclides are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay
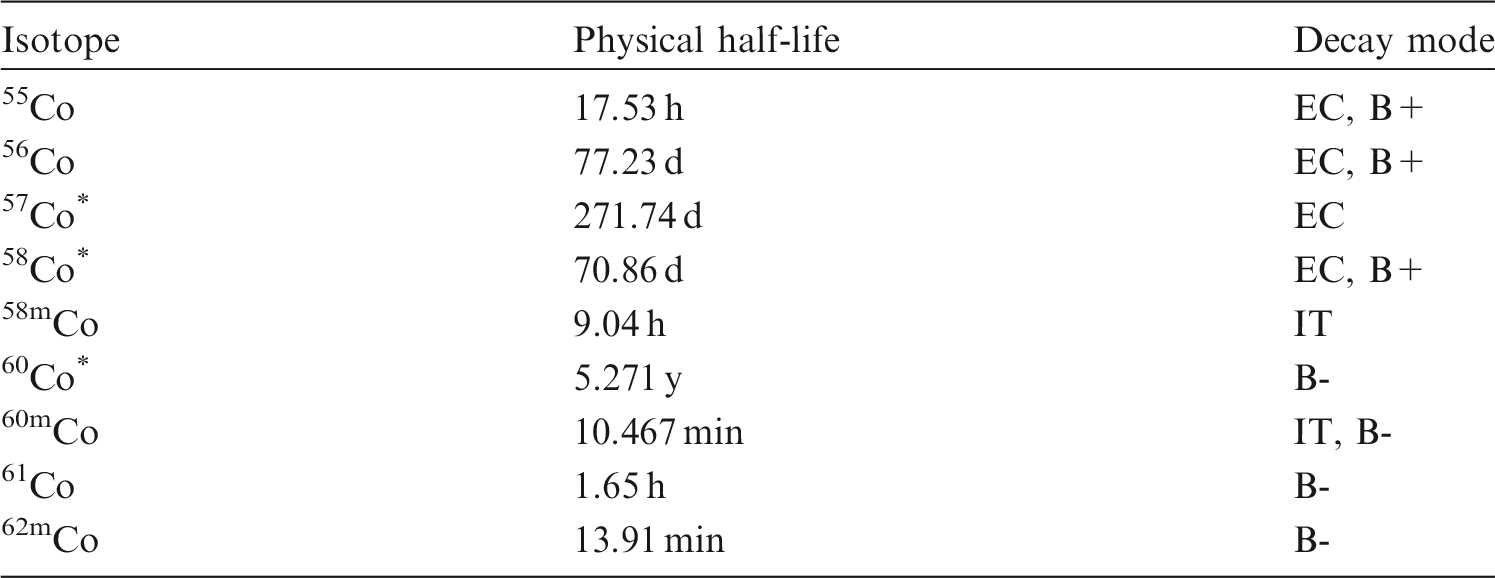
8.2. Routes of intake
8.2.1. Inhalation
(304) 60Co is relatively easy to measure, and there have been a number of reported studies of its lung retention following accidental inhalation, usually of an oxide. Information on absorption from the respiratory tract is available from experimental studies of cobalt in a variety of forms, including nitrate, chloride, and oxides. (305) Absorption parameter values and types, and associated fA values for particulate forms of cobalt are given in Table 8.2. Absorption parameter values for inhaled and ingested cobalt. FAP, fused aluminosilicate particles; PSL, polystyrene. It is assumed that, for cobalt, the bound fraction fb is 0.03, and an uptake rate sb=0.002 d−1 is applied to material in the alveolar-interstitial region and thoracic lymph nodes. It is assumed that fb=0.0 for material in the posterior nasal passage, pharynx, and larynx; bronchial and bronchiolar regions; and extrathoracic lymph nodes. The values of sr for Type F, M, and S forms of cobalt (1 d−1) are element-specific. Materials (e.g. cobalt nitrate) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied; i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of cobalt (0.1). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the highest value for any form of the radionuclide (fA=0.1).

8.2.1.1. Particulate materials
(a) Cobalt nitrate [Co(NO3)2]
(306) Kreyling et al. (1986) followed the biokinetics of 57Co for 1000 d after inhalation of 57Co-labelled Co(NO3)2 by dogs. Most of the ILD was rapidly cleared from the lungs and excreted from the body, mainly in urine. Lung retention was described by a three-component exponential function with biological half-times of 0.8 d (89%), 27 d (8%), and 400 d (3%). From the results of a complementary gavage experiment with Co(NO3)2, it was calculated here (i.e. by the Task Group) that fractional absorption from the alimentary tract was fA = 0.3. [In carrying out assessments here, the systemic model for cobalt described by Leggett (2008) was used, but to fit the nitrate data, it was necessary to increase the transfer rates from blood to urine and intestine.] Assuming that the cobalt retained in the lungs was bound, rather than particulate (see below), and hence that fr = 1, the analysis here gave parameter values of sr = 1 d−1, fb = 0.03, and sb = 0.0017 d−1, giving assignment to Type F. The estimated value of sb reflects the biological half-time of the slowest term in the three-exponential representation of lung retention. (307) Although specific parameter values for cobalt nitrate based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, cobalt nitrate is assigned to Type F. However, the data are used as the basis for the default rapid dissolution rate for cobalt, and with the data on cobalt chloride (see below) are used as the basis for bound-state parameter values for cobalt. Hence, specific parameter values for cobalt nitrate would be the same as default Type F cobalt parameter values.
(b) Cobalt chloride (CoCl2)
(308) Morrow et al. (1968) followed lung retention for 7 d after inhalation of 58CoCl2 by dogs. Few details are given, but a lung retention half-time of 0.01 d was reported, giving fr∼1, sr = 70 d−1, and assignment to Type F. (309) Menzel et al. (1989) followed lung retention for 6 d after inhalation of stable CoCl2 by rats. By that time, approximately 5% of the amount present at the end of exposure remained, but the authors recognised that some clearance took place during exposure. Assuming that the cobalt retained in the lungs was bound, rather than particulate, and hence that fr = 1.0, the analysis here gave parameter values of sr = 4 d−1 and fb ≤ 0.1. sb could not be determined because of the short duration of the measurements. (310) Kreyling et al. (1987) followed the biokinetics of 57Co for 120 d after intratracheal instillation of 57CoCl2 into hamsters to investigate the retention of cobalt in the lungs and extrapulmonary airways observed by Kreyling et al. (1986, see above). Additional information on this experiment is provided by Patrick et al. (1994). Most of the ILD cleared rapidly; approximately 1% ILD was present in the body after 1 month, with high concentrations of 57Co in tracheal and bronchial cartilage, and 0.15% ILD was present in the lungs after 120 d. From results of a complementary gavage experiment with CoCl2, it was calculated here that fA = 0.08. At 1 month after administration, the concentration of 57Co in the lungs was approximately 4 and 40 times the average in the body for gavage and instillation, respectively. Thus, there was some systemic uptake into the lungs following gavage. However, assuming a similar fraction was transferred from blood to lungs after instillation, it would account for only a small fraction of that retained in lungs in the instillation experiment. Assuming that the cobalt retained in the lungs was bound, rather than particulate, and hence fr = 1, the analysis here gave parameter values of sr = 1.4 d−1, fb = 0.015, and sb = 0.015 d−1. (311) Patrick et al. (1994) conducted an interspecies comparison of the lung clearance of ionic cobalt, primarily to determine whether differences in absorption of 57Co following inhalation of 57Co3O4 (Bailey et al., 1989; Kreyling et al., 1991, see below) could be explained by differences in binding of dissolved cobalt. To complement the studies by Kreyling et al. (1986, 1987) in dogs and hamsters (see above), the biokinetics of 57Co was followed for 100 d after intratracheal instillation of 57CoCl2 into guinea pigs, rats (two strains), and a baboon. Autoradiography of the tracheas of rats and a guinea pig 30 d after instillation of 57CoCl2 into the lungs showed that 57Co was mainly concentrated in cartilage rings. For one strain of rat, data are available to show that the proportion of 57Co retained in the lungs at 21 d after systemic injection was 1.2% of the whole-body content (Patrick et al., 1989), compared with 20% at 30 d after 57CoCl2 was instilled into the lungs. This indicates that while some of the 57Co retained in the lungs was from the systemic circulation, most came directly from deposition in the lungs. Assuming that the cobalt retained in the lungs was bound, rather than particulate, and hence fr = 1, analysis here gave values of sr in the range 0.6–0.9 d−1, and the following parameter values for the bound state: Guinea pig, fb = 0.06, sb = 0.013 d−1; HMT rat, fb = 0.03, sb = 0.009 d−1; F-344 rat, fb = 0.016, sb = 0.012 d−1. (312) Although specific parameter values for cobalt chloride based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, cobalt chloride is assigned to Type F. Estimates of the default rapid dissolution rate cover a wide range (from ∼1 to 70 d−1), but the lower values, which are based on more detailed information, are similar to the default rapid dissolution rate chosen for cobalt (see below). The data are used, with data on cobalt nitrate (see above), as the basis for bound-state parameter values for cobalt. Hence, specific parameter values for cobalt chloride would be similar to default Type F cobalt parameter values.
(c) Cobalt oxide
(313) Barnes et al. (1976) followed the biokinetics of 60Co in dogs for 128 d after inhalation of cobaltosic oxide (Co3O4), and for 64 d after inhalation of cobaltous oxide (CoO). The oxides were produced from cobalt nitrate aerosol heated at 850℃ and 1400℃, respectively, before inhalation. Lung clearance of CoO was faster than that of Co3O4; after 8 d, 10% vs 85% ILD remained in the lungs, and after 64 d, 4% vs 60% ILD, indicating Type F and Type M behaviour, respectively. For both oxides, there was high faecal excretion of 60Co during the first 3–4 d, which represented material cleared from the upper respiratory tract, while urinary excretion exceeded faecal excretion after 5 d, reflecting the greater importance of dissolution than particle transport as a clearance mechanism. The authors considered it noteworthy that the 60CoO formed at 1400℃ was more soluble than the 60Co3O4 formed at 850℃, because aerosols formed at higher temperatures are generally less soluble than aerosols formed at lower temperatures. (314) Detailed studies have been conducted of the lung clearance kinetics of various physical forms of cobaltosic oxide (Co3O4), which has been used extensively as a test material to investigate factors that affect particle dissolution in the lungs (e.g. Kreyling et al., 1986, 1988). Kreyling et al. (1986) also found that cobalt oxide aerosols formed at higher temperatures are more soluble than aerosols formed at lower temperatures; the in-vivo dissolution/absorption of a mixed cobalt oxide consisting of Co3O4 and CoO (formed at 950℃) was significantly faster than that for pure Co3O4 particles (formed at 800℃) of similar size. (315) These studies included two direct intercomparisons of clearance in different mammalian species, one of which involved human volunteers, baboon, dog, guinea pig, rat, hamster, and mouse (Bailey et al., 1989), and the other involved baboon, dog, and rat (Kreyling et al., 1991). In these numerous experiments, different parameters were varied, including the specific surface area, which influences the dissolution rate of the compound (ranging from 0.6 to 30 m2 g−1), the AMAD (ranging from 0.8 to 3.5 µm), and the ILD (ranging from 1 to 2000 kBq, depending on species). (316) Generally, lung retention was longer in humans and baboons than in the other species (dogs, guinea pigs, three strains of rats, hamsters, and mice). Absorption from the human lung was consistent with assignment to Type M, as, in that study (Bailey et al., 1989), the test material was designed by means of its physical and chemical parameters to be moderately soluble (specific surface area > 6 m2 g−1); ss ranged from 0.0013 to 0.005 d−1. When the test material was selected to be less soluble (specific surface area < 6 m2 g−1), absorption in baboons and dogs was consistent with assignment to Type S (Kreyling et al., 1988, 1991); ss ranged from 0.0008 to 0.03 d−1. The in-vivo rate of dissolution/absorption in dogs was linearly related to the specific surface area of the particles ranging from 0.6 to 30 m2 g−1 (Kreyling, 1990). Human and baboon data followed the same linear correlation (Kreyling, 1992). The rate-determining step was shown to be intracellular particle dissolution in alveolar macrophages in all species (Kreyling et al., 1990; Kreyling, 1992). The results of two in-vitro dissolution tests with lung serum simulant (Collier et al., 1992) gave ss ranging from 0.0002 to 0.0036 d−1. (317) In more recent studies, 57Co3O4 (inhaled by dogs) was used as a moderately soluble test particle to investigate the effects of chronic exposure to sulphur-related environmental air pollution on respiratory defence mechanisms, including particle dissolution (Kreyling et al., 1992a, 1999; Heyder et al., 2009). It was found that the in-vivo dissolution rate decreased during exposure to the acidic sulphate component, but increased during exposure to the sulphite component, and also during combined exposure to the acidic sulphate component (6 h daily) and sulphite component (18 h daily). (318) Newton and Rundo (1971) followed retention of 60Co in the chest and/or whole body in five men for 0.4–11 y after accidental inhalation of the irradiated metal or its oxide. Estimated half-lives for the long-term clearance of cobalt from the chest were up to 17 y. Using the updated HRTM with the new particle transport model for the AI region (Gregoratto et al., 2010) for three subjects (followed for 2.5–9 y), good fits to the data were obtained here with Type S absorption. For the subject followed for 11 y, the analysis here showed that a slow dissolution rate lower than that of Type S was needed to fit the data; the best estimate was ss = (0 ± 5) × 10–5 d−1. (319) Gupton and Brown (1972) followed retention of 60Co for 4 y in the chest of a man who was exposed to 60Co oxide by inhalation during a period of approximately 6 months prior to the initial count, and following which there was no subsequent exposure. The analysis here showed that retention is predicted adequately by assuming Type S absorption, but a better fit is obtained with a higher dissolution rate ss = (8±2) × 10–4 d−1. (320) Beleznay and Osvay (1994) followed whole-body retention of 60Co in six workers for approximately 4 y, starting 1 d after a short exposure to an aerosol leaking from a hot cell in which a high-activity 60Co source was being manipulated. The authors considered that the aerosol was probably composed of metallic cobalt and cobaltic or cobaltosic oxide formed at 300–400℃ on the surface of the high-activity cobalt wire. Longitudinal profile scans on one subject showed that on the 15th day, a major part of the deposited activity was in the chest, but on the 80th day, this had decreased considerably, with an increase in systemic activity. The authors interpreted the long-term retention of 60Co in the body as mainly systemic. The analysis here showed agreement with the data for model predictions assuming Type M absorption (ss = 0.005 d−1).
(d) Fused aluminosilicate particles
(321) Fused aluminosilicate particles (FAP) or ‘fused clay’ particles have been used extensively as relatively insoluble particles in inhalation studies, both of biokinetics and of radiation effects. A natural clay mineral (montmorillonite) is labelled by ion exchange, and the labelled clay particles are heated to approximately 1100℃ to form aluminosilicate glass microspheres in which the label is incorporated. It has been demonstrated that when cobalt is incorporated into FAP, only a small fraction may be absorbed rapidly. The rest is retained within the particles and is absorbed slowly. Kreyling et al. (1988) followed the lung clearance of 57Co for 3 y after inhalation of 57Co FAP by dogs, and estimated a dissolution rate, ss, of 5 × 10−4 d−1. Kreyling et al. (1992a) followed the biokinetics of 60Co for 600 d after inhalation of 60Co FAP by dogs and estimated a dissolution rate of (9 ± 4) × 10−4 d−1. From measurements following inhalation of 57Co FAP in rats, the long-term dissolution rate, ss, was estimated to be 8 × 10−4 d−1, while an in-vitro dissolution test gave ss = 1.8 × 10−4 d−1 (Collier et al., 1988, 1992). Most of these results give assignment to Type S.
(e) Polystyrene
(322) As with FAP, it has been demonstrated that when cobalt is incorporated into a polystyrene matrix, most of it is retained within the particles and is absorbed extremely slowly, making it an exceptionally useful material for studying long-term particle transport from the lungs. Kreyling et al. (1992b) estimated a rate of dissolution of < 3 × 10−5 d−1 for 57Co-labelled polystyrene inhaled by dogs, but few details were given. Kreyling et al. (1999) and Heyder et al. (2009) used 58Co- and 60Co-labelled polystyrene as insoluble test particles to investigate the effects of chronic exposure to sulphur-related environmental air pollution on respiratory defence mechanisms in dogs, including particle clearance from the alveolar region. Kreyling et al. (1999) estimated dissolution rates of (1 ± 2) × 10−5 d−1 and (2 ± 2) × 10−5 d−1, respectively. All these results give assignment to Type S.
(f) Contaminated dusts (‘residues’) formed at nuclear power plants
(323) Raghavendran et al. (1978) followed retention of 60Co in four workers at the Bhaba Atomic Research Centre for between 400 and 1250 d. Profile scans showed most activity to be in the chest. Retention in the chest was fit by a one- or two-component exponential function, with long-term half-lives in the range 500–18,000 d, indicating Type S behaviour. (324) Hegde et al. (1979) reported information on chest measurements up to approximately 400 d for five inhalation cases of 60Co in boiling water reactor power station workers. Results for four workers were summarised with an average value of 664 d for the biological half-time. Predictions assuming Type S behaviour are in good agreement with the data. (325) Ramsden (1984) followed two cases of lung retention of 60Co for approximately 1500 d after inhalation of mixed corrosion oxide products from water reactor circuitry. The analysis here, using the updated HRTM, showed that a slow dissolution rate lower than that of Type S was needed to fit the data; the best estimate was ss = (1±0.5) × 10–5 d−1. (326) Davis et al. (2007) and Gregoratto et al. (2010) analysed the results of measurements (urine and faeces during the first 2 weeks, and whole body to 15 y) of 60Co in seven workers who inhaled particles of unknown form in the same incident at a nuclear power plant. The dataset is extraordinary in that a group of workers had a simultaneous, brief single inhalation exposure, and they have been followed for so long. In order to account for the later whole-body retention data in each subject, it was necessary to assume slower particle transport from the alveolar region than that assumed in the original HRTM (ICRP, 1994). This study is one of those on which the AI model in the revised HRTM is based (ICRP, 2015). Specific absorption parameter values were fit to the results for each subject by both Davis et al. (2007) and Gregoratto et al. (2010). Most were similar to those for default Type S, but to fit the early urine data, the fractional absorption in the alimentary tract could be no more than approximately 0.1%, and a slow dissolution rate lower than that of Type S was needed to fit the data; the best estimate was ss < 1 × 10−4 d−1. (327) The biokinetics of 60Co was followed for 6 months after intratracheal instillation into rats of a complex radionuclide bearing dust (72% 60Co activity) from the ventilation grid of a nuclear power plant reactor fuel hall (Stradling et al., 1996, 1997). Absorption parameter values (fr = 0.30, sr = 1.5 d−1, and ss = 5 × 10–4 d−1) derived by ICRP (2002a, Section E4.4) are consistent with assignment to Type M. However, as several human studies following intakes at nuclear power plants indicate Type S behaviour, these specific values do not seem representative and are not recommended for use in preference to default Type S. (328) The biokinetics of 60Co was followed for 280 d after intratracheal instillation into rats of a suspension of corrosion ‘crud' particles (oxide-bearing debris, 60% 60Co activity) from the primary containment of a water-cooled reactor (Collier et al., 1994). Few details are given, but it was assessed here that the results are consistent with assignment of the 60Co present to Type S. (329) Molokanov et al. (2010) reported in-vivo lung measurements of 60Co up to 200 d, and several urine and faecal data at approximately 200 d, for four nuclear power plant workers who accidentally inhaled a cobalt compound. No early data are available, but the slow clearance and the small amount in the urine indicate that the material was insoluble. A good fit to the data was obtained here with default Type S absorption but with an increased value for the slow absorption rate, ss = 3 × 10−4 d−1.
(g) Other compounds
(330) Clearance studies of cobalt in the rat after inhalation of neutron-activated fly ash (Griffis et al., 1981) or volcanic ash (Wehner et al., 1984) indicated leaching of cobalt out of the particle matrix, consistent with assignment to Type M. (331) Although numerous studies have been carried out on the toxicity of inhaled cobalt-containing alloys, no data are available from them on the clearance kinetics of cobalt. However, data obtained from diamond polishers (Van den Oever et al., 1990) or after exposure of rats (Brune and Beltesbrekke, 1980) suggest long-term retention in the lungs indicative of Type M or S behaviour.
8.2.1.2. Rapid dissolution rate for cobalt
(332) Most of the estimated values of the rapid dissolution rate, sr, from studies involving inhalation or instillation of cobalt nitrate and chloride into the lungs were in the range 0.6–4 d−1. The exception was the value of 70 d−1, based on a reported lung retention half-time of 0.01 d following inhalation of 58CoCl2 by dogs (Morrow et al., 1968); however, few details were given. Based on the other studies, a value of sr of 1 d−1 is applied here to all Type F forms of cobalt. As it is lower than the general default value of 3 d−1 for Type M and S materials, it is also applied to Type M and S forms of cobalt.
8.2.1.3. Extent of binding of cobalt to the respiratory tract
(333) Experimental evidence, described in the above sections on cobalt nitrate and chloride, consistently shows long-term retention of a few percent of the ILD of cobalt deposited in the lungs in soluble form. (334) Studies of the kinetics of cobalt following inhalation of cobalt nitrate (soluble) and oxides (moderately soluble) by dogs, and following instillation of cobalt chloride into the lungs of hamsters, showed much larger amounts in the tracheobronchial airways than expected for material transiting the tracheobronchial airways following clearance by particle transport from the alveolar region (Kreyling et al., 1986, 1987). Furthermore, the relative amount in the tracheobronchial airways within the lungs increased with the solubility of the material. Cobalt was also found to be distributed in the lungs after intravenous injection of oxide particles (Co3O4) in dogs (Kreyling et al., 1986). Measurements showed decreasing activity in liver with time, and increasing activity in lungs (and other soft tissues and bones). This suggests that it was not particles injected into blood which were directly absorbed by the lungs, but non-particulate cobalt, released into blood from liver (where particulate matter is incorporated and digested by Kupffer cells) and then absorbed in the lungs. (335) Studies were conducted to localise further the distribution of the cobalt retained in the lungs. A study of the detailed location of cobalt in the lungs of dogs at 14 d after instillation of Co(NO3)2 into one lung lobe showed that the retained cobalt was mainly located in the airway cartilage (Godleski and Kreyling, 1990). Autoradiographs of rats and guinea pigs at 100 d after instillation of CoCl2 (Patrick et al., 1994) showed the highest concentrations of cobalt to be in cartilaginous structures of the trachea and bronchi. (336) There is therefore strong evidence for a bound state for cobalt which can be quantified (although the location of the bound cobalt in cartilaginous structures is different from that assumed in the HRTM). Based on this evidence, retention and excretion data for cobalt nitrates and chlorides were analysed assuming that the cobalt retained in the lungs was bound, rather than particulate, and hence fr = 1.0. For cobalt chloride instilled into the lungs of rats and guinea pigs, and followed for 100 d, values of fb averaged 0.03 (range 0.016–0.06), and values of sb averaged 0.011 d−1 (range 0.009–0.013 d−1). For cobalt nitrate inhaled by dogs and followed for a much longer period (up to 1500 d), the bound fraction was estimated here to be fb = 0.03, clearing at a rate sb = 0.0016 d−1. (337) On the basis of these results, a bound fraction with fb = 0.03 and an uptake rate of sb = 0.002 d−1 is adopted here for cobalt. No experimental evidence was found to show that cobalt in soluble form deposited in the conducting airways is retained in a bound state. There is evidence that much of the cobalt deposited in the lungs in soluble form that is not absorbed rapidly is retained in airway cartilage. However, this is located some distance below the epithelial tissue that forms the designated source region for material bound in the airway regions (BB and bb). Locating the bound activity in the source region within the epithelium could substantially overestimate doses to the BB and bb regions. It is therefore assumed here that these bound-state parameter values only apply in the AI region and thoracic lymph nodes.
8.2.2. Ingestion
(338) Human volunteer studies with 60Co chloride (Paley and Sussman, 1963; Smith et al., 1972) showed that when cobalt was present in trace quantities (<1 µg Co), absorption was 0.05 or less but when larger amounts of cobalt were administered (1–12 mg), absorption was 0.1–0.3. A higher value of 0.44 (from 1.2 mg Co) was recorded by Valberg et al. (1969), and this was increased to 0.7 in volunteers suffering from iron deficiency. Similarly, Paley and Sussman (1963) noticed that fasting for 3 h or longer increased the absorption of cobalt by a factor of 2. (339) The absorption of cobalt in forms encountered in the workplace may be considerably lower than these values for relatively soluble inorganic forms. Chevalier and Gonin (1993) estimated the absorption of 60Co ingested as large particles of stellite following their inhalation; large particles deposited in the upper airways are swallowed rapidly, and absorption was assumed to take place solely from the gastrointestinal tract. The absorption values obtained for five subjects were in the range of approximately 10–3 to 10–4. Bailey et al. (1989) measured the absorption of 57Co as cobaltosic oxide (Co3O4) as part of a comparison of the behaviour of inhaled materials in different mammalian species. Estimates of absorption after intragastric administration of oxide particles with geometric mean diameters of 0.8 µm or 1.7 µm were in the range of approximately 0.01–0.05 for mice, hamsters, rats, guinea pigs, and baboons. Comparing the behaviour of 57Co nitrate and a mixed oxide containing Co3O4 and CoO in dogs, Kreyling et al. (1986) obtained results for urinary excretion of 57Co after intravenous injection and ingestion that suggested absorption of approximately 0.3 for the nitrate and 0.06 for the oxide. Collier et al. (1991) compared whole-body retention and urinary excretion of 57Co in rats from 3 weeks to 48 weeks of age after intravenous injection as the nitrate or intragastric administration as Co3O4 (1 µm particles). The results suggested absorption in the range 0.004–0.04 with the greatest values in the youngest animals. (340) In Publication 30 (ICRP, 1979), an f1 of 0.05 was recommended for oxides, hydroxides, and all other inorganic forms ingested in trace quantities. For inorganic forms other than oxides and hydroxides ingested in the presence of carrier material, a value of 0.3 was recommended, although the ingestion of large masses of soluble material would only be expected in exceptional circumstances. In Publication 67 (ICRP, 1993), a value of 0.1 was adopted for dietary intakes by adult members of the public. In this report, an fA value of 0.1 is adopted for direct ingestion of all chemical forms except insoluble oxides, for which an fA value of 0.05 is recommended.
8.2.3. Systemic distribution, retention, and excretion
8.2.3.1. Summary of the database
(a) Data for human subjects
(341) Smith et al. (1972) studied the behaviour of cobalt in 11 healthy adult subjects (10 males and one female) after intravenous injection with 60CoCl2. More than 90% of the injected amount was removed from plasma during the first 30 min. Over the next 30 h, activity in plasma declined with a half-time of approximately 1 d. The concentration of 60Co in plasma was one to two orders of magnitude higher than that in RBCs, but the investigators suggested that the measurement techniques may have underestimated 60Co in RBCs. Measurements of urinary and faecal excretion in six of the subjects during the first 2–8 d after administration revealed that activity was eliminated primarily in urine. The ratio of faecal to urinary excretion during the study period averaged approximately 0.15. Long-term retention in the whole body was estimated for three subjects by external measurements. Average retention for two subjects followed over 1000 d could be described reasonably well by a four-exponential function with the following biological half-times and component sizes: 0.5 d (44%), 6 d (32%), 60 d (13%), and 800 d (11%). External measurements on one subject soon after injection indicated that the liver accumulated approximately one-third of the injected amount. External measurements for eight subjects indicate that the liver contained approximately 20% (10–30%) of the whole-body burden at times from a few days up to 1000 d after injection. (342) Letourneau et al. (1972) used external whole-body measurements to estimate the rate of loss of 58Co from each of 16 male subjects over approximately 1 y (305–386 d) following intravenous injection of 58CoCl2. Estimated retention was slightly lower, on average, than determined in the study by Smith et al. (1972), although there was overlap in the range of retention data found in the two studies. On average, approximately 35–40% of the injected activity was lost with a biological half-time of a few hours, 25% with a half-time < 2 d, 20% with a half-time of approximately 8 d, 10–15% with a half-time of approximately 50 d, and 9% with a half-time of approximately 600 d. The size of the long-term component ranged from 5% to 13%, compared with 9% to 16% in three subjects of Smith et al. (1972) studied for at least 275 d. (343) Jansen et al. (1996) used positron emission tomography to study the early biokinetics of 55Co in two adult males, aged 26 and 30 y, after intravenous injection with 55CoCl2. Whole-body scans were made immediately (∼0.5 h), at 24 h, and at 48 h after injection. The liver and urinary bladder were estimated to contain approximately 50% and 40%, respectively, of the administered activity in the first scan. These values are qualitatively consistent with other human or animal studies in that they indicate rapid transfer of cobalt to the liver and urinary bladder, but are higher than estimated in most studies. (344) Newton and Rundo (1971) studied the behaviour of 60Co in five men for periods up to 11 y after accidental inhalation of the irradiated metal or its oxide. They estimated a long-term clearance half-time on the order of 7 y for systemic cobalt. Measurements on one of the subjects approximately 3 y after intake established the presence of 60Co in the skeleton. Activity was not detectable in the liver. (345) Beleznay and Osvay (1994) measured retention of 60Co in six workers from 10 to 1850 d after they accidentally inhaled 60Co aerosols during manipulation of a high-activity source. A retention component of 25–78 d was interpreted as activity leaving the deep lungs. A long-term component of retention determined in five of the workers followed for extended periods was interpreted as the slowest component of systemic retention of cobalt. The biological half-time of the long-term component varied from approximately 500 d to approximately 1200 d, and averaged approximately 900 d. (346) The collective data for human subjects indicate that the long-term half-time for cobalt taken into the body in inorganic form tends to increase with the length of the observation period: 600 d for observations over 305–386 d (Letourneau et al., 1972); 800 d for observations over approximately 1000 d (Smith et al., 1972); 900 d for observations up to 5 y (Beleznay and Osvay, 1994); and 7 y for observations up to 11 y (Newton and Rundo, 1971). This suggests that there is a component of retention with a biological half-time of many years. As described later, animal studies indicate that the skeleton retains a small portion of deposited cobalt for an extended period.
(b) Data on laboratory animals
(347) The biokinetics of cobalt has been studied in mice, rats, hamsters, guinea pigs, dogs, monkeys, and baboons. Differences between species are indicated. For example, Thomas et al. (1976) compared the biokinetics of cobalt in the mouse, rat, monkey, and dog following intravenous, intragastric, and oral administration of 60CoCl2. The long-term retention half-time was longer in the mouse (495 d) than in the rat (309 d), monkey (183 d), or dog (180 d). The investigators noted that the pattern was different than normally encountered in retention of trace metals, in that larger animals usually have longer retention times. (348) In dogs exposed by inhalation to 60Co aerosols (Barnes et al., 1976), the kidneys and liver showed much higher concentrations of 60Co than the skeleton at early times, but the relative concentration in the skeleton increased over a period of months. The contents of the liver, skeleton, and kidneys decreased in the order liver > skeleton > kidneys at early times, and in the order skeleton > liver > kidneys after 2–4 months. In dogs exposed to 57Co aerosols (Kreyling et al., 1986), the skeleton and muscle each contained several times more activity than the liver, and the kidneys contained approximately the same amount as the liver 1–5 y after exposure. (349) In rats given a single dose of 60CoCl2 by gastric intubation, the liver was initially the main repository, but by 2–4 months, the main measured repository was the skeleton, followed by muscle, liver, and kidney (Smith et al., 1971). In rats chronically exposed to 60Co in drinking water, the liver remained the dominant repository over 170 d, followed by the skeleton and muscle (Smith et al., 1971). Retention of 60Co by rats continuously exposed to 60Co in drinking water was consistent with the long-term whole-body retention component derived from single administration studies (Smith et al., 1971). (350) At 8 d after ingestion of 57Co3O4 particles by baboons, the skeleton and kidneys contained 0.6–1.1 times and 0.09–0.15 times, respectively, as much activity as the liver. At 6 months after inhalation of 57Co3O4 by baboons, the skeleton and kidneys contained 0.6–3 times and 0.1–0.3 times as much activity as the liver, respectively (André et al., 1989). (351) Animal studies reveal that the systemic biokinetics of cobalt depends on the chemical form injected into blood (Nishimura et al., 1976; Inaba et al., 1982). Nishimura et al. (1976) compared the behaviour of intravenously injected 60CoCl2 and 58Co cyanocobalamin in rats. At 21 d after administration of 60CoCl2, 26.4% of the body burden was found in the liver and 13.1% in the kidneys, and cumulative excretion was mainly in urine. At 21 d after intravenous administration of 58Co cyanocobalamin, the kidneys contained 38.8% of the body burden and the liver contained 14.6%; excretion of 58Co was mainly in faeces; and loss from the body was considerably slower than for inorganic cobalt. (352) In studies involving various animal species, more than half of 57Co injected as Co(NO3)2 was excreted in urine in the first 24 h, and more than two-thirds was excreted in urine during the first week (André et al., 1989; Bailey et al., 1989; Collier et al., 1989; Talbot and Morgan, 1989). Cumulative faecal excretion over the first week accounted for approximately 4–28% of the injected cobalt. Other animal studies also indicate that urine is the primary route of excretion of injected cobalt (Comar and Davis, 1947; Barnaby et al., 1968; Onkelinx, 1976; Thomas et al., 1976; Gregus and Klaassen, 1986; Kreyling et al., 1986). Excretion of cobalt in bile amounting to 2–7% of the initial systemic burden has been observed in dogs and rats (Sheline et al., 1945; Cikrt and Tichy, 1981; Gregus and Klaasen, 1986). (353) The distribution of 60Co was examined by autoradiography in tissues of pregnant mice intravenously injected with 60CoCl2 (Flodh, 1968). Sacrifice times were 1 h, 4 h, 24 h, 4 d, and 16 d after injection. Except where otherwise indicated, the following description refers to the mother rather than the fetus. At 1 h, the concentration of 60Co in blood was only approximately one-eighth of that in the liver. Disappearance from blood was gradual after 1 h but largely complete by 24 h. Cartilage showed a high concentration of activity at 1 h. The concentration of 60Co in the cartilage increased with time and was four times higher than in the liver by 4 d. From 24 h onwards, the cartilage in the trachea and larynx had the highest concentration. Bones of the skull, the periosteum of the vertebrae, and the pelvic bone also accumulated cobalt. The liver showed a high concentration at all times studied. Accumulation was high in the kidneys, with a peak at 4 h. Activity was localised mainly in the inner parts of the cortex. After 4 d, the kidney concentration was still as high as the liver. Accumulation in the mammary glands was high, approximately the same concentration as in the liver and kidneys. In the fetus, the radioactivity was localised mainly in the skeleton, with relatively high uptake in hyaline cartilage and cranial bones. According to the investigators, the distribution of inorganic cobalt in the mother was different from that seen in autoradiographic studies involving 58Co-labelled vitamin B12. (354) In animal studies involving administration of inorganic compounds of radiocobalt, relatively high concentrations of cobalt have generally been found in the liver, kidneys, skeleton, and skeletal muscle. The skeleton typically contains more than any other single organ or tissue by a few months after acute intake, indicating tenacious retention of a portion of the deposited activity. Following intraperitoneal, intravenous, or oral administration of 60CoCl2 to rats, the skeletal content decreased by a factor of 6–12 between days 1 and 30, and then showed little decline over the next few months (Barnaby et al., 1968; Thomas et al., 1976). Skeletal muscle showed a longer average retention time than most soft tissues, including the liver and kidneys. (355) In hamsters, rats, and guinea pigs, the liver and kidneys contained approximately 20–40% and 3–4%, respectively, of total body activity at 3 weeks after intravenous injection of 57Co(NO3)2 (Collier et al., 1989). In rats, the liver, skeleton, and muscle each contained approximately 20–25% and the kidneys contained approximately 7–8% of total body activity over 10–72 d after intraperitoneal injection of 58CoCl2 (Hollins and McCullough, 1971). At 386 d after intraperitoneal injection of 58CoCl2, the skeleton, liver, and kidneys contained approximately 65%, 7%, and 2%, respectively, of total body activity (Hollins and McCullough, 1971). (356) The systemic distribution of 57Co-labelled cobalt at 100 d after intraperitoneal injection of CoCl2 into rats depended strongly on the administered mass (Edel et al., 1994). After administration of 5 µg cobalt, the highest concentrations of 57Co were found in the spleen and pancreas, followed by the skull and femur. After administration of 1 mg cobalt, the skull and femur showed far higher concentrations than other tissues.
8.2.3.2. Biokinetic model for systemic cobalt
(357) A systemic biokinetic model for cobalt proposed by Leggett (2008) is applied in this publication. The model structure is shown in Fig. 8.1. Transfer coefficients are listed in Table 8.3. (358) Transfer coefficients were based, where feasible, on data from controlled human studies involving administration of inorganic forms of cobalt. Model predictions of whole-body retention, including different phases of loss from the body, were required to be consistent with central estimates based on combined data of Smith et al. (1972) and Letourneau et al. (1972) for human subjects injected with 60CoCl2 and 58CoCl2, respectively. Parameter values for blood were set for consistency with blood retention data of Smith et al. (1972) for subjects injected with 60CoCl2. Urinary and faecal excretion rates, and uptake and retention by the liver were based mainly on measurements by Smith et al. (1972) and Jansen et al. (1996) for subjects injected with 60CoCl2 and 55CoCl2, respectively. The data for human subjects were supplemented with information on the time-dependent distribution of cobalt among the liver, kidneys, skeleton, and other tissues in laboratory animals receiving inorganic forms of radiocobalt by inhalation, ingestion, or injection. For example, the initial distribution of systemic cobalt and the shift with time in its distribution were modelled after general patterns indicated by data on several animal species. Derivations of parameter values describing uptake and retention in specific repositories are summarised below. Structure of the systemic model for cobalt. ST, soft tissue. Transfer coefficients (d−1) for systemic cobalt. ST, soft tissue; surf, surface; vol, volume; SI, small intestine.
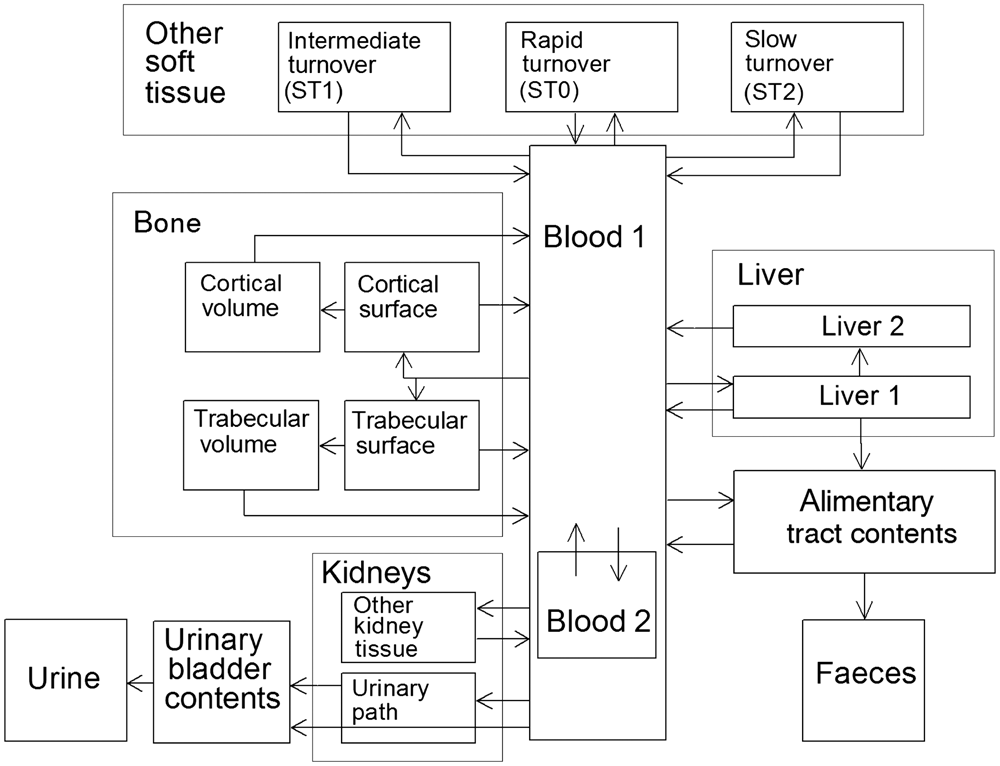

(a) Blood
(359) Blood is divided into two compartments: Blood 1 and Blood 2. Cobalt atoms entering blood are assigned to Blood 1, which is a rapid-turnover plasma pool. Blood 2 is a more slowly exchanging pool that contains the preponderance of activity in blood except for a short period soon after acute uptake of radiocobalt. These compartments are used to reproduce observed rates of disappearance of cobalt from blood, and are difficult to identify with specific components of blood. The relatively slow loss of a portion of injected cobalt from blood may be associated with retention by certain plasma proteins and RBCs, although data of Smith et al. (1972) indicate that RBCs contained, at most, a few percent of the blood content of 60Co during the first 30 h after intravenous injection of 60CoCl2 into human subjects. (360) Activity leaves Blood 1 at a rate of 200 d−1, corresponding to a half-time of approximately 5 min, with 6% of outflow going to Blood 2 and the remaining 94% divided among tissue compartments, urinary bladder contents, and colon contents. Activity moves from Blood 2 back to Blood 1 with a half-time of 1 d.
(b) Liver and faecal excretion
(361) The liver is represented as two compartments, Liver 1 and Liver 2, representing short- and long-term retention, respectively. Liver 1 receives 35% of activity leaving Blood 1. Activity is removed from Liver 1 with a half-time of 1.5 d, with 20% going to the small intestine contents in bile, 5% going to Liver 2, and 75% returning to blood. Activity transfers from Liver 2 to Blood 1 with a half-time of 1 y. Endogenous faecal excretion of cobalt arises from biliary secretion as indicated above, plus secretion from Blood 1 to the right colon. The latter transfer amounts to 2% of cobalt leaving Blood 1.
(c) Kidneys and urinary excretion
(362) The kidneys are divided into two compartments: Kidneys 1 and Kidneys 2. Kidneys 1 receives cobalt from blood after filtration through the glomerulus, representing 4.5% of outflow from Blood 1, and loses cobalt to the urinary bladder contents with a half-time of 1.5 d. The urinary bladder contents receive an additional 30% of outflow from Blood 1 that is filtered at the glomerulus but not retained in the kidneys. Kidneys 2 is a slow-turnover pool that receives 0.5% of outflow from Blood 1, and returns cobalt to Blood 1 with a half-time of 1 y.
(d) Skeleton
(363) Uptake and retention of cobalt in the total skeleton can be modelled on the basis of data from animal studies, but the distribution of cobalt between cortical and trabecular bone or between bone surfaces and bone volume has not been established. It is assumed that 3% of cobalt atoms leaving Blood 1 deposit on trabecular bone surfaces, and 3% deposit on cortical bone surfaces. Cobalt leaves bone surfaces with a half-time of 7 d, with 15% going to the corresponding bone volume compartment and 85% returning to Blood 1. Cobalt is removed from trabecular or cortical bone volume at the rate of bone turnover. Reference values for bone turnover rates are given in Publication 89 (ICRP, 2002b).
(e) Other tissues
(364) Remaining soft tissues are divided into three compartments: ST0, ST1, and ST2, with relatively fast, intermediate, and relatively slow turnover, respectively. These compartments receive 9%, 5%, and 2% of outflow from Blood 1, and return cobalt to Blood 1 with half-times of 7 d, 50 d, and 2 y, respectively. (365) The above parameters yield reasonable consistency between model predictions of retention and excretion, and observations in controlled human studies. Model predictions are also consistent with the following aspects of the biological behaviour of inorganic cobalt indicated by radiocobalt studies on human subjects and laboratory animals:
The peak content of liver is approximately one-third (model prediction ∼35%) of the intravenously injected amount, and occurs during the first hour after injection. A high rate of urinary excretion of cobalt occurs during the first hour or two after absorption or intravenous injection into blood (Apostoli et al., 1994; Jansen et al., 1996). The liver contains approximately 20% (model predictions 15–27%) of the total body burden at times from a few days up to 1000 d after injection. The kidneys and liver initially show similar concentrations of cobalt, but the kidney concentration is approximately twice that of liver at times remote from injection. The skeleton contains less cobalt than the liver during the early weeks after injection, but gradually becomes the dominant systemic repository for cobalt.
8.2.3.3. Treatment of radioactive progeny
(366) Radioactive progeny considered in the derivation of dose coefficients for cobalt isotopes are isotopes of cobalt or iron. Cobalt produced in vivo by radioactive decay is assigned the characteristic model for cobalt, i.e., the systemic model for cobalt as a parent. The model for iron produced in vivo is a modification of the characteristic model for iron described in this report. The compartment in the characteristic model for iron called Other plasma is identified with the compartment in the cobalt model called Blood 1. Iron produced in any compartment of the cobalt model other than Blood 1 is assigned a transfer rate to Other plasma and assumed to follow the characteristic model for iron after reaching Other plasma. The following transfer coefficients from compartments of the cobalt model to Other plasma are assigned: Blood 2, 1000 d−1; soft tissue and bone surface compartments, 1.39 d−1 (the highest transfer rate from a tissue compartment to Other plasma in the characteristic model for iron); bone volume compartments, the rate of bone turnover.
8.3. Individual monitoring
8.3.1. 57Co
(367) 57Co is a high-energy gamma emitter. Monitoring of 57Co is generally accomplished through whole-body measurement (Table 8.4). Urine bioassays are also used in monitoring for 57Co. If needed, lung monitoring may be performed. Monitoring techniques for 57Co. Detection limit values presented in this table refer to shielded room.

8.3.2. 58Co
(368) 58Co is a high-energy gamma emitter. Monitoring of 58Co is generally accomplished through whole-body measurement (Table 8.5). Urine bioassays are also used in monitoring for 58Co. If needed, lung monitoring may be performed. Monitoring techniques for 58Co. Detection limit values presented in this table refer to shielded room.

8.3.3. 60Co
(369) 60Co is a high-energy gamma emitter. Monitoring of 60Co is generally accomplished through whole-body measurement (Table 8.6). Urine bioassays are also used in monitoring for 60Co. If needed, lung monitoring may be performed. Monitoring techniques for 60Co. Detection limit values presented in this table refer to shielded room.

8.4. Dosimetric data for cobalt
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 57Co, 58Co, and 60Co compounds.

AMAD, activity median aerodynamic diameter; FAP, fused aluminosilicate particles; PSL, polystyrene.
Dose per activity content of 57Co in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.

Dose per activity content of 58Co in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.

Dose per activity content of 60Co in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work
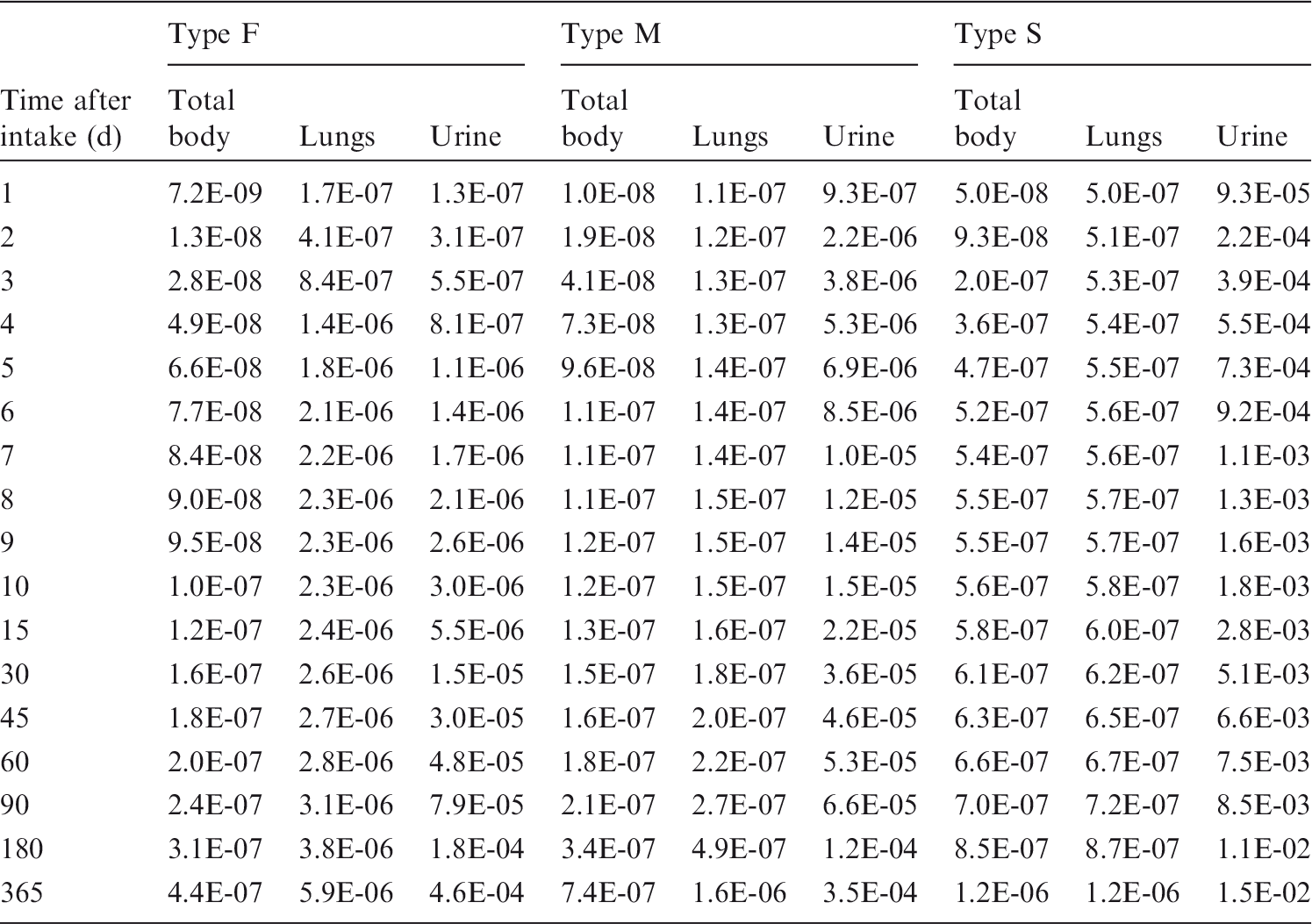

Total body and lung contents, and daily urinary excretion of 57Co following inhalation of 1 Bq Type F.

Total body and lung contents, and daily urinary excretion of 57Co following inhalation of 1 Bq Type M.
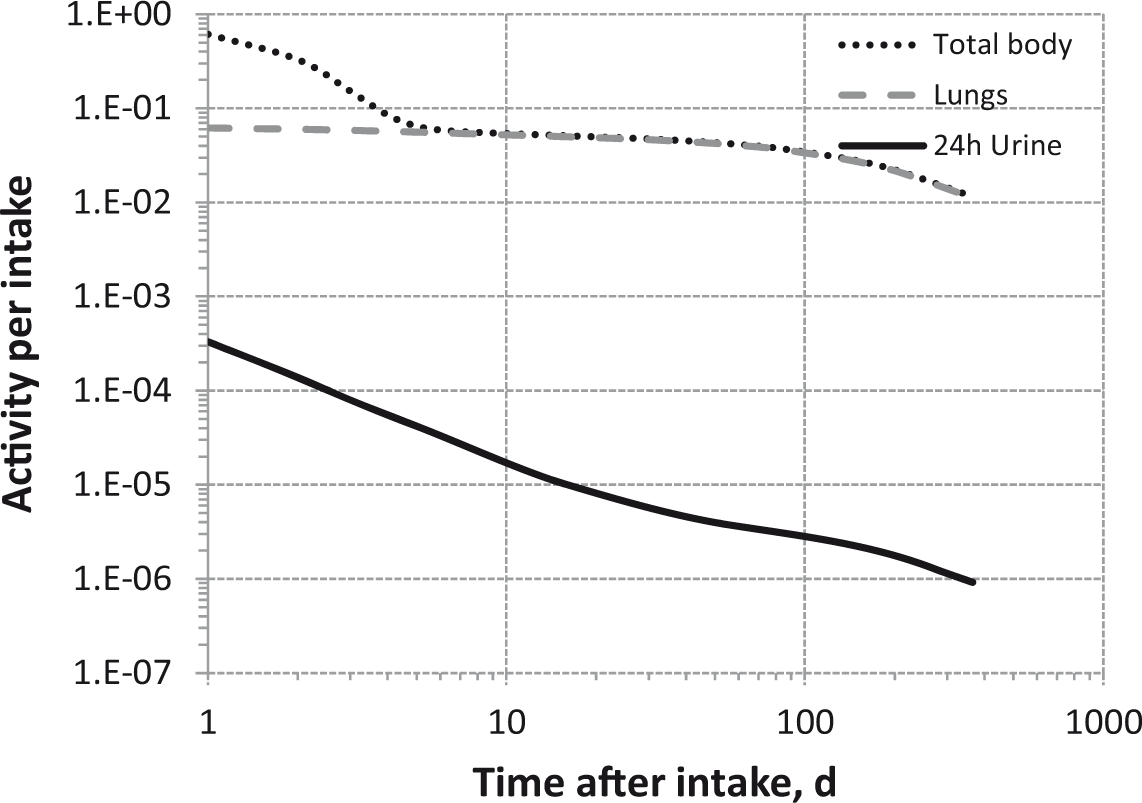
Total body and lung contents, and daily urinary excretion of 57Co following inhalation of 1 Bq Type S.

Total body and lung contents, and daily urinary excretion of 58Co following inhalation of 1 Bq Type F.
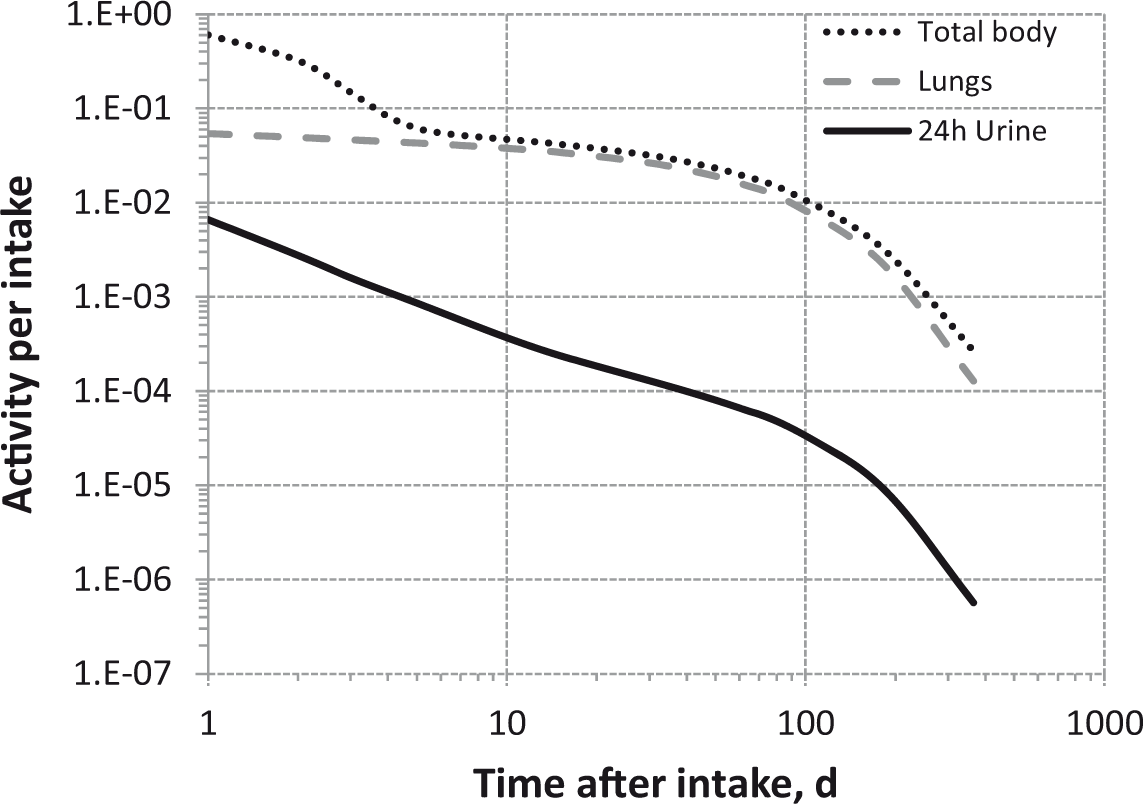
Total body and lung contents, and daily urinary excretion of 58Co following inhalation of 1 Bq Type M.
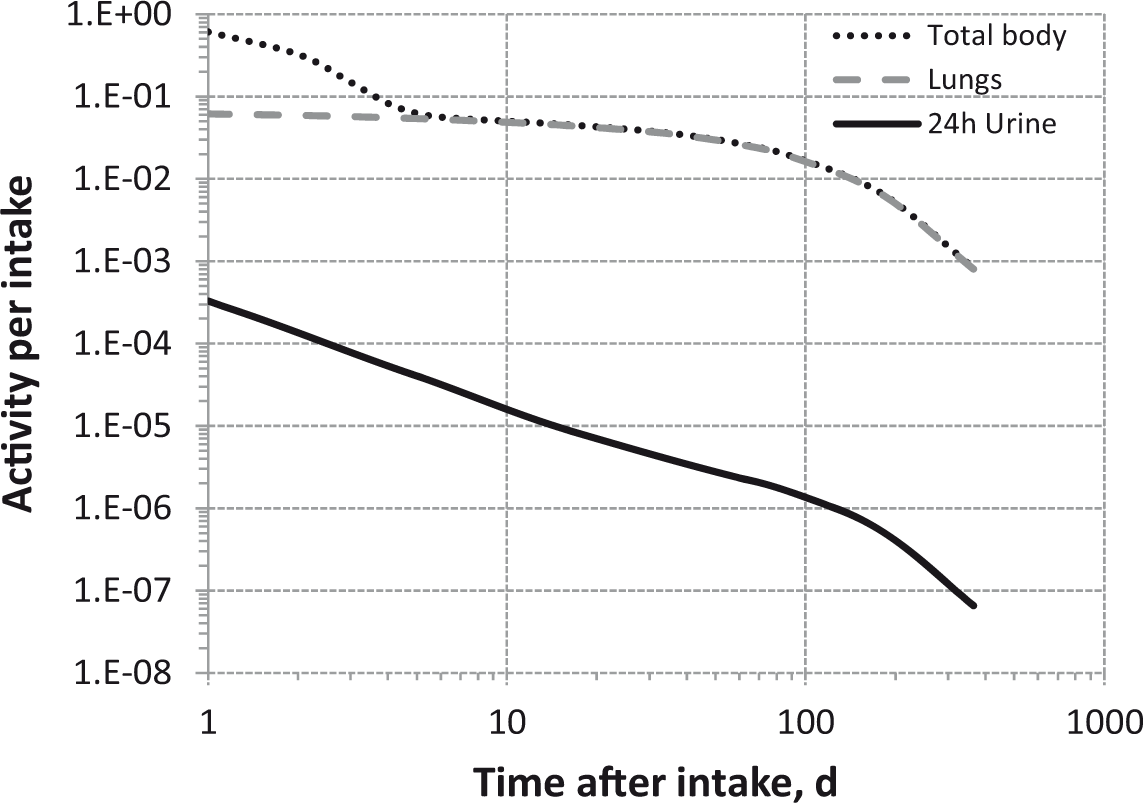
Total body and lung contents, and daily urinary excretion of 58Co following inhalation of 1 Bq Type S.

Total body and lung contents, and daily urinary excretion of 60Co following inhalation of 1 Bq Type F.
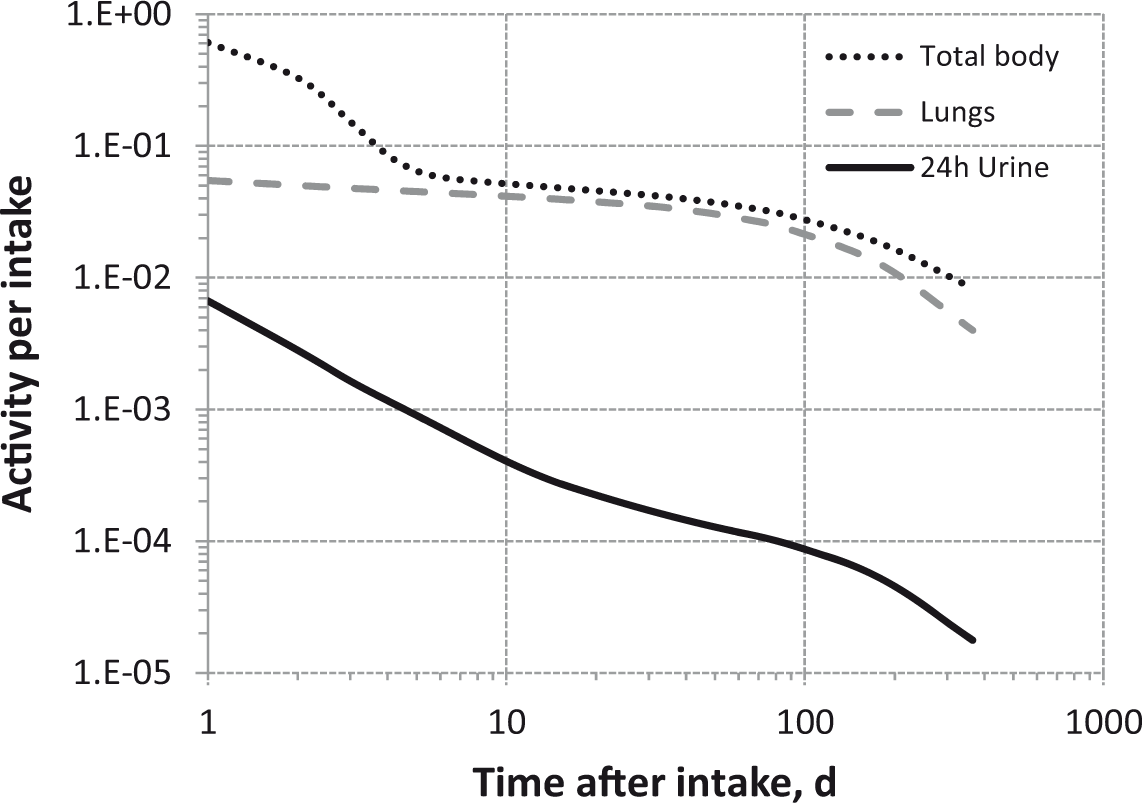
Total body and lung contents, and daily urinary excretion of 60Co following inhalation of 1 Bq Type M.
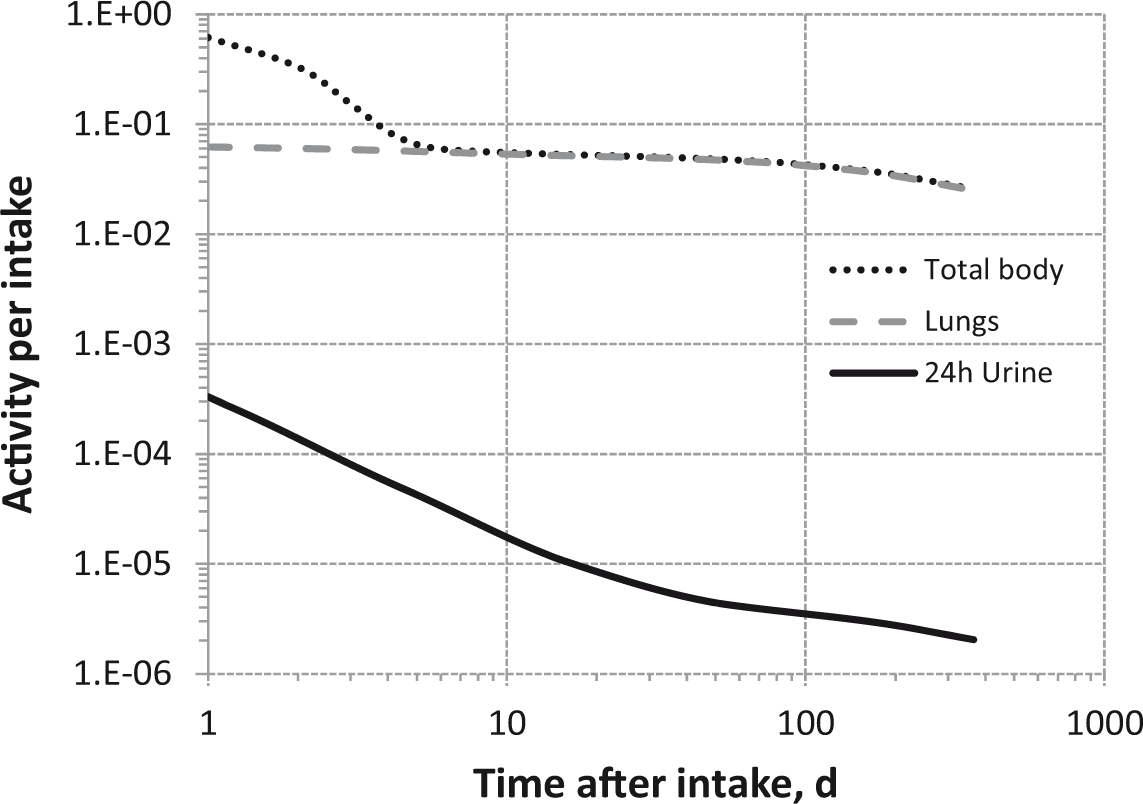
Total body and lung contents, and daily urinary excretion of 60Co following inhalation of 1 Bq Type S.
8.5. References
9. ZINC (Z = 30)
9.1. Chemical forms in the workplace
(370) Zinc is a transition metal that occurs mainly in oxidation state II. Zinc may be encountered in industry in a variety of chemical and physical forms, including metal dusts, oxides, phosphates, sulphides, soluble salts (sulphates, nitrates, chlorides), and chromates. Table 9.1 shows the isotopes of zinc addressed in this publication. (371) 65Zn is a major activation product in nuclear power plants and could be present in corrosion particles. Isotopes of zinc addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay.

9.2. Routes of intake
9.2.1. Inhalation
(372) Little information was found on the behaviour of inhaled zinc in man, and it is difficult to estimate the contribution of absorption to lung clearance in such cases because the systemic excretion of zinc is predominantly via the faecal route. Information on absorption from the respiratory tract is available from experimental studies of several compounds of zinc, or associated with corrosion products. (373) Absorption parameter values and types, and associated fA values for particulate forms of zinc are given in Table 9.2. Absorption parameter values for inhaled and ingested zinc. It is assumed that the bound state can be neglected for zinc, i.e. fb=0. The values of sr for Type F, M, and S forms of zinc (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. zinc oxide) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of zinc (0.5). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.5) for ingestion of the radionuclide.

9.2.1.1. Particulate materials
(a) Zinc oxide
(374) Following inhalation of zinc oxide by rats, Oberdörster et al. (1979) observed a lung retention half-time of approximately 6 h, with 7% ILD retained at 24 h. Rosaminth and Breining (1974) administered zinc oxide to rats by instillation five times over 14 d, and less than 2% of the total ILD was retained 7 d later. Hirano et al. (1989) also administered zinc oxide to rats by instillation, and observed a lung retention half-time of approximately 15 h, with negligible retention after 5 d. The results of all three studies (with stable zinc oxide) are consistent with the assignment to Type F.
(b) Zinc chromate
(375) Following intratracheal instillation of zinc 51Cr chromate to rats, 25% ILD remained at 30 min, and the retention half-time was 1.9 d from 30 min to 6 d, consistent with assignment to Type F (Bragt and van Dura, 1983).
(c) Zinc nitrate
(376) Morrow et al. (1968) followed lung clearance of 65Zn for 70 d after inhalation of 65Zn(NO3)2 by dogs and rats, but few details are given. Lung retention in dogs was described by a two-component exponential function with half-times of 4 d (53%, clearance rate 0.17 d−1) and 120 d (clearance rate 0.0058 d−1), giving lung retention at 30 d of 40% ILD, consistent with assignment to Type M.
(d) Zinc phosphate
(377) Morrow et al. (1968) followed lung clearance of 65Zn for 65 d after inhalation of 65Zn3(PO4)2 by dogs and rats, but few details are given. Lung retention in dogs was described by a two-component exponential function with half-times of 7 d (58%, clearance rate 0.099 d−1) and 330 d (clearance rate 0.0021 d−1), giving lung retention at 30 d of 42% ILD, consistent with assignment to Type M.
(e) Corrosion products (contaminated dusts or ‘residues’ formed at nuclear power plants
(378) The biokinetics of 65Zn was followed for 280 d after intratracheal instillation into rats of a suspension of corrosion ‘crud' particles (oxide-bearing debris, 11% 65Zn activity) from the primary containment of a water-cooled reactor (Collier et al., 1994). Few details are given, but it was assessed here (i.e. by the Task Group) that the results are consistent with assignment of the 65Zn present to Type S.
(f) Other compounds
(379) In one case of accidental human exposure to dust from an experimental hole in a reactor, 65Zn was cleared rapidly from the lungs except for a small component that was retained for a period of several months, indicating Type F behaviour (Newton and Holmes, 1966). Measurements have also been reported following accidental intakes of 65Zn from metallic zinc (Andrasi and Feher, 1967) and reactor graphite dust (Sedlet and Fairman, 1970), but there is insufficient information to assign the material to absorption types as excretion of systemic zinc is predominantly faecal.
9.2.1.2. Rapid dissolution rate for zinc
(380) There is insufficient experimental information to estimate the rapid dissolution rate for zinc. There is therefore no justification for choosing a rate different from the general default value of 30 d−1, which is applied here to all Type F forms of zinc.
9.2.1.3. Extent of binding of zinc to the respiratory tract
(381) Evidence from the zinc oxide studies outlined above suggests that there is probably little binding of zinc. It is therefore assumed that the bound state can be neglected for zinc, i.e. fb = 0.0.
9.2.2. Ingestion
(382) Studies in which 69mZn was administered as chloride to three fed volunteers showed gastrointestinal absorption of zinc of approximately 0.2 (Molokhia et al., 1980). (383) Zinc absorption in humans is influenced by numerous factors including fasting, meal composition, the amount of daily dietary zinc, and the state of health. Experiments performed on five fasting volunteers showed fractional absorption values ranging from 0.4 to 0.8 (Molokhia et al., 1980). Similar experiments performed on 75 fasting subjects given carrier-free 65Zn showed similar fractional absorption values, ranging from 0.4 to 0.86 (Aamodt et al., 1981). (384) When stable or radioactive zinc isotopes were incorporated into meals fed to normal adult subjects, the mean absorption values ranged between 0.05 and 0.5, with a value of approximately 0.3 being typical (ICRP, 1993). It has been suggested that some foods, such as milk and beef, may enhance dietary zinc uptake (Evans and Johnson, 1980; Solomons et al., 1982), while bran and phytate reduce it (Sandstrom and Cedarblad, 1980; Turnland et al., 1984). (385) Experiments performed with eight healthy subjects showed that when the amount of dietary zinc intake decreased from 15 to 2 mg d−1, this resulted in an increase of fractional zinc absorption from 0.6 to approximately 0.9 (Istfan et al., 1983). Similarly, studies performed with 68Zn or 70Zn sulphate given to eight fed volunteers, together with doses of aqueous zinc decreasing from 30 to 2 mg, showed that fractional absorption values increased from 0.37 to 0.73 (Tran et al., 2004). (386) Zinc absorption has been reported to be reduced in the elderly (Turnlund et al., 1982) and in the cirrhotic (Mills et al., 1983). (387) In Publication 30 (ICRP, 1980), an absorption value of 0.5 was recommended for all forms of zinc. The same value was adopted in Publication 67 (ICRP, 1993) for dietary intakes. An fA of 0.5 is also used in this publication for all chemical forms.
9.2.3. Systemic distribution, retention, and excretion
9.2.3.1. Overview of zinc biokinetics and balance in adult humans
(388) Zinc is an essential trace element required for normal growth, protein production, and function of numerous enzymes in mammals (NAS, 1979; Walravens, 1979; Vallee and Falchuk, 1993; Lowe et al., 2009). Dietary intake of zinc by adults is generally in the range 7–20 mg d−1 (Buchet et al., 1983; van Dokkum et al., 1989; Bro et al., 1990; Anke et al., 1991; Becker and Kumpulainen, 1991; Ysart et al., 2000; Hunt and Meacham, 2001; Jaiswal et al., 2002; Noel et al., 2003; Suzuki et al., 2003). Gastrointestinal uptake averages approximately 30–35% but varies with the level of zinc in diet, timing of intake relative to meals, and other factors (Hambidge et al., 1998; Krebs and Hambidge, 2001; Lowe et al., 2009). (389) Faecal loss is the primary route of excretion of zinc. Endogenous faecal excretion appears to arise largely from pancreatic secretions into the small intestine contents, with smaller amounts transferred into the gastrointestinal contents in liver bile, saliva, and other secretions (McClain, 1990; Hambidge et al., 1998). Daily excretion in urine is typically approximately 0.3–0.5 mg (Spencer et al., 1973; Elinder et al., 1978; Wastney et al., 1991; Schuhmacher et al., 1994; Scott and Turnlund, 1994). The amount of zinc lost in sweat under normal conditions appears to be of the same order as losses in urine (Jacob et al., 1981; Johnson et al., 1993). (390) Following acute entry of labelled zinc into blood, 60% or more of the label accumulates rapidly in the liver (Siegel et al., 1961; Spencer et al., 1965; Aamodt et al., 1979). Relatively high concentrations are also seen in the kidneys and pancreas at early times (Siegel et al., 1961; Spencer et al., 1965). Over a period of weeks, the label shifts largely to skeletal muscle and bone, which have low rates of accumulation but long retention of zinc (McKenney et al., 1962; Khristov, 1970; Aamodt et al., 1982). (391) External measurements of 65Zn in human subjects following intravenous or oral administration indicate two main components of systemic retention with half-times on the order of 1–3 weeks (15–30%) and 300–450 d (70–85%) (Richmond et al., 1962; Spencer et al., 1965; Aamodt et al., 1982). Biokinetic studies on human subjects have not been sufficiently long to identify small components of retention with extremely long half-times that may arise (e.g. from binding of zinc to bone mineral). (392) The mass of stable zinc in the whole body of adult humans is on the order of 2 g (ICRP, 1975; NAS, 1979; Zhu et al., 2010). Muscle contains approximately 55–65% of the body’s zinc and bone contains approximately 20–30%.
9.2.3.2. Summary of the database
(a) Human studies
(393) Siegel et al. (1961) measured 65Zn concentrations in tissue samples taken at autopsy 1–174 d after intravenous injection of 65Zn as chloride into 14 terminal patients with various malignancies. The liver, pancreas, spleen, prostate, seminal vesicles, lung, urinary bladder, and skeletal muscle were sampled. Widely differing concentrations of 65Zn were found in different tissues. The highest levels were found in the liver, in which the concentration reached approximately 0.05% of the administered activity per gram of tissue in the first few days after administration. This value was approximately two to eight times that in the pancreas, which contained the second highest concentrations at early times, and approximately 10–30 times that in muscle, which contained the lowest concentrations at early times. Turnover was relatively slow in the liver and relatively fast in the pancreas. The concentration in the pancreas was reduced by approximately two-thirds within 1 week, while the concentration in the liver remained high after 81 d. (394) Richmond et al. (1962) measured uptake, excretion, and whole-body retention of acutely ingested 65Zn in one healthy female subject (A) aged 31 y and three healthy male subjects (B, C, D) aged 29, 45, and 48 y, respectively. Measurements for Subjects A–D were continued up to 431, 664, 416, and 579 d after intake, respectively. Excretion of absorbed activity was primarily in faeces. Whole-body retention in each subject could be represented as the sum of three exponential terms representing fast, intermediate, and slow turnover. Assuming that the term with fast turnover (t½ < 30 h) represented faecal excretion of unabsorbed activity, approximately 20% (range 16–27%) of absorbed activity was lost with a mean biological half-time of 16 d (4.5–26 d), and 80% (73–84%) was lost with a mean half-time of 420 d (387–478 d). (395) Spencer et al. (1965) investigated the biokinetics of intravenously injected 65Zn in 19 patients, at least 11 of whom had terminal cancers. Whole blood of a subject described as representative contained approximately 22% of the injected amount at 13 min, 11% at 1 h, 5% at 2 h, 4% at 10 d, and 3% at 40 d. Measurements on three subjects indicated that 75–90% of the activity in total blood was contained in cellular components at 2–29 d after administration of 65Zn. The main pathway of excretion was via the gastrointestinal tract. In two subjects followed over 45 d, cumulative faecal and urinary excretion averaged 19.2% and 2.1%, respectively, of the administered amount. Urinary excretion of activity became extremely low after the first few days, while a small but nearly constant fraction was excreted daily in faeces for an extended period. Whole-body retention measurements made on each of two subjects for approximately 1 y could be closely approximated as the sum of two exponential terms representing fast and slow components of turnover. The biological half-times of the fast component, representing approximately one-quarter of the injected amount, were 13.1 and 11.8 d in the two subjects. The half-times of the slow components were 334 and 308 d, respectively. In tissue samples obtained at autopsy from 11 subjects dying from metastatic cancers at 1–71 d after administration of 65Zn, the activity concentration was higher in the liver than other tissues over the entire period. The kidney showed the next highest concentration, averaging approximately half of that in liver, over the entire observation period. Relatively high concentrations were also seen in the pancreas, spleen, and adrenals over the early days or weeks after administration of 65Zn. The concentration in the liver at 71 d was still approximately one-quarter of that at 1 d. Concentrations of 65Zn in samples of bone and skeletal muscle were relatively low. The activity concentrations in samples from the vertebrae, ribs, and sternum were substantially higher than in samples from the femur of the same subject. (396) In a case of accidental inhalation of 65Zn, whole-body measurements indicated that 27% of the inhaled activity was retained in the body with a half-time of 18 d, and 73% was retained with a half-time of 453 d (Newton and Holmes, 1966). Similar half-times were estimated from time-dependent activity in faeces. A widespread distribution of activity with a relatively high concentration in the liver was apparent throughout the study. An estimated 20–30% of the total daily excretion of 65Zn was in urine. (397) Hawkins et al. (1976) studied the biokinetics of orally administered 65Zn in nine subjects with skin diseases. The study was motivated by reported findings that some skin diseases respond dramatically to treatment with zinc, and that low plasma zinc concentrations are associated with some skin diseases. Whole-blood and plasma concentrations of 65Zn were measured up to 192 d, and whole-body retention was measured externally up to 231 d. Whole-body retention measurements indicated that average absorption of 65Zn from the gut in these subjects exceeded 70%. Whole-body retention R(t) of absorbed activity as a function of time t (d) in each subject could be represented reasonably well as the sum of two exponential terms: R(t) = A1exp(-0.693t/B1) + A2exp(-0.693t/B2), where the terms represent short- and long-term components of retention, respectively. The coefficients A1 and A2 represented, on average, approximately 16% and 84% of the absorbed amount, respectively. The biological half-times B1 and B2 averaged approximately 23 d and 399 d, respectively. These results are reasonably consistent with the findings of Richmond et al. (1962) for healthy subjects. A subgroup with venous leg ulcers showed a smaller component of long-term retention and a shorter long-term biological half-time than the other subjects. External measurements indicated a high concentration of 65Zn in the liver at early times. (398) Aamodt et al. (1979) and Foster et al. (1979) studied the short-term biokinetics of orally or intravenously administered 69mZn (t½ = 13.8 h) in 17 subjects with taste or smell dysfunction. Activity was measured over the first 5 d in the whole body, urine, faeces, total blood, plasma, and RBCs, and externally over the liver and thigh. The biokinetics of zinc did not appear to be affected by the mode of administration. Biological clearance from blood plasma as a function of time t (d) following intravenous administration was described as a four-exponential retention function, R(t) = 0.79exp(-176t) + 0.175exp(-73.4t) + 0.022exp(-5.87t)+0.013exp(-0.053t). The liver accumulated approximately 50% of the intravenously injected activity during the first 15 min, and reached a peak content of approximately 60% at 2 h. Activity measured over the thigh increased with a doubling time of approximately 5.7 d after both oral and intravenous injection. The rate of build-up in the thigh corresponded approximately with the rate of loss from the liver. Activity in RBCs increased over the 5 d observation period to 6.4% of the injected amount and 2.4% of the ingested amount. (399) Aamodt et al. (1982) studied the effects of oral zinc loading on the biokinetics of zinc in 50 patients with taste or smell dysfunction for up to 440 d following acute ingestion of 65Zn (t½ = 244 d). The study was conducted in three phases: (1) all patients were studied for 21 d after oral intake of 65Zn as ZnCl2; (2) from 21 to 290–440 d (mean 336 d), all 50 subjects received placebo for ZnSO4, which was later used for zinc loading; and (3) over the next 112–440 d (mean 307 d), 14 patients continued on placebo while 36 ingested high levels of stable zinc (100 mg d−1) as ZnSO4. Prior to zinc loading, retention of absorbed zinc could be represented as the sum of two exponential terms with biological half-times of 18.2 d (32%) and 380 d (68%). Retention during the second (placebo) phase was not significantly different for the 36 subjects subsequently treated with ZnSO4 and the 14 subjects who were continued on placebo through the third phase of the study. Subjects receiving ZnSO4 during the third phase showed accelerated loss of 65Zn (t½ = 235 +/- 8 d). Accelerated loss of 65Zn from the thigh, presumably representing mainly loss from muscle, was apparent immediately in these 36 subjects. Accelerated loss from the liver began after a mean delay of 107 d. There was no apparent effect of zinc loading on loss of activity from RBCs. (400) Wastney et al. (1986) studied zinc metabolism in 32 normal subjects after oral (n = 25) or intravenous (n = 7) administration of 65Zn. Activity was measured in blood, urine, faeces, whole body, liver, and thigh over a 9 month period of normal intake of stable zinc (∼10 mg d−1) and an additional 9 month period with supplemental zinc intake of 100 mg d−1. Comparison of kinetic data derived during periods of normal and high intake of zinc suggested up to five sites of regulation of zinc concentrations in the body: absorption from the gut, endogenous secretion into the gut, urinary excretion, exchange between plasma and RBCs, and release by muscle. (401) Wastney et al. (1992) assessed changes in zinc metabolism with age based on biokinetic studies of intravenously or orally administered 65Zn in 26 healthy men and 21 healthy women in the age range 20–84 y. The studies covered a 9 month period when dietary intake of stable zinc was approximately 10 mg d−1, followed by a 9-month period in which intake was approximately 110 mg d−1. 65Zn kinetics was analysed by compartmental analysis using measurements of zinc isotopes in plasma, RBCs, urine, faeces, liver, thigh, and whole body. Significant changes with age in 65Zn kinetics were determined for urinary excretion, exchange between plasma and RBCs, absorption, and endogenous secretion. (402) Miller et al. (1994) described a four-compartment approximation of the model of Wastney et al. (1986). The simplified model consists of a plasma compartment and three satellite compartments representing fast, intermediate, and slow turnover of tissue zinc. The transfer coefficients from plasma to the fast, intermediate, and slow pools and to excretion pathways derived from the collective injection data are 85, 40, 4, and 2.4 d−1, respectively. Removal half-times from the fast, intermediate, and slow pools back to plasma based on the injection data are approximately 112 min, 18 h, and 108 d, respectively. The plasma clearance curve based on these parameter values closely approximates the curve determined in the studies by Aamodt et al. (1979) and Foster et al. (1979) described above. (403) Zinc metabolism and balance were studied in 11 healthy men with adequate or low levels of dietary zinc (Johnson et al., 1993). In terms of the mass of zinc excreted daily, urinary zinc decreased with decreasing zinc intake, while surface losses, presumably representing mainly losses in sweat, were unaffected by the level of zinc in diet. On average, urinary losses represented 6–7% of dietary zinc during periods of adequate zinc intake and 13–16% during periods of low intake. Faecal excretion represented approximately two-thirds of dietary zinc during periods of adequate dietary zinc and 39–48% during periods of low intake. Surface losses represented 4–6% of dietary intake during periods of adequate zinc intake and 12–36% during periods of low intake. The estimated surface losses during periods of adequate dietary zinc are reasonably consistent with results of a study by Jacobi et al. (1981), in which an effort was made to collect whole-body sweat from 13 male subjects living in a controlled environment for several months. (404) Lowe et al. (1997) developed a model of the short-term biokinetics of zinc based on stable isotope studies on six healthy women with a mean age of 30 y. Oral and intravenous tracers enriched in 67Zn and 70Zn, respectively, were administered simultaneously following a 7 d zinc equilibration period involving a controlled diet. Plasma and urine samples were collected over the first 7 d and faecal samples over the first 11 d. A seven-compartment model was developed to describe the kinetics of both tracers, as well as that of naturally occurring zinc. The model structure was used to derive the following central estimates from the measurements: fractional absorption from the gastrointestinal tract, 0.28; daily endogenous secretion, 2.8 mg; daily endogenous excretion, 2.0 mg; fractional turnover rate of the plasma pool, 131 d−1; sizes of extravascular compartments representing fast and slow equilibration with plasma, 7.2 mg and 77 mg, respectively; fractional turnover rates of these rapidly and slowly equilibrating pools, 22 d−1 and 1.5 d−1, respectively; and size and turnover rate of an extravascular pool with very slow turnover, 1083 mg and 0.014 d−1, respectively. Extrapolation of model predictions to infinity based on average parameter values indicated that cumulative faecal and urinary excretion represented 97.3% and 2.7%, respectively, of the oral tracer, and 91.4% and 8.6%, respectively, of the intravenous tracer. (405) King et al. (2001) used stable zinc tracers to compare the biokinetics of zinc in five men, aged 21–35 y, during normal zinc intake and following acute zinc depletion. The study was divided into two metabolic periods: a 16 d baseline period with dietary zinc of 12.2 mg d−1 and a 41 d depletion period with intake of 0.23 mg d−1. Stable isotope tracers of zinc were administered on day 6 or 7 of the baseline period and at the end of the depletion period (day 35). Baseline kinetic data indicated average gastrointestinal absorption of approximately 26%, a plasma zinc concentration of 0.71 µg ml−1, faecal excretion of 9.8 mg d−1 (approximately 80% of dietary zinc), urinary excretion of 0.46 mg d−1 (approximately 4% of dietary zinc), and whole-body content of approximately 1600 mg. The modelled rate of transfer of zinc from plasma to other compartments was approximately 144 d−1. After zinc depletion, gastrointestinal absorption was virtually complete, plasma zinc fell by 65% on average, and faecal and urinary excretion fell by 96% and 74%, respectively. (406) Pinna et al. (2001) studied the effects of low dietary zinc (4.6 mg d−1) on the mass of exchangeable zinc pools and its turnover time in seven healthy men confined during a 20 week clinical study. The estimated mass of exchangeable zinc was maintained when dietary zinc was reduced to approximately one-third of the recommended daily allowance over a 10 week period. Data analysis based on a three-compartment model indicated that the masses of plasma zinc and total exchangeable zinc were 3.25 and 148 mg, respectively, over the different phases of the study. Plasma zinc turned over 5.3 times per hour on average. There was a modest reduction in plasma zinc at 3 weeks after the start of the low zinc diet period, but plasma zinc returned to baseline values after 10 weeks of zinc restriction. (407) The concentration of stable zinc in autopsy samples of ribs from Japanese subjects increased with age from early adulthood to age 60 y (Yoshinaga et al., 1989). There was no clear change with age after age 60 y. (408) Aitken (1976) measured the zinc content of trabecular and cortical bone from 16 male and 12 female cadavers. The mean zinc to calcium ratio was 0.63 µg mg−1 for trabecular bone and 0.45 µg mg−1 for cortical bone. There was a significant increase with age in the zinc to calcium ratios of both trabecular and cortical bone. (409) Alhava et al. (1977) determined the concentration of zinc in cancellous bone of the iliac crest from 66 male and 28 female cadavers. The concentration was statistically related to age despite large variability in subjects of nearly the same age. The concentration reached a maximum during the fifth decade of life in both men and women. Men who died suddenly had a higher concentration than those with a chronic disease. (410) Typical (reference) contents of zinc in the whole body and specific tissues and fluids of adult humans are listed in Table 9.3. Concentrations in plasma and RBCs are based on analyses of samples from living subjects (NAS, 1979; Wastney et al., 1991; Scott and Turnlund, 1994). The other listed concentrations are rounded values based on a review of reported measurements of zinc in tissues collected post mortem, in many cases from subjects who had apparently been in good health up to the time of sudden accidental death (Tipton and Cook, 1963; Tipton and Shafer, 1964; Tipton et al., 1965; Strehlow and Kneip, 1969; Soman et al., 1970; Forssén, 1972; Hamilton et al., 1972; McBean et al., 1972; Evenson and Anderson, 1975; Sumino et al., 1975; Zhu et al., 2010). Median concentrations determined by Tipton and Cook (1963) and Tipton et al. (1965) for soft tissues other than liver were judged to be typical of reported values and were used in Table 9.3. Central estimates for liver reported by Tipton and Cook (1963) and Tipton et al. (1965) are lower than most reported values, and were replaced by the median of reported values from 14 studies of the zinc concentration in adult human liver tissue [see Table 6 of Evenson and Anderson (1975)]. The zinc concentration in bone listed in Table 9.3 is based on measurements reported by Tipton and Shafer (1964), Strehlow and Kneip (1969), and Aitken (1976), which together address zinc concentrations in bone tissue sampled from several skeletal sites. Conversions of concentrations to total contents were based on reference masses of tissues and fluids given in Publication 89 (ICRP, 2002). Reference zinc contents in tissues and whole body of adult humans.

(b) Animal studies
(411) The biokinetics of zinc has been studied in different animal species following acute or chronic administration of zinc tracers. Although some species differences are indicated, the animal studies provide insights into aspects of the biokinetics of zinc not clearly defined by kinetic studies on humans, such as its skeletal behaviour. Species-specific biokinetic models for zinc have been developed from isotopic studies on rats (House et al., 1982; Dunn and Cousins, 1989; House and Wastney, 1997), mice (Wastney and House, 2008), and pigs (Serfass et al., 1996). (412) Following intravenous injection of 65Zn into mice, the highest activity concentration over the first 7 d was found in the pancreas followed by the liver and kidney (Sheline et al., 1943). As much as 50% of the administered activity was eliminated in faeces during the first 7 d. The rate of elimination in urine was substantially lower than that in faeces. (413) Following intravenous injection of 65Zn into dogs, approximately 25% of the administered activity was eliminated in faeces during the first 2 weeks (Montgomery et al., 1943). Substantially less was lost in urine. The liver contained approximately 38% of the administered amount at 3 h and approximately 3.5% at 7 d. A maximum of 0.4% of the administered activity appeared in bile in the first 8 d. As much as 11% of the injected amount was secreted in pancreatic juice in the first 14 d. Activity was also found in large amounts in the juices obtained from an isolated loop of the duodenum. (414) The concentration of 65Zn was measured in rat tissues over 42 d following intravenous injection (Wakeley et al., 1960). At 1 d after administration, the highest concentration was found in pancreas followed by prostate and liver. Thereafter, the concentration in prostate was at least twice that in any other tissue. Bone showed the next highest concentration after the first week. Initial biological half-times for pancreas, liver, kidneys, and muscle were 0.8 d, 1.25 d, 1.7 d, and 40 d, respectively. (415) Ballou and Thompson (1961) investigated the biokinetics of 65Zn administered to rats by intravenous injection, acute oral intake, or chronic feeding. Following intravenous administration, the highest activity concentrations were found in liver, kidneys, and pancreas at early times, and in bone at late times. After chronic feeding for 200–400 d, the highest concentrations were found in hair, bone, and prostate. The concentration did not reach steady state in these tissues during the feeding studies. (416) Taylor (1961) measured the retention of 65Zn in the femur, pelvis, and humerus of rats over a period of 630 d following its intravenous injection into 7-week-old animals. Retention in each bone could be described as a single exponential function. The mean removal half-time was 738 d. Measurements of the specific activities of 65Zn in these three bones and in the ribs at 7 d after injection indicated that the 65Zn was distributed nearly uniformly throughout the zinc content of the skeleton. (417) Haumont (1961) used histochemical methods to examine the distribution of zinc in bones of young adult dogs and immature rats. High concentrations of zinc were found at sites undergoing calcification. Zinc was detected in the haversian systems of compact bone at the borderline between calcified and uncalcified tissue, in the cartilaginous partitions of hypertrophic cells, and in endochondral bone recently deposited in the metaphysis. (418) Calhoun et al. (1970) observed significantly increased uptake of 65Zn in healing bones of rats compared with control rats following its intravenous administration. Uptake of 65Zn at the injured site appeared to be correlated with bone formation. No statistically significant difference was found in the uptake of 85Sr or 45Ca in the injured bones and bones of control animals. (419) Bergman et al. (1972) examined the importance of zinc to cell proliferation in endochondral growth sites of bone in white rats using zinc-deficient feeding and autoradiography. The results of the study suggest that zinc is required in bone formation, especially in the synthesis of the organic matrix. (420) The time-dependent distribution and excretion of 65Zn was studied in rats following a single subcutaneous, intratracheal, or intraperitoneal administration (Khristov, 1970). The relative contents of tissues as a function of time were similar for all modes of administration. Highest initial activity concentrations were found in the pituitary, pancreas, and liver. At 25 d, the highest concentrations were found in pituitary and bone. Excluding activity found at the injection site, whole-body retention following subcutaneous injection was approximately 65% at 1 d, 44% at 10 d, and 37% at 25 d post injection. The liver, muscles, and bones contained, respectively, approximately 24%, 22%, and 32% of the retained activity at 1 d; 7%, 34%, and 31% at 10 d; and 4%, 36%, and 52% at 25 d. (421) The uptake and distribution of 65Zn were measured in rams at 5, 10, and 20 d after single oral or intravenous injection, and in pregnant ewes and a ram 2 weeks after the start of daily feeding (McKenney et al., 1962). The liver and kidney cortex initially contained the highest concentrations of activity. After 20 d, bone and muscle had substantially higher concentrations than the liver and kidney cortex. The relative concentrations in tissues at 20 d after single intake were independent of the route of administration. After daily feeding, the highest concentrations were found in decreasing order in liver, kidney cortex, mammary tissue, pancreas, and spleen. (422) Richmond et al. (1962) measured uptake and retention of 65Zn after a single oral uptake of 65ZnCl2 by dogs, rats, and mice and after intravenous injection of 65Zn into rats and mice. Maximum observation periods were 137, 164, and 540 d for mice, rats, and dogs, respectively. Faecal excretion represented the primary mode of elimination in all animals. Detailed studies of the tissue distribution in rats indicated that rates of loss were similar for tissues other than bone and pelt, which retained zinc more tenaciously than other tissues. (423) Studies on weanling and 7-week-old mice were conducted to investigate whether bone serves as a reservoir of available zinc (Murray and Messer, 1981). The results indicated that availability of bone zinc depended on the rate of bone resorption but not on zinc status, and that the skeleton does not serve as an available reservoir for zinc. Redeposition of zinc in the skeleton following resorption was extensive and independent of the rate of bone mineral deposition. In calcium deficiency, there was increased deposition of zinc, suggesting limited substitution of zinc for calcium in bone mineral. (424) Feaster et al. (1954) studied the behaviour of 65Zn in steers over the first 6 d following acute oral or intravenous administration. Tissue concentrations at 6 d decreased in the order pancreas > liver > pituitary, kidneys, rib sternal end, adrenals > mandible > rib shaft, incisors > whole blood. Accumulation in different bones or portions of bone paralleled their metabolic activity, with highest accumulation in sites with highest blood flow and trabecular bone accumulating more zinc than cortical bone per gram of tissue. (425) At 7 and 14 d after intravenous injection of 65Zn into young horses, the tissue concentrations decreased in the order liver > pancreas > spleen, kidney, heart, lung > rib, femur, skeletal muscle, skin > whole blood, adipose tissue, tibia, metatarsus (Schryver et al., 1980). Tissue samples from the wall of the gastrointestinal tract contained higher concentrations of 65Zn than sampled contents of the tract. Addition of stable zinc to the diet increased the rate of elimination of 65Zn from the body. (426) House et al. (1982) studied zinc metabolism in male rats by combining nutritional balance methods with an analysis of 65Zn kinetics. Disappearance of zinc from plasma was described by a four-exponential retention function. Measurement of zinc in tissues at different times indicated that plasma zinc exchanged more rapidly with zinc in liver and kidneys than it did with zinc in testes, skeletal muscle, or bone. The whole-body zinc content was approximately nine times higher than estimates of exchangeable zinc in the body. (427) Lowe et al. (1991, 1993, 1995) found that intravenously injected zinc isotopes followed similar two-compartment kinetics in rats, dogs, and human subjects over the first few hours after administration. Investigation into the location of the two metabolic pools in the rat indicated that the smaller pool consisted mainly of plasma zinc, and the larger pool resided largely within the liver. In normal human subjects, the fractional turnover rate of the smaller pool was five-fold faster than that of the larger pool. (428) House and Wastney (1997) determined zinc kinetics in 15 tissues of rats, and analysed the data using modelling techniques. The study revealed the existence of slow and fast pools of zinc in muscle and bone.
9.2.3.3. Biokinetic model for systemic zinc
(429) The biokinetic model for systemic zinc is taken from a paper by Leggett (2012). The model structure is shown in Fig. 9.1. Baseline transfer coefficients for workers are listed in Table 9.4. (430) The model includes three groups of tissues representing rapid (minutes to hours), intermediate (days), and slow (weeks to years) exchange with plasma, as indicated by a number of studies of the behaviour of zinc tracers in human subjects. Rapid exchange occurs between plasma and liver, and between plasma and ST0. The kidneys, pancreas, RBCs, and ST1 have intermediate rates of exchange with plasma. Also, part of the zinc entering the liver moves to Liver 2 that returns zinc to plasma with a half-time of a few days. Muscle, bone, and ST2 exchange zinc slowly with plasma. Each of the soft tissue compartments ST0, ST1, and ST2 is assumed to be distributed uniformly in ‘other soft tissues’, which represents all soft tissues except liver, kidneys, pancreas, and muscle. (431) Bone is divided into four compartments: trabecular bone surface, trabecular bone volume, cortical bone surface, and cortical bone volume. Bone surface exchanges zinc slowly with plasma. A small portion (5%) of zinc depositing on bone surfaces transfers to bone volume, from which it is removed to plasma at the rate of bone remodelling, assumed to be 18% y−1 for trabecular bone and 3% y−1 for cortical bone (ICRP, 2002). (432) Systemic zinc is assumed to be removed from the body in faeces, urine, and surface loss representing mainly sweat. Urinary excretion is represented as a transfer from plasma to the urinary bladder contents followed by transfer to urine. Surface loss is represented as a direct transfer from plasma to the environment. Endogenous faecal excretion of zinc is assumed to arise mainly (80%) from secretion into the gastrointestinal contents in pancreatic juice, represented as a transfer from pancreas to small intestine contents. The remaining endogenous faecal excretion is assumed to be equally divided between biliary secretion, represented as a transfer from liver to small intestine contents, and all other secretions into the alimentary tract combined, represented as a direct transfer from plasma to the small intestine contents. (433) All secretions into the alimentary tract are assumed to be subject to re-absorption to blood with the same fractional absorption as dietary zinc. (434) Transfer coefficients between plasma and the liver, kidneys, pancreas, and RBCs were set for consistency with observations of accumulation and loss of zinc tracers by these tissues in tracer studies on human subjects (Siegel et al., 1961; Spencer et al., 1965; Aamodt et al., 1979, 1982; Wastney et al., 1986). Transfer coefficients between plasma and other compartments (excluding the generic removal rates from bone volume to plasma, which represent bone turnover rates) were set for reasonable consistency with results of tracer data where available; the typical distribution of stable zinc in adult humans, as estimated in Table 9.3, assuming long-term ingestion of zinc at a constant rate; and data for laboratory animals where needed to fill gaps in the database for human subjects. (435) The total rate of loss of zinc from the body along all excretion pathways combined was set for consistency with observations of whole-body retention of 65Zn in human subjects following acute uptake to blood (Richmond et al., 1962; Spencer et al., 1965; Hawkins et al., 1976; Aamodt et al., 1982). Transfer coefficients describing removal of zinc in faeces, urine, and surface loss were set so that these pathways account for approximately 80%, 10%, and 10% of total endogenous excretion of zinc during chronic intake, assuming that 35% of endogenous secretion of zinc into the alimentary tract is re-absorbed to blood (Leggett, 2012). The relative quantities of zinc predicted by the model to be excreted in faeces, urine, and surface loss vary, to some extent, with the assigned absorption fraction because this affects the level of re-absorption of secreted zinc to blood, and hence the amount available for excretion along each pathway. For example, if the absorption fraction is assumed to be 0.5 (the reference value applied to zinc in this report), the model predicts that faeces, urine, and surface loss account for approximately 76%, 12%, and 12%, respectively, of total excretion of zinc during chronic intake. Structure of the biokinetic model for systemic zinc. ST, soft tissue; RBC, red blood cells; SI, small intestine; ST0, ST1, and ST2 represent fast, intermediate, and slow turnover, respectively, in soft tissues other than muscle, liver, kidneys, and pancreas; surface and volume refer to bone surface and bone volume, respectively. Transfer coefficients in the biokinetic model for zinc. RBC, red blood cells; ST, soft tissue.


9.2.3.4. Treatment of radioactive progeny
(436) Three isotopes of zinc addressed in this publication have progeny that are considered in the derivation of dose coefficients for the parent radionuclide: 69mZn (t½ = 13.8 h) decays to 69Zn (56.4 min), 62Zn (9.19 h) decays to 62Cu (9.67 min), and 72Zn (46.5 h) decays to 72Ga (14.1 h). 69Zn presumably behaves the same as the parent radionuclide from the time it is produced in the body. 62Cu produced by decay of 62Zn is assumed to decay at its site of production. (437) The systemic model for gallium as a progeny of zinc was based on observations of the behaviour of gallium in human subjects (Nelson et al., 1972; MIRD, 1973; ICRP, 1981; Priest et al., 1995; Bernstein, 1998), particularly autopsy data for patients administered radio-gallium during terminal illness (Nelson et al., 1972; MIRD, 1973), and results of a biokinetic study of intravenously administered 67Ga in a healthy adult (Priest et al., 1995). The gallium model includes compartments representing blood, liver, kidneys, spleen, pancreas, muscle, trabecular bone surface, trabecular bone marrow, cortical bone surface, and cortical bone marrow, and two compartments representing other soft tissue. Gallium is assumed to leave blood at a rate of 5 d−1, with 20% depositing on bone surface, 10% in marrow, 6% in liver, 8% in kidneys, 4% in muscle, 1% in spleen, 0.1% in pancreas, 3% in right colon contents, 10% in a soft tissue compartment with relatively slow transfer back to blood (t½ = 1 y), and the remainder (37.9%) in a soft tissue compartment with relatively fast transfer back to blood (t½ = 0.5 d). The bone and marrow deposits are assumed to be equally divided between trabecular and cortical bone. Gallium is removed from liver, spleen, pancreas, and muscle to blood with a half-time of 5 d; from kidneys to urinary bladder contents with a half-time of 0.5 d; and from bone surface and marrow to blood with a half-time of 2 d. Blood in the gallium model is identified with the plasma compartment of the zinc model. Gallium produced in compartments of the systemic model for zinc (Fig. 9.1) other than plasma are assumed to be transferred to the blood compartment of the gallium model with the following half-times: 1 min for RBCs; 5 d for liver, spleen, pancreas, and muscle; 0.5 d for kidneys and compartments of other soft tissue; 2 d for bone surface compartments; and the bone turnover half-time for bone volume compartments. The subsequent behaviour of gallium that reaches the blood compartment is determined by the gallium model described above.
9.3. Individual monitoring
(438) 65Zn is a gamma emitter. Monitoring of 65Zn is generally accomplished through whole-body measurement or/and urine bioassays (Table 9.5). Monitoring techniques for 65Zn.

9.4. Dosimetric data for zinc
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 65Zn compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 65Zn in total body and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Total body content and daily urinary excretion of 65Zn following inhalation of 1 Bq Type F.

Total body content and daily urinary excretion of 65Zn following inhalation of 1 Bq Type M.
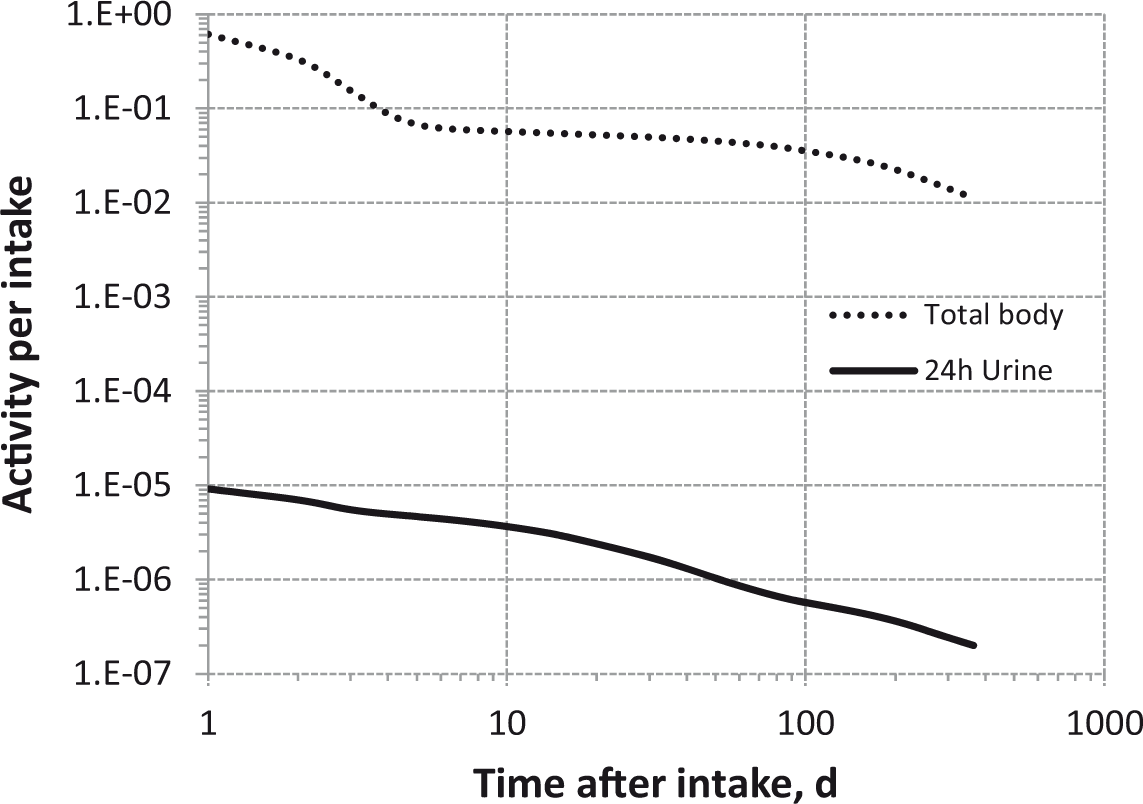
Total body content and daily urinary excretion of 65Zn following inhalation of 1 Bq Type S.
9.5. References
10. STRONTIUM (Z = 38)
10.1. Chemical forms in the workplace
(439) Strontium is an alkaline earth element that mainly occurs in oxidation state II. It is a chemical analogue of calcium. A variety of chemical and physical forms are encountered in industry, including chlorides, sulphates, carbonates, and titanate (SrTiO3). 85Sr, 89Sr, and 90Sr are the three main fission products which may be encountered in the nuclear industry. Strontium can also be present in fragments of irradiated fuels. Table 10.1 shows the isotopes of strontium addressed in this publication. Isotopes of strontium addressed in this publication. Dose coefficients and bioassay data for these radionuclides are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay.

10.2. Routes of intake
10.2.1. Inhalation
(440) Some information is available on the behaviour of inhaled strontium in man following accidental intakes of several compounds. Information on absorption from the respiratory tract is available from experimental studies of strontium as chloride, sulphate, titanate, irradiated fuel fragments, or in FAP. (441) Absorption parameter values and types, and associated fA values for particulate forms of strontium are given in Table 10.2. Absorption parameter values for inhaled and ingested strontium. FAP, fused aluminosilicate particles; PSL, polystyrene. It is assumed that the bound state can be neglected for strontium, i.e. fb=0. The values of sr for Type F, M, and S forms of strontium (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. strontium chloride) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of strontium (0.25). These calculated values are not rounded for purposes of consistency. Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract.
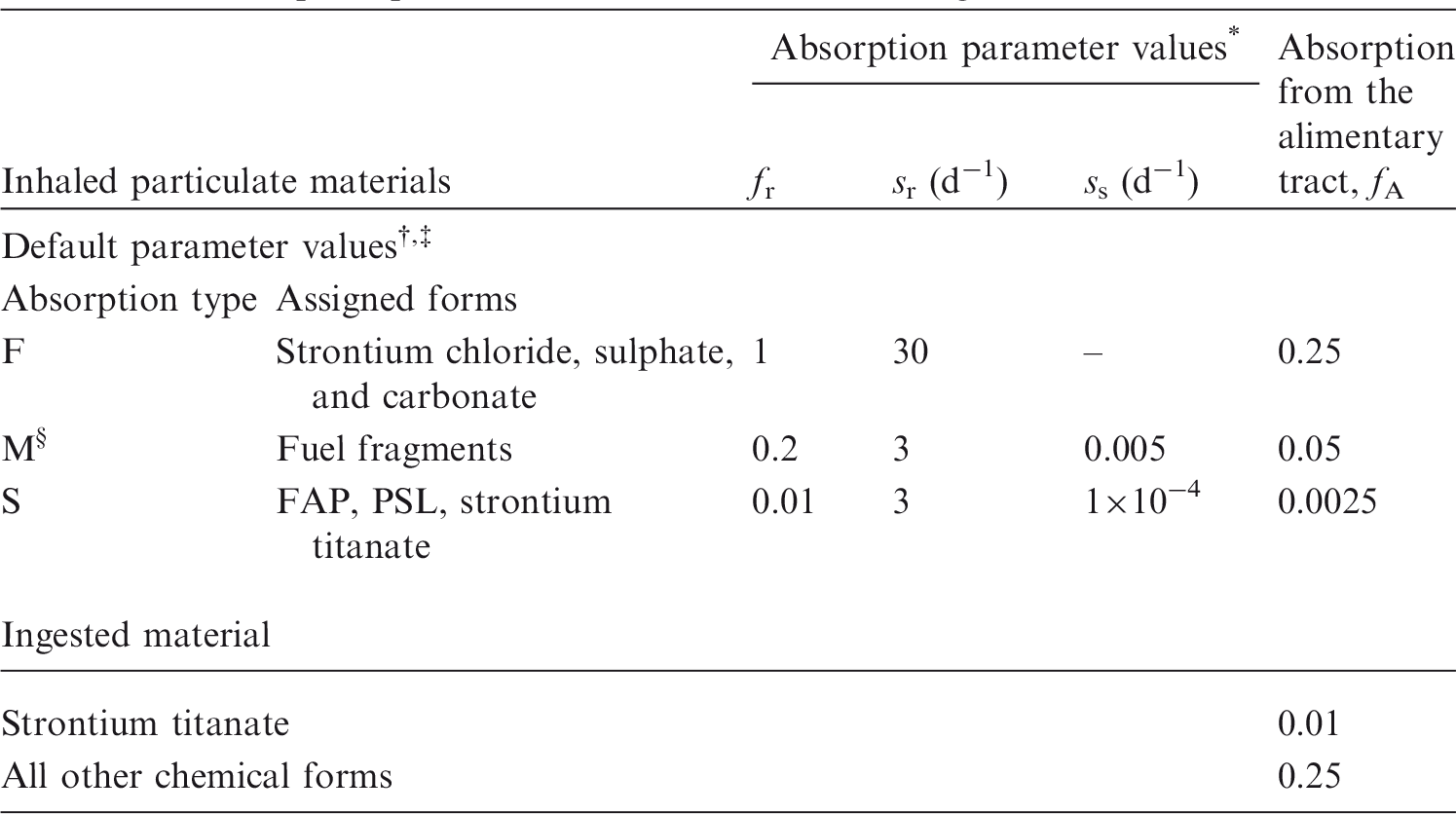
10.2.1.1. Particulate materials
(a) Strontium chloride (SrCl2)
(442) Petkau and Pleskach (1972) measured urinary and faecal excretion of 90Sr for 800 d after a worker’s presumed accidental inhalation of strontium chloride 13 d before the first measurement. The lack of information about the intake, or of measurements during the first week or so after it, limits the conclusions that can be drawn about absorption of the material. The results of measurements made during the first few months suggest that a large fraction (>0.5) was readily soluble, but the later data suggest continuing transfer from the lungs, and hence a low (<0.001 d−1) slow dissolution rate. (443) Animal experiments have shown that following administration of strontium chloride, most of the strontium is cleared rapidly from the respiratory tract. It was reported that at 12 h after inhalation of 85SrCl2 by dogs, the 85Sr remaining in the lungs was less than 1% of the total 85Sr in the body (McClellan and Rupprecht, 1967; McClellan et al., 1972), giving fr of approximately 1. It was calculated here (i.e. by the Task Group) that sr was greater than 8 d−1. However, it was also noted that a large amount of 85Sr was excreted in faeces in the first few days post exposure, apparently as a result of clearance from the upper respiratory tract, ingestion, and only partial gastrointestinal absorption. This shows that the rate of absorption to blood in the upper airways is probably less than the rate of particle transport to the gut (∼100 d−1). Morrow et al. (1968) measured a lung retention half-time of 0.02 d following inhalation of 85SrCl2 by dogs, giving sr = 35 d−1. Naményi et al. (1986) followed the biokinetics of 85Sr for 45 d after intratracheal instillation of 85SrCl2 into rats. Lung retention in healthy control rats was 3.9% ILD at 3 h, from which it was calculated here that sr = 26 d−1, and approximately 0.3% ILD at 24 h. Cuddihy and Ozog (1973) deposited 85SrCl2 directly on to the nasal membranes of Syrian hamsters. From the results, it was calculated here that fr = 1 and sr = 8 d−1. This is somewhat slower than in the other strontium chloride experiments, possibly because of the techniques used, including the anaesthetic, or because clearance from the nasal passage was slower than from the lungs. Similar observations were made for caesium and barium chlorides which were also administered by Cuddihy and Ozog (see caesium and barium inhalation sections of the forthcoming report Occupational Intakes of Radionuclides: Part 3). (444) Based on the results of the experiments outlined above, specific absorption parameter values for strontium chloride were estimated here to be fr = 1 and sr = 30 d−1 (consistent with assignment to default Type F). However, although specific parameter values for strontium chloride based on in-vivo data are available, they are not adopted here because inhalation exposure to it is so unlikely. Instead, strontium chloride is assigned to Type F. However, the data are used as the basis for the default rapid dissolution rate for strontium. Hence, specific parameter values for strontium chloride would be the same as default Type F strontium parameter values.
(b) Strontium sulphate (SrSO4)
(445) Following inhalation of 90SrSO4 by mice and dogs, most of the strontium was cleared rapidly from the lungs, indicating Type F behaviour (Bair, 1961).
(c) Strontium carbonate (SrCO3)
(446) Measurements following accidental inhalation by man of 90SrCO3 indicate Type F behaviour (Rundo and Williams, 1961).
(d) Strontium titanate (SrTiO3)
(447) Strontium titanate was shown to be retained tenaciously in the human lungs (Fish et al., 1967), and was assigned to Class Y in Publication 30 (ICRP, 1979). Bradley et al. (1964) reported external and excreta measurements following inhalation (and possibly also ingestion) of 90SrTiO3. Excretion was predominantly faecal, indicating that the material was relatively insoluble. In-vitro dissolution tests performed with various forms of 90SrTiO3 from high-level radioactive waste facilities (Anderson et al., 1999) showed that 97% of the strontium remained undissolved at 181 d, indicating assignment to Type S. Absorption parameter values calculated here were fr = 0.009, sr = 0.7 d−1, and ss = 1.2×10−4 d−1. In a parallel in-vivo study, the biokinetics of strontium and titanium were followed for 30 d after intratracheal instillation of stable SrTiO3 in rats. Uptake of strontium by the skeleton was below the detection limit. Lung retention showed a slow component, accounting for 15% of the instilled material, with a half-time of 133 d. It was assessed that 85% of the material deposited in the AI region was retained at 30 d, indicating Type S behaviour. A case of accidental inhalation from a source containing 90SrTiO3 was well fitted with the Publication 30 strontium model, and led the authors to the assumption of a 10 µm AMAD and the assignment of this compound to Inhalation Class Y (Navarro and Lopez, 1998). Studies on ingested strontium titanate on rats (see below) suggest fA ∼ 0.01. As specific lung absorption parameter values are only available from in-vitro tests, strontium titanate is assigned to Type S.
(e) Irradiated fuel fragments
(448) Measurements following the accidental inhalation of a mixture of fresh fission products indicate Type M behaviour of the strontium present (Johnson et al., 1983). Results of an in-vitro study on airborne fission products from the Three Mile Island reactor accident are consistent with assignment to Type F (Kanapilly et al., 1980). An in-vitro study on aerosols generated during transfer, cutting, storage, and shipment of nuclear reactor fuel (Dua et al., 1987) gave absorption parameters of fr = 0.4, sr = 0.57 d−1, and ss = 0.0045 d−1, consistent with assignment of the strontium present to Type M.
(f) Fused aluminosilicate particles
(449) FAP or ‘fused clay’ particles have been used extensively as relatively insoluble particles in inhalation studies, both of biokinetics and of radiation effects. A natural clay mineral is labelled by ion exchange, and the labelled clay particles are heated to approximately 1100℃ to form aluminosilicate glass microspheres in which the label is incorporated. It has been demonstrated that when strontium is incorporated into FAP, only a small fraction may be absorbed rapidly, while the remainder is retained within the particles and absorbed slowly. Estimates of the rate of dissolution of Sr FAP were in the range 0.0005–0.002 d−1 (Snipes et al., 1972; Kanapilly and Goh, 1973; Bailey et al., 1985a,b), and indicate Type S behaviour.
(g) Polystyrene
(450) As with FAP, it has been demonstrated that when strontium is incorporated into a polystyrene matrix, only a small fraction may be absorbed rapidly, while the rest is retained within the particles and is absorbed slowly. Bohning et al. (1982) used 85Sr polystyrene to follow lung retention in man for approximately 1 y after inhalation. Although absorption of the label into blood was not measured directly, lung retention at 300 d (37% and 64% ILD in smokers and non-smokers, respectively) is consistent with assignment to Type S.
10.2.1.2. Rapid dissolution rate for strontium
(451) The value of sr estimated for strontium chloride above, 30 d−1, which is the same as the general default value, is applied here to all Type F forms of strontium.
10.2.1.3. Extent of binding of strontium to the respiratory tract
(452) Evidence from the strontium chloride studies outlined above suggests that there is little binding of strontium. It is therefore assumed that the bound state can be neglected for strontium, i.e. fb = 0.0.
10.2.2. Ingestion
(453) Due to the presence of strontium isotopes in fallout material and its long-term retention in bone as a calcium analogue, the metabolism of strontium has been the subject of a number of human volunteer studies. Similar fractional absorption values were obtained from studies in which inorganic forms of radiostrontium were administered orally in solution (Spencer et al., 1960; Suguri et al., 1963; Shimmins et al., 1967; Sips et al., 1996), and from experiments where known quantities of radiostrontium incorporated in food were ingested (Fujita et al., 1966; Carr, 1967). In most cases, mean values were between 0.1 and 0.4, averaging approximately 0.2. (454) Likhtarev et al. (1975) measured the absorption of 85Sr (chemical form not specified) in nine young adult male volunteers, and obtained a mean value of 0.28 with a range 0.1–0.5. LeRoy et al. (1966) measured the absorption of strontium from real and simulated fallout, and after administration of 85Sr chloride. Ten volunteers ingested samples of local fallout, largely comprising silicaceous soil constituents (40–700 µm particles). The estimated absorption averaged 0.03 with a range 0–0.09. For simulated fallout prepared as glass microspheres (30–40 µm), estimated absorption was 0.16 (range 0.06–0.25), compared with 0.17 (0.08–0.34) after administration as the chloride. (455) Most of these data have been re-analysed and summarised in a recent review (Apostoaei, 2002). This author showed that the probability distribution function of f1 values is well represented by a lognormal curve with a geometric mean of 0.22 and a geometric standard deviation of 1.44. (456) A number of factors have been found to increase strontium absorption, including fasting; low dietary levels of calcium, magnesium, and phosphorus; milk diets; and vitamin D (Gruden, 1984; Moon, 1994; Sips et al., 1996; Bianchi et al., 1999). (457) Sips et al. (1996) investigated the gastrointestinal absorption of strontium chloride in eight healthy male volunteers under fasting conditions, and obtained a mean value of 0.25 (range 0.13–0.41). Spencer et al. (1972) showed that overnight fasting increased absorption from approximately 0.25 to 0.55. McAughey et al. (1994) also reported an f1 value of 0.55 (range 0.38–0.72) for four volunteers after an overnight fast compared with 0.11 in a single volunteer ingesting strontium after breakfast. Höllriegl et al. (2006) and Li et al. (2006) reported absorption of stable strontium in 13 human volunteers after an overnight fast, and found f1 values of approximately 0.6 (range 0.25–0.97) when strontium was given as chloride, diluted in aqueous solutions. (458) Similarly, a decrease in the calcium content of the diet from 30–40 to 0–10 mg d−1 kg−1 increased strontium absorption from an average of 0.2 to 0.4 (Shimmins et al., 1967). In contrast, sex, age at exposure in adult groups (Apostoaei, 2002; Höllriegl et al., 2006), smoking, exercise, and use of oral contraceptives in young females (Zitterman et al., 1995) do not seem to change the intestinal absorption of strontium. (459) In a study of stable strontium absorption in 47 normocalciuric volunteers (29 men and 18 women), Vezzoli et al. (1998) reported no clear evidence of sex on strontium absorption. Results from animal studies are generally similar to those from volunteer studies (Coughtrey and Thorne, 1983), although effects of sex on strontium absorption are controversial. Dahl et al. (2001) reported higher plasma strontium levels in male rats and monkeys, compared with females, and concluded that there were no clear sex differences in the gastrointestinal absorption of strontium. (460) Results for the absorption of strontium administered as the titanate (SrTiO3) to rats show low levels of absorption of approximately 0.01 (McClellan and Bustad, 1964). An open literature publication by McClellan et al. (1965) stated that, following ingestion of 85Sr titanate by miniature pigs, absorption ‘was less than a fiftieth of that of the chloride, and reached 0.29% of the dose only in the youngest pig used which was 5 months old’. (461) Radioactive strontium has been shown to accumulate in teeth (Neuzil and Dysart, 1984; Kulev et al., 1994; O’Donnell et al., 1997). Most of this deposit comes from gastrointestinal absorption and subsequent systemic distribution, but a small part may also be adsorbed directly from the oral cavity on to the dental plaque and enamel during mastication. Ex-vivo experiments performed with enamel removed from rat teeth and transferred to culture medium containing 90Sr (chemical form not given) showed rapid and large deposition on the enamel surface (White et al., 1980). Similarly, experiments performed with adult participants rinsing their mouths twice a day for 2 weeks with a strontium chloride solution showed that strontium was incorporated into dental plaque and was retained for at least 6 weeks (Spets-Happonen et al., 1998). In-vitro uptake of strontium directly into plaque-free bovine enamel and, to a lesser extent, human enamel has also been shown after experiments where enamel was agitated for 10 min per day for 7 d in a solution containing 2000 ppm of strontium (Curzon and Spector, 1983). Unfortunately, none of these studies provide enough information to derive robust parameters for strontium adsorption and retention on teeth. (462) In Publication 30 (ICRP, 1979), the recommended absorption values were 0.01 for SrTiO3 and 0.3 for all other compounds. In Publication 67 (ICRP, 1993), a value of 0.3 was recommended for dietary intake by adults. However, due to the strong link between strontium and calcium absorption, and the known discrimination in favour of calcium, a default fA value of 0.25 is adopted here for all chemical forms but strontium titanate, for which a lower fA value of 0.01 is retained.
10.2.3. Systemic distribution, retention, and excretion
10.2.3.1. Summary of the database
(463) Strontium is a chemical and physiological analogue of calcium but has different biokinetics from calcium due to discrimination between these elements by biological membranes and hydroxyapatite crystals of bone. For example, strontium is absorbed less effectively from the intestines, and excreted more effectively by the kidney than calcium, and is lost from bone at a higher rate than calcium over the first few months after deposition in bone (Bauer et al., 1955; Spencer et al., 1960; Barnes et al., 1961; Cohn et al., 1963; Decker et al., 1964; Harrison et al., 1967). (464) The biokinetics of strontium has been studied extensively in human subjects and laboratory animals. A large database related to the transfer of 90Sr from food and milk to the human skeleton was developed in the 1950s and 1960s. Interpretation of these environmental data is complicated by the fact that measured skeletal burdens were accumulated over an extended period, and depend on assumptions concerning fractional uptake of 90Sr from the gastrointestinal tract. More easily interpreted data are available from controlled studies on human subjects. Data on the behaviour of strontium in laboratory animals, particularly dogs, help to clarify the behaviour of strontium at early times after intake. As strontium is a close physiological analogue of calcium, data from controlled studies of calcium in humans provide supporting information for selection of parameter values for strontium, particularly for paths of movement for which comparative information on strontium and calcium transport is available. (465) Reviews of the biokinetic database for systemic strontium can be found in Publication 20 (ICRP, 1973), Publication 67 (ICRP, 1993), and an article by Leggett (1992). More recent human studies are described in articles by Shagina et al. (2003) and Li et al. (2008). The primary datasets underlying specific parameter values in the model for systemic strontium used in this publication are summarised below. Structure of the biokinetic model for systemic strontium. ST, soft tissue; exch, exchangeable; nonexch, non-exchangeable.
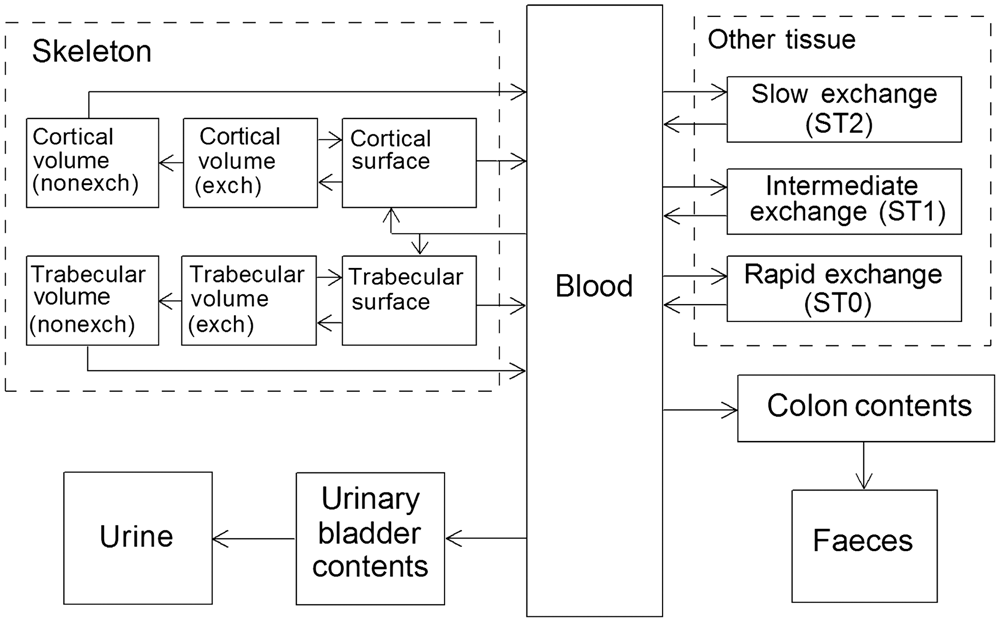
10.2.3.2. Biokinetic model for systemic strontium
(466) The structure of the model for systemic strontium is shown in Fig. 10.1. This is a simplified version of the generic model for bone-volume seekers. Blood is treated as a uniformly mixed pool that exchanges activity with soft tissues and bone surfaces. Soft tissues are divided into three compartments, corresponding to fast, intermediate, and slow exchange of activity with blood (ST0, ST1, and ST2, respectively). The liver and kidneys are not addressed separately in the model for strontium, but are included implicitly in the soft tissue compartments. Bone is divided into cortical and trabecular bone, and each of these bone types is further divided into bone surfaces and bone volume. Bone volume is viewed as consisting of two pools: one that exchanges with activity in bone surface for a period of weeks or months, and a second, non-exchangeable pool from which activity can only be removed by bone-restructuring processes. Activity depositing in the skeleton is assigned to bone surface. Over a period of days, a portion of the activity on bone surfaces moves to exchangeable bone volume and the rest returns to blood. Activity leaves exchangeable bone volume over a period of months, with part of the activity moving to bone surfaces and the rest to non-exchangeable bone volume. The rate of removal from non-exchangeable bone volume is assumed to be the rate of bone turnover, with different turnover rates applying to cortical and trabecular bone. Strontium is assumed to be lost from the body by urinary or faecal excretion alone.
10.2.3.3. Parameter values
(467) The systemic biokinetic model for strontium given in Publication 67 (ICRP, 1993) is reasonably consistent with later information on the biokinetics of strontium and related elements in adult humans (e.g. Shagina et al., 2003; Li et al., 2008). For example, the model predicts that 2.8–3.2% of whole-body 90Sr is eliminated each year at times 25–45 y after acute uptake to blood, compared with average values of 2.7–3.2%, depending on age, in adult males of a Russian population exposed to high levels of 90Sr (Shagina et al., 2003). Average rates of loss for adult females in that population were estimated as 3.2–3.5% up to 45 y of age and 4.4–5.8% at higher ages. The model of Publication 67 is independent of age and sex after age 25 y. (468) The parameter values for strontium applied in Publication 67 (ICRP, 1993) to an adult member of the public are adopted in this publication for application to workers. These values are listed in Table 10.1. The basis for each of the parameter values is summarised below. (469) Results of controlled studies involving adult humans indicate that whole-body retention, presumably representing primarily skeletal retention, is higher in young adults (<25 y) than in middle-aged or elderly persons (Likhtarev et al., 1975; Leggett, 1992). This is thought to be associated with age differences in the bone formation rate, which determines the level of deposition of calcium and related elements in bone, and remains elevated until approximately the middle of the third decade of life. The baseline parameter values for strontium given in this publication apply to ages 25 y or greater. Model predictions for younger adult ages can be derived from the age-specific parameter values given in Publication 67 (ICRP, 1993), interpolating linearly with age between values provided in that document for 15 and 25 y of age. (470) Kinetic analysis of blood disappearance curves for normal subjects intravenously injected with calcium or strontium tracers indicates that these elements initially leave blood at a rate of several hundred plasma volumes per day, and equilibrate rapidly with an extravascular compartment approximately three times the size of the plasma pool (Heaney, 1964; Harrison et al., 1967; Hart and Spencer, 1976). At times greater than 1–2 h after injection, a transfer rate from blood of approximately 15 d−1 yields a reasonable fit to blood disappearance curves for strontium or calcium tracers. The model for strontium used in this publication does not depict the extremely rapid removal of activity during the early minutes, but assigns a removal rate from blood of 15 d−1. (471) Uptake and retention of radiostrontium in soft tissues and bone have been measured in several seriously ill human subjects (Comar et al., 1957; Schulert et al., 1959). The data indicate that soft tissues initially contain approximately as much strontium as bone, but the soft tissue content falls off sharply after a few weeks while the bone content declines slowly over the first few months. (472) Soft tissue contents of 85Sr and 45Ca were measured in postmortem tissues of several human subjects injected with these radionuclides during late stages of terminal illnesses, from a few hours to 4 months before death (Schulert et al., 1959). The fraction of injected activity remaining in soft tissues after clearance of the rapid-turnover pool was approximately the same for the two radionuclides. It appeared that strontium was removed more slowly than calcium from the intermediate-term pool. No information on the presumably small, long-term retention compartment (ST2) could be gained from this relatively short-term study. (473) The rates of transfer of strontium between blood and the soft tissue compartments are set as follows. It is assumed that 50% of strontium leaving blood moves to the rapid-turnover soft tissue compartment ST0; this is the balance after deposition percentages in other compartments are assigned. The corresponding transfer rate from blood to ST0 is 0.50 × 15 d−1 = 7.5 d−1. Based on the assumed relative amounts of strontium in ST0 and blood, the transfer rate from ST0 to blood is set at one-third of the transfer rate from blood to ST0, or 2.5 d−1. Fractional transfer from blood to ST1 is assumed to be 0.1, the same as for calcium; the corresponding transfer rate is 0.1 × 15 d−1 = 1.5 d−1. The removal half-time from ST1 to blood is set at 6 d for strontium [transfer rate = ln(2)/6 d = 0.116 d−1], compared with 4 d for calcium, to account for the slower decline in soft tissue activity for strontium than calcium indicated by human injection data. Fractional deposition in the relatively non-exchangeable soft tissue pool, ST2, is set at 0.0002 (transfer rate = 0.0002 × 15 d−1 = 0.003 d−1) compared with 0.00005 for calcium. This is consistent with the estimate that soft tissues of the adult contain 1% of the body's natural strontium (Schlenker et al., 1982), assuming the removal half-time from ST2 to blood is the same as that used in the model for calcium (5 y, corresponding to a transfer rate of 0.00038 d−1). (474) Data from laboratory animals indicate that fractional deposition on bone surfaces is similar for calcium, strontium, barium, and radium (Bligh and Taylor, 1963; Kshirsagar et al., 1966; Domanski et al., 1969, 1980). This is consistent with limited data from controlled studies on human subjects, including measurements of radiocalcium and radiostrontium in bone samples from subjects injected 3 h or longer before death (Schulert et al., 1959); and external measurements of the build-up of radiocalcium (Anderson et al., 1970; Heard and Chamberlain, 1984) and radiobarium (Korsunskii et al., 1981) after intravenous injection. Based on these data, 25% of calcium, strontium, barium, or radium leaving blood is assigned to bone surfaces. The transfer rate from blood to cortical and trabecular surfaces combined is 0.25 × 15 d−1 = 3.75 d−1. (475) The initial distribution between cortical and trabecular bone appears to be similar for calcium, strontium, barium, and radium (Ellsasser et al., 1969; Wood et al., 1970; Liniecki, 1971; Stather, 1974; Lloyd et al., 1976). Relative deposition on cortical and trabecular bone surfaces is based on the estimated calcium turnover rate of each bone type. As an average over adult ages, deposition on trabecular bone is estimated to be 1.25 times that on cortical bone (Leggett et al., 1982). The transfer rate from blood to trabecular bone surface is (1.25/2.25) × 3.75 d−1 = 2.08 d−1, and the transfer rate from blood to cortical bone surface is (3.75–2.08) d−1 = 1.67 d−1. (476) The residence time on human bone surfaces has not been determined with much precision for any of the alkaline earth elements. The removal half-time of 1 d is estimated for all four elements. This value is consistent with autoradiographic measurements of surface activity in human and canine bone samples taken at times ranging from a few hours to a few days after intravenous injection of 45Ca (Riggs et al., 1971; Groer et al., 1972; Groer and Marshall, 1973; ICRP, 1973). It is also reasonably consistent with measurements of the early decline in whole-body retention of intravenously injected radioactive calcium, strontium, barium, and/or radium in human subjects (Bishop et al., 1960; Spencer et al., 1960; Heaney, 1964; Harrison et al., 1967; Phang et al., 1969; Carr et al., 1973; Likhtarev et al., 1975; Malluche et al., 1978; Henrichs et al., 1984; Newton et al., 1990, 1991) coupled with measurements of soft tissue retention as described earlier. A removal half-time of 1 d refers to the half-time that one would observe theoretically if recycling of activity to bone surfaces was eliminated. Given the considerable amount of recycling from blood to bone surfaces, the corresponding net or apparent half-time would be 3 d or more. (477) Parameter values for exchangeable bone volume are estimated from whole-body measurements for human subjects using data for times after bone surfaces and soft tissues have largely cleared of activity, but before loss from bone resorption becomes an important consideration. Based on analysis of whole-body retention data for human subjects injected with radioisotopes of calcium, strontium, barium, or radium (Bishop et al., 1960; Spencer et al., 1960; Heaney, 1964; Harrison et al., 1967; Maletskos et al., 1969; Phang et al., 1969; Carr et al., 1973; Likhtarev et al., 1975; Malluche et al., 1978; Henrichs et al., 1984; Newton et al., 1990, 1991), the fraction of activity that moves from bone surfaces back to blood is assumed to be the same for all four elements. Specifically, five-sixths of activity leaving bone surfaces is assumed to return to blood, and one-sixth is assumed to transfer to exchangeable bone volume. The transfer rate from trabecular or cortical bone surface to the corresponding exchangeable bone volume compartment is (1/6) × ln(2)/1 d = 0.116 d−1, and the transfer rate from trabecular or cortical bone surface to blood is (5/6) × ln(2)/1 d = 0.578 d−1. (478) Element-specific removal half-times from the exchangeable bone volume compartments are based, in part, on fits to the intermediate-term retention data from human injection studies. It is also considered that the assigned half-times should increase approximately in proportion to the likelihood of the element entering non-exchangeable sites in bone mineral, as suggested by data from in-vitro experiments with hydroxyapatite crystals and whole-body retention patterns for alkaline earth elements in human subjects. A removal half-time of 80 d is assigned to strontium, compared with 100 d for calcium, 50 d for barium, and 30 d for radium (Leggett, 1992). As the data do not allow the derivation of removal half-times as a function of bone type, the same half-time is applied to cortical and trabecular exchangeable bone volume compartments. (479) Discrimination between alkaline earth elements by bone is accounted for by fractional transfer of activity from exchangeable to non-exchangeable bone volume. It is assumed that calcium, strontium, barium, and radium are all equally likely to become temporarily incorporated in bone mineral after injection into blood, but that the likelihood of reaching a non-exchangeable site in bone crystal decreases in the order calcium > strontium > barium > radium. Fractional transfers of calcium, strontium, barium, and radium from exchangeable to non-exchangeable bone volume are set at 0.6, 0.5, 0.3, and 0.2, respectively, for consistency with whole-body and skeletal retention data on these elements (Bishop et al., 1960; Spencer et al., 1960; Heaney et al., 1964; Harrison et al., 1967; Maletskos et al., 1969; Phang et al., 1969; Carr et al., 1973; Likhtarev et al., 1975; Malluche et al., 1978; Henrichs et al., 1984; Newton et al., 1990, 1991), as well as results of in-vitro measurements on hydroxyapatite crystals (Neuman, 1964; Stark, 1968). The derived rate of transfer of strontium from exchangeable trabecular or cortical bone volume to the corresponding non-exchangeable bone volume compartment is 0.5 × ln(2)/80 d = 0.0043 d−1, and the transfer rate to the corresponding bone surface compartment is 0.5 × ln(2)/80 d = 0.0043 d−1. (480) Biological removal from the non-exchangeable bone volume compartments of cortical and trabecular bone is assumed to result from bone turnover. The average bone turnover rates during adulthood are estimated as 3% y−1 and 18% y−1 for cortical and trabecular bone, respectively (ICRP, 2002). The corresponding transfer rates from the non-exchangeable bone volume compartments of cortical and trabecular bone to blood are 0.0000821 d−1 and 0.000493 d−1, respectively. Age-specific rates of bone turnover, including changes with age during adulthood, are provided in the paper by Leggett (1992) for application of the model to specific cases. (481) Clearance of strontium from blood to urine and faeces has been determined in several human studies (Spencer et al., 1960; Barnes et al., 1961; Fujita et al., 1963; Cohn et al., 1963; Samachson, 1966; Harrison et al., 1967; Likhtarev et al., 1975; Wenger and Soucas, 1975; Newton et al., 1990). Based on central estimates derived from results of these studies, it is assumed that 11.5% of strontium leaving blood is transferred to the urinary bladder contents and subsequently to urine, and 3.5% is transferred to the right colon contents and subsequently to faeces. Therefore, the transfer rate from blood to the urinary bladder contents is 0.115 × 15 d−1 = 1.73 d−1, and the transfer rate from blood to the right colon contents is 0.035 × 15 d−1 = 0.525 d−1.
10.2.3.4. Treatment of radioactive progeny
(482) Dosimetrically significant radioactive progeny of strontium isotopes considered in this publication include isotopes of rubidium, krypton, and yttrium. Results of animal studies (Arnold et al., 1955; Lloyd, 1961; Mueller, 1972; Stevenson, 1975) indicate that 90Y produced by decay of 90Sr in soft tissues tends to migrate from the parent and distribute similarly to intravenously injected yttrium, but shows little if any migration from 90Sr when produced in bone volume (see the section on yttrium in this publication for summaries of reported data (Section 11.2.3.)). No information was found on the behaviour of rubidium produced in the body by decay of a strontium parent. The noble gas krypton produced by serial decay of strontium and rubidium isotopes presumably migrates from these radionuclides over a period of minutes to hours, and escapes from the body to an extent determined by the half-life of the krypton isotope. (483) The model used in this publication for yttrium as a progeny of strontium is based on the model for yttrium as a parent described elsewhere in this publication, but additional assumptions are made to address structural differences in the strontium and yttrium models. Yttrium produced in a compartment of bone is assumed to follow the same kinetics as if deposited in the compartment as a parent radionuclide. No distinction is made between the exchangeable and non-exchangeable bone volume compartments of the strontium model when applied to yttrium, i.e. each compartment is treated simply as the bone volume compartment for the corresponding bone type in the yttrium model. Yttrium produced in a soft tissue compartment of the strontium model (ST0, ST1, or ST2) is assumed to transfer to blood with a half-time of 3 d (the shortest removal half-time from compartments of other soft tissue in the model for yttrium as a parent), and then to follow the kinetics of yttrium as a parent radionuclide. (484) The model for rubidium as a progeny of strontium is a condensed version of a proposed model for rubidium as a parent radionuclide (Leggett and Williams, 1988). The model is based on the same principles as the model for caesium, a chemical and physiological analogue of rubidium, described elsewhere in the OIR series. That is, the biokinetics of systemic rubidium is predicted on the basis of the distribution of cardiac output, experimentally determined tissue-specific extraction fractions, and the steady-state distribution of stable rubidium in the body. The reference division of cardiac output in the adult male tabulated in Publication 89 (ICRP, 2002) is applied here. The present version of the model depicts blood plasma as a central compartment that exchanges rubidium with RBCs, trabecular bone surface, cortical bone surface, muscle, and a compartment representing all other soft tissue. Rates of transfer of rubidium from plasma are as follows: 6 d−1 to RBCs, 255 d−1 to muscle, 5.6 d−1 to cortical bone surface, 8.4 d−1 to trabecular bone surface, 855 d−1 to other tissue, 3.9 d−1 to urinary bladder contents, 1.2 d−1 to right colon contents, and 0.1 d−1 to excreta (loss in sweat). Transfer rates from RBCs or tissues to plasma are as follows: 0.35 d−1 from RBCs, 1.14 d−1 from muscle, 1.68 d−1 from bone surface compartments, and 10.3 d−1 from other tissue. Rubidium produced by decay of strontium in blood is assigned to plasma. Rubidium produced in exchangeable or non-exchangeable bone volume compartments of the strontium model are transferred to plasma at the rate of bone turnover. Rubidium produced in soft tissue compartments of the strontium model (ST0, ST1, or ST2) are transferred to plasma at a rate of 10.3 d−1. The subsequent behaviour of rubidium that reaches plasma is determined by the model for rubidium described above. (485) The model for krypton produced by serial decay of strontium and rubidium in systemic compartments is similar to the model applied in the OIR series to radon produced in vivo by decay of a parent radionuclide (ICRP, 2017, see radon section in OIR Part 3). Krypton is assumed to follow the bone model for radon introduced in Publication 67 (ICRP, 1993), but is assigned a higher rate of removal from soft tissues to blood than is assumed for radon. Specifically, krypton produced in non-exchangeable bone volume, exchangeable bone volume, or bone surface transfers to blood at rates of 0.36 d−1, 1.5 d−1, or 100 d−1, respectively. Krypton produced in a soft tissue compartment transfers to blood with a half-time of 15 min, compared with an assumed half-time of 30 min for radon produced by radioactive decay in soft tissues. Krypton entering blood is assumed to be removed from the body (exhaled) at a rate of 1000 d−1, corresponding to a half-time of 1 min. Recycling of krypton to tissues via arterial blood is not depicted explicitly but is considered in the assignment of effective half-times in tissues. The model is intended to yield a conservative average residence time of krypton atoms produced in systemic pools by decay of a parent radionuclide. It is recognised that the residence time of krypton in the body following production in tissues depends on the distribution of the parent radionuclide.
10.3. Individual monitoring
10.3.1. 85Sr
(486) 85Sr monitoring techniques include in-vivo techniques (whole body and, if necessary, lung counting) as well as urine bioassay (Table 10.4). Transfer coefficients for systemic strontium. ST, soft tissue; Exch, exchangeable; Nonexch, non-exchangeable. ST0, ST1, and ST2 are compartments within other soft tissues with fast, intermediate, and slow turnover, respectively. Monitoring techniques for 85Sr.
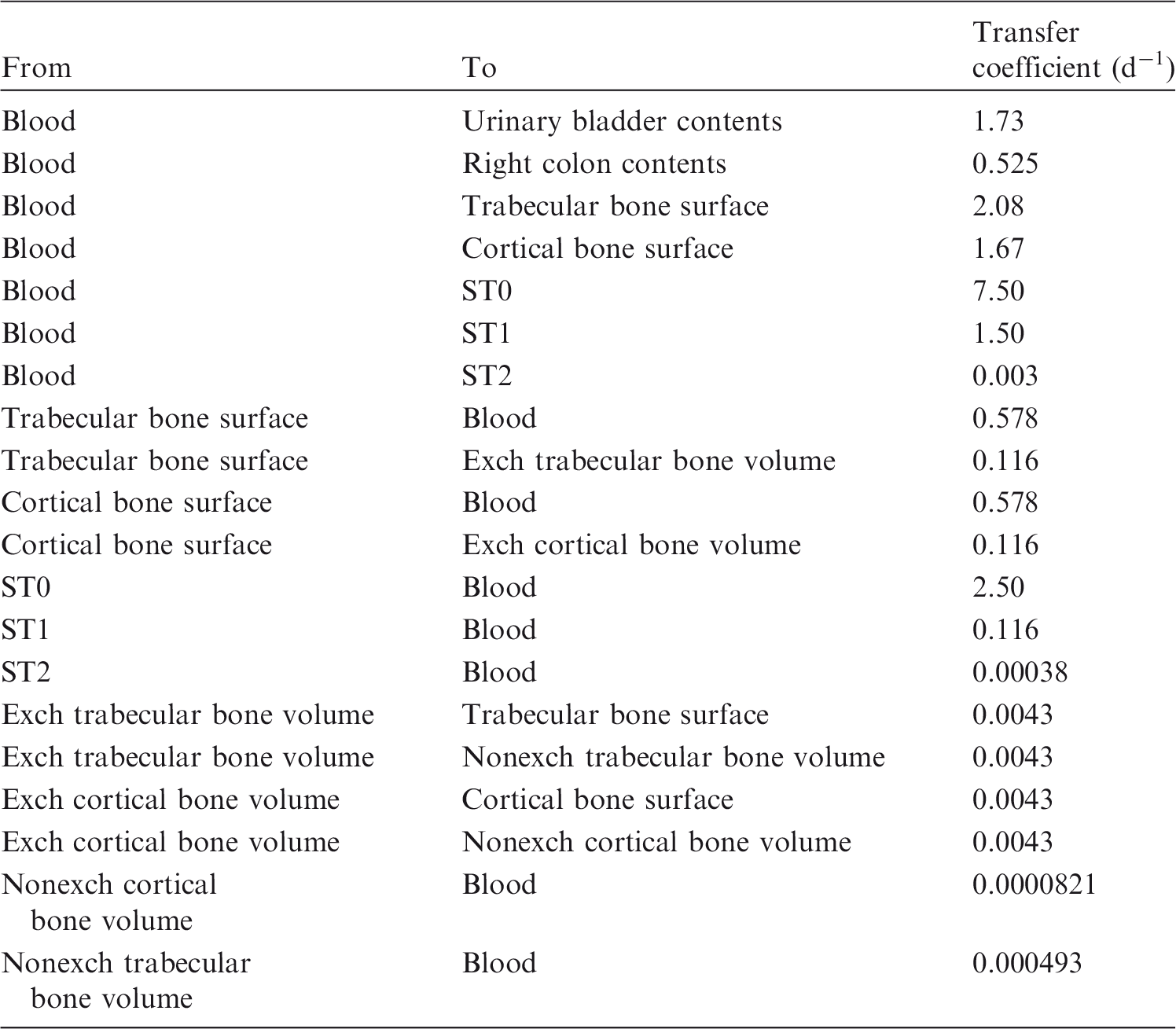

10.3.2. 89Sr
(487) 89Sr is determined by urine bioassay by beta counting following chemical separation. Monitoring techniques for 89Sr.

10.3.3. 90Sr
(488) 90Sr intakes are generally estimated by beta counting of urine excreta samples after chemical separation. 90Sr is determined directly when liquid scintillation counting is used. When beta proportional counting is used, 90Sr content is commonly determined based on 90Y content after a delay of at least 7 d to allow for 90Y ingrowth. Monitoring techniques for 90Sr.

10.4. Dosimetric data for strontium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 85Sr, 89Sr, and 90Sr compounds.

AMAD, activity median aerodynamic diameter; FAP, fused aluminosilicate particles; PSL, polystyrene.
Dose per activity content of 85Sr in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.

Dose per activity content of 89Sr in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
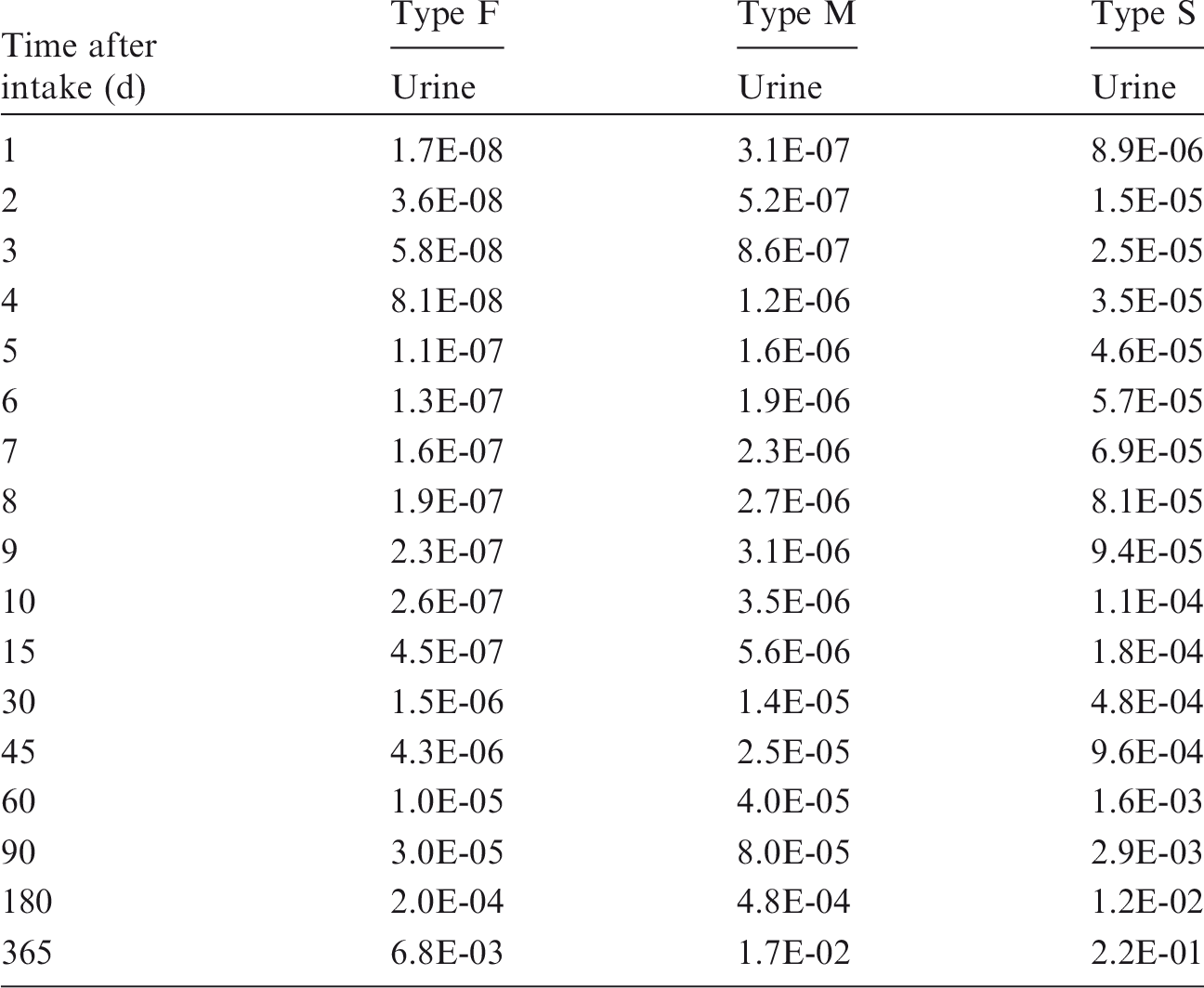
Dose per activity content of 90Sr in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
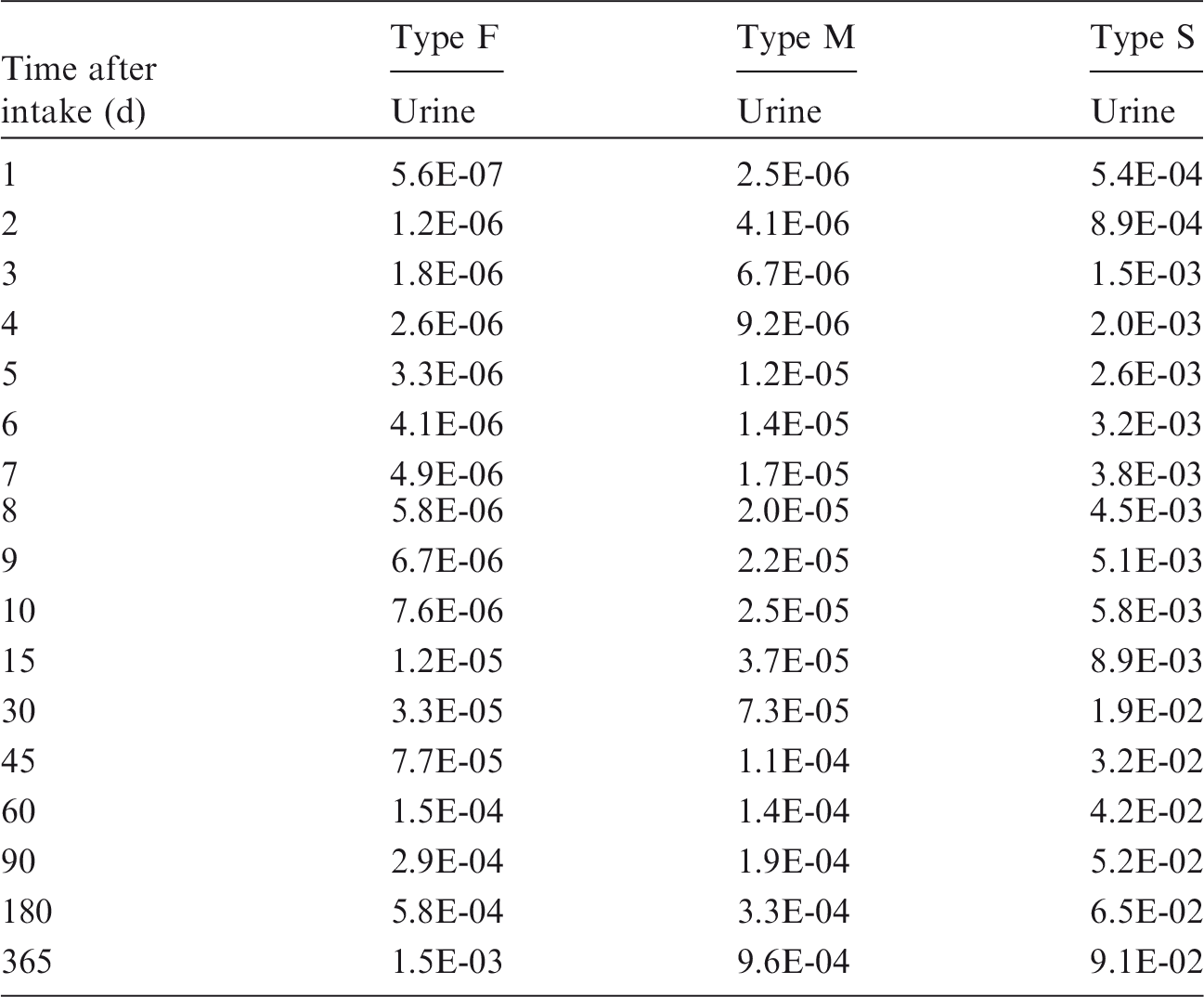
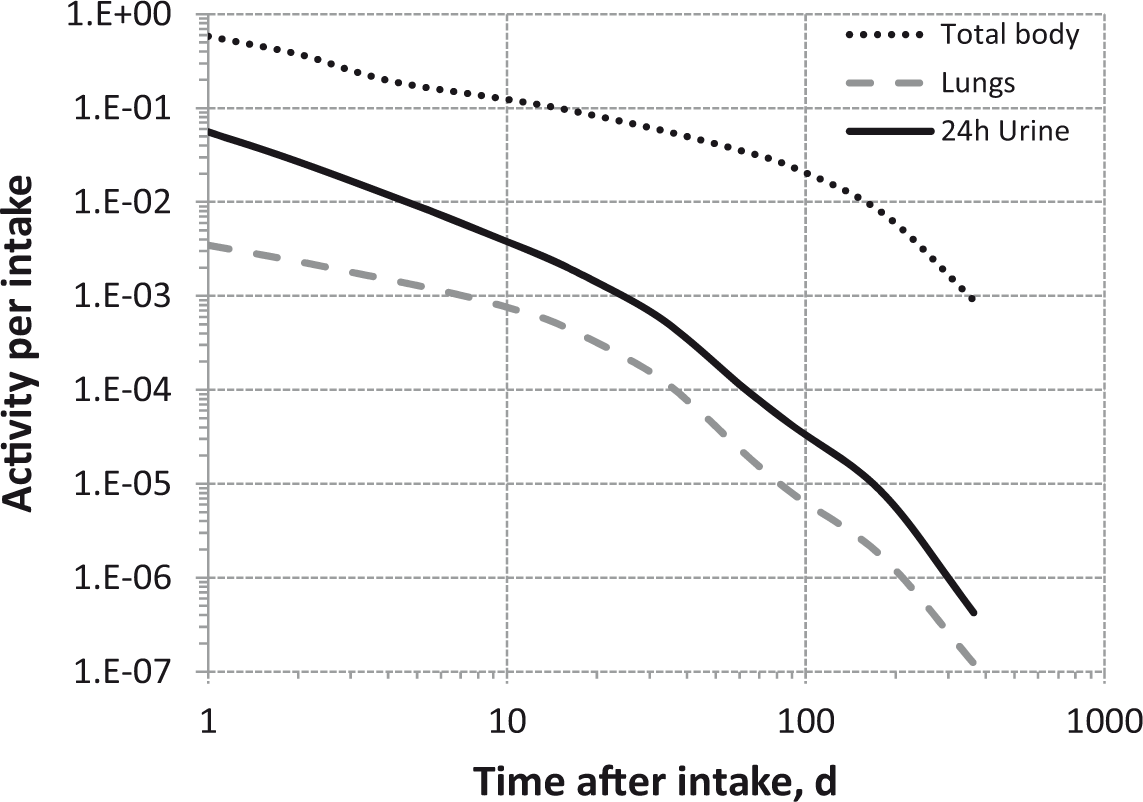
Total body and lung contents, and daily urinary excretion of 85Sr following inhalation of 1 Bq Type F.

Total body and lung contents, and daily urinary excretion of 85Sr following inhalation of 1 Bq Type M.

Total body and lung contents, and daily urinary excretion of 85Sr following inhalation of 1 Bq Type S.
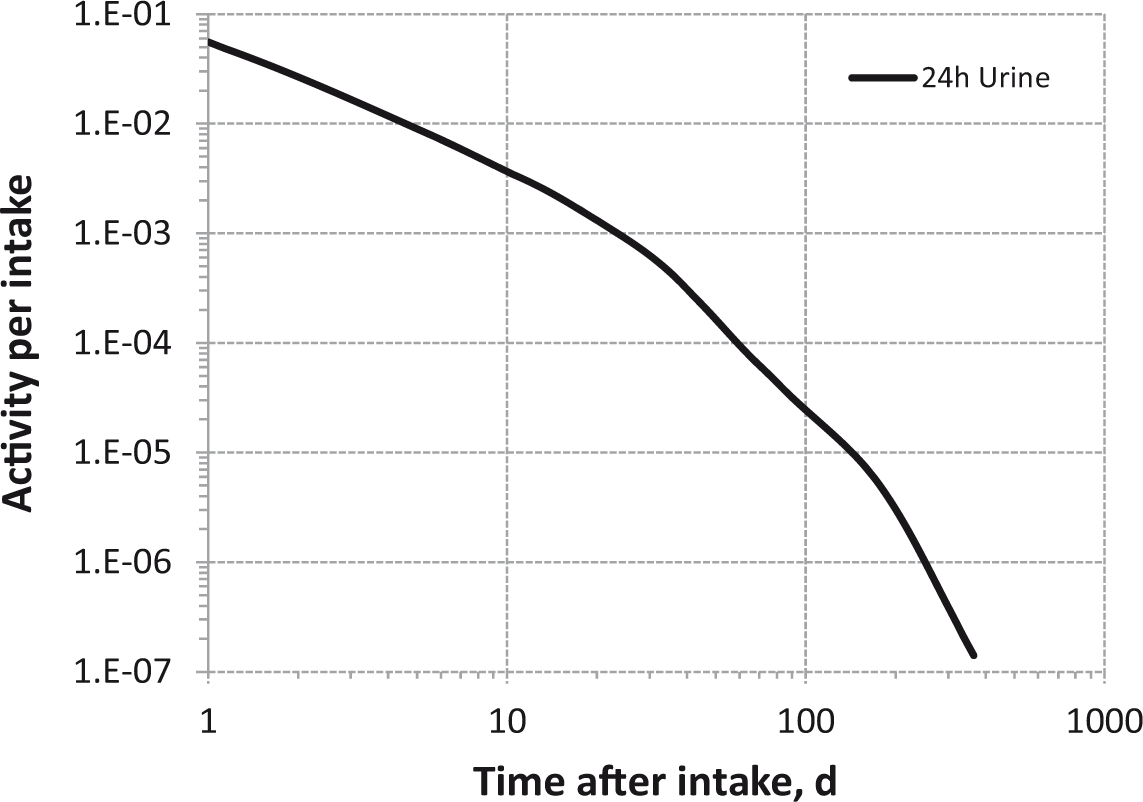
Daily urinary excretion of 89Sr following inhalation of 1 Bq Type F.
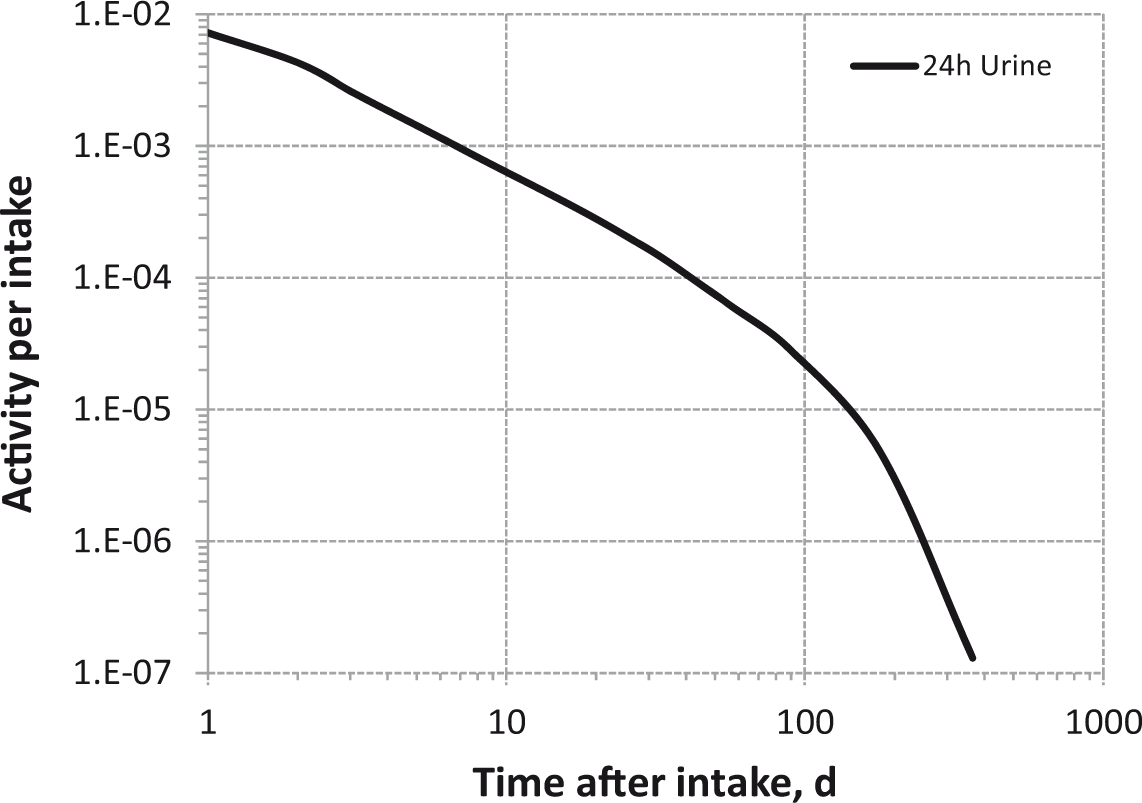
Daily urinary excretion of 89Sr following inhalation of 1 Bq Type M.

Daily urinary excretion of 89Sr following inhalation of 1 Bq Type S.

Daily urinary excretion of 90Sr following inhalation of 1 Bq Type F.

Daily urinary excretion of 90Sr following inhalation of 1 Bq Type M.

Daily urinary excretion of 90Sr following inhalation of 1 Bq Type S.
10.5. References
11. YTTRIUM (Z = 39)
11.1. Chemical forms in the workplace
(489) Yttrium is a rare earth element that occurs mainly in oxidation state III. Yttrium may be encountered in a variety of chemical and physical forms, including oxides (Y2O3), hydroxides, chlorides, fluorides, sulphates, nitrates, and oxalates. Table 11.1 shows the isotopes of yttrium addressed in this publication. (490) 90Y and 91Y are the main fission products that may be encountered in the nuclear industry. 90Y is used in nuclear medicine for the treatment of various cancers with labelled drugs. Isotopes of yttrium addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay.

11.2. Routes of intake
11.2.1. Inhalation
(491) Information on absorption from the respiratory tract is available from experimental studies of yttrium mainly as chloride or in FAP. Analysis of the results to estimate absorption parameter values is facilitated by the close correspondence of faecal excretion to particle transport from the respiratory tract; absorption of yttrium in the alimentary tract is low, and systemic yttrium is excreted mainly in urine. (492) Absorption parameter values and types, and associated fA values for particulate forms of yttrium are given in Table 11.2. Absorption parameter values for inhaled and ingested yttrium. FAP, fused aluminosilicate particles. It is assumed that the bound state can be neglected for yttrium, i.e. fb=0. The values of sr for Type F, M, and S forms of yttrium (1 d−1, respectively) are element-specific. Materials (e.g. Chloride) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of yttrium (1×10−4). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (1×10−4) for ingestion of the radionuclide.

11.2.1.1. Particulate materials
(a) Yttrium chloride (YCl3)
(493) Extensive studies have been conducted on the biokinetics of yttrium following deposition of the chloride in the lungs of dogs, guinea pigs, rats, and mice. Most of the studies involved small masses of radiolabelled yttrium, and showed a similar pattern. Initially, most of the excretion was to faeces, indicating that there was little absorption from the upper respiratory tract. Nevertheless, subsequent clearance of most of the lung deposit was rapid with corresponding systemic uptake: mainly deposition in skeleton and excretion in urine. Similar lung dissolution kinetics was observed in the different species, and the distribution of yttrium absorbed systemically was similar to that observed after intravenous injection. (494) In a detailed low-level study undertaken to complement a lifespan study of the effects of inhaled 91YCl3, the biokinetics of 91Y was followed for 270 d after inhalation of 91YCl3 (in caesium chloride solution) by dogs (McClellan and Rupprecht, 1967; Muggenburg et al., 1998). On average, approximately 60% of whole-body 91Y cleared during the first few days after administration. It was inferred that 91Y deposited in the upper respiratory tract was mainly cleared by mucociliary transport and subsequent swallowing and faecal excretion. This suggests that the rapid dissolution rate is low compared with particle transport rates from these airways. Nevertheless, there was significant deposition in liver and skeleton immediately after inhalation, with the lung content falling to approximately 15% ILD by 4 d, and to approximately 2% ILD by 64 d. Studies of the distribution of activity retained in the respiratory tract provide evidence for the formation of particulate material. Autoradiographs were made using tissues from dogs in the lifespan study that died in the first few weeks after exposure (McClellan and Rupprecht, 1967). Within the respiratory tract, aggregates of radioactivity were observed on bronchial mucosal surfaces and in recesses of the mucosal lining. Smaller particles were also found in alveolar ducts and alveoli. Some of the material had been phagocytised, absorbed into the lymphatic system, and could be seen in the lymphatic spaces beneath the bronchial epithelium. Large amounts of 91Y were found in bronchial cartilage plates but attributed to systemic 91Y, with similar deposition in skeletal cartilage. Muggenburg et al. (1998) reported concentrations in a wide range of tissues at 32 d after inhalation. The concentration in tracheobronchial lymph nodes was similar to that in liver, and higher than in other soft tissues, suggesting some transfer in particulate form. Modelling conducted here (i.e. by the Task Group) showed a good fit to the data with fr = 0.94, sr = 0.74 d−1, and ss = 0.013 d−1 (consistent with assignment to default Type F). As this is the most comprehensive and longest duration dataset for yttrium chloride, it probably provides the best estimates of sr and ss, and these values were used in analysis of some other datasets below. (495) Schiessle et al. (1963) followed the biokinetics of 91Y for 180 d after inhalation of 91YCl3 (carrier free) by guinea pigs. There are comprehensive measurements at seven time points up to 28 d, but few results at later times. Modelling conducted here gave parameter values of fr = 0.81, sr = 1.07 d−1, and ss = 0.016 d−1 (consistent with assignment to default Type F), in broad agreement with those based on the study by Muggenburg et al. (1998). Schmidtke et al. (1963) followed the biokinetics of 91Y for 56 d after inhalation of 91YCl3 with added stable yttrium by guinea pigs. Compared with the behaviour of carrier-free 91YCl3 (Schiessle et al., 1963), lung retention and faecal clearance were somewhat higher, and skeletal uptake and urinary excretion were lower. Schmidtke et al. (1964) performed complementary autoradiographic studies on respiratory tract tissues obtained 21 d after inhalation of 91YCl3 by guinea pigs. Schmidtke (1964) investigated the effect of DTPA on the biokinetics of 91Y for 8 d after inhalation of 91YCl3 (carrier free) by guinea pigs. Unusually, the tissue distribution was measured at several time points during the first day. Modelling conducted here on results from control animals (using a fixed value of ss = 0.013 d−1, derived above, because of the short duration of measurements in this study) gave parameter values of fr = 0.83 and sr = 1.3 d−1 (consistent with assignment to default Type F), in good agreement with those based on the study by Schiessle et al. (1963). Treatment with DTPA caused rapid clearance of 91Y from the lungs and excretion from the body. (496) Wenzel et al. (1969) followed the biokinetics of 88Y for 32 d after inhalation of 88YCl3 by rats, either carrier-free or with added stable yttrium. Lung retention was higher, and skeletal uptake and urinary excretion were lower, in rats exposed to 88Y with stable yttrium than in those that inhaled carrier-free 88Y. Faecal clearance was also higher, suggesting that the additional lung retention was in particulate form, rather than bound. Using fixed values of sr = 0.74 d−1 and ss = 0.013 d−1, derived above, modelling conducted here gave values of fr = 0.94 (consistent with assignment to Type F) for the 88YCl3 inhaled in carrier-free form, and fr = 0.7 (consistent with assignment to Type M) for the 88YCl3 inhaled with added stable yttrium. (497) Bailey et al. (1978) followed the biokinetics of 88Y for 9 d after intratracheal instillation of 88YCl3 into rats. By 2 d, approximately 20% ILD remained in the lungs, 50% ILD had been excreted in faeces, and 30% was deposited in systemic sites or excreted in urine, again suggesting little absorption from the upper airways but considerable absorption from the deep lung. They also developed a systemic compartment model for 88Y in the rat based on an intravenous injection study. With only two time points, there are insufficient data to define all three dissolution parameter values. Using fixed values of sr = 0.74 d−1 and ss = 0.013 d−1, derived above, modelling conducted here showed a good fit to the data with fr = 0.7 (consistent with assignment to default Type M). (498) Hirano et al. (1990) followed the lung retention and distribution of yttrium for 162 d after intratracheal instillation into rats of 100 µg of stable yttrium as chloride. The retention half-time of approximately 170 d is far greater than observed in the studies with 88YCl3 or 91YCl3 reviewed here. There was also relatively little systemic uptake, but few details are given. The authors concluded that the yttrium was retained in the lungs in an insoluble form. The clearance was considerable slower than would be expected for insoluble particles in rats (ICRP, 2002), suggesting that there was considerable binding of yttrium to lung structures. Yttrium was detected in alveolar and interstitial macrophages and in basement membranes, supporting this inference. However, dose-related inflammatory responses were seen over the range of masses (10–200 µg) administered in complementary short-term experiments, and so the kinetics may well differ from those pertaining at tracer levels. Marubashi et al. (1998) reported that 30 d after intratracheal instillation into rats of 50 µg of stable yttrium as chloride, approximately 67% ILD remained; again, much slower clearance than observed in the radiotracer studies. (499) Gensicke and Nitschke (1964) showed that treatment with hexametaphosphate increased the clearance of 91Y after inhalation of 91YCl3 by mice. There is insufficient information in the paper to enable dissolution parameter values to be derived reliably, but the biokinetics in the controls appears to be broadly similar to that in the other radiotracer studies outlined above, with activity in the skeleton exceeding that in the lungs by approximately 1 week after inhalation. (500) Based on the results of the experiments outlined above, approximate absorption parameter values were derived here of fr = 0.9, sr = 1 d−1, ss = 0.01 d−1 (consistent with assignment to default Type F). Although specific parameter values for yttrium chloride based on in vivo data are available, they are not adopted here, because inhalation exposure to it is unlikely. Instead, yttrium chloride is assigned to Type F. However, the data are used as the basis for the default rapid dissolution rate for yttrium.
(b) Yttrium oxide (Y2O3)
(501) Newton et al. (1971) measured tissue retention of 91Y at 8 and 64 d after inhalation of 91Y2O3 by dogs. At 8 d, the activity in the skeleton was approximately 30% of that in the lungs, and at 64 d, they were approximately equal. From results of a complementary gavage experiment, it was calculated here that fractional absorption from the alimentary tract was fA = 3×10−4. Using a fixed value of sr = 0.74 d−1 derived above for yttrium chloride, modelling conducted here gave values of fr = 0.45 and ss = 0.006 d−1 (consistent with assignment to Type M). Given the relatively sparse information, specific parameter values are not recommended here for yttrium oxide; instead, it is assigned to Type M.
(c) Yttrium phosphate (YPO4)
(502) Newton et al. (1971) measured tissue retention of 91Y at 8 and 64 d after inhalation of 91YPO4 by dogs. The activity in the skeleton was approximately 20% of that in the lungs at 8 d, and 45% at 64 d. [The authors noted that following both inhalation and gavage of 91YPO4, the ratio of deposition in the skeleton to that in the liver (∼3:1) was lower than following inhalation of other forms of 91Y (∼6:1 for chloride, oxide, and FAP), but that this observation needed confirmation.] From results of a complementary gavage experiment, it was calculated here that fractional absorption from the alimentary tract was fA = 0.0004. Using a fixed value of sr = 0.74 d−1 derived above for yttrium chloride, modelling conducted here gave values of fr = 0.33 and ss = 0.002 d−1 (consistent with assignment to Type M). Given the relatively sparse information, specific parameter values are not recommended here for yttrium phosphate; instead, it is assigned to Type M.
(d) Fused aluminosilicate particles
(503) FAP or ‘fused clay’ particles have been used extensively as relatively insoluble particles in inhalation studies, both of biokinetics and of radiation effects. A natural clay mineral is labelled by ion exchange, and the labelled clay particles are heated to approximately 1100℃ to form aluminosilicate glass microspheres in which the label is incorporated. It has been demonstrated that when yttrium is incorporated into FAP, only a small fraction is absorbed rapidly, and the remainder is retained within the particles and absorbed slowly. (504) In a detailed low-level study performed to complement a lifespan study of the effects of inhaled 91Y FAP (Hahn et al., 1994), the biokinetics of 91Y was followed for 320 d after inhalation of 91Y FAP by dogs (Hobbs et al., 1971). By 8 d after inhalation, 97% of 91Y remaining in the body was in the lungs, with < 1% in the skeleton, but the latter had increased to approximately 10% by 256 d. Using a fixed value of sr = 0.74 d−1 derived above for yttrium chloride, modelling conducted here gave values of fr = 0.004 and ss = 0.0009 d−1 (consistent with assignment to Type S). In a similar low-level study performed to complement a lifespan study of the effects of inhaled 90Y FAP (Hahn et al., 1983), the biokinetics of 90Y was followed for 12 d after inhalation of 90Y FAP by dogs (Hobbs et al., 1970; Barnes et al., 1972). The shorter duration reflects the 64 h half-life of 90Y. During this period, the activity distribution was similar to that seen in the more extensive 91Y FAP study. Estimates of the rate of dissolution of Y FAP following inhalation of 88Y FAP by rats and men were in the range (1.5–5) x10−4 d−1 (Bailey et al., 1981, 1985), indicating assignment to Type S. Rates of dissolution of 91Y FAP measured in vitro varied considerably, depending on particle size and conditions, in the range 1×10−5–0.001 d−1 (Kanapilly and Goh, 1973), and indicate Type M or S behaviour.
11.2.1.2. Rapid dissolution rate for yttrium
(505) Studies with yttrium chloride give values of sr of approximately 1 d−1, and this is applied here to all Type F forms of yttrium. As it is lower than the general default value of 3 d−1 for Type M and S materials, it is also applied to Type M and S forms of yttrium.
11.2.1.3. Extent of binding of yttrium to the respiratory tract
(506) The results of autoradiographic studies of the distribution of 91Y after inhalation of 91YCl3 suggest that the 91Y retained in the lungs was in particulate form rather than bound to lung structures. It is therefore assumed that the bound state can be neglected for yttrium, i.e. fb = 0.0.
11.2.2. Ingestion
(507) There is little information on absorption of ingested yttrium. Studies performed on dogs and goats suggested that yttrium absorption from the gastrointestinal tract is very low (Nold et al., 1960). One other study performed on rats with 91Y used to label solid and liquid food showed that the total recovery of yttrium in the gastrointestinal tract between 30 min and 12 h after ingestion was approximately 98% (Marcus and Lengemann, 1962). (508) A study performed with rats fed daily with 90Y in drinking water showed that, after a 60 d period of ingestion, the skeleton contained less than 0.01% of the total ingested activity (Sullivan et al., 1963). This poor absorbability of yttrium has also been noticed in studies using fowl, and has led to the designation of yttrium as a non-absorbed reference substance (Sklan et al., 1975). (509) Recent studies performed in rats (Damment and Pennick, 2007) and human subjects (Pennick et al., 2006) with lanthanum carbonate can provide an indirect estimate of yttrium absorption because of their chemical analogies. Results in rats showed that 0.004% of the administered dose was recovered in the urine over a period of 7 d (Damment and Pennick, 2007), and results in humans showed an absolute bioavailability of lanthanum of approximately 0.0013% (Pennick et al., 2006). (510) In Publication 30 (ICRP, 1980), an absorption value of 1×10−4 was recommended. As no relevant additional data on the gastrointestinal absorption of yttrium are available, an fA value of 1 × 10−4 is adopted here for all chemical forms.
11.2.3. Systemic distribution, retention, and excretion
11.2.3.1. Summary of the database
(a) Overview
(511) The biokinetics of systemic yttrium varies with the mode of administration and the administered form and mass, due, in part, to the tendency of yttrium compounds to form colloids (Lloyd, 1961; Rosoff et al., 1961; Spencer, 1968). Colloidal yttrium deposits largely in the liver, spleen, or bone marrow, with the distribution depending on particle size (Dobson et al., 1948). Yttrium that is absorbed to blood across membranes or injected intravenously in non-colloidal form initially clears with a half-time of 1 h or less (Ekman and Aberg, 1961; Kawin, 1963; Schmidtke, 1964), and transfers mainly to bone surfaces, liver, kidneys, and urinary bladder contents (Hamilton, 1949; Durbin, 1960; Herring et al., 1962; Ando et al., 1989; Muggenburg et al., 1998). A few percent of the absorbed or injected amount clears more slowly from blood, presumably due mainly to attachment to plasma proteins (Rosoff et al., 1958; Hirano and Suzuki, 1996). (512) Yttrium is retained tenaciously by bone, and a substantial portion of that deposited in soft tissues also shows relatively slow return to blood. After intravenous administration of 88Y as citrate to human subjects, approximately one-fifth of the injected amount was excreted within a few days, primarily in urine, and the remainder was retained with a projected half-time of years (Etherington et al., 1989a,b).
(b) Data for human subjects
(513) Rosoff et al. (1961) and Spencer (1968) studied the rate of excretion of 90Y in elderly hospital patients after intravenous injection of different forms of yttrium, and the effects of chelating agents on the excretion rate. Less than 0.5% of the administered amount was excreted in urine during the first 24 h after administration of 90YCl3. Approximately 5% of the administered activity was excreted in urine during the first day after administration of 90Y as nitrilotriacetate (90Y-NTA), a form thought to prevent the formation of yttrium hydroxy colloids. The chelating agents EDTA and DTPA were found to be effective in removing 90Y from the body if administered in the first day or two after intake of 90Y. (514) Retention, distribution, and urinary and faecal excretion of yttrium were studied in two healthy adult male volunteers who received 88Y as citrate (t½ = 107 d) by intravenous injection (Etherington et al., 1989a,b). The behaviour of 88Y as determined by in-vivo measurement and bioassay was similar in the two subjects. An estimated 22% of the injected amount was excreted in the first few days, with urinary excretion accounting for 94% and 93% of the excreted amount in Subjects A and B, respectively, over 5 d, and 91% in Subject B over 14 d. The combined retention data for the subjects could be approximated by a two-exponential function to time t (d) after injection:
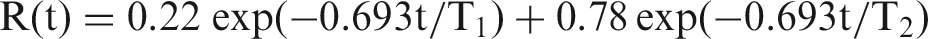
(c) Data for laboratory animals
(515) For comparison with findings summarised above for their two human subjects, Etherington et al. (1989a,b) determined the tissue distribution of 88Y in rats intravenously injected with similar 88Y solutions. The findings for rats were broadly consistent with the systemic biokinetics estimated for the human subjects, the main difference being that removal from the liver was faster and faecal excretion rate was higher in rats. On average, urinary and faecal excretion accounted for 26.1% and 8.4%, respectively, of injected activity after 4 d in rats. The contents of liver, kidneys, gastrointestinal tract, and carcass (including skeleton) accounted for 4.4%, 1.4%, 0.9%, and 58.6%, respectively. (516) In rats receiving 91YCl3 by parenteral injection, 55–65% of the administered amount was deposited in the skeleton, and little of this was lost over the next 2–3 months (Hamilton, 1949; Durbin, 1960). At 4 d after administration, the liver contained approximately 12% of the administered activity, and excreta (primarily urine) accounted for approximately 26% (Durbin, 1960). Data of Ando et al. (1989) indicate that the liver contained a major portion of the systemic activity between 3 h and 2 d after intravenous injection of 90YCl3 into rats. (517) Watanabe et al. (2005) studied the effectiveness of CaNa3DTPA in removing 90Y from the body in rats contaminated with 90Y chloride via a puncture wound. In control animals, the concentration of 90Y in bone was, on average, approximately 10 times that in liver, six times that in kidney, and 60 times that in blood during the first 24 h. At 7 d, the concentration in bone was approximately 39 times that in liver, 17 times that in kidney, and 1900 times that in blood. Prompt treatment of the wound with CaNa3DTPA was found to be more effective than systemic treatment in minimising accumulation of 90Y in bone. (518) A goat receiving 91Y by intravenous injection excreted approximately 20% of the injected amount in urine and 4% in faeces over the first 10 d (Ekman and Aberg, 1961). The concentration of 91Y in blood serum declined by a factor of approximately 8 from a few minutes to 3 h after injection, and a factor of approximately 2.5 from 3 to 24 h after injection. Approximately half of the total 10 d urinary losses occurred on the first day and approximately one-quarter occurred on the second day. Faecal losses were approximately 0.7% on day 1, 2% on day 2, and 0.4% on day 3, and declined monotonically thereafter. Examination of cartilage from the trachea and ribs indicated that 91Y may have been bound to chondroitinsulphuric acid. (519) After brief inhalation of 91YCl3 by guinea pigs, approximately 28% of the deposited activity was absorbed to blood over the first 8 d (Schmidtke, 1964). At that time, the skeleton, liver, kidneys, and blood of animals not receiving chelation therapy contained approximately 65%, 5%, 1%, and 0.15%, respectively, of the absorbed activity. Urinary excretion during the first 8 d accounted for approximately 22% of the absorbed amount. (520) The biokinetics, dosimetry, and radiological effects of 91Y have been studied in dogs exposed to different 91Y aerosols (McClellan and Rupprecht, 1967; Barnes et al., 1972; Muggenburg et al., 1998). Detailed systemic data were obtained for dogs exposed to relatively soluble 91YCl3 aerosols. A sharp drop in whole-body 91Y occurred during the first few days after exposure, presumably due to clearance of activity deposited in the upper respiratory tract by mucociliary transport, and subsequent swallowing and faecal excretion. After approximately 3 weeks, the rate of decline of the body burden approximated the radiological half-life of 91Y. Daily losses in urine and faeces were measured in three dogs through 64 d post exposure. On average, approximately 15% of the initial body burden was removed in urine, and approximately 45–50% in faeces during the first week. Faecal excretion was the dominant route of excretion during the first 4 d, but beyond 2 weeks, post injection daily urinary excretion was 1.5–4 times greater than daily faecal excretion. Tissue concentrations of 91Y measured in three dogs at 32 d after intake indicated that the skeleton, liver, and kidneys contained approximately 75%, 15%, and 1%, respectively, of the systemic burden. Autoradiographs were made using tissue collected at necropsy of dogs dying in the early postexposure period. In bones, activity was prominent on bone surfaces. The concentration in long bones was higher near the ends than in the shaft. Activity was generally diffuse in the liver and spleen. Absorbed 91Y was found in bronchial cartilage. (521) In studies on young dogs receiving 91Y by intravenous or intraperitoneal injection, activity depositing in the skeleton was found to concentrate on non-growing, highly calcified surfaces and resorbing surfaces of bone (Jowsey et al., 1958; Herring et al., 1962). No deposition was found in osteoid tissue. It was suggested that the mechanism of binding of yttrium to bone surfaces may be different from that of plutonium or americium despite the general similarities in the skeletal behaviour of these elements (Herring et al., 1962). (522) Weanling rabbits were injected intravenously with 91Y, 90Sr free from 90Y, or 90Sr and 90Y in equilibrium to compare the relative distributions of strontium and yttrium, and to determine whether 90Y produced in vivo from decay of 90Sr behaves differently from yttrium introduced as a parent radionuclide (Lloyd, 1961). A qualitative similarity in the two chemically dissimilar radionuclides 90Y and 90Sr was observed in that the tissues containing the highest concentration of 90Sr were also those containing the highest concentration of 91Y (i.e. bone, pituitary, cartilage, and kidney). The distributions of 90Sr and 91Y differed quantitatively. For example, kidney, liver, and spleen concentrated 91Y to a much greater extent than 90Sr. The rate of disappearance of 91Y from the soft tissues was much lower than the rate of disappearance of 90Sr. At 9 d, the 91Y concentration in the liver was 150 times that of 90Sr. When 90Sr was injected, there was a secondary uptake of 90Y in the liver, spleen, and kidneys after the initial distribution of 90Sr. (523) Stevenson (1975) studied the influence of age and sex on the relative behaviours of 90Y and 90Sr in rats over a period of 32 d following administration of solutions with 90Sr and 90Y in equilibrium. The activity ratio 90Y:90Sr in bone depended to some extent on age and sex, but was typically 1.0–1.6 at 1 d, increased by approximately 30% over the next 3 d, and then declined to near-equilibrium levels over the next month. The ratio 90Y:90Sr in the liver rose from approximately 10 at 30 min after injection to approximately 400 by the fourth day. During the same period, the ratios for the kidney and spleen rose from approximately 3 to approximately 100–150, and the ratio for the heart rose from 1.5 to 14–22. (524) By measuring the relative activities of 90Sr and 90Y in various tissues of a beagle, Arnold et al. (1955) concluded that 90Y does not separate from 90Sr in bone volume. Their conclusion was based mainly on the observation that 90Y did not become more concentrated than 90Sr at sites where migrating 90Y would have tended to accumulate. (525) Mueller (1972) studied the relative behaviour of strontium and yttrium in mice, and intraperitoneal injection of 90Sr and 90Y in radioactive equilibrium or 90Sr freshly purified from 90Y. At 7 d after injection of equilibrium activities, the concentration ratio 90Y:90Sr was approximately 150 for liver and spleen, and near 1 for bone. At 7 d after injection of purified 90Sr, the activity ratio was approximately 3 for liver and spleen, and 0.9 for bone.
11.2.3.2. Biokinetic model for systemic yttrium
(526) The structure of the systemic model for yttrium is shown in Fig. 11.1. Transfer coefficients are listed in Table 11.3. The transfer coefficients describing movement of yttrium between bone compartments and removal from bone are default values applied in the OIR series to bone-surface seekers. Other transfer coefficients in the model are based on deposition fractions and biological half-times summarised below. Deposition fractions and half-times describing uptake and retention by the liver and rates of urinary and faecal excretion were selected for consistency with yttrium injection data for healthy human subjects described earlier. The remaining deposition fractions and half-times were based on animal data described earlier, with preference given to data for large animals. (527) Blood is divided into compartments Blood 1 and Blood 2, representing fast and slow clearance, respectively. Yttrium leaves Blood 1 at a rate of 16.6 d−1, corresponding to a biological half-time of 1 h. Outflow from Blood 1 is divided as follows: 3% moves to Blood 2; 15% moves to the urinary bladder contents; 1% moves to the small intestine contents; 40% moves to bone surfaces, equally divided between trabecular and cortical surfaces; 10% moves to a fast-turnover liver compartment called Liver 0; 1% moves to the kidneys; 22% moves to a fast-turnover soft tissue compartment called ST0; and 8% moves to a slow-turnover soft tissue compartment called ST1. Activity is removed from Liver 0 with a biological half-time of 3 d. Activity leaving Liver 0 is divided among a slow-turnover liver compartment called Liver 1, Blood 1, and small intestine contents (representing biliary secretion) in the ratio 0.5:0.4:0.1. Activity is removed from Blood 2 to Blood 1 with a half-time of 1.5 d; from ST0 to Blood 1 with a half-time of 3 d; and from Liver 1, kidneys, and ST1 to Blood 1 with a half-time of 1 y. The fate of yttrium deposited on bone surfaces is described by the generic model for bone-surface seekers, except that yttrium biologically removed from bone is assumed to return to blood rather than to be channelled through bone marrow. Thus, yttrium is removed from cortical or trabecular bone surfaces at a rate proportional to (1.5 times) the turnover rate of that bone type. The assumed bone turnover rates are 3% y−1 for cortical bone and 18% y−1 for trabecular bone. One-third of activity removed from bone surfaces is buried in bone volume, and two-thirds transfers to Blood 1. Activity is removed from cortical or trabecular bone volume to Blood 1 at the rate of turnover of that bone type. (528) Model predictions are compared with the human injection data of Etherington et al. (1989a,b) in Figs 11.2–11.4. In these two subjects, urinary excretion accounted for 93–94% of the excreted amount over 5 d and 91% over 14 d. Model values are 92% over 5 d and 89% over 14 d. Structure of the biokinetic model for systemic yttrium. ST, soft tissue; SI, small intestine. Parameter values in the systemic model for yttrium. ST, soft tissue; SI, small intestine. Model predictions of whole-body retention of intravenously injected yttrium, compared with observations of Etherington et al. (1989a,b) for two human subjects intravenously injected with 88Y as citrate (The line is the model prediction, whilst the symbols represent data from Etherington et al., 1989a,b). Model predictions of liver content of yttrium as a function of time after intravenous injection, compared with observations of Etherington et al. (1989a,b) for two human subjects intravenously injected with 88Y as citrate (The line is the model prediction, whilst the symbols represent data from Etherington et al., 1989a,b). Model predictions of urinary excretion of yttrium as a function of time after intravenous injection, compared with observations of Etherington et al. (1989a,b) for two human subjects intravenously injected with 88Y as citrate.

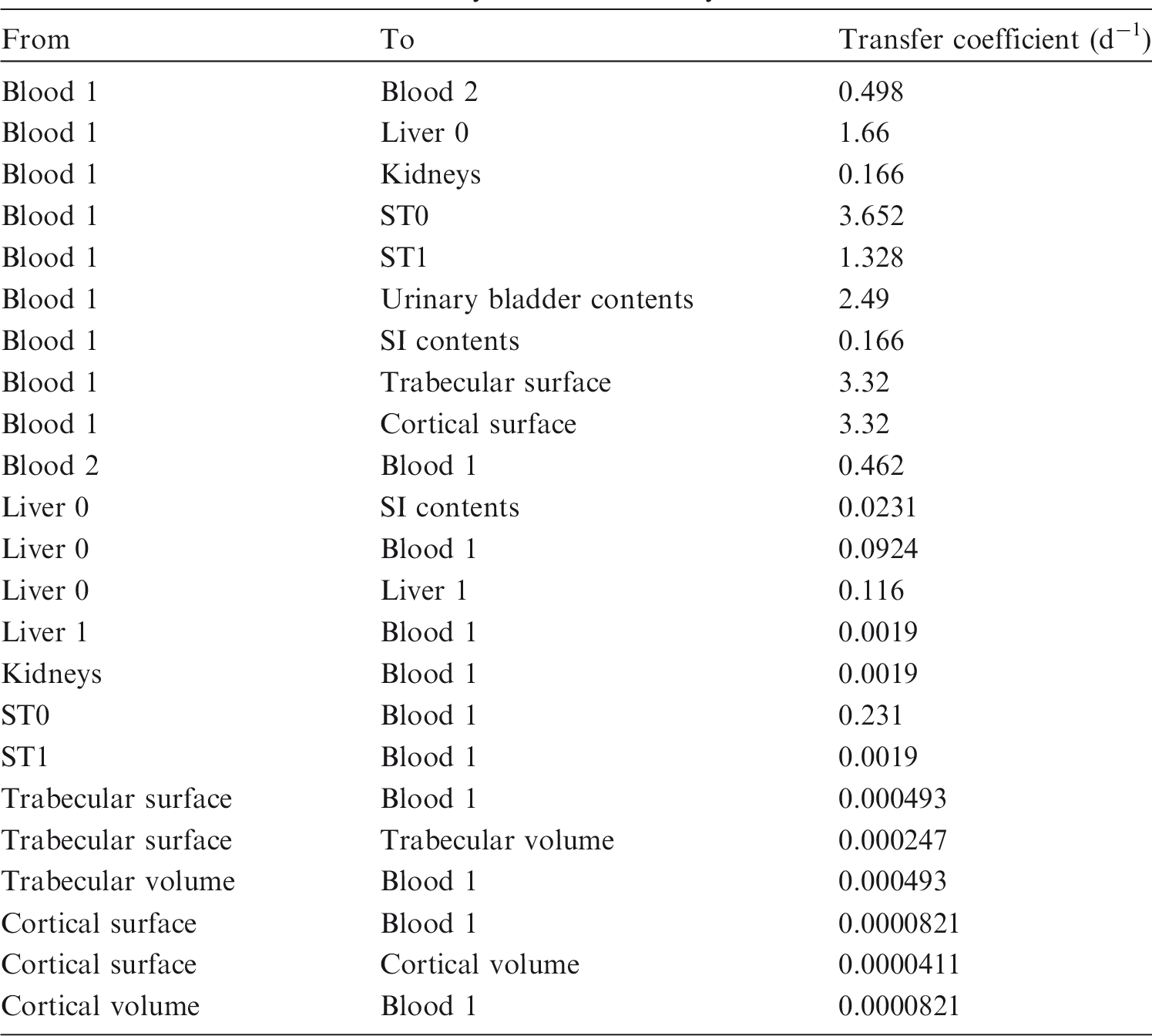
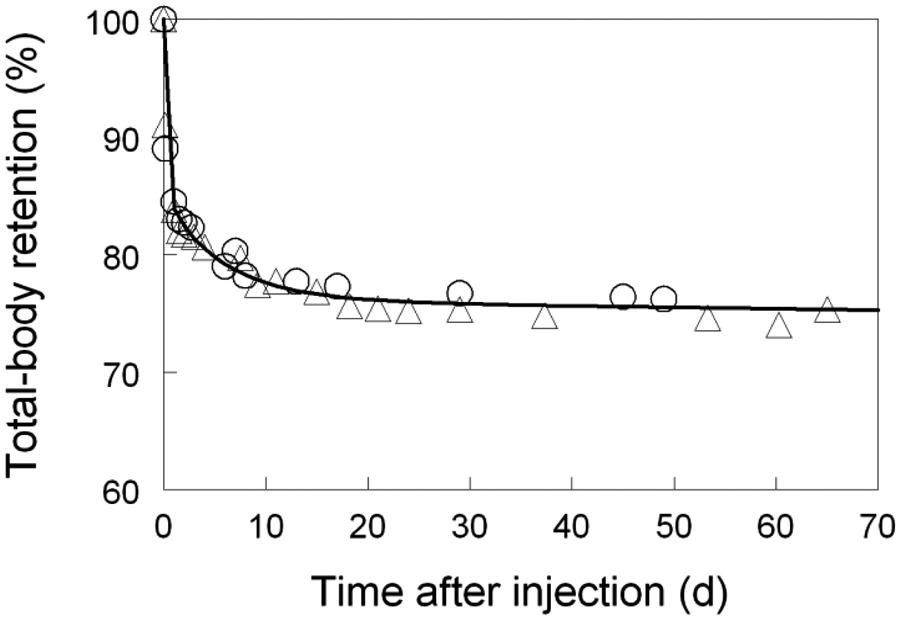

11.2.3.3. Treatment of radioactive progeny
(529) Chain members addressed in the derivation of dose coefficients for internally deposited yttrium isotopes include isotopes of yttrium, strontium, zirconium, and niobium. An yttrium isotope produced in the body after uptake of an yttrium parent is assumed to have the same systemic biokinetics as the parent. Isotopes of zirconium and niobium produced in systemic compartments after intake of an yttrium parent are assigned the characteristic systemic models for zirconium and niobium, respectively, described elsewhere in this publication. The characteristic systemic models for yttrium, zirconium, and niobium all have the same model structure. A zirconium or niobium atom produced in a compartment by radioactive decay is assumed to behave as if it had entered that compartment as a parent radionuclide. This includes subcompartments of ‘other soft tissues’. (530) The model for strontium produced in systemic compartments after intake of an yttrium parent is an extension of the characteristic model for strontium described elsewhere in this publication. That model is extended for application to strontium as a progeny of yttrium by adding individual compartments representing liver and kidneys, which are represented explicitly in the model for yttrium. Each of these compartments is assumed to exchange strontium with blood. Parameter values describing rates of uptake and removal of strontium by liver and kidneys are set for reasonable agreement with postmortem measurements from human subjects injected with 85Sr during late stages of various terminal illnesses (Schulert et al., 1959). The transfer coefficients from blood to liver and kidneys are both set at 0.05 d−1. The transfer coefficient from blood to the intermediate-term soft tissue compartment in the characteristic model for strontium is reduced from 1.5 d−1 to 1.4 d−1 to leave the total outflow rate from blood unchanged. The removal half-times from liver and kidneys to blood are set at 6 d and 2 d, respectively. (531) The blood compartment of the strontium model (named Blood) is identified with the compartment Blood 1 of the yttrium model (Fig. 11.1). Thus, strontium produced in Blood 1 by decay of yttrium is assumed to be produced in Blood in the strontium model. Strontium produced by radioactive decay in compartments of the yttrium model that are not identifiable with compartments of the strontium model is treated as follows. Strontium produced in Blood 2 of the yttrium model is assumed to transfer to Blood in the strontium model at a rate of 1000 d−1 (t½ ∼ 1 min). Strontium produced in either of the two liver compartments of the yttrium model is assumed to transfer to Blood in the strontium model with a half-time of 6 d, which is the removal half-time of strontium from the liver in the strontium model described above. Strontium produced in either of the two compartments of other soft tissues in the yttrium model is assumed to transfer to Blood in the strontium model at a rate of 2.5 d−1, which is the shortest removal half-time from the soft tissue compartments in the characteristic model for strontium. Strontium reaching Blood in the strontium model subsequently follows the model for strontium described above. The single kidney compartment in the model for strontium as a progeny of yttrium is identified with the single kidney compartment in the model for yttrium. Strontium produced in that compartment by decay of yttrium is assumed to behave as if entering the compartment as a parent radionuclide.
11.3. Individual monitoring
(532) Monitoring of 90Y is generally accomplished by measuring its beta emission in urine, either using liquid scintillation or beta proportional counting (Table 11.4). Monitoring techniques for 90Y.

11.4. Dosimetric data for yttrium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 90Y compounds.

AMAD, activity median aerodynamic diameter; FAP, fused aluminosilicate particles.
Dose per activity content of 90Y in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Daily urinary excretion of 90Y following inhalation of 1 Bq Type F and water soluble forms, including chloride.

Daily urinary excretion of 90Y following inhalation of 1 Bq Type M.

Daily urinary excretion of 90Y following inhalation of 1 Bq Type S.
11.5. References
12. ZIRCONIUM (Z = 40)
12.1. Chemical forms in the workplace
(533) Zirconium is a transition metal that mainly occurs in oxidation state IV. It may be encountered in industry in a variety of chemical and physical forms, including oxides, carbonates, oxalates, and zircon (ZrSiO4). Zirconium radioisotopes such as 93Zr and 95Zr are encountered in the nuclear industry. 95Zr is a high-yield fission product that can be associated with irradiated fuel or corrosion products; it decays to 95Nb, another high-yield fission product. They could be present in fragments of irradiated fuel, and in acidic fission product solutions. Both 95Zr and 95Nb also occur as neutron activation products derived from zirconium-based fuel (Zircalloy) cladding. Table 12.1 shows the isotopes of zirconium addressed in this publication. Isotopes of zirconium addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay.
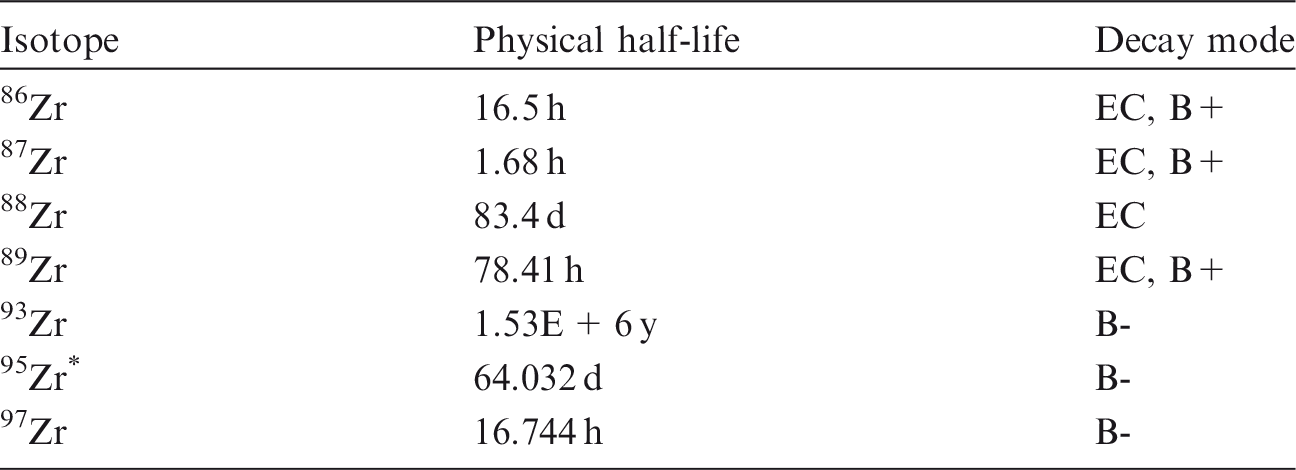
12.2. Routes of intake
12.2.1. Inhalation
(534) In all the studies noted below, the zirconium isotope followed was 95Zr (t½ = 64 d), which decays to 95Nb (t½ = 35 d). In most studies, both radionuclides were deposited in the respiratory tract, and the combined activity of the two radionuclides followed. Thus, in interpreting the results, it has to be assumed that their behaviour was similar. Furthermore, the 95Nb measured was partly that which deposited, and partly that formed from the in-situ decay of 95Zr. Due to the relatively short half-lives of these radionuclides, few studies are of sufficient duration to distinguish Type M and S behaviour based on the Publication 71 (ICRP, 1995) criteria of lung retention or total absorption up to 180 d after intake. (535) Some information was found on the behaviour of inhaled zirconium in man, mainly associated with irradiated fuel. Information on absorption from the respiratory tract is available from experimental studies of zirconium as oxalate, oxide, and irradiated uranium dioxide. (536) Absorption parameter values and types, and associated fA values for particulate forms of zirconium are given in Table 12.2. Absorption parameter values for inhaled and ingested zirconium. It is assumed that the bound state can be neglected for zirconium, i.e. fb=0. The values of sr for Type F, M, and S forms of zirconium (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. zirconium oxalate) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of zirconium (0.002). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.002) for ingestion of the radionuclide.

12.2.1.1. Particulate materials
(a) Zirconium oxalate
(537) Following inhalation by guinea pigs of carrier-free 95Zr oxalate, the activity in the lungs immediately after the 30-min exposure, and 1 and 28 d later was approximately 24%, 10%, and 5% of the ‘recovered dose’, respectively. Amounts in the skeleton at these times were 8%, 15%, and 9% respectively. Similar results were obtained using 95Zr oxalate with added zirconium oxychloride (ZrOCl2) (Schiessle et al., 1964; Schmidtke et al., 1964). The large uptake in the skeleton at the first measurement suggests a rapid dissolution rate, sr, of the order of 100 d−1. However, approximately 10% of the activity deposited in the lungs was not cleared rapidly (fr ∼ 0.9). The decrease in lung content between 4 and 28 d did not give any obvious increase in activity in the skeleton, and hence no indication of a significant ‘bound state’ from which clearance is by absorption alone. The amount retained in the lungs at 28 d suggests assignment to Type M, but is very close to the criterion for assignment to Type F. (538) Thomas et al. (1971) studied the biokinetics of 95Zr–95Nb following inhalation by mice of aerosols formed by heating droplets of zirconium oxalate solution to various temperatures. In-vitro dissolution tests were conducted on similar materials by Kanapilly and Goh (1973) and Kanapilly et al. (1973). Immediately after inhalation of the aerosols formed at 100℃ and 250℃ (both zirconium oxalate, but mainly droplets and solid particles, respectively), the skeleton contained approximately 20% of the body content, and the lungs contained 2% and 25%, respectively. It was noted that the ratio of 95Nb to 95Zr in the lungs was lower than in the aerosol, indicating a pronounced differential loss of 95Nb. Nevertheless, the results suggest that at the lower temperature, most of the material deposited in the lungs was absorbed rapidly: fr ∼ 0.9 and sr of the order of 100 d−1. For both materials, these results indicate Type F behaviour, as do those of the in-vitro dissolution tests. (539) As rapid absorption is incomplete, the results are difficult to interpret, all the more so because of the radionuclide mixture present. Furthermore, absorption of 95Nb from the lungs following deposition of the oxalate is also complex (see niobium inhalation section (Section 13.2.1.)). Hence specific parameter values are not adopted in this publication for zirconium oxalate. The information above suggests assignment to Type F, but also that absorption is slower than for niobium oxalate, for which there is more comprehensive information, which gives assignment to Type M. Zirconium oxalate is therefore also assigned to Type M.
(b) Zirconium oxide and carbonate
(540) As noted above, Thomas et al. (1971) studied the biokinetics of 95Zr–95Nb following inhalation by mice of aerosols formed by heating droplets of zirconium oxalate solution. The aerosols formed at 600℃ [Zr(CO3)2 and ZrOCO3] and at 1100℃ (ZrO2 and ZrOCO3) gave very similar results in vivo (with no differential loss of niobium). From 10 to 130 d after inhalation, the lungs contained more than 90% of the sacrifice body burden while the skeleton content increased from 2% sacrifice body burden at 2 d to 6% sacrifice body burden at 130 d. These results indicate Type S behaviour. In-vitro tests on similar materials by Kanapilly and Goh (1973) and Kanapilly et al. (1973) confirmed low dissolution rates, but their duration was too short to distinguish Type M from Type S. (541) Cuddihy (1978) applied simulation modelling to measurements of 95Nb following inhalation of similar 95Nb-labelled zirconium aerosols (formed at 1000℃) by dogs to obtain an absorption function (fractional absorption rate):

(c) Zirconium tritide
(542) For details, see the hydrogen inhalation section in this publication (Section 2.2.1.). Measurements of tritium following intratracheal instillation of zirconium tritide into rats were consistent with assignment to Type S.
(d) Nuclear weapons fallout
(543) During the early 1960s, measurements were made of 95Zr–95Nb activities in human lungs due to fallout from atmospheric nuclear weapons tests. Most were made post mortem (Schönfeld et al., 1960; Osborne, 1963; Wrenn et al., 1964; Dutailly et al., 1966), but in-vivo measurements were also made, enabling the variation with time in individual subjects to be determined (Rundo and Newton, 1962, 1965). Several authors compared their measurements with those predicted from measured air concentrations using a single exponential model (ICRP, 1959). Biological lung retention half-times were estimated to be between approximately 70 d (Wrenn et al., 1964) and more than 120 d (Rundo and Newton, 1965). Wrenn et al. (1964) noted that little 95Zr–95Nb activity was found in other tissues, and Wegst et al. (1964) showed that 95Zr–95Nb activity in the lungs was present in particulate form. Overall, this indicates Type M or S behaviour.
(e) Irradiated fuel
(544) Following an accidental release, zirconium could be present in fragments of irradiated fuel where the matrix is predominantly uranium oxide. The results of a study on one person following accidental inhalation of irradiated fuel indicate Type M behaviour of the zirconium present (Rundo, 1965). In another, measurements of 95Zr–95Nb made on a worker for 6 months following an accidental intake, probably of irradiated fuel (UO2), indicate Type S behaviour (Thind, 1995). (545) Mirell and Blahd (1989) made whole-body measurements of activity on seven people from approximately 2 weeks to several months after exposure to the initial Chernobyl reactor accident plume in Kiev, Ukraine. Biological retention half-times were similar for different radionuclides (49 d for 95Zr–Nb) and different from those expected for systemic retention, indicating that they were trapped in particles and metabolically inert, and thus indicating Type M rather than Type F behaviour. (546) Tissue distribution and retention of several radionuclides were followed for 3 months after intratracheal instillation of irradiated UO2 powder into rats (Lang et al., 1994). For 95Zr, the total amounts absorbed by 1 and 3 months were estimated to be approximately 1% and 3% ILD, respectively, indicating values of fr < 0.01 and ss ∼ 0.001 d−1, and assignment to Type S. (547) The in-vitro dissolution of samples of particles released from the Chernobyl accident was measured for up to 60 d (Cuddihy et al., 1989). For all radionuclides, including 95Zr–95Nb, 10% dissolved in a few hours, and the rest with a half-time of 160 d. Hence, fr = 0.1, sr ∼ 10 d−1, and ss = 0.004 d−1, consistent with assignment to Type M.
(f) Other compounds
(548) Measurements of 95Zr–95Nb in the lungs of a person for 5 months following an accidental intake of unspecified material indicate Type M or S behaviour (Cofield, 1963).
12.2.1.2. Progeny radionuclides of zirconium formed in the respiratory tract
(549) The general approach to treatment of progeny radionuclides formed in the respiratory tract is described in OIR Part 1, Section 3.2.3 (ICRP, 2015). In summary, it would be expected that the rate at which a particle dissociates is determined by its matrix, and hence the physico-chemical form of the inhaled material, but that the behaviour of soluble (Type F) material in the respiratory tract would depend on its elemental form, i.e. that of the progeny radionuclide. Nevertheless, for simplicity, in the OIR series, it is assumed that progeny radionuclides formed in the respiratory tract have the same dissolution parameter values as the parent inhaled. (550) Of particular importance in the case of zirconium is the formation of 95Nb (t½ = 35 d) from 95Zr (t½ = 64 d). Some experimental results were found from which the absorption of 95Nb could be compared directly with that of 95Zr under the same conditions. However, the 95Nb in the respiratory tract would have been partly administered with the 95Zr, and partly formed in the respiratory tract by decay of the 95Zr parent. (551) Thomas et al. (1971) studied the biokinetics of 95Zr–95Nb following inhalation by mice of aerosols formed by heating droplets of zirconium oxalate solution to various temperatures (see above). For the aerosols formed at 100℃ and 250℃ (both zirconium oxalate), the ratio of 95Nb to 95Zr in the lungs was lower than in the aerosol, indicating a pronounced differential loss of 95Nb. The aerosols formed at 600℃ [Zr(CO3)2 and ZrOCO3] and at 1100℃ (ZrO2 and ZrOCO3) showed no differential loss of niobium. (552) Lang et al. (1994) followed the tissue distribution and retention of several radionuclides for 3 months after intratracheal instillation of irradiated UO2 powder into rats (see above and niobium inhalation section (Section 13.2.1.)). For 95Zr, the estimated total amounts absorbed by 1 and 3 months were approximately 1% and 3% ILD, whereas for 95Nb, they were approximately 5% and 9% ILD. (553) Thus, there is evidence that for some, especially soluble, forms of zirconium, the niobium progeny is absorbed from the lungs more rapidly than the zirconium parent. However, as there is insufficient information to estimate element-specific rapid dissolution rates for either element, the general default value of 30 d−1 is applied to both, and so their dissolution parameter values are the same.
12.2.1.3. Rapid dissolution rate for zirconium
(554) Evidence from the zirconium oxalate studies outlined above suggests a rapid dissolution rate of the order of 100 d−1, but only of part of the ILD (fr < 1). There is therefore no justification for choosing a rate different from the general default value of 30 d−1, which is applied here to all Type F forms of zirconium.
12.2.1.4. Extent of binding of zirconium to the respiratory tract
(555) Evidence from the zirconium oxalate studies outlined above suggests that, following the rapid phase of absorption, approximately 10% ILD clears slowly from the lungs. Clearance of this material does not appear to be mainly by absorption to blood, as assumed for material in the ‘bound state’, and therefore does not give evidence for significant binding of zirconium. Moreover, the results available are difficult to interpret (see above). It is therefore assumed that the bound state can be neglected for zirconium, i.e. fb = 0.0.
12.2.2. Ingestion
(556) Few human data are available on the absorption of zirconium from the gastrointestinal tract. In a study using stable tracer 96Zr chloride given to a healthy male volunteer, the absorption of zirconium was estimated to be 2.5×10−3 (Veronese et al., 2003a,b). A broader study was conducted with stable tracers in a total of 14 volunteers who received zirconium in the form of oxalate or citrate (Greiter et al., 2011). The fractional absorption was found to be equal to (7.4 ± 1.5) × 10−3 for oxalate and (1.10 ± 0.23) × 10−3 for citrate. (557) These values are similar to those found in animals. Fletcher (1969) reported values ranging from 3 × 10−4 to 2 × 10−3 for the fractional absorption of 95Zr in young adult rats after administration of a number of chemical forms, including the chloride, sulphate, and organic complexes with lactate and oxalate. Similar values were reported by Shiraishi and Ichikawara (1972) for zirconium oxalate in adult rats, de Bartolo et al. (2000) for zirconium sulphate in rabbits, and Sirotkin et al. (1970) for zirconium chloride in cows. Taylor et al. (1983) obtained values ranging from 1.5 to 8 × 10−4 for the fractional absorption of the chemically similar radionuclide 181Hf in rats and hamsters. (558) Reference values used previously were 0.002 in Publication 30 (ICRP, 1979) and 0.01 for intake from members of the public (ICRP, 1989). However, this latter value was adopted for taking account of the biologically incorporated form of the element present at low concentration in the diet. On the basis of the recent human and animal data, an fA value of 0.002 is adopted here for all chemical forms.
12.2.3. Systemic distribution, retention, and excretion
12.2.3.1. Summary of the database
(a) Human subjects
Mealey (1957) studied the biokinetics of 89Zr (t½ = 78.4 h) following its intravenous administration as citrate to a comatose subject with brain cancer but with vital signs, electrolyte levels, and renal function within normal limits. Activity cleared slowly from plasma, apparently due to binding of 89Zr to plasma proteins. Approximately 10% of the injected amount (corrected for decay) remained in plasma at 7 d. There was little, if any, accumulation of 89Zr in RBCs. Urinary excretion accounted for 2.5% of the administered amount over the first 24 h and 7.6% over 7 d. Intravenously administered 89Zr was also measured in biopsy samples from two patients undergoing neurological surgery. In one of the subjects, the 89Zr concentrations in bone (skull) and muscle were 1.2 and 4.8% of injected 89Zr kg−1 tissue, respectively, at 90 min after administration. In the other subject, concentrations of 89Zr in bone, muscle, and normal brain tissue were 0.9, 7.6, and 0.8% kg−1, respectively, at 3 h. High accumulation of 89Zr in muscle was also indicated by external measurements from other patients. External measurements from one subject over three successive days indicated a sustained high concentration of activity in muscle but a substantial decrease in the concentrations in the skull and brain during this period.
(559) The biokinetics of zirconium was studied in three healthy subjects (one male and two females in the age range 27–60 y) following oral or intravenous administration of stable zirconium isotopes (Veronese et al., 2003a,b). Clearance of injected zirconium from plasma could be characterised by a relatively fast component representing approximately half of the administered amount, followed by a slower component. The half-time associated with the faster component was estimated as 3.6 h in two subjects and 0.8 h in the third subject. The investigators derived a half-time of approximately 3 d for the slower component after combining their findings with longer-term measurements of plasma clearance of zirconium reported by Mealey (1957). (560) Relatively long-term studies of the biokinetics of orally or intravenously administered stable zirconium isotopes were later conducted on seven male and six female subjects in the age range 26–60 y (Greiter et al., 2011). The zirconium isotopes were prepared either in citrate or oxalate solution. Blood plasma and urine were sampled up to 100 d after administration. Mean fractional absorption of zirconium was seven-fold higher after oral intake of zirconium oxalate than after intake of zirconium citrate. The derived urinary excretion data are difficult to interpret in terms of typical excretion rates due to the high variability of the measurements and a relatively high detection limit. Approximately 20% and 40% of the urinary measurements were below the detection limit in the injection and oral tracer studies, respectively. Taken at face value, the data indicate that urinary losses over the first week averaged approximately 6% of the intravenously injected amount. The investigators’ proposed biokinetic model for zirconium with expected transfer coefficients based on results of the study predicts total urinary losses of approximately 2% at 7 d and 8% at 100 d after intravenous injection.
(b) Laboratory animals
(561) Bone was found to be the main systemic repository for zirconium tracers following their administration by various routes to rats (Durbin, 1960; Fletcher, 1969), guinea pigs (Schiessle et al., 1961), and mice (Bäckström et al., 1967; Thomas et al., 1971; Abou et al., 2011). Autoradiographic studies on rats (Hamilton, 1947) indicated that skeletal zirconium was confined largely to bone surfaces. (562) At 4 d after intramuscular administration of 95Zr as citrate to rats, the liver, kidneys, and bone contained approximately 6.6%, 4.9%, and 35%, respectively, of the administered activity (Durbin, 1960). Approximately 18% of administered activity had been excreted by that time, mainly in faeces. Nearly two-thirds of the administered amount remained in the body after 2–4 months. (563) Autoradiographic studies following intravenous administration of 95Nb or 95Zr–95Nb to mice indicated qualitatively similar distributions of activity in the two cases (Bäckström et al., 1967). These distributions were also similar to that observed by the investigators in an earlier study of 103Ru. All of these radionuclides showed an affinity for connective tissue as well as bone. The affinity for bone increased in the order 103Ru < 95Nb < 95Zr–95Nb (Bäckström et al., 1967). (564) Following intraperitoneal administration of 95Zr citrate to rats, approximately 60% of the injected amount was retained after 1 month and approximately 50% was retained after 3 months (Richmond et al., 1960). In a similar study on mice conducted by the same investigators (Furchner et al., 1964), nearly half of the injected 95Zr was lost rapidly from the body, and approximately two-thirds of the administered amount was lost within a few weeks. Measurements up to 420 d after injection indicated that the remaining one-third was removed with a biological half-time of several years. (565) Fletcher (1969) studied the behaviour of 95Zr and 95Nb in rats following oral or intravenous administration of 95Zr–95Nb or pure 95Nb as oxalates. Whole-body retention of 95Zr over 80 d was determined by external counting and correction for counts for simultaneously injected 95Nb and 95Nb formed in vivo by radiological decay of 95Zr. The correction was based on the assumption that 95Nb formed in vivo behaves as if it had been injected intravenously at the time of formation. This assumption was consistent with the measured distributions of 95Nb and 95Zr at 80 d. Whole-body retention of injected 95Zr was greater in males than females at all measurement times. As an average over both sexes, approximately 90% of intravenously administered 95Zr was retained in the body after 8 d, 80% was retained after 30 d, and 60% was retained after 80 d. The concentration of 95Zr in tissues following administration of a mixture of 95Zr and 95Nb was determined using physical decay measurements or beta scintillation counting of their distinctive beta emissions. At 8 d, an estimated 89–92% of whole-body 95Zr was in bone, and the kidneys, spleen, and liver each contained a few tenths of 1% of the administered amount. (566) The relative behaviours of 95Zr and 95Nb were studied in mice following inhalation of these radionuclides at near-equilibrium conditions in aerosols produced at various temperatures (Thomas et al., 1971). Comparison of the activity ratios 95Nb:95Zr in the aerosols, lung, bone, and liver indicated different systemic biokinetics of these radionuclides. Bone was the main systemic repository for both 95Zr and 95Nb, but 95Zr showed higher accumulation in bone and lower accumulation in liver than 95Nb. (567) Shiraishi and Ichikawara (1972) studied the gastrointestinal absorption, retention, and distribution of 95Zr–95Nb following a single oral administration to rats of different ages. Similar rates of loss of absorbed activity were seen for all age groups following an initially rapid decline in the whole-body content, presumably representing removal of unabsorbed activity from the body. At 40 d after administration to adult rats, approximately 63% of the retained activity was in bone, 3.8% was in the liver, 20% was in muscle, and 2.9% was in the kidneys. (568) Razumovski˘ et al. (1966) studied the effects of various complex-forming agents on the biokinetics of 95Zr and 95Nb in rats. At 3 d after intraperitoneal administration of 95Zr–95Nb oxalate to control animals, the liver, spleen, kidneys, and femur contained approximately 4.2%, 0.56%, 1.4%, and 0.6% of the administered activity, respectively. (569) Ando and Ando (1986) examined the early distribution of 95Zr in soft tissues of tumour-bearing rats following its intravenous administration as oxalate or nitrate. At 3, 24, and 48 h after administration of either form of 95Zr, the liver contained approximately 3–4%, the kidneys contained approximately 1–1.5%, and skeletal muscle contained approximately 13–17% of the administered amount. (570) Abou et al. (2011) investigated the behaviour of 89Zr in mice following its intravenous administration as oxalate, chloride, phosphate, citrate, or desferrioxamine. Concentrations were determined in blood, liver, kidneys, bone, marrow, muscle, heart, lungs, spleen, and gastrointestinal tissues at 4 h, 8 h, and 6 d. After 6 d, the total excretion of 89Zr amounted to approximately 20% for the chloride or oxalate, but only approximately 5% for the phosphate. Mice injected with the citrate excreted approximately 30% after 1 d and 35% after 6 d. Virtually all 89Zr administered as desferrioxamine was excreted the first day. For administration of 89Zr as phosphate, the highest concentrations were found in the liver and spleen at all times. For administration of 89Zr as oxalate, chloride, or citrate, the concentration in bone was generally more than twice that in other tissues at early times, and more than 10 times that in other tissues at 6 d. Bone marrow cells showed little activity compared with calcified tissues. The epiphysis, consisting mainly of cartilage, contained most of the bone activity. The authors concluded that weakly bound zirconium is a bone seeker, and likely binds to phosphate constituents of mineralised bone and epiphysis. (571) Results of studies on rats indicate that a substantial portion of 95Nb formed in vivo from decay of systemic 95Zr is free to redistribute. For example, the distribution of 95Nb formed in vivo from decay of ingested or intravenously injected 95Zr in rats was similar to the distribution of administered 95Nb, and considerably different from the distribution of 95Zr (Fletcher, 1969). Following oral administration of 95Zr–95Nb to suckling rats, the ratio of 95Zr to 95Nb was 4–5 in bone and close to 1 in other tissues (Shiraishi and Ichikawa, 1972). Measurements of activity in blood and tissues of rats following intraperitoneal injection of 95Zr–95Nb as oxalate indicated preferential accumulation of 95Zr in bone (Rama Sastry et al., 1964).
12.2.3.2. Biokinetic model for systemic zirconium
(572) The systemic model for zirconium used in this publication depicts the following general behaviour of zirconium. Approximately half of zirconium atoms entering blood transfer to tissues and excretion pathways within a few hours, and the remainder combine with plasma proteins and are cleared much more slowly from blood. More than 95% of zirconium atoms leaving blood deposit in tissues and < 5% enter excretion pathways, primarily the urinary bladder contents. Soft tissues initially contain a substantial portion of extravascular zirconium, but bone eventually contains > 90% of the systemic burden due to a relatively high deposition fraction and much slower turnover than soft tissues. Zirconium atoms that reach blood have a long residence time in the body due to a low excretion rate and a high level of accumulation in bone. (573) The structure of the systemic model for zirconium is shown in Fig. 11.1. Transfer coefficients are listed in Table 12.3. These values were derived from primary parameter values in the form of deposition fractions and biological half-times. The parameter values were set to yield blood disappearance curves and urinary excretion rates for zirconium consistent with those observed in human subjects, a relatively high zirconium content in soft tissues at early times as observed in human subjects, and a time-dependent systemic distribution of zirconium suggested by animal studies. The comparative biokinetics of zirconium and niobium as observed in animal studies has been taken into account. Niobium shows qualitatively similar systemic behaviour to that of zirconium, but a lower rate of transfer to bone, higher urinary clearance, and apparently greater uptake or retention or both by soft tissues than zirconium. It was convenient to derive transfer coefficients for zirconium in soft tissues, in particular, by scaling values developed from more easily interpreted soft tissue data for niobium, to which the same model structure (Fig. 12.1) is applied in this publication. Except where there are over-riding considerations, the assigned deposition fractions and removal half-times describing uptake and retention of zirconium in soft tissue compartments are one-half of the values used in the model for niobium. (574) In the systemic model for zirconium, atoms that are absorbed or injected into blood initially enter a blood compartment called Blood 1. Zirconium leaves Blood 1 at a rate of 5 d−1, corresponding to a removal half-time of approximately 3.3 h. Outflow from Blood 1 is divided as follows: 40% goes to a slow-turnover blood pool representing plasma proteins (Blood 2 in Fig. 12.1); 40% goes to a soft tissue pool with relatively fast turnover (ST0); 15% transfers to bone surfaces and is equally divided between cortical and trabecular bone; 1.5% goes to the liver; 0.25% goes to the kidneys; 0.75% transfers to a soft tissue compartment with relatively slow turnover (ST1); 2% enters the urinary bladder contents; and 0.5% is secreted into the small intestine contents. The deposition fractions for Blood 2 and ST0 are the same as assumed in the model for niobium; the fraction for bone surfaces is five times greater than for niobium; the fraction for the urinary bladder contents is approximately one-fifth of the value for niobium; and values for other repositories are one-half of the values applied to niobium. (575) Zirconium is assumed to transfer from Blood 2 back to Blood 1 with a half-time of 1.5 d, from ST0 to Blood 1 with a half-time of 1.5 d, from ST1 to Blood 1 with a half-time of 35 d, and from kidneys to Blood 1 with a half-time of 70 d. The transfer coefficients derived from these and other half-times given below are rounded values. Zirconium entering the liver is assigned to a compartment called Liver 0. Zirconium is removed from Liver 0 with a half-time of 1 d, with two-thirds going to a long-term retention compartment of liver called Liver 1 and the other one-third equally divided between small intestine contents (representing biliary secretion) and Blood 1. Zirconium transfers from Liver 1 to blood with a half-time of 70 d. The removal half-times from Blood 2 and ST0 to Blood 1 were set for consistency with the blood retention patterns observed in healthy human subjects. The removal half-times from other soft tissue compartments were set to one-half of the values for niobium. The fate of zirconium depositing on bone surface is described by the generic model for bone-surface-seeking radionuclides, except that zirconium removed from bone is returned directly to blood rather than channelled through bone marrow. (576) Model predictions of retention of zirconium in blood are compared in Fig. 12.2 with central values for healthy human subjects following intravenous injection with stable isotopes of zirconium (Veronese et al., 2003b; Greiter, 2008). For the case of intravenous injection of zirconium, the model predicts cumulative urinary excretion of approximately 2.3% of the injected amount over the first 24 h, 5.5% over the first 7 d, and 11% over the first 100 d. These predictions are reasonably consistent with values observed in human subjects following intravenous injection of zirconium tracers (Mealey, 1957; Greiter, 2008; Greiter et al., 2011). Structure of the biokinetic model for systemic zirconium. ST, soft tissue; SI, small intestine. Comparison of model predictions of blood retention of zirconium with central values for healthy human subjects following intravenous administration of stable zirconium isotopes (Veronese et al., 2003b; Greiter, 2008) (the line is the model prediction and circles represent data from Veronese et al., 2003b and Greiter, 2008). Parameter values in the systemic model for zirconium. ST, soft tissue; SI, small intestine.



12.2.3.3. Treatment of radioactive progeny
(577) Chain members addressed in the derivation of dose coefficients for internally deposited zirconium isotopes include isotopes of yttrium, strontium, and niobium. The characteristic systemic models for yttrium, zirconium, and niobium all have the same model structure. An yttrium or niobium atom produced in a given compartment by radioactive decay after intake of a zirconium parent is assumed to behave as if it had entered that compartment as a parent radionuclide. The model for strontium produced in systemic compartments after intake of a zirconium parent is the same as the model for strontium produced after intake of an yttrium parent, as described in the section on yttrium (Section 11.2.3.).
12.3. Individual monitoring
(578) 95Zr is a gamma emitter. Monitoring of 95Zr is generally accomplished through whole-body measurement or/and urine bioassays (Table 12.4). Monitoring techniques for 95Zr. Lung measurement of 95Zr is not generally used in routine monitoring of workers. Monte Carlo program Visual Monte Carlo was used to simulate the photon emission, to calculate the calibration factor for the geometry and radionuclide, and to calculate the detection limit in the lung (Hunt et al., 2012).

12.4. Dosimetric data for zirconium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 95Zr compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 95Zr in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Total body and lung contents, and daily urinary excretion of 95Zr following inhalation of 1 Bq Type F.

Total body and lung contents, and daily urinary excretion of 95Zr following inhalation of 1 Bq Type M.

Total body and lung contents, and daily urinary excretion of 95Zr following inhalation of 1 Bq Type S.
12.5. References
13. NIOBIUM (Z = 41)
13.1. Chemical forms in the workplace
(579) Niobium is a transition metal that occurs mainly in oxidation states III and V. It may be encountered in industry in a variety of chemical and physical forms, including oxides and oxalates. Minerals that contain niobium often contain tantalum and thorium. Table 13.1 shows the isotopes of niobium addressed in this publication. (580) 95Nb is a high-yield fission product that may be associated with irradiated fuel or corrosion products. 95Nb also arises as the progeny of 95Zr, another high-yield fission product, that also occurs as a neutron activation product derived from zirconium-based fuel cladding. It could also be present in fragments of irradiated fuel. Isotopes of niobium addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay

13.2. Routes of intake
13.2.1. Inhalation
(581) Cuddihy (1978) reviewed information on the lung clearance of inhaled niobium compounds. He noted that the chemistry of niobium is complex as it can exist in any oxidation state between I and V. It does not form simple soluble compounds in aqueous solution but tends to hydrolyse and form hydrophilic colloids. Niobium oxalate complexes are stable in acids up to pH 5.5. Niobium oxides, the most common being Nb2O5, are sparingly soluble in mineral acids and almost inert in solutions of approximately neutral pH, as are most biological fluids. (582) In all the studies noted below, the niobium isotope followed was 95Nb (t½ = 35 d), the progeny of 95Zr (t½ = 64 d). In most studies, both radionuclides were deposited in the respiratory tract, and thus the 95Nb followed was partly that which deposited, and partly that formed from the in-situ decay of 95Zr. In most studies, the combined activity of the two radionuclides was measured, and thus in interpreting the results, it has to be assumed that their behaviour is similar. Furthermore, the inhaled material was only a pure niobium compound in a few studies. Due to the relatively short half-lives of these radionuclides, few studies are of sufficient duration to distinguish between Type M and S behaviour based on the Publication 71 (ICRP, 1995) criteria of lung retention or total absorption up to 180 d after intake. (583) Some information was found on the behaviour of inhaled niobium in man, mainly associated with irradiated fuel. Information on absorption from the respiratory tract is available from experimental studies of niobium as oxalate, oxide, and irradiated uranium dioxide. (584) Absorption parameter values and types, and associated fA values for particulate forms of niobium are given in Table 13.2. Absorption parameter values for inhaled and ingested niobium. It is assumed that the bound state can be neglected for niobium, i.e. fb=0.0. The values of sr for Type F, M, and S forms of niobium (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. niobium oxalate) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of niobium (0.01). Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.01) for ingestion of the radionuclide.

13.2.1.1. Particulate materials
(a) Niobium oxalate
(585) The oxalate has been studied extensively as a form that is relatively soluble in biological fluids (see above). In probably the most detailed study (Cuddihy, 1978), retention was followed in 27 dogs up to 128 d after inhalation of 95Nb-labelled zirconium oxalate by dogs. Cuddihy (1978) applied simulation modelling to obtain a time-dependent absorption function (fractional absorption rate):
(586) The same function was used to model particle transport of relatively insoluble niobium oxide administered to dogs in the same study (see below). This suggests that ‘binding’ to lung tissues was not a significant factor in the time-dependent absorption. The absorption can be broadly approximated using the HRTM dissolution model with fr = 0.6, sr = 1 d−1, and ss = 0.007 d−1, consistent with assignment to Type M. A good fit is obtained by using three dissolution compartments: 0.57 at 2.5 d−1, 0.17 at 0.13 d−1, and 0.26 at 0.0041 d−1. [An intake of material with these characteristics could be simulated with software that implements the HRTM by assuming an intake of two materials: 57% with fr = 1 and sr = 2.5 d−1; and 43% with fr = (0.17/0.43), sr = 0.13 d−1, and ss = 0.0041 d−1.] (587) In other studies with dogs, rats, and mice, the observed behaviour was broadly similar, but variable, indicating assignment to Type F in some and Type M in others. At 30 d after inhalation of 95Nb oxalate by three dogs, the lungs contained approximately 15% ILD, indicating assignment to Type M (Kanapilly et al., 1969). After inhalation of 95Nb oxalate by rats in one study (Moskalev et al., 1964), approximately 85% ILD was absorbed within a day (fr ∼ 0.85 and sr > 10 d−1) and the rest with a half-time of approximately 10 d, indicating assignment to Type F. In another study (Thomas et al., 1967), approximately 30% ILD was absorbed within a day (fr ∼ 0.3 and sr > 10 d−1), and relatively little thereafter, indicating assignment to Type M. (588) Thomas et al. (1971) studied the biokinetics of 95Zr–95Nb following inhalation by mice of aerosols formed by heating droplets of zirconium oxalate solution to various temperatures. In-vitro dissolution tests were conducted on similar materials by Kanapilly and Goh (1973) and Kanapilly et al. (1973). Immediately after inhalation of the aerosols formed at 100℃ and 250℃ (both zirconium oxalate, but mainly droplets and solid particles, respectively), the skeleton contained approximately 20% of the body content, the lungs contained 2%, and 25% of the body content, respectively, and the skeleton contained approximately 20% in both cases. This suggests that most of the material deposited in the lungs was absorbed rapidly at the lower temperature: fr of approximately 0.9 and sr of the order of 100 d−1. For both materials, niobium was absorbed faster than zirconium, especially that formed at 100℃. These results indicate Type F behaviour, as do those of the in-vitro dissolution tests. (589) Although specific parameter values for niobium oxalate based on in-vivo data are available, they are not adopted in this report because inhalation exposure to it is unlikely, and because a wide range of absorption was reported from different studies. Instead, niobium oxalate is assigned to Type M.
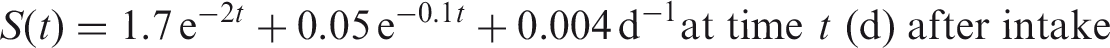

(b) Zirconium oxide and carbonate
(590) As noted above, Thomas et al. (1971) studied the biokinetics of 95Zr–95Nb following inhalation by mice of aerosols formed by heating droplets of zirconium oxalate solution. The aerosols formed at 600℃ [Zr(CO3)2 and ZrOCO3] and at 1100℃ (ZrO2 and ZrOCO3) gave very similar results in vivo, with no differential loss of niobium. From 10 to 130 d after inhalation, the lungs contained more than 90% of the sacrifice body burden, while the skeleton content increased from 2% sacrifice body burden at 2 d to 6% sacrifice body burden at 130 d. These results indicate Type S behaviour. In-vitro tests on similar materials by Kanapilly and Goh (1973) and Kanapilly et al. (1973) confirmed low dissolution rates, but their duration was too short to distinguish Type M from Type S. (591) Cuddihy (1978) applied simulation modelling to measurements of 95Nb following inhalation of similar 95Nb-labelled zirconium aerosols (formed at 1000℃) by dogs to obtain an absorption function (fractional absorption rate):
(592) Although specific parameter values for niobium oxide based on in-vivo data are available, they are not adopted here because inhalation exposure to it is unlikely. Instead, niobium oxide is assigned to Type S.

(c) Nuclear weapons fallout
(593) During the early 1960s, measurements were made of 95Zr–95Nb activities in human lungs due to fallout from atmospheric nuclear weapons tests. Most were made post mortem (Schönfeld et al., 1960; Osborne, 1963; Wrenn et al., 1964; Dutailly et al., 1966), but in-vivo measurements were also made, enabling variation with time in individual subjects to be determined (Rundo and Newton, 1962, 1965). Several authors compared their measurements with those predicted from measured air concentrations using a single exponential model (ICRP, 1959). Biological lung retention half-times were estimated to be between approximately 70 d (Wrenn et al., 1964) and more than 120 d (Rundo and Newton, 1965). Wrenn et al. (1964) noted that little 95Zr–95Nb activity was found in other tissues, and that Wegst et al. (1964) had shown that 95Zr–95Nb activity in the lungs was present in particulate form. Overall, this indicates Type M or S behaviour.
(d) Irradiated fuel
(594) Following an accidental release, niobium could be present in fragments of irradiated fuel where the matrix is predominantly uranium oxide. The results of a study on one person following accidental inhalation of irradiated fuel indicate Type M behaviour of the 95Zr–95Nb present (Rundo, 1965). In another, measurements of 95Zr–95Nb made on a worker for 6 months following an accidental intake, probably of irradiated fuel (UO2), indicate Type S behaviour (Thind, 1995). (595) Mirell and Blahd (1989) made whole-body measurements of activity on seven people from approximately 2 weeks to several months after exposure to the initial Chernobyl reactor accident plume in Kiev, Ukraine. Biological retention half-times were similar for different radionuclides (49 d for 95Zr–Nb) and different from those expected for systemic retention, indicating that they were trapped in particles and metabolically inert, and thus indicating Type M rather than Type F behaviour. (596) Tissue distribution and retention of several radionuclides were followed for 3 months after intratracheal instillation of irradiated UO2 powder into rats (Lang et al., 1994). For 95Nb, the total amounts absorbed by 1 and 3 months were estimated to be approximately 5% and 9% ILD, respectively, indicating values of fr < 0.05 and ss ∼ 0.002 d−1, and assignment to Type M. (597) The in-vitro dissolution of samples of particles released from the Chernobyl accident was measured for up to 60 d (Cuddihy et al., 1989). For all radionuclides, including 95Zr–95Nb, 10% dissolved in a few hours, and the rest with a half-time of 160 d. Hence, fr = 0.1, sr ∼ 10 d−1, and ss = 0.004 d−1, consistent with assignment to Type M.
(e) Other compounds
(598) Measurements of 95Zr–95Nb in the lungs of a person for 5 months following an accidental intake of unspecified material indicate Type M or S behaviour (Cofield, 1963).
13.2.1.2. Rapid dissolution rate for niobium
(599) As noted above, the oxalate has been studied extensively as a form of niobium that is relatively soluble in biological fluids. The results show rather complex behaviour with more than one phase of absorption, perhaps reflecting the complex chemistry of niobium. Where measurements have been made soon after administration, there is evidence of very rapid uptake (sr ∼ 100 d−1) but only of part of the ILD (fr < 1). There is therefore no justification for choosing a rate different from the general default value of 30 d−1, which is applied here to all Type F forms of niobium.
13.2.1.3. Extent of binding of niobium to the respiratory tract
(600) As described above, the oxalate has been studied extensively as a form of niobium that is relatively soluble in biological fluids. The results show more than one phase of absorption. However, Cuddihy (1978) applied simulation modelling to the results of 95Nb measurements following inhalation by dogs of niobium oxalate and relatively insoluble niobium oxide. The same function was used to model particle transport of both materials, which suggests that ‘binding’ to lung tissues was not a significant factor in the time-dependent absorption of the oxalate because it is assumed, in the HRTM, that material in the bound state is not cleared by particle transport, only by absorption to blood. It is therefore assumed that the bound state can be neglected for niobium, i.e. fb = 0.0.
13.2.2. Ingestion
(601) Information on the concentration of stable niobium in human diet and urine has been published by Schroeder and Balassa (1965), but these values were considered to be insufficient for estimating the absorption of niobium from the human gastrointestinal tract (ICRP, 1989). (602) Data on the absorption of niobium are available from a number of animal studies. A first set of values has been determined by Fletcher (1969), who quoted a range of fractional absorption from 4 × 10−4 to 2 × 10−3 for 95Nb administered to rats in various chemical forms. (603) Further studies have been performed on 95Nb given as oxalate. They show that fractional absorption of 95Nb given to rats varied from approximately 10−3 (Mraz and Eisele, 1977a) to 2 to 5×10−2 (Thomas et al., 1971). These values may vary according to the species as shown by Furchner and Drake (1971), who measured whole-body retention of 95Nb given as oxalate, and estimated levels of absorption of approximately 0.02 in mice and dogs, 0.008 in rats, and 0.009 in monkeys. However, these values may overestimate the true absorption because the retention of 95Nb rapidly fell to less than detection limits. (604) Fasting is known to increase uptake by the gut. Harrison et al. (1990) measured absorption of 0.008 for 95Nb administered as the citrate to normally fed guinea pigs, and 1.4 × 10−2 for animals fasted 24 h before and 2 h after administration. Paquet et al. (1998) investigated the fractional absorption of niobium given to fed rats, and obtained values of approximately 0.01, 0.04, and 0.03 for the citrate, oxalate, and chloride forms, respectively. (605) In Publication 30 (ICRP, 1979), an absorption value of 0.01 was recommended. This value was adopted in Publication 56 (ICRP, 1989) for dietary intakes, and is also adopted here as a default value for all chemical forms (fA = 0.01).
13.2.3. Systemic distribution, retention, and excretion
13.2.3.1. Summary of the database
(606) There is little information on the systemic behaviour of niobium in humans. Data for laboratory animals indicate broadly similar systemic biokinetics of niobium for different animal species, different routes of exposure, and different chemical forms of niobium taken into the body. Typically, 50% or more of niobium entering blood transfers to tissues and excretion pathways within a few hours, and the remainder clears much more slowly due to binding with plasma proteins. Excretion is mainly in urine. Niobium distributes somewhat uniformly throughout the body, but is retained for much longer in bone than in other tissues, so that bone eventually contains a large portion of the whole-body content. Niobium depositing in bone appears to be retained largely on bone surfaces. Whole-body retention has generally been described as the sum of two retention components of approximately equal size. The short-term component typically has a biological half-time of a few days, and the long-term component has a half-time of a few months. Reported biokinetic studies have not been sufficiently long to characterise longer-term components of retention such as may be present in bone. (607) Hamilton and coworkers (Hamilton, 1948; Durbin et al., 1957; Durbin, 1960) studied the biokinetics of 95Nb in rats following intramuscular injection of relatively soluble niobium compounds. A substantial portion of the absorbed activity apparently combined with plasma proteins and was slowly removed from blood to tissues and excretion pathways. Activity distributed throughout the body and was removed more slowly from bone, kidney, and lymphatic tissue than from other repositories. Activity was excreted mainly in urine over the first 2 weeks, but the faecal to urinary excretion ratio increased over time. At 4 d after administration of 95Nb citrate, the mean contents of bone, liver, kidneys, and blood were 16%, 8.4%, 2.9%, and 7.7% of the administered activity, respectively, and approximately 39% of the administered amount had been excreted by that time. Autoradiographic studies indicated that skeletal 95Nb was located largely on bone surfaces. (608) The distributions of 90Nb and 95Nb were studied in rats over a 4 d period following their intravenous administration in a solution of oxalic acid (Matthews and Gartside, 1965). Comparison with blood retention of 131I-labelled plasma proteins suggested that a substantial portion of the injected activity combined with plasma proteins. Retention in blood was approximately 30% of the injected amount at 1 d, 16% at 2 d, 11% at 3 d, and 5% at 4 d after correction for radiological decay. Whole-body retention fell to approximately 65% at 4 d. Bone contained approximately one-quarter of the injected amount at the end of the study, based on extrapolation of data for the femur. The liver content was in the range 4.0–5.4% from 1.2 h to 4 d after injection. Activity in most tissues decreased with time, but activity in the kidneys increased from approximately 2% after 1.2 h to approximately 4% at 3–4 d. (609) Semenov et al. (1966) investigated the distribution of 95Nb in rats following its intravenous or subcutaneous administration as the oxalate. Similar behaviour was seen for the two modes of exposure. Niobium in blood combined with plasma proteins, primarily albumin. Little activity was accumulated by RBCs. Following intravenous injection, the blood contained approximately 17% of the administered activity at 1 d, 2.9% at 4 d, and 0.12% at 64 d; the liver contained approximately 5–7% during the first day, 9% at 2–8 d, and 2% at 64 d; the kidneys contained approximately 1–2% during the first day and 2–3% during days 2–64; and the muscles contained 13–24% during the first 8 d, 9% at 16–32 d, and 4% at 64 d. The concentration in bone increased steadily for several days after injection and then remained at approximately the same level for the remainder of the 64 d study. The concentration in bone was higher than that in most other tissues at 32 and 64 d after injection. Approximately 23% of the administered amount was excreted in urine, and approximately 10% was excreted in faeces over the first 20 d after intravenous injection. A substantial portion of activity entering the gastrointestinal contents appeared to arise from secretions other than liver bile. (610) Razumovski˘ et al. (1966) studied the effects of various complex-forming agents on the biokinetics of 95Zr and 95Nb in rats. At 3 d after intraperitoneal administration of 95Nb oxalate to control animals, the liver, spleen, kidneys, and femur contained approximately 3.1, 0.62, 0.89, and 0.23% of the administered activity, respectively. (611) Autoradiographic studies on mice demonstrated high concentrations of 95Nb in bone and connective tissue during the first 4 d after its intravenous administration as oxalate (Bäckström et al., 1967). The distribution of activity was similar to that observed after intravenous administration of 95Zr–95Nb, but bone appeared to accumulate a smaller portion of the administered activity following injection of pure 95Nb. (612) Fletcher (1969) studied the behaviour of 95Nb in rats following its administration as oxalate. Approximately 30% of intravenously administered activity deposited in the skeleton, 18% in muscle, 2.5% in liver, and 2.5% in kidneys. Whole-body retention declined more slowly in males than in females. Retention was approximately 70% of the injected amount at 8 d, 50% at 40 d, and 40% at 80 d as an average for males and females. (613) Furchner and Drake (1971) studied retention and excretion of 95Nb after oral and intravenous administration as oxalate to mice, rats, monkeys, and dogs, and after intraperitoneal administration as oxalate to mice and rats. The duration of individual studies ranged from 4 d to 192 d. Little difference in retention was seen following intravenous and intraperitoneal administration. Whole-body retention of intravenously injected 95Nb was described as the sum of three exponential terms for mice and rats, and the sum of two exponential terms for monkeys and dogs. The cumulative urinary:faecal excretion ratio over the first 3 d was approximately 9 for mice, 3 for rats and dogs, and 6 for monkeys. Estimated long-term biological half-times were approximately 100 d for monkeys, 150 d for dogs, 180 d for rats, and 460 d for mice. The long-term half-time represented approximately one-half of the administered amount in monkeys, dogs, and rats, and approximately one-quarter of the administered amount in mice. Rats receiving 95Nb by intraperitoneal injection were killed at 1, 4, 7, 14, 23, 35, and 45 d for tissue distribution studies. The percentage of total body activity in bone in these animals increased from approximately 16% at 1 d to approximately 27% at 23 d, and remained near 27% thereafter. The muscle, pelt, and liver contained approximately 33–37%, 17–21%, and 4–5%, respectively, of total body activity over the entire observation period. The kidney content increased from approximately 1.5% of total body activity at 1 d to more than 3% after 35 d. (614) 95Nb oxalate was administered orally or intravenously to sheep and swine 6–18 h after birth or 3 weeks after weaning (Mraz and Eisele, 1977b). At 3 d after intravenous administration, the mean skeletal content was approximately 67% of the injected amount in newborn sheep compared with 43% in weaned sheep, and 66% in newborn swine compared with 51% in weaned swine. The mean contents in the liver, kidneys, and muscle at 3 d varied little, if any, with age. The liver contained 1.7% of the injected amount in newborn and weaned sheep, and 3.4–3.5% in newborn and weaned swine; the kidneys contained 0.7–1.1% in newborns and weanlings of both species; and muscle contained 6.4–7.3% in newborns and weanlings of both species. (615) Cuddihy (1978) measured the distribution, retention, and excretion of 95Nb in beagle dogs following its inhalation as oxalate or oxide aerosols, and used the results to model the respiratory, gastrointestinal, and systemic biokinetics of the inhaled activity. Frequent whole-body measurements were made, and urine and faecal samples were collected daily throughout the study. Dogs were killed for tissue distribution studies at 1 h and 2, 4, 8, 16, 32, 64, and 128 d. An estimated 60% of the initial lung burden was absorbed into the systemic circulation after inhalation of the oxalate aerosols, compared with <1% after inhalation of the oxide. Daily urinary excretion of 95Nb was two to three times greater than daily faecal excretion following early rapid clearance of activity from the upper respiratory tract. As predicted by Cuddihy’s model, whole-body retention was 44% at 8 d and 28% at 128 d following acute input of stable niobium to blood. The predicted bone contents at these two times were approximately 14% and 16%; the liver contents were 9% and 8%; contents of other soft tissues were 17% and 6%; cumulative urinary losses were 45% and 60%; and cumulative faecal losses were 5% and 10%. (616) Following intravenous administration of 95Nb oxalate to pregnant rats, there was a slow decrease in the activity concentrations in blood and liver during the first day and a simultaneous increase in kidneys and bone (Schneidereit et al., 1985). Whole-body retention over the first 20 d after injection into dams was described as the sum of two exponential terms with biological half-times of 1.3 d (∼30%) and 46 d (∼70%). Only a small portion of the injected activity was transferred to the fetus. (617) The effects of various chelating agents on retention and elimination of 95Nb were tested in mice following its intraperitoneal administration as oxalate (Gachalyi et al., 1987). Whole-body retention of 95Nb in control animals was described as the sum of two exponential terms with mean biological half-times of 1.1 d (∼50%) and 54 d (∼50%). The mean concentrations in liver, kidneys, and bone of control animals were, respectively, 3.9, 0.50, and 2.0% g−1 at 4 d, and 2.7, 0.54, and 2.4% g−1 at 14 d. Desferrioxamine was shown to be an effective chelating agent for 95Nb, particularly when combined with diethylenetriaminepentaacetic acid (DTPA). (618) Harrison et al. (1990) measured retention of 95Nb following its oral or intraperitoneal administration in a citrate solution to adult and newborn guinea pigs. Whole-body retention following intraperitoneal injection was slightly lower in newborns than in adults, with approximately 50% of the injected activity excreted by newborns during the first day compared with approximately 40% in adults. The remaining activity cleared with a half-time of approximately 30 d in both age groups as estimated from measurements through day 7. Urinary excretion accounted for more than 90% of total losses in adults over the 7 d observation period. (619) The distribution of 95Nb formed in vivo from decay of ingested or intravenously injected 95Zr in rats was similar to the distribution of administered 95Nb and considerably different from the distribution of 95Zr (Fletcher, 1969). Following oral administration of 95Zr–95Nb to suckling rats, the ratio of 95Zr to 95Nb was 4–5 in bone and approximately 1 in other tissues (Shiraishi and Ichikawa, 1972). Measurements of activity in blood and tissues of rats following intraperitoneal injection of 95Zr–95Nb as oxalate indicated preferential accumulation of 95Zr in bone (Rama Sastry et al., 1964).
13.2.3.2. Biokinetic model for systemic niobium
(620) The structure of the systemic model for niobium is shown in Fig. 13.1. Transfer coefficients are listed in Table 13.3. These transfer coefficients are rounded values derived from the deposition fractions and removal half-times summarised below. (621) The transfer coefficients were set, in part, for reasonable consistency with predictions of the systemic model of Cuddihy (1978) of the contents of the whole body (Fig. 13.2), bone, liver, and total soft tissues over the first few months after acute input of niobium to blood. The Cuddihy model was used as a guide for modelling the early behaviour of niobium because it was based on detailed measurements of the fate of absorbed niobium in beagle dogs, which have proven to be a useful laboratory model for the behaviour of bone seekers; and its predictions are reasonably representative of biokinetic data for niobium from other animal studies. The present blood retention model was designed for reasonable consistency with observed blood clearance of the related element zirconium in human subjects over the first few days after intravenous injection (Veronese et al., 2003; Greiter, 2008), as well as the blood clearance curve predicted by the Cuddihy model for niobium. Parameter values for the kidneys, which are not addressed explicitly in the Cuddihy model, were set for reasonable agreement with collective data on the kidney contents of 95Nb over the first few months after intravenous or intraperitoneal administration to rats (Semenov et al., 1966; Fletcher, 1969; Furchner and Drake, 1971). The fate of niobium depositing on bone surface is described by the generic bone model for bone-surface-seeking radionuclides used in this report, except that niobium removed from bone is assumed to return to Blood 1 rather than channelled through bone marrow. (622) In the present model, niobium initially entering the systemic circulation is assigned to a compartment called Blood 1. Niobium leaves Blood 1 at a rate of 8 d−1, corresponding to a removal half-time of approximately 2 h. Outflow from Blood 1 is divided as follows: 40% transfers to a slow-turnover blood compartment called Blood 2, representing plasma proteins; 3% transfers to liver; 0.5% transfers to kidneys; 3% transfers to bone surfaces and is equally divided between cortical and trabecular surfaces; 40% transfers to ST0, a soft tissue compartment with relatively fast turnover; 1.5% transfers to ST1, a soft tissue compartment with relatively slow turnover; 11% transfers to urinary bladder contents; and 1.0% transfers to small intestine contents. Activity transfers from Blood 2 back to Blood 1 with a half-time of 0.5 d, from ST0 to Blood 1 with a half-time of 0.5 d, from ST1 to Blood 1 with a half-time of 70 d, and from kidneys to Blood 1 with a half-time of 140 d. Niobium entering liver is assigned to a compartment called Liver 0. Niobium is removed from Liver 0 with a half-time of 2 d, with two-thirds going to a long-term retention compartment of liver called Liver 1 and the other one-third equally divided between Blood 1 and small intestine contents (representing biliary secretion). Relative transfer rates from Blood 1 and Liver 0 into small intestine contents are set so that biliary secretion accounts for one-third and other endogenous secretions (represented as transfer from Blood 1 to small intestine contents) account for two-thirds of total faecal excretion. Niobium transfers from Liver 1 to blood with a half-time of 140 d. As indicated earlier, parameter values describing the fate of niobium depositing on bone surface are generic values applied in this publication to bone-surface-seeking radionuclides. Structure of the biokinetic model for systemic niobium. ST, soft tissue; SI, small intestine. Whole-body retention of niobium after acute uptake to blood. Values indicated by closed circles are based on a model developed by Cuddihy (1978) as a fit to inhalation data for dogs. Values indicated by other symbols are based on curve fits to observations of Furchner and Drake (1971) for intravenously injected 95Nb. Parameter values in the systemic model for niobium. ST, soft tissue; SI, small intestine.



13.2.3.3. Treatment of radioactive progeny
(623) Chain members addressed in the derivation of dose coefficients for internally deposited niobium isotopes include isotopes of yttrium, zirconium, and niobium. The characteristic systemic models for yttrium, zirconium, and niobium all have the same structure. An atom of any of these elements produced in a compartment by radioactive decay after intake of a niobium parent is assumed to behave as if it had entered that compartment as a parent radionuclide.
13.3. Individual monitoring
(624) Monitoring of 95Nb is generally accomplished through whole-body measurement or/and urine bioassays.
13.4. Dosimetric data for niobium
Monitoring techniques for 95Nb.

Lung monitoring of 95Nb is not generally used in routine monitoring of workers. The Monte Carlo program Visual Monte Carlo was used to simulate the photon emission, to calculate the calibration factor for the geometry and radionuclide, and to calculate the detection limit in the lung (Hunt et al., 2012).
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 95Nb compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity contentof 95Nb in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
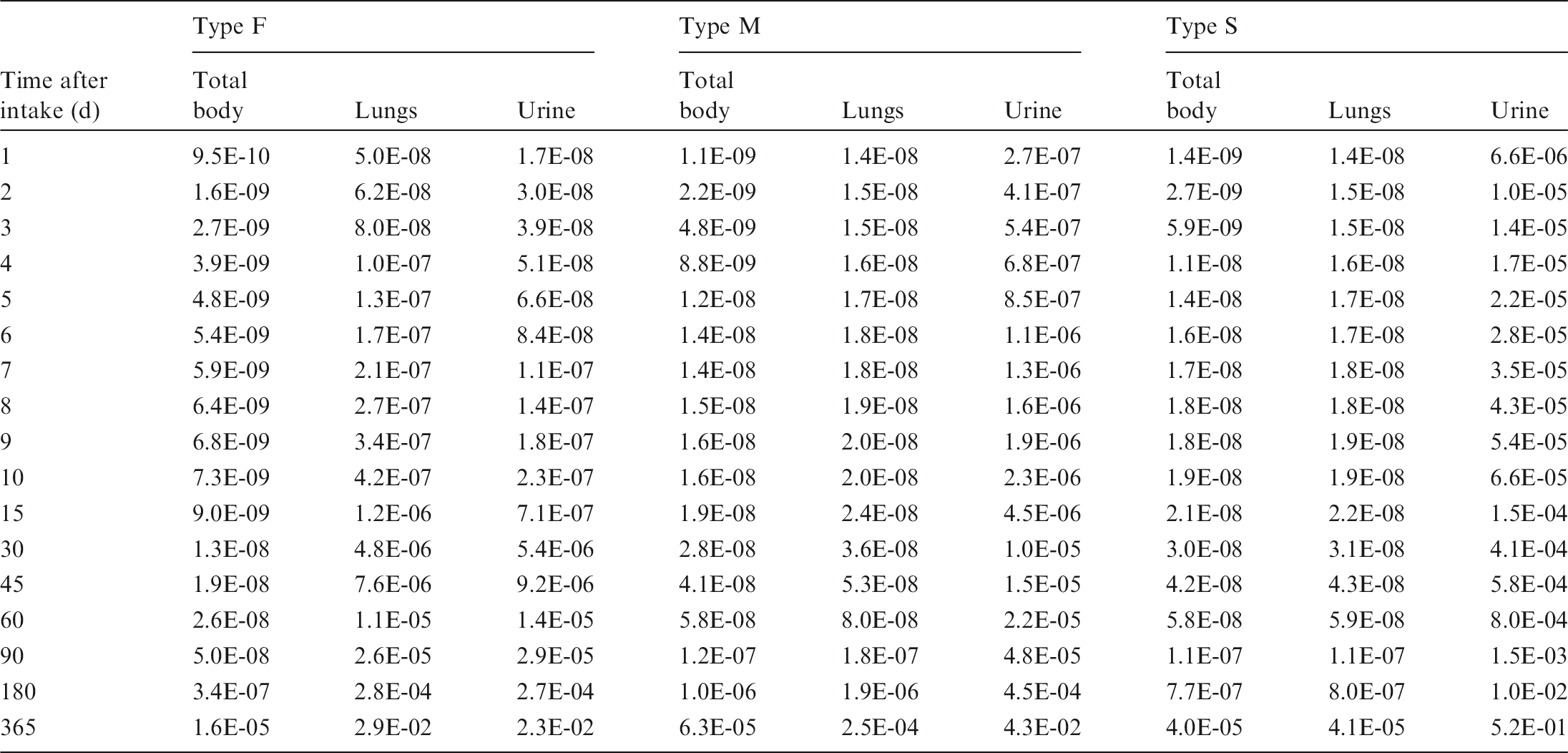

Total body and lung contents, and daily urinary excretion of 95Nb following inhalation of 1 Bq Type F.

Total body and lung contents, and daily urinary excretion of 95Nb following inhalation of 1 Bq Type M.

Total body and lung contents, and daily urinary excretion of 95Nb following inhalation of 1 Bq Type S.
13.5. References
14. MOLYBDENUM (Z = 42)
14.1. Chemical forms in the workplace
(625) Molybdenum is a transition metal that mainly occurs in oxidation states IV and VI. It is an essential element for plants, animals, and humans, present in two groups of enzymes: the nitrogenases and the molybdopterins. Molybdenum may be encountered in industry in a variety of chemical and physical forms, including oxides, halides, sulphides, nitrates, and ammonium molybdate. In the nuclear industry, 99Mo is a fission product and could be encountered in fragments of irradiated fuel. Large activities of 99Mo are used in 99mTc generators in nuclear medicine. Table 14.1 shows the isotopes of molybdenum addressed in this publication. Isotopes of molybdenum addressed in this publication. Dose coefficients and bioassay data for this radionuclide are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. B+, Beta-plus decay; B-, Beta-minus decay; EC, electron-capture decay; IT, isomeric transition decay

14.2. Routes of intake
14.2.1. Inhalation
(626) Little information is available on the behaviour of inhaled molybdenum in man following accidental intakes, or from experimental studies in animals. (627) Absorption parameter values and types, and associated fA values for particulate forms of molybdenum are given in Table 14.2.
14.2.1.1. Particulate materials
(a) Ammonium molybdate
(628) Cuddihy et al. (1969) measured the tissue distribution of 99Mo in three dogs at 8 d after inhalation of a solution of ammonium molybdate. Approximately 2% of the sacrifice body burden was in the lungs, compared with 79% sacrifice body burden in systemic organs (liver, skeleton, muscle, and kidney), showing that most of the molybdenum deposited in the lungs had been absorbed, and giving assignment to Type F.
(b) Molybdenum chloride
(629) Cuddihy et al. (1969) measured the tissue distribution of 99Mo in three dogs at 8 d after the inhalation of molybdenum chloride (MoCl4) with 2.5 µm AMAD. Approximately 6% sacrifice body burden was in the lungs, compared with 68% sacrifice body burden in systemic organs, giving assignment to Type F.
(c) Molybdenum oxide
(630) Cuddihy et al. (1969) measured the tissue distribution of 99Mo in three dogs at 8 d after the inhalation of molybdenum oxide (MoO3) with 1.5 µm AMAD. Approximately 46% sacrifice body burden was in the lungs, compared with 39% sacrifice body burden in systemic organs, giving assignment to Type M.
(d) Other compounds
(631) Measurements of 99Mo and 99mTc whole-body retention and excretion in urine were made from 1.3 d up to approximately 10 d after intake of an aerosol released during handling of a 99Mo source (99Mo alkaline solution) by workers at a company manufacturing 99mTc generators for use in nuclear medicine (Alvarez et al., 1994; Navarro et al., 1995). Navarro et al. showed good agreement between Publication 30 (ICRP, 1979) model predictions (Lung Class D), and measured whole-body retention and urinary excretion for two workers representative of Group 1 (workers who were in the facility where the accident happened, and exposed directly to the source aerosol) and Group 2 (workers who were in a nearby laboratory and were contaminated by the aerosols dispersed through the air-conditioning system). A critical analysis of the data (Giussani et al., 2004) showed different biokinetic behaviours between workers in Group 1 and Group 2. This seems to suggest that the aerosol composition was different in the two environments. Analysis of the data for several workers
1
conducted here confirmed good agreement assuming Type F absorption, and less good agreement for Type M (with a correspondingly lower value of fA). However, with the first measurement made more than 1 d after intake and a large contribution to systemic uptake from absorption in the alimentary tract, it was not possible to estimate a specific value for sr from the data.
Data kindly provided by Dr M.A. Lopez, CIEMAT.
14.2.1.2. Rapid dissolution rate for molybdenum
(632) There is insufficient experimental information to estimate the rapid dissolution rate for molybdenum. There is therefore no justification for choosing a rate different from the general default value of 30 d−1, which is applied here to all Type F forms of molybdenum.
14.2.1.3. Extent of binding of molybdenum to the respiratory tract
(633) Cuddihy et al. (1969) observed that 8 d after inhalation of ammonium molybdate or molybdenum chloride by dogs, the amounts of 99Mo associated with the nasal turbinates were similar to those in the lungs. This suggests that there could be some binding of molybdenum. However, the experimental information is insufficient to estimate the extent of any bound state, and it is assumed by default that fb = 0.
14.2.2. Ingestion
(634) Human investigations with stable isotopes have shown that fractional absorption of molybdenum in inorganic form (chloride and ammonium molybdate) is greater than 0.85 (Turnlund et al., 1995a,b; Giussani et al., 1998, 2006). These studies also showed that intestinal absorption of molybdenum is usually complete within the first 4 h after administration (less than 2 h if administered in liquid form), indicating that the absorption is only from the upper part of the alimentary tract (Giussani et al., 2006). (635) A large number of studies have been conducted in ruminants in order to investigate the metabolism of molybdenum after ingestion, and the potentially lethal effects of an imbalance between the contents of molybdenum, copper, and sulphur in the diet (Huising and Matrone, 1976; Price et al., 1988). Those effects were due to interactions of those elements in the rumen of the animals (production of thiomolybdates), and they were not observed in non-ruminants, except when thiomolybdates were administered to them directly (Mills et al., 1978; Mills, 1985; Chen et al., 1988); therefore, data from studies with ruminants will not be further considered here. Molybdenum is readily absorbed by non-ruminants when ingested as salts of molybdic acid, such as MoO3 or CaMoO4 (Mills and Davis, 1987). In contrast, the highly insoluble compound molybdenum disulphide is only poorly absorbed (Underwood, 1971). The absorption of molybdenum is considered to be dependent on its concentration in diet, the amounts of copper and sulphur present, and the age of the animals (Comar et al., 1949; Nederbragt, 1983). (636) In Publication 30 (ICRP, 1979), the recommended absorption values were 0.05 for the sulphide and 0.8 for all other compounds of the element. The value of 1 was adopted in Publication 67 (ICRP, 1993) for dietary intakes. The fA values proposed in this publication are 0.05 for sulphide and 0.9 for all other compounds.
14.2.3. Biokinetics of systemic molybdenum
14.2.3.1. Summary of the database
(a) Human subjects
(637) In recent years, the biokinetics of molybdenum in healthy volunteers was investigated in a series of studies using stable isotopes as tracers. (638) A study referred to here as the ‘GSF study’ was conducted by the Institute of Radiation Protection of GSF (now Helmholtz Zentrum München) in Munich, Germany, in collaboration with the Department of Physics of the State University of Milano, Italy (Cantone et al., 1995; Giussani et al., 1998, 2006, 2007; Werner et al., 2000; Tavola, 2004). Intestinal absorption, plasma clearance, and urinary excretion of molybdenum were studied in a series of investigations on healthy volunteers (six males and 11 females, age ranging from 27 to 63 y) by simultaneous oral and intravenous administration of two independent tracers. Repeated studies on the same subjects were conducted to investigate whether and how the amount and form of administration affect the biokinetic profiles. (639) The clearance of molybdenum from blood plasma was rapid in all subjects and could be described with a bi-exponential function with mean characteristic half-times of 30 min (median 29 min, range 4–70 min) and 6.6 h (median 4.4 h, range 2.6–30 h). The mean transit time in plasma was calculated to be approximately 150 min, and the average mass of the distribution compartment was evaluated to be in the range 7–19 kg, indicating that molybdenum was at least partially homogeneously distributed between blood plasma and interstitial fluids. (640) Urinary excretion in the first day after intake ranged between 30% and 80% of the intake, depending on the total mass of molybdenum present in the circulation; the higher the content of circulating molybdenum, the higher the fraction excreted. The excretion process was rapid; most of the molybdenum was excreted in the first 8–12 h after administration. It was also shown that administration of elevated dietary molybdenum mobilised molybdenum stored in the body and increased its excretion rate. No significant dependence of the results on age or sex was observed. (641) Another large study, referred to here as the ‘USDA study’, was conducted at the metabolic research unit of the Western Human Nutrition Research Center of the US Department of Agriculture (USDA), Presidio of San Francisco (Turnlund and Keyes, 2004; Turnlund et al., 1995a,b). In the first set of investigations, four healthy male subjects were kept on a low molybdenum diet for 24 d, and the metabolic fate of infused molybdenum in plasma was followed. In the second series of investigations, four healthy male subjects were kept on a low molybdenum diet for 102 d (depletion regime, daily intake 22 µg molybdenum), followed by an 18 d repletion period (daily intake approximately 500 µg molybdenum). A further investigation was structured in five dietary regimes, each with a duration of 24 d (dietary intake in each of the five periods: 22, 72, 121, 467, and 1490 µg molybdenum d−1, respectively). In all dietary regimes except the depletion regime, the basic diet (containing, on average, 22 µg molybdenum d−1) was supplemented with molybdenum taken from a liquid formula, and the behaviour of systemic molybdenum was studied by injection of the stable isotope 97Mo. (642) Analyses of the blood plasma samples showed a correlation between daily intake and the plasma level of molybdenum. It was also observed that the intravenous administration of even low amounts of tracer (33 µg 97Mo) affected the metabolism of endogenous molybdenum. Initial clearance from plasma was slightly faster than in the GSF studies; the published data could be described with a bi-exponential function with half-times of 8 and 40 min. (643) Molybdenum turnover as reflected by urinary excretion was faster with higher dietary molybdenum intakes, similar to what was observed in the GSF study. The percentage of oral tracer excreted in the urine over 6 d increased from 18% during the depletion period to 82% at the higher dietary regime. Similarly, the percentage excretion of the infused tracer increased from 33% to 87%. Faecal excretion of systemic molybdenum was negligible, as less than 2% of the infused tracer was excreted over 6 d. The faecal to urinary excretion ratio ranged from 1:20 to 1:62 depending on the total mass of circulating molybdenum. (644) Rosoff and Spencer (1964) injected 99Mo (as ammonium molybdate) into four seriously ill human patients and observed fast elimination from blood plasma (less than 4% of the tracer was present 1 h after injection), similar to the pattern observed by Turnlund and Keyes (2004). Ten percent of the injected amount was eliminated in urine after 24 h, and 25% was eliminated in urine after 6 d. (645) In studies conducted in the 1960s using 99Mo (molybdate) as a liver scanning agent (Sorensen and Archambault, 1963, 1964; Henning et al., 1965), the level of 99Mo in blood after 6 h was approximately 1/300 to 1/600 of the original level. In these studies, the whole-body retention half-time was reported to be of the order of 20–40 d; however, the estimates were highly uncertain due to the short half-life of 99Mo (2.75 d). Elimination in the urine amounted to 8% after 6 h, 20% after 24 h, and 30–60% after 2 weeks. (646) Recently reported concentrations of stable molybdenum in human organs and tissues are generally lower than values reported in older studies, suggesting that improvements in the measuring techniques have led to greater precision and to the elimination of contaminating factors. Most reported values for the molybdenum concentration in whole blood fall between 0.4 µg l −1 and 1.2 µg l−1, and around 0.6 µg l−1 for blood plasma (Iyengar et al., 1978; Versieck et al., 1988; Vanhoe et al., 1989, 1994; Schramel and Wendler, 1995; Rodushkin et al., 1999; Heitland and Köster, 2006; Yoshida et al., 2006). Blood concentrations appear to be enhanced in people living in regions with higher daily intakes or suffering from particular diseases. (647) Autopsy determinations of molybdenum in human organs and tissues (Tipton and Cook, 1963; Tipton et al., 1965; Schroeder et al., 1970; Sumino et al., 1975; Iyengar et al., 1978; Coughtrey and Thorne, 1983; Versieck, 1983; Zeisler et al., 1988; Yoo et al., 2002) consistently demonstrate highest concentrations in the liver and kidneys, and show that the liver is the most important storage site for molybdenum in the body. Reported concentrations in liver peak around 1 µg g−1. Based on the reference organ masses given in ICRP (2002), these values correspond to 1.8 mg molybdenum in the liver of males (range 0.9–2.7) and 1.4 mg molybdenum in the liver of females (range 0.7–2.1). Values for kidneys peak around 0.3 µg g−1, corresponding to 90 µg (range 60–120) in the kidneys of males and 80 µg (range 55–110) in the kidneys of females (ratio liver:kidneys = 20:1). The preference of molybdenum for liver is confirmed by the findings of the studies with 99Mo in nuclear medicine, with reported uptake by the liver to be as high as 80% of the administered activity (Sorensen and Archambault, 1963; Henning et al., 1965; Colombetti et al., 1974; Shearer et al., 1988). (648) In previous ICRP publications, bone was reported ‘… to be a major store of molybdenum’, based on data presented by Coughtrey and Thorne (1983) and recalculated on the basis of measurements of molybdenum concentration in bone ashes made by Nusbaum et al. (1965). These values, however, have not been confirmed by any other study (Schroeder et al., 1970; Sumino et al., 1975; Yoo et al., 2002). Furthermore, none of the studies concerning the distribution of 99Mo administered to patients either as an agent for liver scanning or accidentally as an impurity in radiopharmaceuticals labelled with 99mTc reported evidence of accumulation of molybdenum in skeletal tissues (Sorensen and Archambault, 1963, 1964; Henning et al., 1965; Colombetti et al., 1974; Shearer et al., 1988).
(b) Laboratory animals
(649) In dogs, molybdenum translocated from the lung following inhalation of various compounds of the element was deposited mainly in liver, skeleton, muscle, and kidney, with liver and kidney containing the highest concentrations (Cuddihy et al., 1969). When 99Mo was administered intravenously as ammonium molybdate to mice, the liver showed the highest uptake with retention of approximately 26% of the administered activity at 1 h and approximately 21% at 1 d. The 99Mo content of the kidney was relatively high, accounting for approximately 3.8% of the administered activity at 1 h and 3.9% at 1 d (Rosoff and Spencer, 1973). When molybdenum was administered to rats as ammonium molybdate, 74% was excreted within 3 h (Ando et al., 1989), and the tissue distribution was similar to that reported for mice. (650) The marked differences between the ruminants and non-ruminants were clearly shown in the study by Bell et al. (1964) comparing absorption and excretion of molybdenum in swine and cattle. Swine showed fast clearance from blood plasma, fast absorption from the gastrointestinal tract, and rapid excretion in the urine (50–80% within 24 h after administration, depending on the total amount of circulating molybdenum). The results for swine are consistent with those observed in the human stable tracer investigations.
14.2.3.2. Biokinetic model for systemic molybdenum
(651) In Publication 30 (ICRP, 1979), on the basis of human data, the whole-body retention R(t) of molybdenum in humans was described by the following equation:
(652) For molybdenum translocated to organs or tissues, fractions of 0.1 and 0.9 were assumed to be retained with half-times of 1 and 50 d, respectively. (653) In Publication 67 (ICRP, 1993), for molybdenum entering the transfer compartment, 10% was assumed to be deposited in the skeleton and to be retained with a biological half-time of 10,000 d. The remaining activity was distributed to liver (25%), kidneys (5%), and all other tissues (60%). A urinary:faecal excretion ratio of 8:1 was assumed for molybdenum that has entered the transfer compartment. (654) In this publication, a recycling model for molybdenum biokinetics is presented. The definition of the model structure and the procedure for the determination of the model parameters were presented elsewhere (Giussani, 2008) and are summarised briefly here. (655) The structure of the model consists of:
two compartments to describe the available data of molybdenum in blood plasma; liver; kidneys; urinary bladder; and generic tissue pool (other tissue). (656) The available data do not indicate that the skeleton is a major repository for molybdenum as was assumed in some previous systemic models for this element. In the present model, the skeleton is pooled together with the rest of the systemic tissues, excluding the liver, in a generic common compartment. (657) The splitting into two subunits of the compartment associated with the systemic circulation was made in accordance with the results of the analysis presented by Giussani et al. (2007). (658) The stable isotope studies showed that the absorption and excretion processes changed for increasing amounts of administered tracers (and, consequently, of circulating molybdenum). The values of the characteristic parameters given in Table 14.1 were therefore determined by fitting the model predictions to a subset of the available data corresponding to the investigations with molybdenum administration lower than or in the same order of the average daily intake. No allowance was made for age- or sex-dependent parameters, as no indication of such dependence was evident from the review of data presented in the previous sections. (659) The Blood 1 compartment receives material from outside (alimentary tract, respiratory tract, wounds), and distributes it to urinary excretion (direct pathway, 19.5%), liver (42.8%), and Blood 2 (37.7%) with a biological half-time of 30 min. The Blood 2 compartment transports material into a kidney compartment that exchanges molybdenum with blood (3.2%), into a generic compartment taken to represent all other tissues (48.8%), and into the urine through the renal urinary path (48.0%), with a half-time of 280 min. The total mass of compartments associated with the extracellular fluids (Blood 1 + Blood 2) amounts to 12 kg. (660) The retention half-times of molybdenum in the kidneys and in the other tissues are equal to 14.6 d and 21.5 d, respectively; from these compartments, molybdenum is transported back to Blood 2. (661) The retention half-time in liver is equal to nearly 41 d; 28% is secreted into the right colon contents and is subsequently lost in faeces, and 72% is transported back to the extracellular fluid compartment represented by Blood 2. The characteristic half-time for transfer from the urinary pathway into the bladder contents is equal to 0.5 d. (662) In the following figures, the model predictions are compared with the corresponding human data from the stable tracer studies.

14.2.3.3. Treatment of radioactive progeny
(663) The radioactive progeny considered in the calculations of dose coefficients for molybdenum isotopes are isotopes of niobium or technetium. The models for niobium and technetium as progeny of systemic molydenium are modifications of the models applied in the OIR series to niobium and technetium as parent radionuclides. (664) External measurements on normal human subjects indicated that 99mTc produced in the liver by decay of 99Mo following intravenous administration of 99Mo as sodium or ammonium molybdate was retained in the liver for an extended period (Sorensen and Archambault, 1963). In contrast, 99mTc depositing in the liver after administration as a parent radionuclide was largely removed with a half-time of a few hours (Sorensen and Archambault, 1963). On the basis of these findings, technetium produced in the liver by decay of a molybdenum parent is assigned to the long-term retention compartment of liver in the characteristic model for technetium described elsewhere in this publication. The removal half-time from that compartment to blood is 22 d. For modelling convenience, the compartment of the molybdenum model called Blood 1 is identified with the blood compartment of the technetium model. Technetium produced in the compartment Blood 2 of the molybdenum model is assumed to transfer to blood in the technetium model at a rate of 1000 d−1 (t½ = 1 min). Technetium produced in a compartment of kidneys or ‘other tissue’ in the molybdenum model is assumed to transfer to blood in the technetium model at a rate of 0.462 d−1; the rate of transfer to blood from the intermediate-term compartment of ‘other tissue’ in the technetium model. After reaching blood, technetium is assumed to follow its characteristic model. (665) No information was found on the behaviour of niobium produced in vivo following intake of a molybdenum parent. For modelling convenience, the compartment of the molybdenum model called Blood 1 is identified with the central blood compartment of the characteristic model for niobium (also named Blood 1; see Fig. 13.1). It is assumed that niobium produced in Blood 2 of the molybdenum model transfers to Blood 1 of the niobium model at a rate of 1000 d−1. Niobium produced in a tissue compartment of the molybdenum model is assumed to transfer to Blood 1 in the niobium model at a rate of 1.39 d−1; the highest rate of transfer to blood from an ‘other tissue’ compartment of the niobium model. After reaching Blood 1 in the niobium model, niobium is assumed to follow its characteristic systemic model. Absorption parameter values for inhaled and ingested molybdenum. It is assumed that the bound state can be neglected for molybdenum, i.e. fb=0. The values of sr for Type F, M, and S forms of molybdenum (30, 3, and 3 d−1, respectively) are the general default values. Materials (e.g. molybdenum chloride) are listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of molybdenum (0.9). These calculated values are not rounded for purposes of consistency. Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the highest value for ingestion of the radionuclide (fA=0.9). Systemic model for molybdenum radionuclides. Parameter values in the systemic model for molybdenum. Includes bone and all soft tissues other than liver and kidneys. Concentration in plasma of injected molybdenum tracer. Data are from 15 investigations in six volunteers (GSF study). Cumulative urinary excretion of the intravenous tracer. Dots, data from the GSF study (one volunteer, error bars: experimental uncertainties); triangles, data from the USDA study, depletion conditions (eight volunteers, mean ± standard error). Cumulative faecal excretion of the intravenous tracer. Dots, data from the USDA study, depletion conditions (eight volunteers, mean ± standard error).

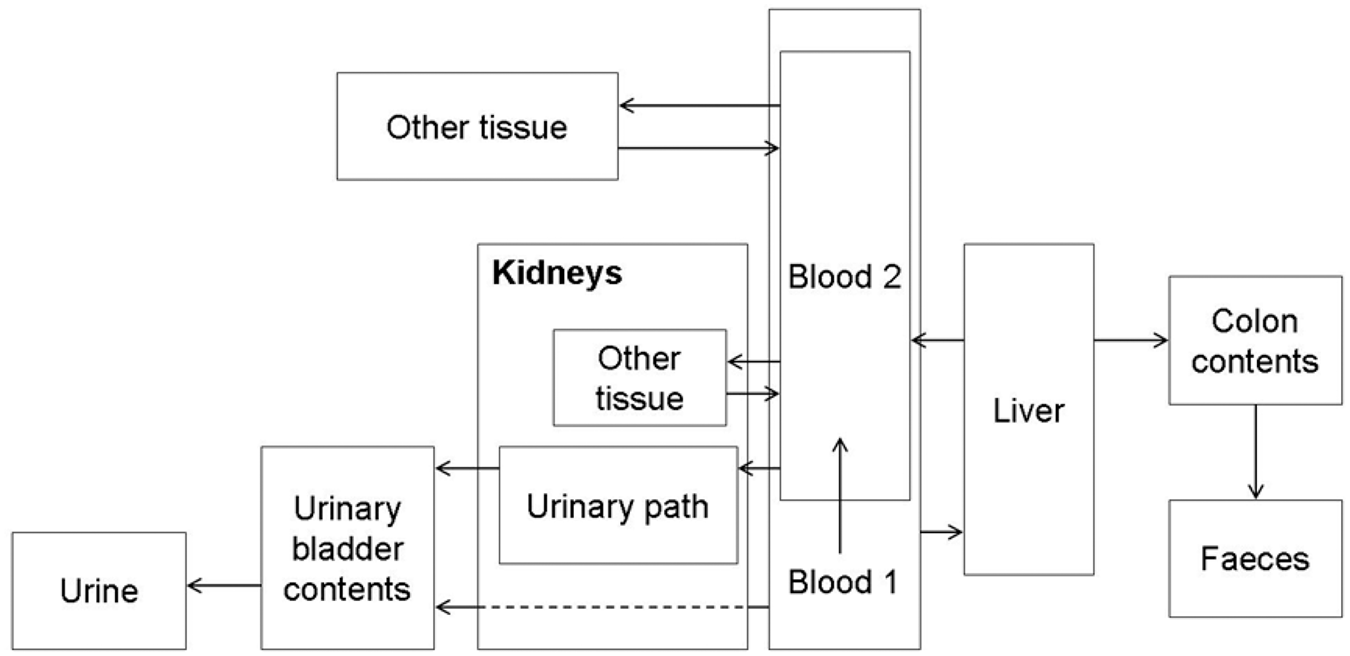




14.3. Individual monitoring
(666) Monitoring of 99Mo is generally accomplished through whole-body measurement or/and urine bioassays (Table 14.4). Monitoring techniques for 99Mo. Not commonly measured in the lung.

14.4. Dosimetric data for molybdenum
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 99Mo compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 99Mo in the total body, lungs and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Total body and lung contents, and daily urinary excretion of 99Mo following inhalation of 1 Bq Type F.

Total body and lung contents, and daily urinary excretion of 99Mo following inhalation of 1 Bq Type M.
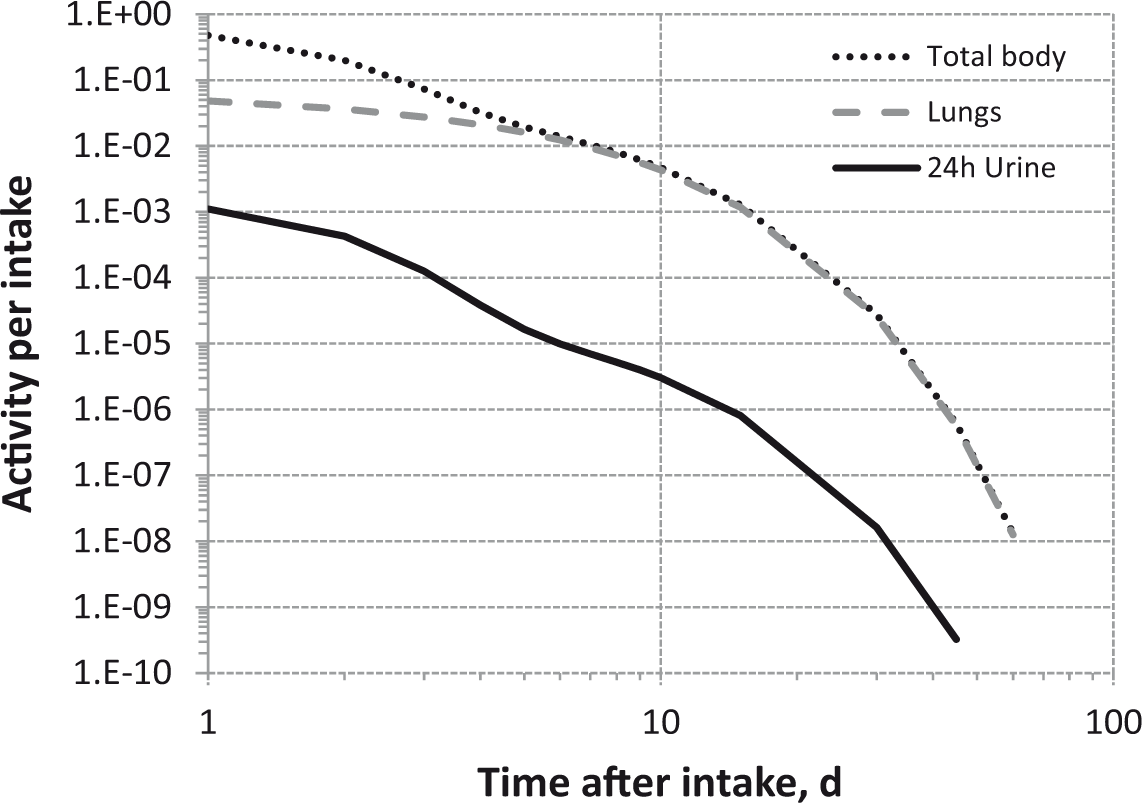
Total body and lung contents, and daily urinary excretion of 99Mo following inhalation of 1 Bq Type S.
14.5. References
15. TECHNETIUM (Z = 43)
15.1. Chemical forms in the workplace
(667) Technetium is a transition metal that occurs mainly in oxidation states IV, VI, and VII. Technetate or pertechnetate (TcO4−) is the most common technetium ion in solution. Technetium may be encountered in industry in a variety of chemical and physical forms, such as oxides (TcO2, Tc2O7), sulphides, halides, and nitrates. Technetium is an artificial element obtained either from uranium fission or after bombarding molybdenum with neutrons. 99Tc is a high-yield product of uranium fission, and with its relatively long half-life is an important component of nuclear waste. 99mTc is frequently used in nuclear medicine for a wide variety of diagnostic tests as a label for different pharmaceuticals. Table 15.1 shows the isotopes of technetium addressed in this publication.
15.2. Routes of intake
15.2.1. Inhalation
(668) Most of the experimental information available on the behaviour of technetium following deposition in the respiratory tract relates to pertechnetate, or materials labelled with 99mTc, especially DTPA. Some information is also available from accidental human intakes. (669) Absorption parameter values and types, and associated fA values for particulate forms of technetium are given in Table 15.2. Isotopes of technetium addressed in this publication. Dose coefficients and bioassay data for these radionuclides are given in the printed copy of this publication. Data for other radionuclides listed in this table are given in the accompanying electronic annex. Absorption parameter values for inhaled and ingested technetium. It is assumed that the bound state can be neglected for technetium, i.e. fb=0.0. The value of sr for Type F forms of technetium (100 d−1) is element specific. The values for Types M and S (3 d−1) are the general default values. Materials (e.g. pertechnetate) are generally listed here where there is sufficient information to assign to a default absorption type, but not to give specific parameter values (see text). For inhaled material deposited in the respiratory tract and subsequently cleared by particle transport to the alimentary tract, the default fA values for inhaled materials are applied, i.e. the product of fr for the absorption type and the fA value for ingested soluble forms of technetium (0.9). These calculated values are not rounded for purposes of consistency. Default Type M is recommended for use in the absence of specific information on which the exposure material can be assigned to an absorption type; for example, if the form is unknown, or if the form is known but there is no information available on the absorption of that form from the respiratory tract. Activity transferred from systemic compartments into segments of the alimentary tract is assumed to be subject to re-absorption to blood. The default absorption fraction fA for the secreted activity is the reference fA (0.9) for ingestion of the radionuclide.


15.2.1.1. Particulate materials
(a) Pertechnetate
(670) The absorption of 99mTc from the lungs following its administration as pertechnetate (TcO4−, molecular mass 163 Da) is very rapid. Barrowcliffe et al. (1986) measured retention half-times of approximately 10 min after intratracheal instillation into rats. Man et al. (1989) measured retention half-times of 3–4 min after inhalation by dogs, several times faster than for 99mTc DTPA (see below) inhaled by the same dogs. Following inhalation of sodium 99mTc-labelled pertechnetate by healthy volunteers, Yeates et al. (1973) and Chopra et al. (1979) measured half-times of absorption of 99mTc from lungs to blood of approximately 10 min, with less than 2% of the ILD retained after 2 h. Chopra et al. (1979) obtained similar results in patients with systemic sclerosis. Rinderknecht et al. (1980) measured retention half-times in healthy volunteers averaging 13 min for inhaled 99mTc-labelled pertechnetate, significantly faster than for 99mTc DTPA (average 44 min), with faster clearance in patients with interstitial lung disease and slower clearance in patients with pulmonary alveolar proteinosis. (671) Based on the results of the experiments outlined above, specific absorption parameter values for pertechnetate were estimated here to be fr = 1 and sr = 100 d−1 (consistent with assignment to default Type F). However, although specific parameter values for pertechnetate based on in-vivo data are available, they are not adopted separately here. The data are used as the basis for the default rapid dissolution rate for technetium. Hence, specific parameter values for pertechnetate would be the same as default Type F technetium parameter values, and therefore pertechnetate is assigned to Type F instead.
(b) 99mTc-labelled DTPA (diethylenetriaminepentaacetic acid)
(672) 99mTc DTPA has been used extensively as a convenient, radiolabelled, low molecular mass (492 Da) solute to study pulmonary epithelial permeability in man. Following inhalation of 99mTc DTPA by healthy non-smokers, lung retention half-times of 99mTc were reported to be 59 min (corresponding to a clearance rate of approximately 17 d−1) by Jones et al. (1980), 72 min (14 d−1) by Braude et al. (1984), 56 min (18 d−1) by Nolop et al. (1987a), and 85 min (12 d−1) by Silveira et al. (2003). ‘Baseline’ clearance rates were reported to be 1.48% min−1 (21 d−1) by Nolop et al. (1987b), 0.7% min−1 (10 d−1) by Köhn et al. (1990), 0.83% min−1 (12 d−1) by Smith et al. (1992), and 0.69% min−1 (10 d−1) by Foster and Stetkiewicz (1996). See also the section on 14C-labelled DTPA (Section 3.2.1). Stather et al. (1983) followed the biokinetics of 14C after administration of 14C-labelled DTPA to healthy volunteers by inhalation, intravenous injection, and ingestion (which indicated that approximately 3% was absorbed from the alimentary tract). Modelling by the authors gave an estimated rate of absorption from lungs to blood of approximately 13 d−1 (fr ∼ 1), similar to that obtained for 99mTc DTPA, suggesting that it is characteristic of DTPA rather than technetium. Nolop et al. (1987a) obtained similar retention half-times for 99mTc DTPA (56 min) and 113mIn DTPA (62 min), indicating that the results were not affected by dissociation of 99mTc DTPA in the lungs. Thin-layer chromatography of 99mTc in urine following inhalation of 99mTc DTPA suggested that 99mTc DTPA did not dissociate during its movement from lungs to urine (Köhn et al., 1990). (673) Jefferies et al. (1984) reported that as premature infants with hyaline-membrane disease recovered, the retention half-time (initially shorter) averaged 56 min (18 d−1), similar to that in healthy adults, which suggests no effect of age on absorption of 99mTc DTPA from lungs to blood. (674) The absorption of 99mTc DTPA following deposition in different regions of the respiratory tract has been investigated. Chopra et al. (1979) measured retention half-times in healthy non-smokers of 35 min (29 d−1) and 65 min (15 d−1) for ‘upper’ and ‘lower’ lung fields measured with a gamma camera. Both fields were peripheral (i.e. predominantly alveolar). Oberdörster et al. (1986) found absorption to be slower in dogs for 99mTc DTPA inhaled with rapid shallow ventilation of large particles to maximise bronchial deposition (1.31% min−1, 19 d−1), than for inhalation with slow deep ventilation of small particles to maximise alveolar deposition (2.29% min−1, 33 d−1). Wolff et al. (1988) measured similar rates of clearance (∼7 d−1) of 99mTc DTPA instilled into the nasal passage, trachea, fifth-generation airway, and peripheral airway (approximately 10th generation) of dogs. Bennett and Ilowite (1989) found clearance of 99mTc DTPA by absorption from the bronchial mucosa to be slower than that from the alveolar region in healthy non-smokers; retention half-times were 296 min (3.4 d−1) and 107 min (9.3 d−1), respectively. Smith et al. (1992) reported clearance of 99mTc DTPA to be faster following deep inhalation to enhance alveolar deposition than following inhalation with normal tidal breathing. (675) The absorption of 99mTc DTPA from the lungs has been found to be faster in smokers and in patients with a wide variety of lung diseases. Due to its potential diagnostic use for detecting pathological changes in lung epithelial function, it has been studied extensively. However, according to a review by Peterson (1989), the long list of conditions that produce similar increases in the clearance rate, including severe lung disease, smoking, exposure to ozone, and even increased lung volume, make it insufficiently specific in diagnosis. For example, Jones et al. (1980) found a significantly shorter lung retention half-time of 99mTc of 20 min (50 d−1) in asymptomatic smokers than in non-smokers (59 min). Similarly, Nolop et al. (1987a) measured ‘baseline’ lung retention half-times of 99mTc of 25 min (40 d−1) in healthy smokers and 56 min (18 d−1) in non-smokers; hyperinflation increased the clearance rate in both groups. Minty et al. (1981) found a rapid, but only partial, increase in retention half-time in smokers who abstained from cigarettes for 3 weeks. (676) Although specific parameter values for 99mTc DTPA based on in-vivo data could be assessed, they are not adopted here because the systemic behaviour of technetium inhaled in this form differs from that of the model adopted here. Instead, it is assigned to Type F.
(c) 99mTc-labelled carbon
(677) An aerosol of ultrafine (<100 nm) 99mTc-labelled carbon particles (‘Technegas’) has been developed for lung ventilation scans in nuclear medicine. Sodium pertechnetate in saline is vaporised in a graphite crucible at approximately 2500℃ in an argon atmosphere, then diluted with air. The condensation aerosol formed consists of primary particles of approximately 5–15 nm diameter, forming agglomerates of approximately 100 nm diameter. Roth et al. (1997) investigated its deposition and clearance following inhalation by healthy volunteers. From total urine collection during 24 h after inhalation, they assessed that approximately 9% of the deposited 99mTc activity dissolved, mostly in the first 6 h. (678) To assess to what extent, and how, inhaled particles from ‘urban combustion’ or ‘soot-like’ particulate matter pass into the systemic circulation, volunteers inhaled ultrafine 99mTc-labelled carbon particles, in most cases produced with a Technegas generator or a modified version (Nemmar et al., 2002; Mills et al., 2006; Wiebert et al., 2006; Möller et al., 2008). Brown et al. (2002), however, used a spark generator (arc between carbon electrodes to which 99mTc pertechnetate had been applied). Nemmar et al. (2002) concluded that inhaled 99mTc-labelled carbon particles pass rapidly into the systemic circulation, based on the estimated liver uptake and the results of thin-layer chromatography of blood samples, which indicated that there was one species present corresponding to pertechnetate, and another which they attributed to 99mTc-labelled carbon particles. The other studies did not support this conclusion. All reported that particle accumulation in the liver was not detectable corresponding to fractions of the 99mTc-labelled carbon particles deposited in the lungs of < 1.5% for Brown et al. (2002) and < 0.5% for Möller et al. (2008). Mills et al. (2006) found (also using thin-layer chromatography) that the 99mTc transferred to blood was associated with pertechnetate rather than with particle-bound 99mTc. (679) With regard to dissolution, Nemmar et al. (2002) observed that activity was detected in blood at 1 min, reached a maximum between 10 and 20 min, and remained at this level up to 60 min. A considerable fraction of 99mTc leached from the particles and distributed as pertechnetate, as indicated by accumulation of 99mTc in the bladder, thyroid, and salivary glands. For a representative subject, activity in the bladder reached approximately 25% of the initial lung activity in 45 min. Brown et al. (2000, 2002) measured leaching in vitro (0.9% saline) to be approximately 10–15% in 5 min and 15–25% in approximately 24 h. Mills et al. (2006) noted that the Technegas generator produces a mixture of 99mTc-labelled particles and soluble oxides of 99mTc pertechnetate in the presence of even minute amounts of oxygen. Wiebert et al. (2006) and Möller et al. (2008) made specific efforts to fix the 99mTc radiolabel firmly to the carbon particles. Wiebert et al. (2006) reported dissolution in vitro (0.9% saline) to be approximately 3% in 70 h, compared with 11% in 24 h for particles produced by the standard Technegas method. Möller et al. (2008) reported dissolution in vitro (0.9% saline) to be approximately 4% in 24 h. In both studies, urinary excretion of 99mTc in 24 h following inhalation by volunteers was approximately 1% of activity deposited in the lungs. (680) These results suggest the fraction of 99mTc leaching rapidly from 99mTc-labelled carbon particles varies from a few percent to tens of percent, depending on the method of formation. The retention measurements made in the inhalation studies suggest that the remaining material is relatively insoluble, and more likely to be Type M or S than Type F, but the short duration of measurements limits the inferences that can be drawn.
(d) Other particulate forms
(681) The use of 99mTc-labelled materials, such as albumin, erythrocytes, ferric oxide, polystyrene, resin Teflon, and sulphur colloid, to study mucociliary clearance from the bronchial tree relies on there being relatively little absorption from the lungs to the body fluids over the first day or so after deposition (Albert et al., 1969; Puchelle et al., 1979; Mossberg and Camner, 1980; Sutton et al., 1981; Matthys et al., 1983; Isawa et al., 1984; Man et al., 1989; ICRP, 1994, Annex E).
(e) Undefined particulate forms
(682) The results of measurements of 99Mo and 99mTc whole-body retention and excretion in urine taken from 1.3 d up to approximately 10 d after intake of an aerosol released during handling of a 99Mo source (99Mo alkaline solution) by workers at a company manufacturing 99mTc generators for use in nuclear medicine (Alvarez et al., 1994; Navarro et al., 1995) are consistent with assignment to Type F (see Section 14.2.1).
15.2.1.2. Rapid dissolution rate for technetium
(683) Evidence from the pertechetate studies outlined above suggests a rapid dissolution rate of the order of 100 d−1, which is applied here to all Type F forms of technetium.
15.2.1.3. Extent of binding of technetium to the respiratory tract
(684) Evidence from the experimental studies outlined above suggests that there is probably little binding of technetium. It is therefore assumed that the bound state can be neglected for technetium, i.e. fb = 0.0.
15.2.2. Ingestion
(685) Technetium administered as 99mTc pertechnetate is generally well absorbed by human subjects. Mean absorption values of approximately 0.9 and 0.95 were obtained by McAfee et al. (1964) and Beasley et al. (1966), respectively, whereas the data presented in Andros et al. (1965) suggest a mean absorption fraction of 0.6. (686) In rats, the fractional absorption seems to range between 0.4 and approximately 0.9 for pertechnetate (Gerber et al., 1989; Archimbaud et al., 1992; Berthol et al., 2003), and were equal to approximately 0.5 for technetium chloride (Hamilton, 1948; Sullivan et al., 1977). (687) In Publication 30 (ICRP, 1980), an absorption value of 0.8 was recommended for all compounds of technetium. A lower value of 0.5 was adopted in Publication 67 (ICRP, 1993) for uptake from food. In this publication, an fA value of 0.9 is used for all chemical forms in the workplace.
15.2.3. Systemic distribution, retention, and excretion
15.2.3.1. Summary of the database
(a) Overview
(688) Most biokinetic studies of technetium in human subjects and laboratory animals have involved its administration as the ion pertechnetate (TcO4−), the most readily available chemical form and the starting point for technetium chemistry. The initial distribution of pertechnetate is similar to that of inorganic iodide. Pertechnetate and iodide are both concentrated selectively in the thyroid, salivary glands, and stomach wall. In contrast to iodide, pertechnetate trapped by the thyroid is not organically bound in the thyroid but is largely released back to blood over a period of hours. In euthyroid subjects, 1–4% of intravenously injected pertechnetate is typically accumulated by the iodide-concentrating mechanism of the thyroid in the first 15 min to 1 h, which is similar to accumulation of radioiodide in the blocked thyroid. Thyroid uptake of both iodide and pertechnetate are increased substantially in hyperthyroid subjects. A significant biological difference between the pertechnetate ion and iodide is their markedly different excretion pattern. Iodide is excreted mainly in urine. After intravenous administration, approximately 30% of administered pertechnetate is excreted in urine over the first 24 h, but thereafter the urinary excretion rate decreases markedly, while cumulative faecal excretion increases to 20% or more of the injected amount at 72 h, and may eventually exceed cumulative urinary excretion. Most of the absorbed or injected pertechnetate is lost from the body within a few days, but a small percentage is retained for a period of weeks or longer. During prolonged intake, relatively high concentrations are found in bone, kidneys, liver, skin, hair, and thyroid.
(b) Data for human subjects
(689) Harper et al. (1962) and Andros et al. (1965) found that intravenously injected 99mTcO4 localised within a few minutes in the thyroid, stomach, and salivary glands in human subjects and a variety of laboratory animals. Blood clearance could be described in terms of two approximately equal components with half-times of 8–12 min and 4–8 h. The first component appeared to represent distribution in the extracellular space. (690) Sorensen and Archambault (1963) developed a liver scanning technique using 99Mo molybdate but based on measurement of gamma radiation emitted by its progeny 99mTc. 99mTc was found to remain in the liver for an extended period after its production by decay of 99Mo already taken up by liver cells. In contrast, 99mTc depositing in the liver after administration as a parent radionuclide was removed with a half-time of a few hours, in parallel with the decrease in the external counts over the head. Approximately 58% of injected 99mTc was recovered in urine, and 24% was recovered in faeces over the first 3 d after its administration as a parent radionuclide. (691) McAfee et al. (1964) examined the tissue distribution or excretion of 99mTc administered as pertechnetate to six healthy male volunteers and 23 patients with suspected brain tumours. The gastrointestinal absorption and tissue distribution of activity resembled that of 131I administered as inorganic iodide. Absorbed activity was concentrated in the thyroid, salivary glands, and stomach. In contrast to iodide, a substantial fraction accumulated in the colon and was excreted in faeces. An abdominal scan performed 3 h after intravenous administration revealed high levels of activity within the stomach and duodenal loop, and higher levels in the splenic flexure and descending colon. Following either oral or intravenous administration, the highest count rates were observed over the stomach and the next highest rates were observed over the liver. For both organs, the effective (biological plus radiological) removal half-time was initially approximately 2 h, but increased to 5–7 h after the first hour, which is approximately the radiological half-life of 99mTc. This suggests near-equilibrium conditions between the rate of secretion into the stomach and removal of the secretion into the small intestine. The thyroid content peaked at 3–4% of the administered amount. In contrast to iodide, pertechnetate was not organified by the thyroid and was largely returned to blood over a period of hours. The thyroid content at 24 h was estimated as 0.5–1% of the administered amount. Approximately 20–25% of intravenously injected activity remained in blood after 1 h, and approximately 0.8–5% remained after 24 h. Blood plasma and RBCs contained, on average, approximately 70% and 30%, respectively, of the total blood content at 1 h after intravenous injection. The rate of urinary excretion of activity closely reflected the plasma concentration. Following oral administration to nine subjects, the average urinary excretion was 25% over the first 24 h, 3% over 24–48 h, and 1% over 48–72 h. Following intravenous administration to 12 subjects, average urinary excretion was 27% at 24 h, 4% over 24–48 h, and 2% over 48–72 h. Total urinary excretion by individual subjects was in the range 15–50% over 24 h and 15–58% over 72 h. Total faecal excretion over 72 h was 30–55% after oral administration and 10–45% after intravenous administration. Total loss in urine plus faeces over 72 h averaged 50% (range 28–68%) following intravenous administration, and 70% (39–88%) following oral administration. (692) Andros et al. (1965) studied the biokinetics of 99mTc over the first 72 h following its oral or intravenous administration as pertechnetate to 86 patients, including 57 euthyroid subjects. Following intravenous administration, the thyroid accumulated up to 2% of the administered amount at 1 h. The serum contained, on average, approximately 0.00045% ml−1 at 24 h, indicating that approximately 2% of the dosage was in blood at that time, assuming equal concentrations in plasma and RBC water. The concentration ratios saliva:plasma and gastric juice:plasma averaged 37.5 (range 11.5–66) and 17.5 (11–28.5), respectively. In seven subjects, average urinary excretion was 35.7% of the intravenously administered amount after 24 h, 6.2% at 24–48 h, and 4.8% at 48–72 h, giving a total of 46.7%. Average faecal excretion in six of these subjects was 8.8% after 72 h. In a normal young adult female, total urinary and faecal excretion at 72 h after intravenous injection accounted for 33.1% and 28.2%, respectively, of the administered amount. (693) Beasley et al. (1966) used 95mTc (t½ = 60 d) and 96Tc (t½ = 4.3 d) to study the relatively long-term biokinetics of technetium in eight normal human volunteers (ages 22–43 y) following its oral or intravenous administration as pertechnetate. The distribution and whole-body retention of activity were monitored externally, and samples of plasma, urine, faeces, sweat, tears, and intestinal mucosa were analysed. By 10 min after intravenous injection, the activity had begun to localise in the bladder. At 2 h, activity was found in relatively high concentrations in the salivary and thyroid glands, stomach, liver, and urinary bladder. The specific activity of the saliva was high, approaching 95% of dosage per litre of saliva at 2–3 h. For several days after oral or intravenous administration, the saliva contained 10–30 times the technetium concentration in plasma. Technetium was not concentrated in lacrimal or sweat glands, but the concentration in nasal secretions was high. There was no indication of localisation in the liver or kidneys at 3 d in a subject who received technetium orally. Biopsies of the stomach, duodenum, and rectal mucosa were performed on selected subjects at 2, 7, and 19 d. No appreciable activity was observed in the rectal mucosa, but concentrations in stomach and duodenum were 40–100 times plasma concentrations at comparable times. On average, approximately 28% of the injected activity was excreted in urine, and approximately 2–3% was excreted in faeces during the first 24 h. Thereafter, the urinary excretion rate declined rapidly, and faecal excretion soon became the dominant excretion pathway. Cumulative urinary and faecal excetion averaged approximately 35% and 55%, respectively, of the injected amount after 8 d. Biological retention R (%) in the whole body could be described as the sum of three exponential terms, R(t) = 76.7exp(-0.693t/1.6)+19exp(-0.693t/3.7)+4.3exp(-0.693t/22), where t is in days. (694) Harden et al. (1967, 1969) investigated the uptake of 99mTc pertechnetate by the stomach wall and salivary glands, and its secretion in saliva and gastric juice following its intravenous administration to human subjects. In 10 subjects with no evidence of diseases of the alimentary tract, the stomach contained, on average, 3.0% of the administered amount at 20 min and 6.8% at 1 h, based on external measurement. In seven male volunteers, the average concentration ratio 99mTc in saliva:99mTc in plasma at 40–70 min after administration was 27.3. The average concentration ratio 99mTc in gastric juice:99mTc in plasma over that time was 11.0. Clearance of 99mTcO4 was approximately half that of 132I in both saliva and gastric juice. (695) Atkins and Richards (1968) studied thyroidal uptake of 99mTc pertechnetate in 143 patients who were hospitalised for reasons other than thyroid disease. Uptake of 99mTc and 131I by the thyroid were positively correlated. Uptake of 99mTc in 120 euthyroid subjects averaged approximately 2% and exceeded 5% in only one subject. Fifteen hyperthryoid subjects had 99mTc uptake in the range 3.5–28.5%. (696) At 20 min after intravenous administration of 99mTc as pertechnetate to 11 euthyroid subjects, the estimated activity in the thyroid ranged from 0.2% to 6.3%, and averaged 2.8% of the injected amount (Shimmins et al., 1969). The estimated mean clearance rate from plasma to thyroid was 22 ml min−1, corresponding to a blood clearance rate of 6–8 d−1 assuming a well-mixed blood pool of volume 3.9–5.3 l (ICRP, 2002). The thyroidal turnover rate ranged from 0.031 to 0.108 min−1, and averaged approximately 0.07 min−1 (100 d−1). (697) In euthyroid subjects, the thyroid typically contained 1.5–4% of administered activity from 1 min to 1 h after intravenous injection of 99mTc as pertechnetate (Sodee, 1966). The estimated median content was approximately 2.7% at 2–15 min, 2.4% at 1 h, 2% at 2 h, and 1.5% at 3 h. (698) Mean (± standard deviation) thyroid uptake of intravenously injected 99mTcO4 in 18 normal volunteers was estimated as 1.6 ± 0.7% (Goolden et al., 1971). Uptake in 20 patients with thyrotoxicosis ranged from 0.8% to 22%. (699) Thyroid uptake of 99mTc pertechnetate was measured 20 min after administration of a tracer dose in seven normal controls and 52 patients with thyroid disease (McGill et al., 1971). The mean uptake was 0.96 ± 0.17% in normal subjects, 2.87 ± 0.39% in patients with non-toxic goitre, 16.7 ± 1.9% in thyrotoxic patients, and 1.94 ± 0.27% in hypothyroid patients. (700) Hays and Berman (1977) investigated the biokinetics of 99mTc pertechnetate during the first 8 h of its continuous intravenous infusion into normal volunteers. A group of nine subjects was studied during hours 0–4, and another group of 10 subjects was studied during hours 4–8. One gram of sodium iodide was administered intravenously to the second group at 6.5 h. Plasma, salivary, and urinary activities were assayed, and external measurements were made over the neck, thigh, and right upper abdomen. The investigators found that pertechnetate was initially distributed much like iodide, and that the administration of iodide markedly reduced transport of pertechnetate into the thyroid, saliva, stomach, and small intestine. In contrast to the systemic behaviour of iodide, the large intestine appeared to play an important role in the retention and excretion of pertechnetate. The investigators developed a biokinetic model for pertechnetate from the results of their study, analogous with iodide biokinetics, and data from previous biokinetic studies of pertechnetate. The model depicts three main subsystems that determine the fate of systemic pertechnetate: the thyroid trap; technetium distributed throughout the body, represented by plasma and two extravascular compartments; and four compartments within the gastrointestinal tract representing the salivary glands, stomach plus upper small intestine, and two lower intestinal pools. One of the latter compartments is identified with the bowel wall on the basis of external measurements.
(c) Data for laboratory animals
(701) Following intravenous administration of 99mTc pertechenate to mice, the organ with the highest accumulation was the stomach, which contained 10% of the administered amount (corrected for radioactive decay) at 1–3 h and 14% at 6 h (McAfee et al., 1964). From 1 to 6 h, the small intestine contents increased from 2% to 6% and the large intestine contents increased from 2% to 9% of administered technetium. (702) Matthews and Mallard (1965) studied the distribution and tumour uptake of 99mTc pertechenate in the first few hours after its intravenous administration to rats, and compared its behaviour with that of other tracers. The distribution was found to be broadly similar to that of 131I administered as iodide. Pertechnetate equilibrated rapidly with the extracellular spaces of several organs. Some observed differences from 131I as iodide were that the liver accumulated three times as much 99mTc as 131I, the kidneys accumulated two to five times as much 99mTc as 131I, and the 99mTc content of the intestines continued to rise for 4.25 h while that of 131I reached a peak relatively quickly and then began to decline. The content of 99mTc in the liver decreased from 8.7% of the administered amount at 0.57 h to 4.2% at 4.25 h. At 3–4 h after injection, the concentration of 99mTc in the liver was approximately 2.5 times that in bone and seven times that in muscle. (703) Yeh and Kriss (1967) compared the biokinetics of 99mTc pertechnetate and a 99mTc citrate complex in mice over the first 24 h after intravenous administration. The pertechnetate showed high concentration in the salivary glands, stomach, thyroid, and colon. The liver content decreased from 8.5% of the administered amount at 0.5 h to 2.8% at 24 h. The kidney content was 1.8% at 0.5 h and below the detection limit at 24 h. Whole-body retention was 70% at 0.5 h and 16.5% at 24 h. The citrate complex showed a much higher urinary excretion rate than pertechenate and, in contrast to pertechnetate, was not localised in the salivary glands, stomach, or thyroid. The liver content was approximately 2.5% of the administered amount from 0.5 to 2 h, and declined to 1.5% at 24 h. The kidney content decreased from 2.3% at 0.5 h to 0.8% at 24 h. Whole-body retention was 20% at 0.5 h and 7% at 24 h. (704) McRae et al. (1974) studied the effects of stannous tin on the distribution of pertechnetate in rats. The following distribution was determined at 1 h after intravenous administration of 99mTcO4 to control animals: liver, 4.3% of administered activity; kidneys, 1.0%; stomach, 17.1%; intestines, 7.2%; skeleton, 7.3%; muscle, 11.3%; and skin, 27.2%. (705) Coffee et al. (1984) studied the biokinetics of intravenously injected 95mTcO4 or 99mTcO4 administered to rats with and without a 99TcO4 carrier. Retention in all organs was reduced substantially by administration of the carrier. The relative concentrations in tissues at 24 h after injection of 99mTcO4 with no carrier were: liver, 0.15; kidneys, 0.82; stomach, 0.40; large intestine, 0.05; and skin, 0.11. (706) Maize containing bound 99Tc was introduced acutely into the rumen of sheep (Kirchmann et al., 1986). The 99Tc concentration in the kidneys over the period 1–28 d after administration was an order of magnitude greater than that in the liver, and three orders of magnitude greater than that in muscle. The biological half-times for 99Tc in kidneys, liver, and muscle based on measurements at 7 and 28 d after administration were approximately 6, 9, and 9 d, respectively. (707) The biokinetics of 99Tc was studied in sheep following its introduction into the rumen as pertechnetate or biologically bound to algae (Bruwaene et al., 1986). Tissue concentrations and urinary and faecal excretion rates were determined up to 3 months after administration. The biokinetics of 99Tc administered in algae appeared to be broadly similar to that for 99Tc administered as pertechnate, except for possible differences in uptake and retention by the thyroid, but variability in the data for 99Tc administered in algae hampered precise characterisation of its biokinetics. Gastrointestinal absorption of 99Tc was low. Urinary excretion amounted to approximately 1% of the dosage. Highest concentrations of 99Tc were found in thyroid tissue, followed by liver and kidney. Relatively high concentrations were also found in the skin and wool. Two components of whole-body retention were observed following administration of 99Tc either as pertechnetate or algae. Two components of retention were also evident for the liver, kidneys, and thyroid following administration of 99Tc as pertechnetate. Following administration as pertechnetate, the size (coefficient) of the first component of retention was approximately 35 times that of the second component for the whole body, six times that of the second component for the kidneys, and two times that of the second component for the thyroid; the size of the second component was not determined for the liver. The estimated biological half-times of the long-term components for the whole body and individual tissues were in the range 20–50 d. (708) Holm and Rioseco (1987) investigated the transfer of 99Tc from lichens to reindeer in a region of central Sweden. Activity was measured in reindeer tissues between 1963 and 1981. Activity concentrations in the liver and kidneys were typically much higher than those in muscle. The mean activity concentration in bone expressed on a wet weight basis was approximately 2.5 times that in liver and 10 times that in muscle. Compact and trabecular bone showed similar concentrations of 99Tc. (709) Gerber et al. (1989) compared the biokinetics of 95mTc in rats (monogastric animals) and sheep (polygastric animals) following its intravenous injection or ingestion as TcO4 or biologically incorporated in maize. The pattern of absorption and excretion and, to some extent, the organ distribution and retention depended on the animal species and the form of administered activity. Pertechnetate given orally was better absorbed by rats than by sheep. Absorption of activity bound to maize was approximately equal to that of TcO4 in sheep, but much less than that of TcO4 in rats. Endogenous excretion of injected activity was primarily in urine for rats and primarily in faeces for sheep. The highest tissue concentration at 3 and 7 d following intravenous administration to sheep, and all modes of administration to rats, was found in the thyroid, followed by the kidneys. Following ingestion of either form of 99mTc by sheep, the kidneys showed the highest tissue concentration. Bone, skin, muscle, and liver contributed significantly to the whole-body burden. Biological half-times for tissues of sheep were estimated from tissue concentrations up to 90 d, and characterised for each tissue as the sum of two exponential terms. The half-time of the first component of retention was approximately 5 d for all tissues. The half-time of the second component was approximately 20 d for kidneys, 40 d for liver, and 50 d or longer for bone, muscle, and skin. (710) Jones (1989) studied the intestinal absorption and systemic biokinetics of 95mTc following its oral or direct gastrointestinal administration to goats and swine. At 200 h after administration, the highest tissue concentration in both species was found in the thyroid, followed by kidney, and liver. In swine, the total content of the liver was approximately three times the content of the kidneys or thyroid. The removal half-time from the thyroid based on single-exponential fits to measurements made over several days was approximately 20 h for swine and 30 h for goats. (711) Ennis et al. (1989) studied the transfer of technetium isotopes to milk and tissues of lactating goats. At 35–40 d after oral administration of 99Tc pertechnetate, the concentration of 99Tc in tissues and fluids decreased in the order thyroid > hair > kidney > mammary gland > liver > lower large intestine > muscle > blood > milk. The concentration of 99Tc in the thyroid was approximately 20 times that in the kidneys, 100 times that in the liver, and 1000 times that in muscle. (712) Zuckier et al. (2004) compared the time-dependent distributions of 125I, 99mTc, and 188Re in mice after their intravenous injection as iodide, pertechnetate (99mTcO4), and perrhenate (188ReO4), respectively. The early distributions of these three radionuclides were remarkably similar. Activity concentrations of all three in salivary glands and stomach were several times higher than the blood concentration, remained elevated over the initial 2 h, and subsequently declined. A broadly similar pattern of accumulation and decline of pertechnetate and perrhenate was observed in the thyroid. In contrast, the concentration of 125I in the thyroid continued to increase through the 19 h time point, presumably due to organification of the iodide. At 20 min, the concentration of 99mTc decreased in the order stomach > salivary glands > thyroid > liver > kidney > spleen > muscle. This order was maintained at 2 h except that the concentration in the thyroid had become slightly greater than that in the salivary glands by this time. (713) Valenca et al. (2005) investigated the effects of cigarette smoke on the initial distribution of intravenously injected 99mTc pertechnetate in mice. The following concentrations (% injected 99mTc g−1) were determined in control animals at 1 h: stomach, 5.7; RBCs, 3.6; lung, 1.7; thyroid, 1.1; kidney, 0.89; spleen, 0.36; bone, 0.26; and testis, 0.25.
15.2.3.2. Biokinetic model for systemic technetium
(714) The structure of the systemic model for technetium used in this publication is shown in Fig. 15.1. Transfer coefficients are listed in Table 15.3. (715) The model structure is a modification of the generic structure for bone-volume-seeking radionuclides. Although technetium is not regarded as a bone seeker, the structure provides a convenient starting place for modelling its systemic kinetics. Compartments representing the thyroid, salivary glands, stomach wall, and right colon wall are added to the model because they have been identified in human or animal studies as important repositories for pertechnetate. The bone, kidneys, liver, thyroid, and other soft tissues are each divided into multiple compartments representing different phases of retention and, in the case of bone, different types of tissue. (716) The model of blood kinetics is based on data for human subjects (McAfee et al., 1964; Andros et al., 1965). Blood is treated as a well-mixed pool. The total outflow rate from blood is 100 d−1, corresponding to a half-time of 10 min. An apparent half-time of a few hours following the initially rapid blood clearance reflects return of technetium to blood from tissues with relatively fast turnover. Fig. 15.2 compares model predictions of blood retention over the first 24 h after intravenous injection of technetium with mean values determined for 10 subjects diagnosed with 99mTc for possible brain tumours (McAfee et al., 1964). (717) The thyroid gland is divided into compartments Thyroid 1 and Thyroid 2, representing fast and slow phases of turnover. Technetium is assumed to transfer from blood to Thyroid 1 at a rate of 7 d−1, and from Thyroid 1 back to blood at 100 d−1, based on flow rates derived by Shimmins et al. (1969) from human studies. Technetium transfers from Thyroid 1 to Thyroid 2 at a rate of 1 d−1, representing 1% of outflow from Thyroid 1, and transfers from Thyroid 2 to blood at a rate of 1 d−1. The model predicts that the thyroid content plateaus at approximately 2.8% of intravenously administered technetium at 15–30 min, and declines to approximately 1.5% at 3 h and approximately 0.5% by 24 h, in reasonable agreement with data for human subjects (McAfee et al., 1964; Sodee, 1966; Meller and Becker, 2002). (718) Parameter values describing transfer of systemic activity to the alimentary tract content via the salivary glands and stomach wall are based on comparison with kinetics of inorganic iodide as represented in the systemic model for iodine applied in the OIR series [see Leggett (2010)]. Data for human subjects indicate that the rate of transfer of pertechnetate from plasma to saliva and gastric fluids is approximately half that of iodide (Harden et al., 1969; Hays and Berman, 1977). On this basis, the transfer coefficients from blood to salivary glands and stomach wall are set at one-half the corresponding values estimated previously for iodine, yielding transfer coefficients for technetium of 2.6 d−1 from blood to salivary glands and 4.3 d−1 from blood to stomach wall. It is assumed that the rate of loss from both salivary glands and stomach wall to the alimentary tract content is 50 d−1, as estimated for iodide. The model predicts that the stomach wall and contents together contain approximately 7.6% of the acute input of technetium to blood after 1 h, compared with a mean of 6.8% based on external measurements from human subjects (Harden et al., 1967). (719) Uptake and retention by the liver are based on collective data for human subjects and laboratory animals which suggest that the liver rapidly accumulates up to 8–9% of intravenously administered technetium, and loses most of this with a half-time of approximately 2 h and the rest at a much slower rate (McAfee et al., 1964; Matthews and Mallard, 1965; Beasley et al., 1966; Yeh and Kriss, 1967). Liver retention is described in terms of compartments Liver 1 and Liver 2, representing fast and slow turnover, respectively. Liver 1 receives 4.5% of outflow from blood. Activity is removed from Liver 1 with a half-time of 2 h, with 99% returning to blood and 1% moving to Liver 2. The removal half-time from Liver 2 to blood is 20 d. This same half-time is applied in this model to all long-term components of soft tissues, and is consistent with the mean long-term retention half-time of 22 d for the whole body determined by Beasley et al. (1966) in a 60 d study on healthy human subjects. The model predicts that the liver content peaks at nearly 9% of intravenously administered technetium in the early hours after injection, and declines to approximately 2% at 1 d after injection. (720) Parameter values for the kidneys are based on findings for laboratory animals. These data indicate that the kidneys rapidly accumulate 1–2% of intravenously administered technetium and lose much of this during the first day, but maintain a higher activity concentration than most other tissues at times remote from administration (Yeh and Kriss, 1967; McRae et al., 1974; Coffee et al., 1984; Kirchmann et al., 1986; Gerber et al., 1989). The kidneys are assumed to consist of a short-term compartment (urinary path in Fig. 15.1) that takes up a portion of technetium filtered at the glomerulus, and loses technetium to the urinary bladder contents, and a long-term compartment (other kidney tissue) that exchanges technetium with blood. It is assumed that 0.7% of outflow from blood deposits in urinary path and 0.04% deposits in other kidney tissue. Activity is removed from urinary path to urinary bladder contents with a half-time of 2 h, and from other kidney tissue to blood with a half-time of 20 d. The model predicts that the kidney content peaks at approximately 1.5% in the early hours after acute input to blood, and that the kidney concentration is initially similar to that of the liver but gradually increases to approximately five times the liver concentration. (721) The remaining outflow from blood is divided among soft tissue compartments called ST0, ST1, and ST2, with fast, intermediate, and slow return to blood, respectively. Parameter values for these compartments are set for consistency with blood disappearance curves and whole-body retention data for human subjects. ST1 and ST2 receive 3% and 0.18%, respectively, of outflow from blood, and return activity to blood with half-times of 1.5 d and 20 d, respectively. ST0 receives 71.88% of outflow from blood, where insignificant digits are retained to achieve mass balance (i.e. so that all outflows from blood add to exactly 100%). The removal half-time from ST0 to blood is 20 min. ST0 is analogous to the compartment called ‘fast exchange’ in the model of Hays and Berman (1977), which receives approximately 72% of outflow from plasma and returns activity to plasma with a half-time of approximately 10 min. (722) The model for bone depicts a low rate of uptake of technetium, but predicts that bone contains a sizable portion of the whole-body content during chronic intake due to long-term retention of a small portion of the deposited technetium. It is assumed that 0.35% of outflow from blood deposits on cortical bone surfaces, and 0.35% deposits on trabecular bone surfaces. Activity is removed from each bone surface compartment with a half-time of 1.5 d, with 99% returning to blood and 1% entering the associated bone volume compartment. Activity is removed from a bone volume compartment at the reference rate of bone turnover for the given bone type (ICRP, 2002). For the case of intravenous administration, the model predicts that the bone content peaks during the first day at approximately 5% of the injected amount. (723) Parameter values describing urinary and faecal excretion are set for agreement with mean cumulative excretion of technetium determined by Beasley et al. (1966) in a relatively long-term study on healthy human subjects. It is assumed that 1.7% of outflow from blood goes directly to the urinary bladder contents, and 0.7% transfers from blood to the urinary bladder contents after a brief retention (2 h removal half-time) in the kidneys (urinary path in Fig. 15.1). Endogenous faecal excretion of technetium is assumed to arise, in part, from the unabsorbed portion of secretions into the alimentary tract in saliva and gastric juice, and, in part, from transfer of outflow from blood to the right colon wall and subsequent transfer to the right colon contents. The rate of transfer from blood to the right colon wall (3.4 d−1, representing 3.4% of the outflow from blood) and the removal half-time from the wall to the contents (0.5 d) are set to produce the portion of the observed cumulative faecal excretion in human subjects (Beasley et al., 1966) not accounted for by activity secreted into higher portions of the alimentary tract. (724) Model predictions of whole-body retention of technetium as a function of time after its acute input to blood are compared in Fig. 15.3 with a curve fit to observed values for human subjects (Beasley et al., 1966). Predictions of cumulative urinary and faecal excretion of technetium after its acute input to blood are compared in Fig. 15.4, with mean values estimated from results from the same study. Structure of the biokinetic model for systemic technetium. ST, soft tissue; St, stomach; SI, small intestine; cont, contents. Parameter values in the systemic model for technetium. ST, soft tissue. Comparison of model predictions of blood clearance of technetium following intravenous injection with observations for 10 human subjects administered 99mTc as pertechnetate (McAfee et al., 1964). The closed circles and error bars represent means and standard deviations, respectively, of observed values. Model predictions of whole-body retention of technetium following its acute input into blood, compared with a fit (circles) to observations for human subjects (Beasley et al., 1966). Model predictions of cumulative urinary and faecal excretion of technetium following its acute input into blood, compared with central estimates based on observations for human subjects (Beasley et al., 1966).

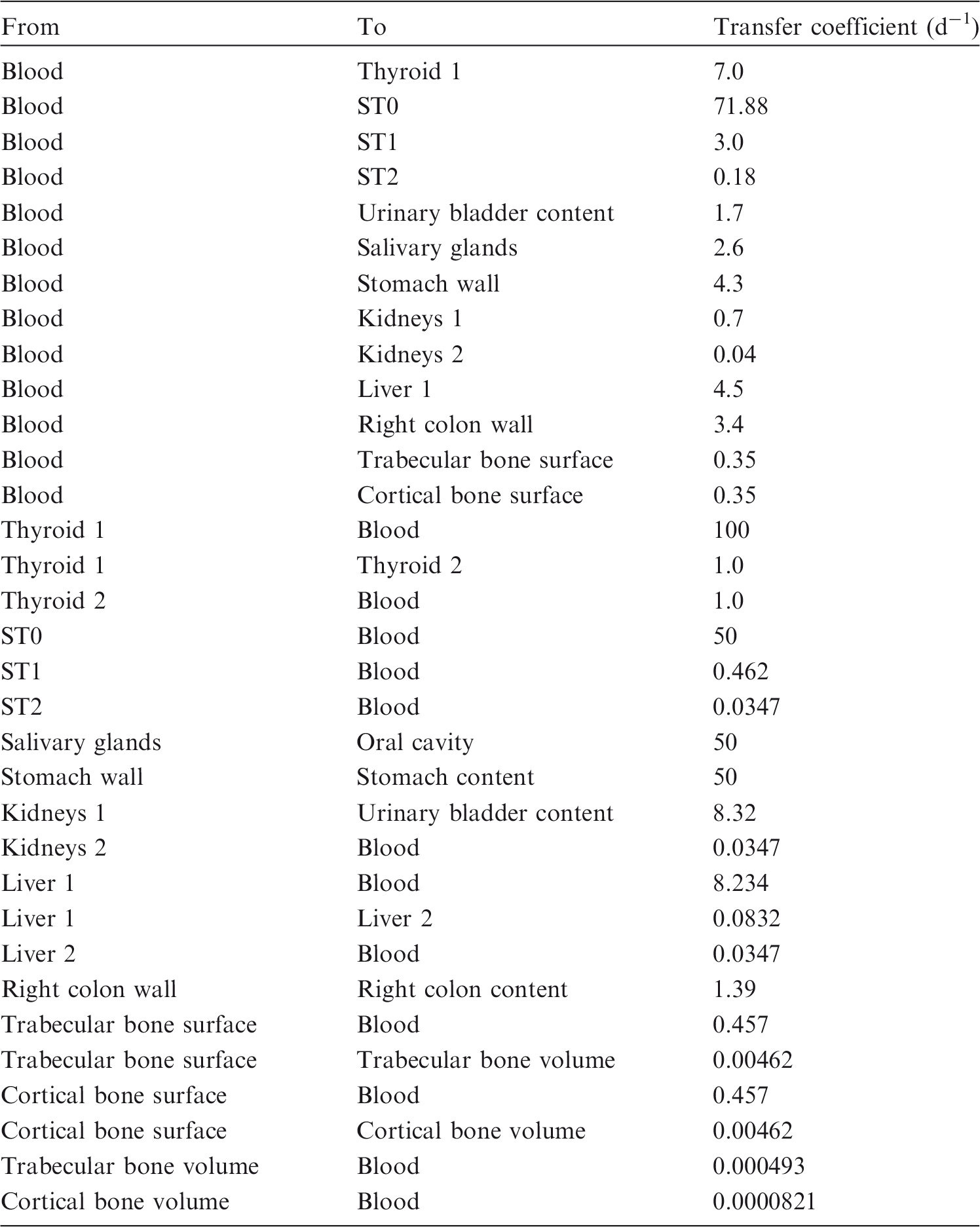

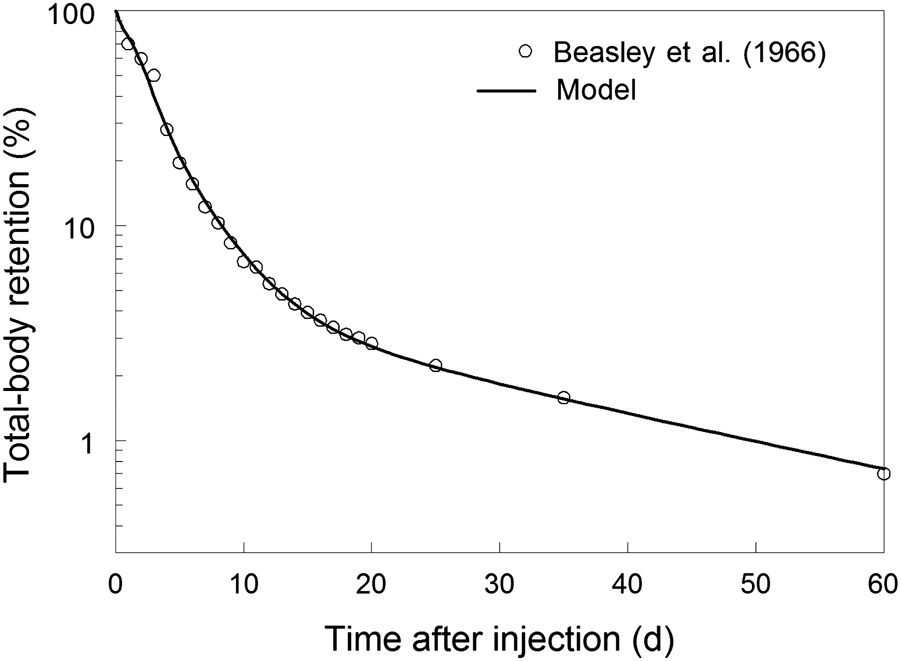
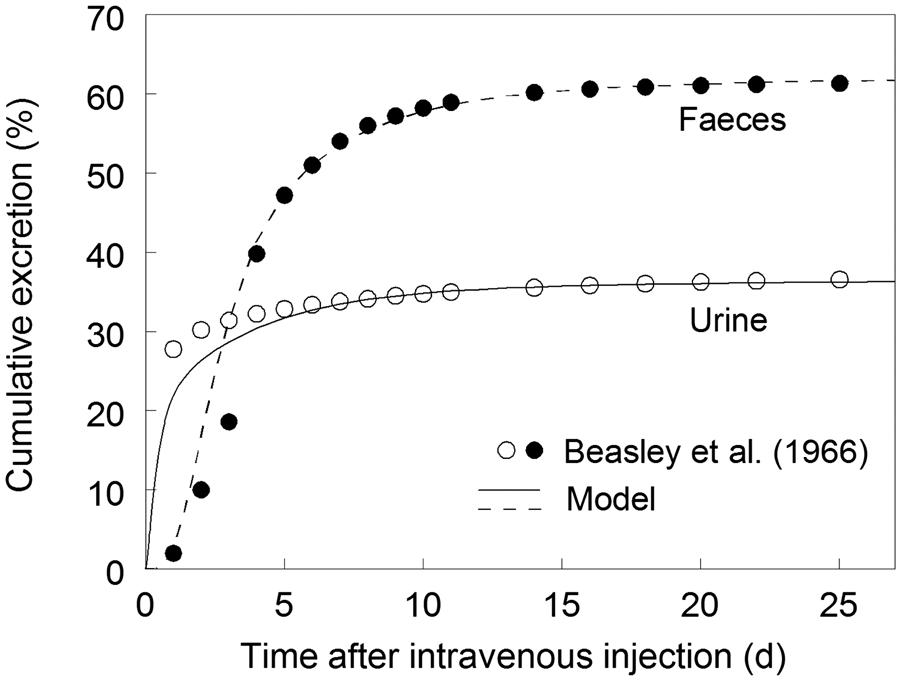
15.2.3.3. Treatment of radioactive progeny
(725) All of the chain members addressed in this publication in the derivation of dose coefficients for internally deposited isotopes of technetium are also isotopes of technetium. These chain members are assigned the biokinetic model for technetium as a parent radionuclide, starting at the time of production of the progeny in the body.
15.3. Individual monitoring
15.3.1. 99Tc
(726) 99Tc is a beta emitter. Monitoring of individuals is done through urine bioassay techniques (Table 15.4). Monitoring techniques for 99Tc.

15.3.2. 99mTc
(727) Monitoring of 99mTc is generally accomplished through whole-body measurement. In addition, 99mTc may be detected through urine bioassay. If needed, lung monitoring may be performed (Table 15.5). Monitoring techniques for 99mTc.

15.4. Dosimetric data for technetium
Committed effective dose coefficients (Sv Bq−1) for the inhalation or ingestion of 99Tc and 99mTc compounds.

AMAD, activity median aerodynamic diameter.
Dose per activity content of 99Tc in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.
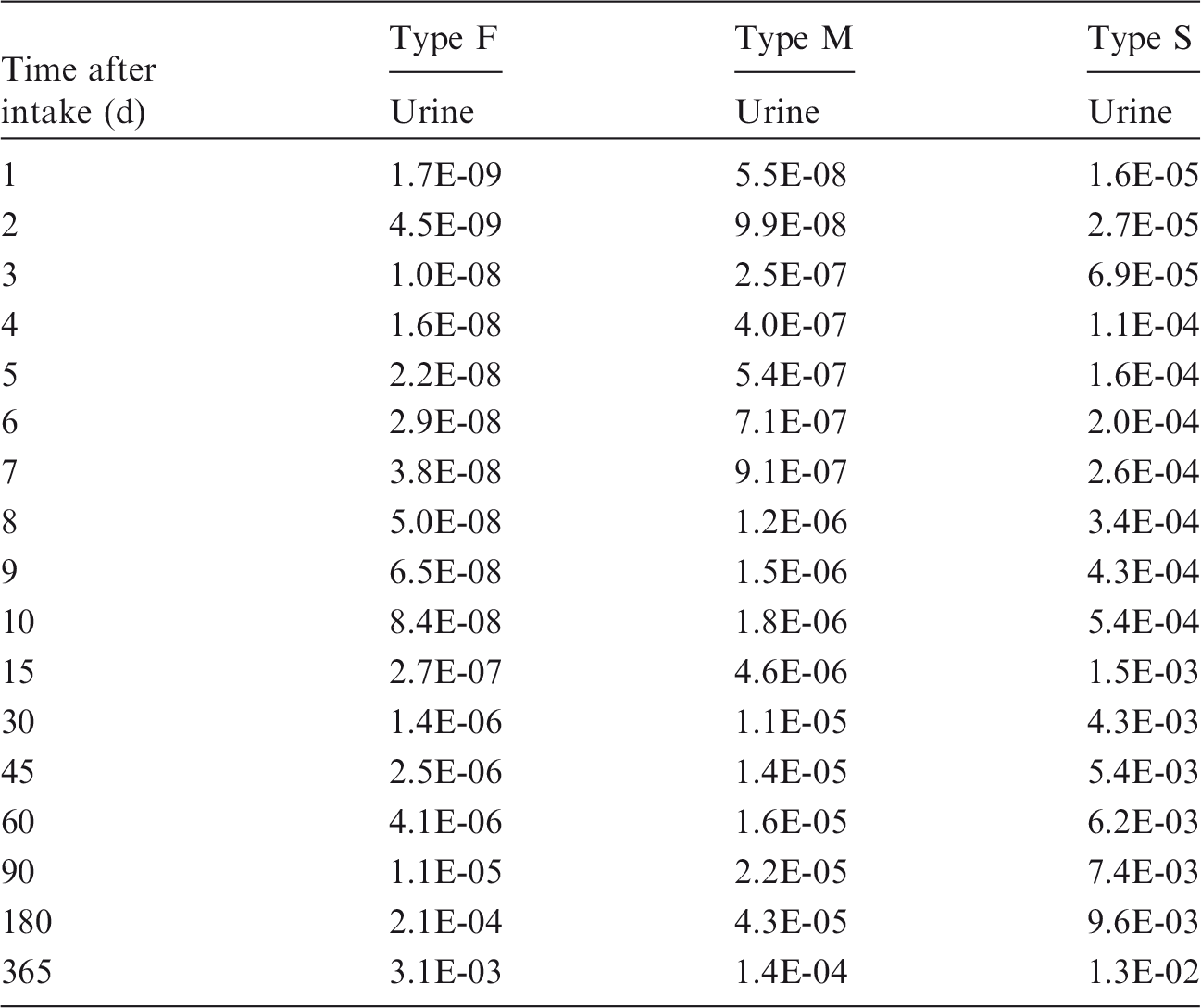
AMAD, activity median aerodynamic diameter.
Dose per activity content of 99mTc in the total body and in daily excretion of urine (Sv Bq−1); 5 µm activity median aerodynamic diameter aerosols inhaled by a reference worker at light work.


Daily urinary excretion of 99Tc following inhalation of 1 Bq Type F.

Daily urinary excretion of 99Tc following inhalation of 1 Bq Type M.
Daily urinary excretion of 99Tc following inhalation of 1 Bq Type S.

Total body content and daily urinary excretion of 99mTc following inhalation of 1 Bq Type F.

Total body content and daily urinary excretion of 99mTc following inhalation of 1 Bq Type M.

Total body content and daily urinary excretion of 99mTc following inhalation of 1 Bq Type S.