
Abstract

ICRP PUBLICATION 128: Approved by the Commission in July 2014
© 2015 ICRP. Published by SAGE.
Keywords: Radiopharmaceuticals; Biokinetics; Dosimetry; Patients
Preface
In 1987, the International Commission on Radiological Protection (ICRP) published a report entitled ‘Radiation dose to patients from radiopharmaceuticals’ (ICRP, 1987). This report contained results from calculations of organ absorbed dose and effective dose equivalent per unit activity administered for some 120 radiopharmaceuticals in regular use at the time. Over the years, ICRP has provided the following radiopharmaceutical reports, amendments, and corrections:
ICRP, 1987. Radiation dose to patients from radiopharmaceuticals. ICRP Publication 53. Ann. ICRP 18(1–4). ICRP, 1991. Radiation dose to patients from radiopharmaceuticals. Addendum 1 to ICRP Publication 53. ICRP Publication 62. Ann. ICRP 22(3). ICRP, 1997. General principles for the radiation protection of workers. Erratum for ICRP Publication 62. ICRP Publication 75. Ann. ICRP 27(1). [Not used in this report] ICRP, 1998. Radiation dose to patients from radiopharmaceuticals. Addendum 2 to ICRP Publication 53. ICRP Publication 80. Ann. ICRP 28(3). ICRP, 1998. Radiation dose to patients from radiopharmaceuticals. Addendum 2 to ICRP Publication 53. Errata: Printing errors in ICRP Publication 53. ICRP Publication 80. Ann. ICRP 28(3). [Not used in this report] ICRP, 2008. Radiation dose to patients from radiopharmaceuticals. Addendum 3 to ICRP Publication 53. ICRP Publication 106. Ann. ICRP 38(1/2). ICRP, 2013. Radiation dose to patients from radiopharmaceuticals. A fourth addendum to ICRP Publication 53. Available at: http://www.icrp.org/docs/Radiation%20Dose%20to%20Patients%20from%20Radiopharmaceuticals%20-%20A%20fourth%20addendum%20to%20ICRP%20Publication%2053.pdf
This report includes a compendium of current information relating to radiation dose to patients for widely used radiopharmaceuticals, and also provides new information for 82Rb-chloride and 123I-, 124I-, 125I-, and 131I-iodide.
The data on effective dose shown in this report are calculated as specified in Publication 60 (ICRP, 1991a). However, work is in progress to develop a new set of dose coefficients calculated in accordance with the Publication 103 methodology (ICRP, 2007).
The data are not intended for therapeutic applications of radionuclides. More detailed and patient-specific dosimetry and dose planning should be applied for therapeutic application of radionuclides.



The membership of Committee 2 during the period of preparation of this report was:


The membership of Committee 3 during the period of preparation of this report was:


The authors wish to thank the former ICRP Assistant Scientific Secretary Michiya Sasaki, former ICRP Interns Ian Steadman, Taylor Whitter, Tudor Dragea, and Robert Martin who contributed to the checking and editing at the early stage of this publication.
1. INTRODUCTION
(1) The administration of radioactive substances to humans for diagnosis, therapy, or research purposes is a well-established and developing branch of medical practice, and is, in most countries, recognised under the name of ‘nuclear medicine and molecular imaging’. New methods and new radiopharmaceuticals are being introduced continually. Reasonably accurate dosimetry for representative groups of patients for each specific investigation is needed to optimise use of the various alternative radiodiagnostic techniques, and to estimate the collective radiation exposure and risk from nuclear medicine investigations. The limited, but increasing, use of radiopharmaceuticals for therapy requires even more detailed and patient-specific dosimetry and dose planning for both tumour and normal tissue. The data presented in this report are intended for diagnostic nuclear medicine and not for therapeutic applications. (2) With regard to dose calculations for diagnostic radiopharmaceuticals, a number of reports have been published by the Commission. In 1987, Publication 53 (ICRP, 1987) was published containing dose coefficients for approximately 120 substances and superseding Publication 17 (ICRP, 1971). In 1991, dose data for six additional substances were published in Publication 62 (ICRP, 1991b), and data for another 10 substances were published in Publication 80 (ICRP, 1998). In 2008, biokinetic information and dose coefficients covering 25 different substances were published in Publication 106 (ICRP, 2008) – a third addendum to Publication 53. This publication also includes recommendations relating to breast feeding for mothers who have undergone nuclear medicine procedures. A fourth addendum including 6 substances has been available on the ICRP's website (www.icrp.org). Further work of the Task Group on Radiation Dose to Patients from Radiopharmceuticals has included 82Rb-chloride, 123I-, 124I-, 125I-, and 131I-iodide as well as 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)-N-(3-fluoropropyl) nortropane (FP-CIT). (3) Information regarding dose calculations from radiopharmaceuticals has also been published in reports from the International Commission on Radiation Units and Measurements (ICRU), notably ICRU Reports 32 and 67 (ICRU, 1979, 2002). At the national level, several absorbed dose catalogues for radiopharmaceuticals and collections of published values have also been issued (Roedler et al., 1978; NCRP, 1982; Johansson et al., 1992; ARSAC, 2014). Of particular importance is the work of the Medical Internal Radiation Dose (MIRD) Committee of the US Society of Nuclear Medicine, and the dosimetry work performed at Oak Ridge National Laboratory (http://crpk.ornl.gov/), at the Radiation Internal Dose Information Center at Oak Ridge Associated Universities in Oak Ridge, TN, USA (now disbanded), and the Radiation Dose Assessment Resource (RADAR) (www.doseinfo-radar.com). (4) A computer software code named ‘MIRDOSE’ was developed (Stabin, 1996) to facilitate automated and standardised internal dose calculations for nuclear medicine applications. This code was completely rewritten and renamed ‘OLINDA’ (Organ Level INternal Dose Assessment) (Stabin et al., 2005). The OLINDA/EXM code (where EXM stands for ‘EXponential Modelling’) allows users to fit data to one, two, or three exponential functions. The OLINDA/EXM code uses the same technical basis (phantoms, organ masses, equations, relationships assumed, and other details) as the MIRDOSE code and the RADAR system. (5) Reference biokinetic and dosimetric models and reference data for workers and members of the public exposed to radionuclides have been published by the Commission, giving dose coefficients for intake of radionuclides by inhalation and ingestion (ICRP, 1973, 1979, 1980, 1981, 1993, 1994, 1996, 2012). (6) The Task Group has made extensive use of the information and material available from these sources.
2. SELECTION OF RADIOPHARMACEUTICALS
(7) Certain general principles were followed in establishing the list of radiopharmaceuticals for inclusion in this report. A radiopharmaceutical that has been described in the literature and proposed for use in humans was included if there is evidence that it has been in, or is coming into, common use, provided that acceptable and sufficient metabolic data for making absorbed dose calculations are available. The list of radiopharmaceuticals covers not only those used in the practice of nuclear medicine, but also some of those used in clinical research. (8) It is important to note that the inclusion of a radiopharmaceutical in this report does not imply any recommendation regarding its use. For this reason, the amounts of administered radiopharmaceutical required for a particular investigation are not given. The list is based on the judgement of the Task Group regarding their past, present, or potential future application in nuclear medicine procedures. Data relating to these substances were obtained from an extensive search of the literature. Some information had been published in scientific journals covering subjects other than nuclear medicine. (9) Complete radionuclide and radiochemical purity is assumed in all absorbed dose calculations.
3. SELECTION OF ORGANS AND TISSUES FOR DOSE CALCULATIONS
(10) Absorbed doses are calculated for most organs and tissues (‘target organs and tissues’). These absorbed doses may arise as a result of radioactive decay occurring in other regions (‘source regions’). Thus, absorbed doses in a particular organ or tissue are typically the sum of contributions from various sources, including the target organ or tissue itself. Two groups of target organs and tissues are included in the calculation of absorbed dose (Table 3.1):
target organs and tissues for which the absorbed dose is always calculated (Group 1); and other organs and tissues that receive significantly higher absorbed doses than the average to the rest of the body, or which are of special interest in the investigation (Group 2). (11) The absorbed dose to organs and tissues not included in Table 3.1 can usually be approximated by using the absorbed dose provided for ‘Other tissues’ (e.g. muscle). The absorbed doses given in the annexes are the mean absorbed doses to an organ or region. In general, these mean absorbed doses are calculated assuming uniform distribution of the radionuclide in the source regions. (12) An exception to the assumption of a uniform dose distribution is made for the kidneys, where a non-uniform distribution of radionuclides may be taken into account. However, even in this case, absorbed doses to other organs and tissues are calculated under the assumption that the radionuclide is distributed uniformly throughout both kidneys; this is justified because, in practice, use of a non-uniform distribution when calculating the absorbed doses to other organs and tissues results in very small changes (<10%) in the results obtained. (13) Discussions were held regarding whether or not to calculate doses to regions of the brain that will receive doses considerably higher than the average dose, such as the putamen and nucleus caudatus from 123I-labelled FP-CIT. As S values for the calculation of regional doses in this case have been published (Bouchet et al., 1999), the decision was made to include the absorbed dose to the region of the brain that receives the highest absorbed dose as a footnote to the dose table. However, this dose is not used in calculation of the effective dose for these radiopharmaceuticals. It is important to stress that the doses are small; even if the central regions of the brain receive doses 10 times higher than average, this is still below levels at which known deterministic effects (‘tissue reactions’) can be observed. (14) The lens of the eye is considered as a tissue at risk in Publication 60 (ICRP, 1991a) because of the possibility of inducing opacities that may interfere with vision. The radionuclides in radiopharmaceuticals currently used in nuclear medicine do not concentrate in the tissues of the healthy human eye, with the possible exception of iodo-amphetamine which is used in the synthesis of melanin (Winchell et al., 1980). For this reason, the lens of the eye is not included in the list of target organs and tissues. Organs and tissues for which absorbed dose is calculated. The absorbed dose to thymus is used as a substitute. Mainly muscle tissues.

4. Biokinetic models and data
(15) The Task Group encountered several problems in finding good biokinetic information from measurements on man. In general, published data are scarce, especially with regard to quantitative measurements. The clinician is often only interested in the initial distribution and metabolism of a test substance, whereas for dosimetry calculations, long-term retention is of prime importance. (16) The Task Group wishes to repeat the requests most recently made in Publication 106 (ICRP, 2008) for securing the maximum information possible from any investigation that involves radiopharmaceuticals. The information needed for dose calculations includes fractional long-term retention of radionuclides and labelled compounds, turnover of the radiopharmaceutical and its metabolites, fractional gastrointestinal absorption values for orally administered compounds, distribution of radionuclides within different organs, and their excretion pathways. Collection of such data should be encouraged by professional and scientific societies and by regulatory authorities, and data should be made available by publication and storage in accessible databases. The editors and referees of scientific journals are encouraged to request such information in papers on new, as well as commonly administered, radiopharmaceuticals. (17) For each radioactive compound, the Task Group has agreed upon a biokinetic model giving quantitative estimates for the distribution and metabolism of the radiopharmaceutical in the body. The literature on which each model is based is referenced. In appropriate cases, the range of pathological variation expected in the metabolic data is also indicated. (18) Some biokinetic models have been developed within a generic framework for application to a class of radiopharmaceutical (e.g. monoclonal antibodies and brain receptor substances). Each of the generic model frameworks is a compromise between biological realism and practical considerations regarding the amount and quality of information that is available to determine parameter values for specific compounds. (19) A realistic maximum model (assuming no biological elimination) have been developed for substances labelled with 11C. (20) For absorbed dose calculations, knowledge of the time–activity curve in different organs and tissues of the body after administration of a radiopharmaceutical is needed. The best way to get this information is by pharmacokinetic analysis, which includes knowledge about mechanisms affecting radionuclide localisation and physiological assumptions regarding its behaviour in body tissues. On the basis of this knowledge, a biokinetic model is defined, delineating the detailed distribution and flow, or transfer, of the radionuclide. (21) This biokinetic model, in turn, allows the derivation of a mathematical model, consisting of differential and/or integral equations for the variation with time of the amounts of radionuclide in different parts of the body. The model may be either compartmental or non-compartmental. Knowledge of the values for compartment sizes, flow rates, and other physiological parameters allows numerical solution of the equations, giving activity–time relationships for all parts of the system which are then integrated to obtain the cumulated activities needed for calculations of absorbed dose. (22) The method outlined above could, in principle, be applied to derive absorbed doses in those disease states leading to quantitative changes in normal physiological processes. However, this is not generally possible because, with some exceptions, there is insufficient information to define a complete model including all pools or compartments, as well as flow rates in or out of the system and between the parts of the system. For absorbed dose calculations, only the time–activity curves are needed; these can be established in alternative ways, as discussed in detail in ICRU Reports 32 and 67 (ICRU, 1979, 2002), the MIRD primer (Loevinger et al., 1991) and the MIRD Pamphlet 21 (Bolch et al., 2009). (23) For example, a simple approach involves modification of the bone dose in younger individuals in whom bone growth is assumed to result in higher uptakes and thus doses. In these tissues, the absorbed dose may be approximately two to five times higher for 99mTc-phosphonates (Gelfand et al., 1983; Kaul et al., 1985) compared with the mean absorbed dose to the bone surfaces, which is the target tissue considered in this report. Similar ratios can be derived for 67Ga-citrate from data reported by Gelfand et al. (1983). Thus, in these cases, the use of the same biokinetic model for both children and adults would underestimate radiation doses to a particular part of the skeleton, although the mean absorbed dose to bone surfaces is not likely to be underestimated substantially. In calculations of absorbed doses to children, age-dependent data are used for organ mass, blood distribution, and S values. (24) The influence of pathological changes on absorbed dose has also been studied. Variations of absorbed dose in disease states can generally be calculated using the same model as for the healthy state, but with appropriate data for organ or tissue mass, uptake, and retention. Separate absorbed dose estimates are presented in cases where such variations lead to significant changes in these absorbed doses. (25) The models and absorbed dose values presented are intended for use in diagnostic nuclear medicine and clinical research with radionuclides, and should not be used in radionuclide therapy. (26) Some radiopharmaceuticals administered to breast-feeding women may be excreted in the breast milk and thus transferred to the breast-fed child. This problem is covered in Annex D of this report. Excretion in breast milk in connection with occupational exposure is covered in Publication 95 (ICRP, 2004). (27) In the case of radionuclides such as 67Ga, 111In, 125I, and 201Tl, administered in forms that result in their uptake in cell nuclei, the minor fraction of the energy carried by Auger electrons may have a disproportionately large effect due to their very short range in tissue (Stepanek et al., 1996; Bingham et al., 2000; Taylor, 2000; Kassis, 2004). The assumption made here, that the absorbed dose is distributed uniformly within the cell, may result in underestimation of the risk. (28) This problem has been discussed in earlier publications (ICRP, 1979, 1991a, 2003), and by many other authors (e.g. Hofer, 1996; Gardin et al., 1999; Bingham et al., 2000; Feinendegen and Neumann, 2004). MIRD has given detailed advice and presented S values for the cellular level (Goddu et al., 1994, 1997). Nonetheless, it is still difficult to establish the intracellular distribution of the radionuclides of interest so that such detailed S values can be used effectively. (29) It is usually assumed that daughter radionuclides produced within the body stay with, and behave metabolically like, their parent nuclide. This may be an oversimplification in some cases, and if specific information to the contrary is available, the dose estimates presented here should be modified appropriately. (30) For some substances, such as iodine-labelled compounds, pertechnetate, and some radiopharmaceuticals used for renal studies, blocking agents may be administered before or simultaneously with the radiopharmaceutical (e.g. to induce competitive inhibition of uptake in specific organs). In such circumstances, including blocking of the thyroid, total inhibition of radionuclide uptake has been assumed, although this may be difficult to achieve in practice. (31) It is often possible to reduce the absorbed dose to a patient by increasing the rate of elimination of the radionuclide from the body, for example by more frequent emptying of the urinary bladder (with hydration, diuretics, and catheterisation), the bowel (with laxatives and enemas), and the gallbladder (with a meal of high fat content and cholecystokinin).
5. METHODS FOR CALCULATING ABSORBED DOSE
5.1. Calculation of absorbed dose
(32) The mean absorbed dose DT to a target organ or tissue T is the sum of the contributions, D(T←S), arising from nuclear transformations of the radionuclide in various source organs S:
(33) Several methods of calculating the absorbed dose to an organ from radioactive sources in the same organ and in other organs have been proposed and used. For a review of these methods, the reader is referred to ICRU Reports 32 and 67 (ICRU, 1979, 2002), Publication 30 (ICRP, 1979), and NCRP Report 84 (NCRP, 1985). The most common method currently in use in nuclear medicine was originally developed from an approach by Loevinger and Berman (1968), using tabulated data on absorbed fractions of energy in a target tissue from a specific source region (Snyder et al., 1969; Loevinger et al., 1991). This method was later improved by Snyder et al. (1975) who introduced the ‘S value’, which also contains all necessary physical information for a specific radionuclide. (34) With this more straightforward method, the absorbed dose in T from a radionuclide in a single source organ S is given by:
(35) The value of S(T←S) depends on the radiation type, the energy emitted per transformation, the mass of the target organ, and the geometry of the mathematical phantoms representing the adult and children of various ages. When the source organ is the total body excluding the organs already listed in the biokinetic data table, a common approximation is to use the S value calculated on the basis of ‘total body’ as a source. However, a formally correct S value for this case can be derived (Cloutier et al., 1973; Roedler and Kaul, 1976; Coffey and Watson, 1979); this latter method is used in this report. (36) If S values are not available, the absorbed dose per nuclear transformation is calculated using the absorbed fraction ϕ, derived from Snyder et al. (1978):


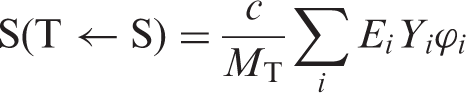
5.2. Calculation of cumulated activity
(37) For a more detailed description of the mathematical analysis of biokinetic models, reference should be made to MIRD Pamphlet No. 12 (Berman, 1977) and ICRU Report 32 (ICRU, 1979). The following text serves as a short account of the calculation of cumulated activity in selected cases. (38) The cumulated activity ÃS in a source organ or tissue S depends on the administered activity, A0, the physical half-life, T, and the biokinetics of the radiopharmaceutical. ÃS, which represents the number of disintegrations occurring in source region S, is obtained by integrating the time-dependent activity:
(39) Although the mechanisms by which radionuclides are distributed within, or excreted from, the body are not necessarily well represented by first-order kinetic models, such models are generally adequate for representing overall uptake and retention of radionuclides in individual organs and tissues. As this is all that is required for dosimetric calculations, these models are used extensively in this report. (40) A general first-order kinetic model can be represented as a system of n compartments, interlinked with constant rate coefficients. In such a system, the rate of change of the amount of material (qi) in compartment i is given by:
(41) A direct correspondence between compartments and anatomical regions of the body does not usually exist. However, for absorbed dose calculations, it is necessary to know the amount of substance in different regions of the body. Therefore, for practical reasons, specific organs and tissues are considered instead of compartments. The activity in an organ or tissue can usually be described sufficiently accurately by a sum of exponentials:
(42) The constants in this equation are often derived directly from measurements. Expressed in terms of fractional distributions to the organ or tissue, and fractions of organ or tissue contents where half-times are given in the biokinetic data tables of this report, AS is given by:
(43) The effective half-time can be calculated from the corresponding biological half-time Ti and the functional physical half-life Tp:
(44) Eq. (5.7) describes the build-up and subsequent decline of activity. If Ti = Tj for some combination of i and j, the corresponding term in the sum in Eq. (5.7) becomes:
(45) A special case, which often occurs, is that immediate uptake in the organ is assumed. Eq. (5.7) then reduces to:
Integrating Eq. (5.7) over time up to infinity gives the normalised cumulated activity:
In cases when the retention function cannot be described by a sum of exponential functions, the cumulated activities are derived directly from the metabolic model. (46) For absorbed dose calculations in nuclear medicine, it has often been assumed that the effective half-time in an organ equals the physical half-life. The reason for this approximation is that the substance, in these cases, is labelled with a radionuclide with a physical half-life that is short in comparison with the biological half-time. For short-lived radionuclides, a slow biological excretion may not be apparent and, for absorbed dose calculations, the approximation is sufficiently accurate. However, this assumption has the consequence that infinite biological half-times are given in the tables and this is not strictly correct. This should be kept in mind when biokinetic data are used.
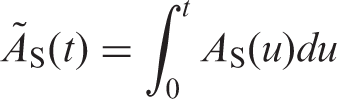


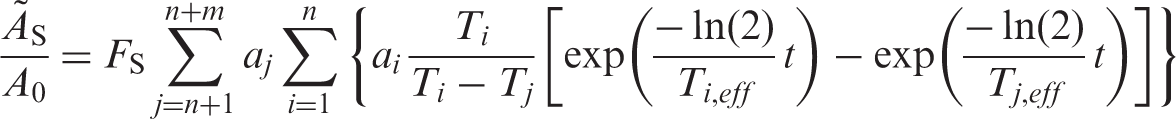





5.3. Uncertainties in absorbed dose estimates
(47) The uncertainty in the estimate of the mean absorbed dose for an organ or tissue in a reference person reflects uncertainties in the cumulated activity and the S value. Differences between planned and actual administered activity are considered to be minor contributors to the total uncertainty if regular quality control is performed (IAEA, 2006). Variation in mass of the target organ and, for photon radiation, variations in the distance between the source and target organs are the major contributors to the uncertainty in S values, whereas physical data (e.g. yield and energy deposition in the target organs) are not considered to be major contributors to the uncertainty. Experimental validation of calculated absorbed doses have indicated agreement within 20–60%; the latter for patients who differed considerably from the body size and shape assumed in the calculations (i.e. the uncertainty for the dose to the reference person would be considerably lower). The reader is referred to Roedler (1980) for a review. (48) Variations in the estimated cumulated activity largely arise from uncertainties in the quantitative description of uptake, distribution, and retention of the radiopharmaceutical in tissues (Norrgren et al., 2003; Jönsson et al., 2005). Functional impairment of an organ can introduce considerable variation in these factors. Variation in the body’s retention of radionuclides administered as radiopharmaceuticals is limited by the short radioactive half-life of these radionuclides: thus, variation in the uptake and distribution of the radiopharmaceutical among the organs and tissues is often the major contributor to uncertainties in cumulated activity. (49) Calculations have shown (Roedler, 1980; Zanzonico, 2000) that estimates of absorbed dose to different organs will not generally deviate from actual absorbed doses in patients by more than a factor of three. The deviation is even less for substances labelled with short-lived radionuclides such as 99mTc. The effective dose is less sensitive to variations in the distribution pattern than organ doses, and may vary within a factor of two.
6. EFFECTIVE DOSE
6.1. Use of effective dose in nuclear medicine
(50) Radiation exposure of the different organs and tissues in the body results in different probabilities of harm and different severities. The Commission uses the term ‘detriment’, meaning health detriment, for the combination of probability and severity of harm. (51) The detriment depends on the type of radiation or, more specifically, the ionisation density. This is accounted for by introducing the concept of equivalent dose. The mean equivalent dose HT in a target organ or tissue T is given by ICRP (1991a):
(52) To reflect the combined detriment from stochastic effects due to the equivalent doses in all the organs and tissues of the body, the equivalent dose in each organ and tissue is multiplied by a tissue weighting factor, and the results are summed over the whole body to give the effective dose. The special name for the SI unit for effective dose is the sievert (Sv). (53) The effective dose was developed primarily for radiation protection of occupationally exposed persons (ICRP, 1977, 1991a). It attributes weighting factors wT to organs or tissues, representing the fraction of the total stochastic risk (i.e. fatal cancer and serious inherited disorders) resulting from the irradiation of that organ or tissue T when the whole body is irradiated uniformly. The effective dose is calculated by adding the weighted organ or tissue mean dose equivalents, HT, i.e.:
(54) If the body is irradiated uniformly, all the HT values are the same and the equivalent dose at any point in the body is numerically equal to the effective dose. (55) The weighting factors used in computing this quantity are applied both to workers and the general population. (56) The Commission has issued its 2007 Recommendations (ICRP, 2007), superseding the 1990 Recommendations (ICRP, 1991a) with updated and amended tissue weighting factors (and radiation weighting factors). Currently, work is in progress within ICRP to generate correspondingly updated dose coefficients for the calculation of doses to workers and members of the public due to intake of radioactive substances. In due course, doses to patients from intake of radiopharmaceuticals will also be calculated. However, pending the availability of such updated information, the present data should be used. (57) Effective dose can be of practical value for comparing doses related to stochastic effects from: different diagnostic examinations and interventional procedures; the use of similar technologies and procedures in different hospitals and countries; and the use of different technologies for the same medical examination, provided that the representative patients or patient populations for which the effective doses are derived are similar with regard to age and gender. However, comparisons of effective doses may be inappropriate when there are significant dissimilarities between the age and gender distributions of the representative patients or patient populations being compared (e.g. children, all females, elderly populations), and the Commission’s reference distribution of both genders and all ages. This is a consequence of the fact that the magnitudes of risk for stochastic effects are dependent on age and gender. (58) Effective dose should not be used to assess risks of stochastic effects in retrospective situations for exposures in identified individuals, nor should it be used in epidemiological evaluations of human exposure. (59) Risk assessment for medical uses of ionising radiation is best evaluated using appropriate risk values for the individual tissues at risk, and for the age and gender distribution of the population groups undergoing the medical procedures. (60) For the exposure of young children, the risk would be higher, perhaps by a factor of two or three (ICRP, 1991a, Annex C). For many common types of diagnostic examination, the higher risk will be offset by the reduction in administered activity relative to that to an adult. For an age at exposure of approximately 60 years, the risk would be lower, perhaps by a factor of three. At higher ages at exposure, the risks are even less (ICRP, 1991a, Annex C). The specific demographics of the medically exposed population present obstacles to applying the concept of effective dose as a tool for comparing doses from medical irradiation with other sources of exposure to humans. Weighting factors for calculation of effective dose E according to the Commission’s 1990 Recommendations (ICRP, 1991a). Adrenals, brain, upper large intestine, small intestine, kidney, muscle, pancreas, spleen, thymus, and uterus.



6.2. Calculation of effective dose
(61) The organs and tissues considered for calculation of effective dose are listed in Table 6.1. Those with specific weighting factors are always included in the calculation. For the gonads, the arithmetic mean of the absorbed doses to ovaries and testes is used in conjunction with the weighting factor of 0.20. Absorbed doses to blood and blood vessels are not included in the calculation. (62) The definition of ‘colon’ or ‘large intestine’ follows that given in Publication 67 (ICRP, 1993, Para. 14). The weighting factor is to be applied to the mass average of the equivalent dose in the walls of the upper and lower large intestine (ULI and LLI) of the gastrointestinal tract. As the ratio between the masses of the walls of the ULI and LLI is independent of age, the equivalent dose to the colon Hcolon is given as:
(63) The biokinetic model presented here contains no information on uptake and retention of radionuclides in the oesophagus. As the transit time of materials through the oesophagus is normally quite rapid in comparison with the physical half-life, only the absorbed dose from penetrating radiation emitted from other source regions is considered. In the absence of absorbed fraction values for the oesophagus, the dose to the thymus has been used previously as a surrogate (ICRP, 1991b), and this method is used in the present report. (64) The weighting factor for the remainder tissues, 0.05, is applied on the mass-weighted average dose of those organs listed in the footnote of Table 6.1. In those cases in which a single remainder tissue or organ receives an equivalent dose that exceeds the dose to any other organ, a weighting factor of 0.025 should be applied to that organ, and 0.025 to the average dose in the rest of the remainder tissues or organs as defined above. This ‘rule’ may also apply for any other organ that is recognised as radiation sensitive. (65) As many radiopharmaceuticals are excreted rapidly in the urine, the absorbed dose to the wall of the urinary bladder is often large compared with the absorbed dose to other organs and tissues in the same study, and may contribute considerably to the effective dose. In cases where the contribution is more than 50%, a note at the foot of the dosimetry table states the actual contribution. (66) The presence of chemical forms of the radionuclide other than that intended may change the distribution and kinetics of the radionuclide. This may lead to a different distribution of the absorbed dose. (67) In this report, complete radiochemical purity has been assumed, unless otherwise stated.

7. DOSE TO EMBRYO AND FETUS
(68) The absorbed dose to the uterus, which is included in the dose tabulations, may be used as a substitute for the absorbed dose to the embryo if the subject is in the first 2–3 months of pregnancy. Similarly, the absorbed dose to the fetus from radioactive substances without placental transfer is expected to be in the same range as the dose to the uterus. For radioactive substances with placental transfer, the absorbed dose to organs and tissues of the mother may, as a first approximation, be taken as representative of the absorbed dose to the corresponding organs and tissues of the fetus. (69) More detailed radiation dose estimates for the fetus from administration of a number of radiopharmaceuticals to women at various stages of pregnancy are given by Russell et al. (1997). Their data illustrate that the majority of studies will probably involve fetal doses <10 mGy. Only studies using 131I-iodide, 201Tl-chloride, and 67Ga-citrate appear to result in fetal doses >10 mGy, according to present knowledge. Therapeutic administrations are routinely contra-indicated in the case of pregnancy or breast feeding as this may result in very high fetal doses. In addition, beyond 10–13 weeks of gestation, the fetal thyroid may receive extremely high doses in cases of therapy using 131I-iodide (Watson et al., 1989; Berg et al., 1998). For substances in their ionic form, a comprehensive compilation of doses to the embryo and fetus is found in Publication 88 (ICRP, 2001).
8. REFERENCES FOR THE MAIN TEXT
Annex A. Special Biokinetic and Dosimetric Models
A.1. Organ and tissue masses for different ages
(A1) The masses of the organs and tissues are inherent in the S values used (Stabin and Siegel, 2003; Stabin et al., 2005). The masses of the phantoms used for calculation of the S values are those presented by Stabin and Siegel (2003) (Table A.1). The phantoms were produced by Cristy and Eckerman (1987), based predominantly on data in Publication 23 (ICRP, 1975). As the masses refer to the phantoms used, they may deviate somewhat from those in Publications 23 (ICRP, 1975) and 89 (ICRP, 2002). Masses (g) of models of selected organs and tissues at different ages.* LLI, lower large intestine; SI, small intestine; ULI, upper large intestine. †‘Remaining tissue’ is defined as the part of the phantom remaining when all defined organs except muscles have been removed. The muscle mass is taken from Publication 23 (ICRP, 1975). ‡Data from Publication 89 (ICRP, 2002).

A.2. Blood volume and blood flow models
(A2) Substances that remain largely in the blood are assumed to be distributed according to the relative blood volume of the different organs. Examples of such substances are labelled blood cells and radionuclides attached to macro-molecules, but this blood distribution model has also been used, where appropriate, for other substances. This model requires information on blood volumes in different organs and tissues. These data were taken from Leggett and Williams (1991) and Williams and Leggett (1989), and were also proposed by the Commission in Publication 89 (ICRP, 2002). The haematocrit, or fractional red cell content of the blood, has been considered constant for blood circulating through all tissues. The data are presented in Table A.2 and refer to adults. The fractional blood volumes used for children have been calculated assuming that the blood content in an organ or tissue per unit mass of tissue relative to that of the total body is independent of age. The total blood volume in children is taken from Publication 89 (ICRP, 2002) and is presented in Table A.1. (A3) In the biokinetic models used in this report, the term ‘uptake’ or ‘content’ of a radionuclide in an organ or tissue usually includes the radioactivity in blood in that organ or tissue. However, when the blood distribution model is used, a specified fraction of the activity is associated with the blood. In this case, the activity in blood in an organ or tissue has been added to the activity in that organ or tissue for purposes of dose calculations. Table A.2 presents the fractional cardiac output to different organs and tissues. These fractions, which are also proposed in Publication 89 (ICRP, 2002), were taken from Leggett and Willams (1995). These data have been applied as a model for the activity distribution of radionuclides with very short physical half-lives (i.e. seconds up to a few minutes). Adult values for blood content and blood flow in different organs.

A.3. Gastrointestinal tract model
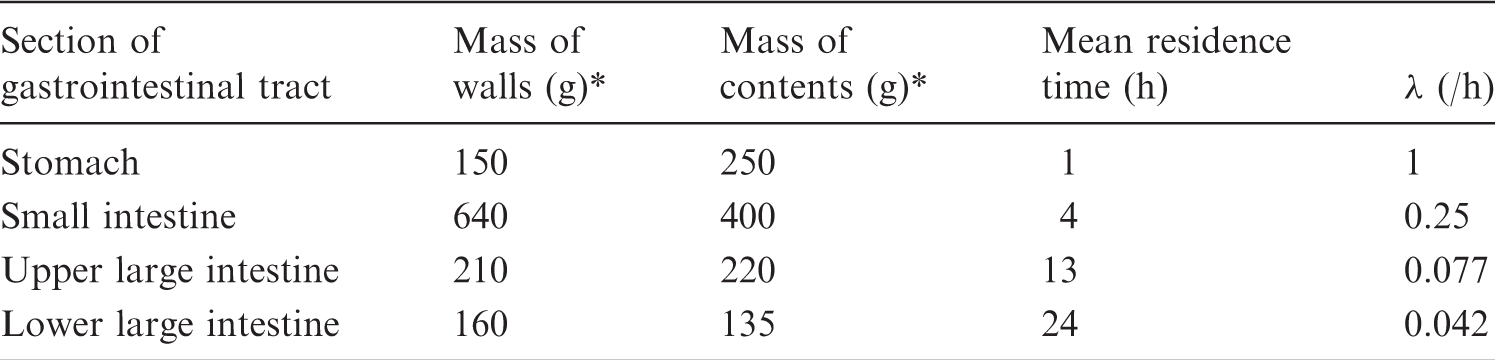
(A4) The model presented in Publication 30 (ICRP, 1979) for the gastrointestinal tract has been used for adults and children aged 1–15 years. The model, shown in Fig. A.1, consists of four compartments: stomach, small intestine, ULI, and LLI. Immediate mixing within each compartment is assumed. The recently introduced model for the human alimentary tract (HAT model; ICRP, 2006) and Publication 89 (ICRP, 2002) present a more detailed model for the gastrointestinal tract, but this has not been implemented for calculations in the present report. (A5) For substances included in this report, the Task Group does not generally consider the deviations in effective dose caused by using the Publication 30 model instead of the HAT model to be significant. An example of a large deviation case arises when the activity is distributed in the stomach. Using the HAT model, the mean residence time in the stomach contents is considerably longer than that using the Publication 30 model. This results in an absorbed dose to the stomach wall that is 30–40% larger with the HAT model compared with the Publication 30 model. (A6) The same mean transit time in small and large intestine (41 h) is used for both children and adults. In fact, the mean transit time is somewhat shorter in children than that in adults (ICRP, 2002, 2006): 36 h compared with 41 h for adults. The assumption of a transit time of 41 h in children will affect estimates of the absorbed dose to different parts of the gastrointestinal tract, depending upon the physical half-life of the radionuclide. Absorbed doses for radionuclides with long half-lives will be overestimated, and those for radionuclides with short half-lives will be underestimated. For newborn babies, however, use of the same gastrointestinal transit time as for adults is not recommended. For substances for which the dose is calculated for newborn babies, further details about data for the transit time through the intestine are given in the biokinetic model, where applicable. (A7) A modified model is used for non-absorbable inert markers intended for studying different aspects of the physiology of the gastrointestinal tract (e.g. gastric emptying, intestinal transport and transit time, abnormal intestinal permeability, etc.). These substances are usually labelled with 99mTc or 111In. Small quantities of non-absorbable markers (i.e. up to a few percent) may be absorbed into the blood. For the purposes of this report, the amounts absorbed are considered to have a negligible effect on the dose calculations. The modification to the standard ICRP model is that the gastric residence time is changed to 0.5 h for fluids and 1.5 h for solids (ICRP, 2002, 2006). Compartment model used to describe the kinetics of radionuclides in the gastrointestinal tract. Parameters used for calculating absorbed dose to the gastrointestinal tract.

A.4. Kidney–bladder model
(A8) This model is applied to all substances used for kidney function tests, and to other substances if urinary excretion results in a significant absorbed dose to the bladder wall. In all of these cases, the bladder is a separate entry in the biokinetic data tables. (A9) It is assumed that the fraction of the total excretion which passes through the kidneys and bladder is known. Activity excreted via this route passes through the kidneys with a transit time established from other clinical studies, and subsequently enters the bladder in urine where it remains until the bladder is emptied and the radioactive contents leave the body. (A10) The rate at which a radionuclide is excreted is determined from knowledge of the amount of activity in the total body, ATB, which is assumed to be described by the sum of a series of exponential functions:
The cumulated activity in the kidneys from the excretion process, (A11) The expression is approximate as fr may differ for the individual components of whole-body clearance. However, for practical application, this approximation is judged to be adequate. The cumulated activity in the kidneys given in the biokinetic data tables for the individual substances is the sum of the cumulated activity from the excretion process and a contribution from activity distributed uniformly in the remaining organs and tissues, which can include the kidneys. (A12) The cumulated activity in bladder contents, (A13) Calculating the radiation absorbed dose to the bladder wall involves consideration of a complex relationship between urine flow rate, voiding period, and urine volume initially present in the bladder when the radiopharmaceutical is administered, and is critically dependent on the model used to describe the geometrical relationships between the wall of the bladder and its contents. Such a model was developed by Snyder and Ford (1976) to investigate the effects of the above physiological variables on absorbed dose to the bladder wall, and was extended by Smith et al. (1982) to examine these effects for any radiopharmaceutical. The MIRD Committee has published a dynamic bladder model (Thomas et al., 1999) incorporating more physiologically realistic features providing for a varying bladder volume, varying initial content and voiding interval, and a night gap in the voiding pattern. (A14) Within the ranges of urine flow rate of 0.5–2 l/day, voiding period of 0.5–8 h, and initial bladder contents of 0–300 ml, the predicted bladder wall dose varies over a range of approximately 25 fold for radiopharmaceuticals that are cleared rapidly by the renal system [e.g. 99mTc-labelled mercaptoacetyl triglycine (MAG3)], reducing to a range of approximately five fold for substances that are cleared more slowly (e.g. 131I-iodide). For voiding periods of ≥3.5 h, the bladder dose predicted by the simplified method used in this report lies within the spread of doses obtained using the above ranges of parameter values, but may be as much as five times lower than the highest values. As the voiding period decreases, the simple method leads to a further underestimate of the dose, which, for a period of 0.5 h, may be of the order of 25 fold. (A15) An age-related bladder voiding model is used. The voiding periods are based on urinary production rates as described in Publication 89 (ICRP, 2002), and volume of the content as described by Stabin and Siegel (2003). The voiding periods are presented in Table A.4. (A16) The S values used for calculation of the absorbed dose to the bladder wall relate to the contents and the wall of the bladder as the source and target tissue, respectively. It should be noted that the S values, which for electrons and beta particles represent a surface dose to the bladder wall, are based on fixed average bladder contents (Table A.4). These S values have been used in the present report in conjunction with cumulated activities in the bladder contents estimated for an age-dependent bladder voiding interval presented in Table A.4. This method does not allow for the variation in dose rate to the wall as the bladder fills with urine containing radionuclides. Parameters used for calculating absorbed dose to the urinary bladder wall. The lower limit of the interval applies for females and the upper limit applies for males. Data from Publication 89 (ICRP, 2002).




A.5. Model for radiopharmaceuticals used to measure glomerular filtration rate
(A17) The following biokinetic model has been used for a variety of labelled inulin and inulin-like radiopharmaceuticals used for the measurement of glomerular filtration rate (GFR). After intravenous administration and initial rapid distribution in extracellular fluid, it is assumed that the radionuclide is excreted exclusively by the kidneys according to the kidney–bladder model. In the normal case, total body retention is described by a mono-exponential function with a half-time of 100 min, fraction excreted by the kidneys of 1.0, and renal transit time of 5 min. (A18) For chelated compounds (DTPA, EDTA), there is evidence of a small degree of in-vivo dissociation of the radioactive label, leading to longer retention of approximately 1% of the administered radionuclide. This fraction is assumed to be distributed uniformly and to be eliminated with a half-time of 7 days. This is a simplifying approximation as the dissociated label will exhibit specific biokinetics depending upon its chemical form. Nevertheless, it is considered adequate for estimating the contributions to absorbed dose from this dissociated label, provided that the examinations are conducted with a blocked thyroid for those radiopharmaceuticals for which the dissociated label would concentrate preferentially in the thyroid. (A19) In the abnormal case, it is assumed that the retention half-time of the major component is increased to 1000 min and that the renal transit time is increased to 20 min. Organ mass (kg): based on Atkins et al. (1975). Normal liver. †Early to intermediate diffuse parenchymal liver disease. ‡Intermediate to advanced diffuse parenchymal liver disease.

A.6. Models for bone-seeking radionuclides administered as radiopharmaceuticals
(A20) For calculation of effective dose, the radiation-sensitive part of bone tissues has been identified as a 10-µm-thick layer on bone surfaces, representing endosteal and periosteal cells (ICRP, 1991a). The Task Group is aware that this figure is subject to revision, and that future dose estimates should be based on an extended bone surface volume (Gössner et al., 2000). However, for the present report, the dose estimates were produced using the earlier established method. (A21) The mean absorbed dose to bone surfaces and red marrow is presented in this report. Calculation of the absorbed dose for these tissues is a complex task as they comprise an intricate mixture of soft tissues and bone. For the present report, the calculations are based on S values derived by Stabin and Siegel (2003), based on methods for calculating the absorbed fraction for the non-penetrating radiation developed by Eckerman and Stabin (2000) and Bouchet et al. (2000). The S values from bone tissues to bone surfaces and red marrow are dependent on the distribution of activity within the bone. Two different cases can be distinguished:
surface-deposited activity in trabecular bone and cortical bone (‘bone surface seekers’); and activity deposited uniformly throughout the entire volume of the mineral bone in trabecular and cortical bone (‘bone volume seekers’). (A22) In Publication 30 (ICRP, 1979), a general rule concerning short-lived radionuclides was introduced and used for various elements: ‘radionuclides with a physical half-life less than 15 days are assumed to be surface deposited’. The same general rule, extended to apply to the effective half-life, is adopted in the present report. Thus, for the absorbed dose calculations, substances with an effective half-time of <15 days have been assumed to be surface deposited, and those with an effective half-time of >15 days have been assumed to be volume distributed, unless otherwise stated. In cases with two or more biological excretion half-times, the different components are considered separately, thus a fraction excreted slowly from the skeleton may be considered to be volume deposited, while the remaining part is surface deposited. (A23) If nothing is known about the distribution of cumulated activity between cortical and trabecular bone, it is assumed to be distributed uniformly on surfaces or throughout the volume, as appropriate. (A24) The distribution of activity thus follows the surface area or mass distribution of mineral bone. For adults, the mass ratio cortical:trabecular bone, according to Publication 89, is 80:20 and the surface area ratio is 40:60 (ICRP, 2002). As no reference values for the distribution between cortical and trabecular bones for children are available, and as the information on this matter in the open literature is very scarce, the Task Group has also adopted these values for 15- and 10-year-old children. For 5- and 1-year-old children, the mass ratio cortical:trabecular bone used for the calculations is assumed to be 60:40 and the surface area ratio is assumed to be 30:70. (A25) A few radiopharmaceuticals are concentrated to a significant extent in the metaphyseal growth plates of children’s bones. This factor is not taken into account in the dose calculations given herein. Thus, radiation doses to this part of the skeleton may be underestimated for children. However, the mean absorbed dose to bone surfaces is not likely to be underestimated substantially. Uptake values (fractions) for large colloids (100–1000 nm diameter).* Examples: 99mTc micro-aggregated albumin, 99mTc-phytate. †Normal liver. ‡Early to intermediate diffuse parenchymal liver disease. §Intermediate to advanced diffuse parenchymal liver disease.

A.7. Model for colloids taken up preferentially in the liver, spleen, and red marrow
(A26) Colloids of 99mTc-sulphur and 198Au were discussed in MIRD Reports No. 3 and No. 4, respectively (Atkins et al., 1975; Cloutier et al., 1975). The colloids were assumed to be taken up preferentially in the liver, spleen, and red marrow, with a uniform distribution of any residue in the remainder of the body. Uptake fractions were given for three patient categories: normal liver condition, early to intermediate diffuse parenchymal liver disease, and intermediate to advanced diffuse parenchymal liver disease. These categories differ not only in biokinetics, but also with regard to liver and spleen mass. In the normal case, the uptake in liver, spleen, and red marrow was set at 85, 7, and 5% for sulphur colloid and 90, 3, and 7% for gold colloid, respectively. These values were estimates based on clinical studies, but no details about the methods used for calculating the percentages were given. However, the values are in good agreement with results obtained from animal studies. (A27) Studies on man have shown decreased uptake of colloids with increasing degree of liver disease, with corresponding increases in uptake for other organs (Herzog et al., 1987; Groshar et al., 2002). The change in uptake depends on the particle size of the administered colloid. (A28) The Task Group has adopted the same view as the MIRD Committee with regard to choice of patient categories, definition of organs with active uptake, organ masses, and biokinetic differences between large and small colloids. The uptake values used are based on the report by Herzog et al. (1987), which contains results of quantitative measurements with conjugate view whole-body counting and double-window regional counting over liver and spleen. For all types of colloid, immediate uptake is assumed. The biological half-time of the radionuclide is assumed to be long compared with the physical half-time, except for iodine-labelled albumin micro-aggregates. For these substances, the metabolic breakdown of the particles is assumed to be represented by biological half-times (fraction) of 3 h (0.8) and 5 days (0.2). (A29) The organ masses for different patient categories and uptake data for different sizes of colloid are presented in Tables A.5–A.7. For further details, the reader is referred to the biokinetic data on the individual substances. Uptake values (fractions) for small colloids (<100 nm diameter).* Example: 99mTc mini-/micro-aggregated albumin. †Normal liver. ‡Early to intermediate diffuse parenchymal liver disease. §Intermediate to advanced diffuse parenchymal liver disease.

A.8. Model for liver and biliary excretion
(A30) This model is intended for substances that are actively taken up in hepatocytes and excreted, via the biliary tract, to the intestine. Typical examples are a large group of technetium-labelled iminodiacetic acid (IDA) derivatives (e.g. BIDA, HIDA, EIDA, PIPIDA, PBIDA, and DISIDA). (A31) Several biokinetic models have been presented in the literature for technetium-labelled IDA derivatives (Ryan et al., 1977; Wistow et al., 1977; Taavitsainen et al., 1980; Brown et al., 1981, 1982; Wu et al., 1984). The substance is assumed to be taken up rapidly in the liver from the blood and then excreted, via the biliary tract, partly to the gallbladder for temporary storage and partly to the intestine. A minor portion of the radiopharmaceutical is excreted in the urine. In pathological states (liver disease, occlusion of the biliary tract, congenital biliary atresia), the same model is used but with different kinetic data (transfer factors). The compartmental model is shown in Fig. A.2. (A32) Similar models have been used for all substances that undergo biliary excretion. For each substance, the fraction and half-time for movement between compartments are specified in the biokinetic data table. Unless otherwise stated in the model, it is assumed that 65% of the activity entering the liver is transferred directly from the liver to the small intestine, and 35% goes to the gallbladder (Wu et al., 1984). (A33) The gallbladder empties at intervals on stimulation by food. It is assumed to empty in an identical manner for all substances. The first emptying is after 3 h, during which time 75% of the radioactive material present in bile is assumed to be excreted to the small intestine. The second emptying is after 9 h, again associated with the excretion of 75% of the radioactive material in bile. For the dose estimation, the third and final emptying is assumed to occur after 24 h when all the radioactive material is excreted. Earlier emptying can be induced by a meal of high fat content or by cholecystokinin. (A34) The final excretion from the body follows the models for the gastrointestinal tract and the kidney–bladder system (see above). Model for liver and biliary excretion. The flows are defined as follows: 1, uptake in liver; 2, uptake in kidney; 3, excretion from liver to gallbladder; 4, excretion from liver directly to small intestine; and 5, emptying of gallbladder to small intestine.

A.9. Model for salivary glands
(A35) Some substances are actively taken up in the salivary glands. In those cases, an approximate absorbed dose in the salivary glands is estimated and included in the dose table. This organ is not included in the presently used tables of S values. Johansson (1996) presented S values for self-irradiation of the salivary glands for 99mTc. The calculation method lined up in this report has also been used by the Task Group for other radionuclides, using the unit density sphere model in the formerly freely available MIRDOSE3 program (Stabin, 1996). The S values have been calculated considering the three pairs of salivary glands (parotid, submaxillary, and sublingual) with masses according to Publication 23 (ICRP, 1975). Those masses do not deviate significantly from those reported in the updated version, Publication 89 (ICRP, 2002). To estimate the absorbed dose from source in other organs, the brain is used as a substitute target organ, except in cases when the brain is also a source.
A.10. References for Annex A
Annex B. Explanations
B.1. Presentation of data
(B1) In Annex C, the data on each substance are presented in three types of information showing: a biokinetic model in text form and references to that, a biokinetic data table, and one or more tables over absorbed dose per unit of activity administered. The Biokinetic data table contains the weghted summary of the information given in the text section and its references. Unless otherwise stated, the model refers to intravenous administration. (B2) The rate of the biological process (e.g. uptake, metabolism, and excretion) is usually given as the half-time of the corresponding exponential function. If the process is assumed to be multi-exponential, the fraction (a) of the organ content belonging to each exponential component is given in the next column. When rates are given as fractions per time unit (k) as reported in cited publications, they are transformed into half-times according to the formula T = 0.693/k. (B3) The following abbreviations have been used:
S, source organ or tissue; Fs, fractional distribution to organ or tissue S; T, biological half-time for an uptake or elimination component; a, fraction of Fs taken up or eliminated with the corresponding half-time; -, uptake; and Ãs/Ao, cumulated activity in organ or tissue S per unit of administered activity. (B4) The tables sometimes contain empty spaces under the headings T and a, usually because the kinetics are described by complex exponential, or non-exponential, expressions, that cannot be defined easily. This is the case for activity in the gastrointestinal tract, the gallbladder, and the urinary bladder. In these cases, the tables only present the cumulated activities together with the fractional distribution. (B5) The relative cumulated activities are presented in hours (h). Average organ or tissue absorbed doses are given as milligrays (mGy) per megabecquerel (MBq). The effective dose is given as millisieverts (mSv) per MBq. All dose values are given in exponential notation (e.g. 2.6E-02 = 2.6 × 10−2 or 0.026 and 4.9E+01 = 4.9 × 10+1 or 49). The calculations have been performed without rounding, but the final result is given with two digits. (B6) Dose calculations have been performed for adults and 15-, 10-, 5-, and 1-year-old children. The organs (or tissues) are presented in alphabetical order except ‘Remaining organs’, which is placed at the end. The dose to organs or tissues not mentioned in the table can usually be approximated with the value given for ‘Remaining organs’.
Annex C. Biokinetic Models and Dose Rates
C.1. 3H-neutral fat and free fatty acids
C.1.1. Biokinetic model
(C1) Orally administered fat is absorbed rapidly and completely from the gastrointestinal tract. Within 3–4 h, all activity has reached the blood via the lymphatic system. After transient uptake and chemical modification in the liver, the fat is transported to the adipose tissue, which occurs principally in subcutaneous tissue, yellow marrow, and the abdominal cavity, and to the muscles. Other organs and tissues receive small amounts. It is then metabolised by β-oxidation, with water and carbon dioxide (CO2) as end products. The turnover rate is highly dependent on the nutritional state, especially the supply of carbohydrates. (C2) Pedersen and Marqversen (1981) measured 14CO2 in expired air in five healthy subjects who were given labelled neutral fat in a test meal after an 8-h fast. Unrestricted food was allowed from 6 h later. After 1 day, 15–33% of ingested fat had been metabolised, and this increased to 25–40% by 10 days. The residue was retained for a much longer time with a calculated half-time of 304–493 days. Malmendier et al. (1974) injected 14C-labelled palmitic acid into four fasting normal subjects and measured expired air for 24 h. They found that 45% of the fatty acid was oxidised directly to CO2. No carbohydrate was given simultaneously, which may explain the larger fraction that was metabolised more rapidly than in the study of Pedersen and Marqversen (1981). Hirsch et al. (1960) studied the turnover of neutral fat incorporated into adipose tissue, and found half-times up to 750 days. (C3) The model adopted here is intended for fat containing unbranched long-chain (13–18 C atoms) fat molecules and labelled with 14C or 3H, administered orally or intravenously. Rapid and complete resorption is assumed. After transient uptake in the liver, the activity is deposited in the adipose tissue (85%), in muscles (10%), and in all other organs and tissues (5%) according to their fat content as given in Publication 23 (ICRP, 1975). Assuming adequate supply of carbohydrates, 30% is metabolised rapidly (T1/2 = 2 days) and 70% is retained for a longer time (T1/2 = 400 days). The half-time of 400 days assumed for the longer-term component of retention of 3H (and 14C) in the body fat is longer than the overall half-time of 40 days assumed for the total body hydrogen (and carbon) in Publication 30 (ICRP, 1979) and Publication 56 (ICRP, 1990). This long-lived component refers only to the fraction of the body fat which is labelled following administration of a single dose of labelled fat (Gunnarsson et al., 2000), and which probably represents only a small fraction of the total body carbon pool. The long-lived component refers in this case to the fraction of body fat which becomes labelled with the administered radiopharmaceutical. This probably only represents a small fraction of the total carbon pool in the body. (C4) This model is intended for adults only. It is possible that the metabolism is significantly different in children, with longer half-times in some tissues (e.g. the nervous system). The absorbed dose per unit activity administered for adults are presented in Table C.2. Biokinetic data for 3H-neutral fat and free fatty acids.

C.1.2. References for 3H-neutral fat and free fatty acids
C.2. [1-11C]-acetate
C.2.1. Biokinetic model
(C5) Acetate labelled with 11C in the carboxyl position, [1-11C]-acetate, is used for dynamic positron emission tomography (PET) studies of myocardial metabolism (Armbrecht et al., 1990; van den Hoff et al., 1996; Sun et al., 1997), and in renal (Shreve et al., 1995), pancreatic (Shreve and Gross, 1997), and nasopharyngeal disease (Yeh et al., 1999). (C6) In most tissues, after extraction from the blood, [1-11C]-acetate is activated to acetyl co-enzyme A (CoA) and enters the tricarboxylic acid (TCA) cycle. From the TCA cycle, the label is lost mainly in the form of 11CO2 (Armbrecht et al., 1990). In resting myocardium, the behaviour of [1-11C]-acetate can be summarised as follows (Armbrecht et al., 1990):
extraction of approximately two-thirds of the activity in a single capillary transit; a very rapid initial washout phase (T1/2 < 5 s); activation of [1-11C]-acetate to [1-11C]-acetyl-CoA within a few seconds; labelling of TCA cycle intermediates takes several minutes; onset of rapid 11CO2 release after 2–3 min; and 11CO2 release is bi-exponential. (C7) In all the tissues studied, peak uptake appears to be reached within less than 3 min. After 3–5 min, 50% of the tissue activity is present as 11CO2, 24% as non-ionised species, and 13% each as acetate and TCA-amino acid intermediates (Sun et al., 1997). The rate of metabolism of the radiopharmaceutical reflects the rate of oxidative metabolism in the tissue, and thus the oxygen supply. (C8) Clinical studies indicate that in both myocardium and kidney parenchyma, the initial uptake is complete by 2.5–3 min post injection, and that between 3 and 30 min, the 11C is lost from the tissues with a half-time of approximately 10 min. In normal pancreas, the uptake is also complete in 3 min, and by 30 min, the activity is lost from the tissue with a half-time of 38 min. In the liver, uptake is again rapid, peaking at approximately 3 min; thereafter, loss of 11C from the tissue follows a tri-exponential clearance, with 35% being cleared with a half-time of 10 min and the remainder with half-times of 1 (30%) and 2 h (35%). (C9) Few data on the fractional deposition of [1-11C]-acetate in human tissues appear to be available in the literature. However, as there is a high extraction rate for [1-11C]-acetate in most tissues and its rate of metabolism reflects the tissue oxygen supply, the rate of blood flow, expressed as a fraction of cardiac output, in the tissue may be used as an approximation of the tissue uptake of [1-11C]-acetate. Leggett and Williams (1995) have tabulated blood flow data for most human tissues, and these values have been used to construct the biokinetic model illustrated in Table C.3 below. In this model, uptake in all tissues is assumed to be rapid, with a half-time of 1 min. Absorbed doses for 3H-neutral fat and free fatty acids. The physical half-life of 3H is 12.35 years.

C.2.2. References for [1-11C]-acetate
C.3. 11C-labelled amino acids (generic model)
C.3.1. Biokinetic model
(C10) The methionine analogue [75Se]-selenomethionine has been used in nuclear medicine for many years (ICRP, 1987), and more recently, a number of other amino acids labelled with 11C or 18F have been used, or proposed, for clinical applications such as l-[methyl-11C]-methionine (Deloar et al., 1998), l-[2-18F]-fluorotyrosine (Cottrall et al., 1973; Taylor and Cottrall, 1973; Coenen et al., 1989), [18F]-p-fluorophenylalanine (Cottrall et al., 1973), 6-[18F]-fluorotryptophan (Atkins et al., 1972; Taylor and Cottrall, 1973), cis-4-[18F]-fluoroproline and trans-4-[18F]-fluoroproline (Wester et al., 1999a,b), and l-3-[18F]-fluoro-α-methyl tyrosine (Inoue et al., 1998). (C11) The Commission has only published biokinetic models for [75Se]-selenomethionine (ICRP, 1987) and l-[methyl-11C]-methionine (ICRP, 2008). Taylor (2000) developed the generic biokinetic model described in Table C.5 below for use in assessment of the internal dose received by human subjects injected intravenously with amino acids labelled with 11C, 18F, or 75Se. Comparison of the radiation doses to adults calculated using this generic model with those calculated using compound-specific models for [11C]-labelled and [18F]-labelled amino acids and [75Se]-selenomethionine indicated that, in general, the effective doses, as well as the organ and tissue doses, calculated using the generic model agreed within a factor of two or less with those calculated using compound-specific models. It was further noted that the generic model tended to overestimate, rather than underestimate, the organ and tissue doses. It was concluded that for [11C]-, [18F]-, and [75Se]-labelled amino acids or their analogues, the generic biokinetic model could be applied for general radiation protection purposes. (C12) The generic model assumes that, following entry of a labelled amino acid into the blood stream, the radiopharmaceutical is taken up instantaneously by the organs and tissues. This is followed by a phase of rapid elimination of that fraction of the injected material which goes directly into the excretory pathways or is excreted following early metabolism, a second phase that represents loss due to metabolic breakdown of labelled proteins and other compounds with relatively rapid turnover times, and a final phase representing elimination of the small fraction of the radionuclide that had been incorporated into structural proteins or other body components with very slow turnover. (C13) In the model, elimination of the radionuclide from the various organs and tissues is assumed to approximate a three-component exponential relationship with biological half-times of 0.5, 50, and 5000 days. The long biological half-time assigned to the small final component of the model reflects the evidence that 14C incorporated into structural tissues such as bone is retained with a very long half-time (Stenhouse and Baxter, 1977; Stenström et al., 1996). (C14) The generic model assumes that 20% of the administered activity is excreted directly from the blood to the urinary bladder with biological half-times of 0.2 h (0.25) and 6 h (0.75) in the blood. It has also been assumed that 3% of the injected activity is excreted into the small intestine; half with a biological half-time of 6 h and half with a biological half-time of 12 h. As labelled amino acids are potentially important for studies of protein synthesis in the brain (Bergmann et al., 1995; Schmidt et al., 1997; Shoup et al., 1999), it is assumed that 1.5% of the injected activity deposits in brain, from where it is released back to the circulation with biological half-times of 50 (70%) and 5000 days (30%). The parameters of this generic model are shown in Table C.5. (C15) Taylor (2000) noted that the biokinetic data from humans or animals that were used to derive both the compound-specific and the generic models are subject to fairly large uncertainties (coefficients of variation ranging from approximately 20% to approximately 80%); therefore, when comparing doses calculated by the generic and compound-specific biokinetic models, differences in individual tissue or organ doses of a factor of two, or even three, should be regarded as good agreement. (C16) This agreement appears to be close enough for the single generic biokinetic model to be used for normal prospective radiation dosimetry, and for general assessment of the risk from the use of amino acids labelled with 11C, 14C, 18F, or 75Se. In situations where compound-specific retrospective dosimetry is necessary (e.g. in the case of accidental intake of a large amount of a radionuclide compound), it might reasonably be expected that some subject- and compound-specific biokinetic information would be available upon which a more accurate person-specific dose assessment could be based. This model is not appropriate for the interpretation of bio-assay data following intake of 14C-labelled amino acids. Biokinetic data for [1-11C]-acetate. This biokinetic model is not applicable for 14C.

C.3.2. References for 11C-labelled amino acids (generic model)
C.4. 11C-labelled brain receptor substances (generic model)
C.4.1. Biokinetic model
(C17) A large number of radiopharmaceuticals labelled with 11C are being developed for PET studies of different types of receptor in the human brain. For most of these agents, the available biokinetic data are insufficient to construct realistic compound-specific biokinetic models for calculating the internal radiation dose delivered to persons undergoing investigation. Table C.7 shows a list of references and available data. A generic model for brain receptor substances that predicts the internal dose with sufficient accuracy for general radiation protection purposes has, therefore, been developed (Nosslin et al., 2002). (C18) Biokinetic data for 13 11C radiopharmaceuticals used clinically for imaging different brain receptors indicate that, despite differences in chemical structure, their uptake and retention in the human brain and other tissues are broadly similar. The proposed model, which is shown in Table C.8, assumes instantaneous deposition of 5% of the injected activity in the brain, with the remaining activity being distributed rapidly and uniformly throughout all other tissues. Elimination from all tissues is assumed to occur with a half-time of 2 h. It is further assumed that 75% of the injected 11C is excreted in the urine and 25% via the gallbladder, with a half-time of 2 h. Absorbed doses for [1-11C]-acetate. The physical half-life of 11C is 20.4 min.

C.4.2. References for 11C-labelled brain receptor substances (generic model)
C.5. l-[methyl-11C]-methionine
C.5.1. Biokinetic model
(C19) The amino acid l-[methyl-11C]-methionine can be applied in tumour diagnosis and in the study of protein synthesis using PET. Deloar et al. (1998) reported quantitative PET studies on the distribution of l-[methyl-11C]-methionine in five healthy, male volunteers aged 22–40 years. The data suggested that approximately 90% of the activity was lost from all tissues during the first 90 min after injection, with biological half-times of approximately 20–30 min. Thereafter, the activity appeared to be lost more slowly, with a half-time that could be considered to be long in relation to the physical half-life of 11C. (C20) The biokinetic model presented in Table C.10 below was developed on the basis of the human data of Deloar et al. (1998b), who estimated the uptake of l-[methyl-11C]-methionine into the brains of the five volunteers to be 2.8 ± 0.7% of the injected activity; some seven times higher than the value of 0.4% previously estimated by Comar et al. (1976). Biokinetic data for 11C-labelled amino acids (generic model). For l-[methyl-11C]-methionine, the compound-specific data (ICRP, 2001) should be used.

C.5.2. References for l-[methyl-11C]-methionine
C.6. 11C-labelled thymidine
C.6.1. Biokinetic model
(C21) 11C-labelled thymidine is a DNA precursor that can be used as an in-vivo marker for cell proliferation in malignant tumours. It also has applications in tumour staging and for monitoring the effectiveness of treatment. It has been used in two forms: labelled with 11C in the methyl group, [methyl-11C]-thymidine, or labelled with 11C on C2 of the pyrimidine ring, [2-11C]-thymidine. The two forms differ in respect of the metabolic fate of the 11C label. [Methyl-11C]-thymidine is metabolised to [11C]-β-amino-iso-butyric acid, while the C2-labelled molecule is metabolised to [11C]CO2. For dosimetric purposes, it is necessary to develop appropriate biokinetic models to describe the fate of the 11C following administration of each of the two compounds.
[Methyl-11C]-thymidine
(C22) PET studies in a small number of patients (Martiat et al., 1988; Thierens et al., 1994) have provided information for the distribution of [methyl-11C]-thymidine over a period of 40 min following intravenous injection. Thierens et al. (1994) observed that 95% of the activity was cleared rapidly from the blood (T1/2 = 1 min) and deposited in the liver (40–45%), skeletal muscle (30–34%), and kidneys (5–6%), with much smaller quantities going to other tissues. At 10 min after injection, less than 15% of the activity remaining in the blood was present as [methyl-11-C]thymidine; this amounts to less than 0.75% of the injected activity. (C23) Martiat et al. (1988) reported ‘substantial’ uptake in lungs, spleen, and intestine, but Thierens et al. (1994) stated that the concentration in spleen and lungs does not exceed that observed in muscle. Using the data of Martiat et al. (1988) to calculate organ contents at 30-min post injection suggests uptake of 40% in liver, 10% in kidneys, 2% in lungs and spleen, and 13% in muscle. Analysis of the tissue retention data reported by Martiat et al. (1988) and Thierens et al. (1994) suggests biological half-times of retention ranging from 60 min in the lungs to 460 min in muscle. (C24) The data of Martiat et al. (1988) and Thierens et al. (1994) have been used to derive the biokinetic model for [methyl-11C]-thymidine.
[2-11C]-thymidine
(C25) Van der Borght et al. (1992) compared the retention of [2-11C]-thymidine and [methyl-11C]-thymidine in a PET study involving five patients. Although the masses of labelled thymidine injected, 3.1 µmol [2-11C]-thymidine and 0.17 µmol [methyl-11C]-thymidine, differed by a factor of 18, they were both small in relation to the plasma levels of non-radioactive thymidine, and mass-related changes in the biokinetics of the two labelled compounds appear unlikely. The initial plasma clearance was very rapid, with more than 99% of the injected activity being removed with a half-time of less than 1 min. Although there were some differences in the retention of the small fraction of the injected activity remaining in the plasma at 10 min, these were quite small. At 10-min post injection, 70% of the plasma activity was in the form of [11C]CO2. The retention of 11C in the liver and kidneys was seven and three times less for [2-11C]-thymidine than for [methyl-11C]-thymidine, respectively. (C26) The dosimetric model for [2-11C]-thymidine, as shown in Table C.12, is based on the assumption that 70% of the injected compound is converted rapidly to [11C]CO2, which then follows the biokinetic model for continuous inhalation of [11C]CO2 proposed in Publication 53 (ICRP, 1987); the remaining activity is assumed to follow a model derived from that for [methyl-11C]-thymidine, but with uptake values for liver and kidneys based on the observations of Van der Borght et al. (1992). Absorbed doses for 11C-labelled amino acids (generic model). The physical half-life of 11C is 20.4 min.

C.6.2. References for 11C-labelled thymidine
C.7. 11C-labelled substances (realistic maximum)
C.7.1. Biokinetic model
(C27) It is assumed that 50% of the decay occurs while the substance passes the urinary bladder, and the remaining 50% of the total disintegration occurs when it is distributed homogeneously throughout the whole body. Brain receptor substances – comparison of 11C retention in brain up to approximately 90 min. n.a., not available.

C.8. 11C-raclopride
C.8.1. Biokinetic model
(C28) Raclopride is a synthetic compound of the salicylamide series with high selectivity and affinity for central D2-dopamine receptors. It can be labelled with 11C and used in PET. The neurotransmitter dopamine may be involved in various neuropsychiatric diseases. 11C-raclopride is cleared rapidly from both plasma and whole blood, and crosses the blood–brain barrier. After intravenous administration, 11C-raclopride localises in the basal ganglia, a region with a high density of dopamine receptors. PET images show the concentration of 11C-raclopride in the region of the putamen relative to the rest of the brain. Images can be taken immediately after injection and continued for approximately 60 min (Glatting et al., 2004; Slifstein et al., 2007). (C29) The proposed biokinetic model shown in Table C.16 is mainly based on experimental data from Ribeiro et al. (2005) (11 measurements from 2 to 112 min after application). The source organs have been grouped according to three retention half-times. Slifstein et al. (2011) used the LLI and cortical bone, but not muscle, as source regions. They also reported a lower kidney uptake. Biokinetic data for 11C-labelled brain receptor substances (generic model).

C.8.2. References for 11C-raclopride
C.9. 14C-neutral fat and free fatty acids
C.9.1. Biokinetic model
(C30) Orally administered fat is absorbed rapidly and completely from the gastrointestinal tract. Within 3–4 h, all activity has reached the blood via the lymphatic system. After transient uptake and chemical modification in the liver, the fat is transported to the adipose tissue, which occurs principally in subcutaneous tissue, yellow marrow, and the abdominal cavity, and to the muscles. Other organs and tissues only receive small amounts. It is then metabolised by β-oxidation, with water and CO2 as end products. The turnover rate is highly dependent on the nutritional state, especially the supply of carbohydrates. (C31) Pedersen and Marqversen (1981) measured 14CO2 in expired air in five healthy subjects, who were given labelled neutral fat in a test meal after an 8-h fast. From 6 h later, unrestricted food was allowed. After 1 day, 15–33% of ingested fat was metabolised, and this increased to 25–40% by 10 days. The residue was retained for a much longer time with a calculated half-time of 304–493 days. Malmendier et al. (1974) injected 14C-labelled palmitic acid into four fasting normal subjects and measured expired air over 24 h. They found that 45% of the fatty acid was oxidised directly to CO2. No carbohydrate was given simultaneously, which may explain the larger fraction metabolised more rapidly than in the study of Pedersen and Marqversen (1981). Hirsch et al. (1960) studied the turnover of neutral fat incorporated into adipose tissue and found half-times up to 750 days. (C32) The model adopted here and shown in Table C.18 is intended for fat containing unbranched long-chain (13–18 C atoms) fat molecules and labelled with 14C or 3H, administered orally or intravenously. Rapid and complete resorption is assumed. After transient uptake in the liver, the activity is deposited in the adipose tissue (85%), in muscles (10%), and in all other organs and tissues (5%) according to their fat content as given in Publication 23 (ICRP, 1975). Assuming adequate supply of carbohydrates, 30% is metabolised rapidly (T1/2 = 2 days) and 70% is retained for a longer time (T1/2 = 400 days). The half-time of 400 days assumed for the longer-term component of retention of 3H (and 14C) in the body fat is longer than the overall half-time of 40 days assumed for the total body hydrogen (and carbon) in Publications 30 and 56 (ICRP, 1981, 1990). This long-lived component refers only to that fraction of the body fat that is labelled following administration of a single dose of labelled fat administered as a radiopharmaceutical, and which probably represents only a small fraction of the total body carbon pool. The long-lived component refers in this case to the fraction of body fat which becomes labelled with the administered radiopharmaceutical. This probably only represents a small fraction of the total carbon pool in the body. (C33) This model is intended for adults only. It is possible that the metabolism is significantly different in children, with longer half-times in some tissues (e.g. the nervous system). Absorbed doses for 11C-labelled brain receptor substances (generic model). The physical half-life of 11C is 20.4 min.
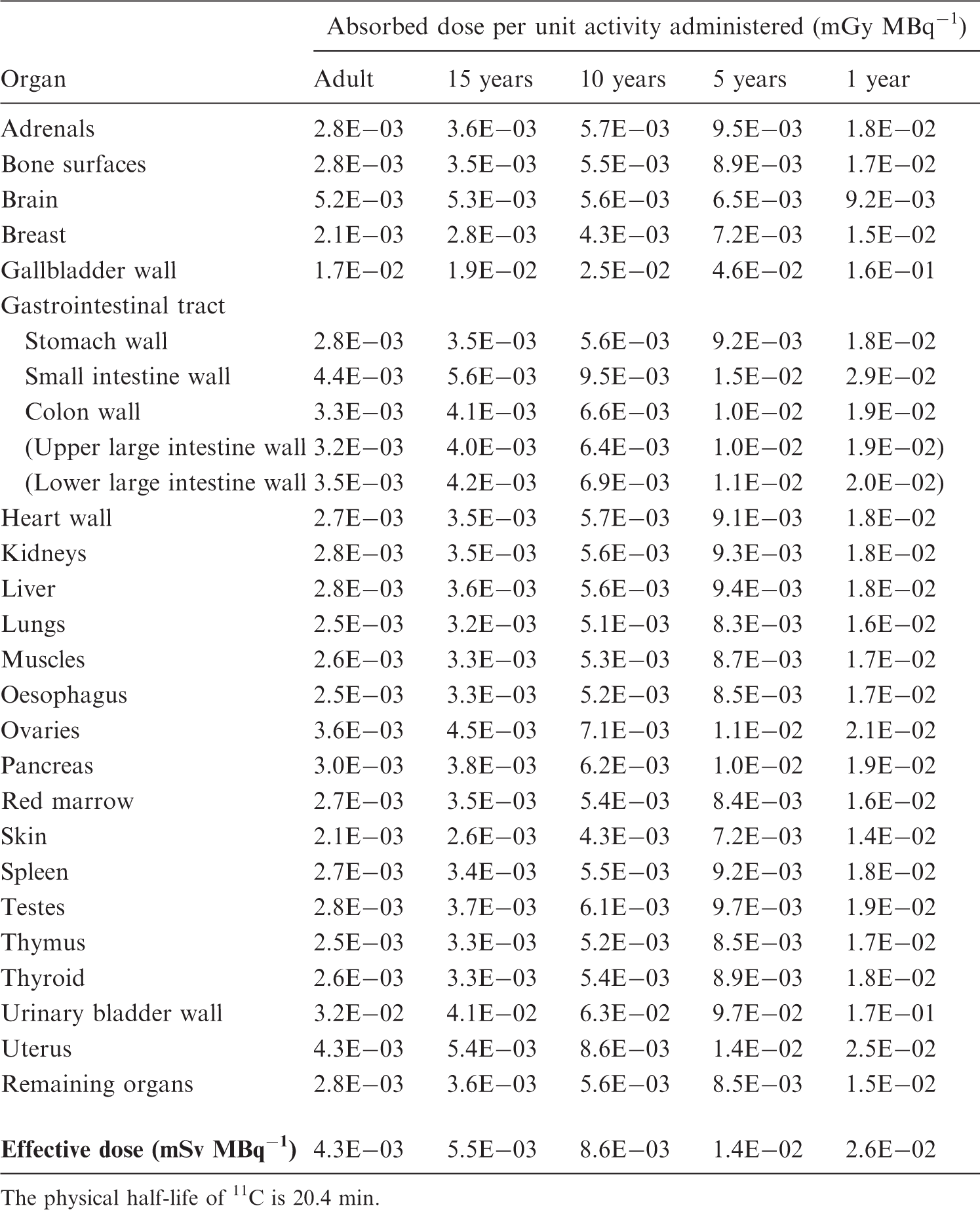
C.9.2. References for 14C-neutral fat and free fatty acids
C.10. 14C-labelled urea
C.10.1. Biokinetic model
(C34) Urea (carbamide, H2NCONH2) is the main end product in the human catabolism of proteins, polypeptides, amino acids, and other nitrogen-containing substances. It is freely water soluble and distributes rapidly into the total body water. The main part is excreted unchanged by the kidneys, while a small part diffuses into the intestinal content where it is broken down by urease-producing bacteria to ammonia and CO2. The CO2 is reabsorbed and equilibrates with bicarbonate, thus entering the CO2/bicarbonate pools in the body and finally being exhaled by the lungs (Walser and Bodenlos, 1959).
14C urea
(C35) A breath test employing per-oral administration of 14C-urea is commonly used to detect the presence of Helicobacter pylori in the stomach of patients with peptic ulcer and other gastric diseases. Normally, the stomach does not contain urease-producing bacteria, so the urea is rapidly absorbed unchanged into body water. H. pylori, on the other hand, produces urease and therefore brings about extensive early expiration of labelled CO2, resulting in a positive breath test (Marshall and Surveyor, 1988; Combs et al., 1999; Walser and Bodenlos, 1959). (C36) In the model for per-oral administration, there is, in the normal case, complete and rapid (T1/2 = 5 min) resorption from the stomach. In the case of H. pylori infection in the stomach, it is assumed that 65% is immediately converted to CO2, which is treated further according to the dosimetric model for CO2/bicarbonate (see below). The remaining 35% is resorbed from the stomach in the same way as in the normal case. (C37) Urea resorbed in the stomach is distributed rapidly in the total body water. Eighty percent is excreted by the kidneys with a half-time of 6 h, and 20% is broken down rapidly in the same way as intravenously administered urea to ammonia and CO2, treated according to the dosimetric model for CO2/bicarbonate.
14C carbon dioxide/bicarbonate
(C38) CO2 is formed continuously in the metabolism of all organic substances in the body. Together with water, it forms carbonic acid (H2CO3), which dissociates and equilibrates with bicarbonate ions (HCO−3). The substances are present in all body fluids. Winchell et al. (1970) presented a kinetic model with two compartments, one (Compartment I) with rapid (within 3 min) equilibration with CO2/HCO−3 in blood and another (Compartment II) with slower equilibration. CO2 leaves the system from Compartment I by exhalation. A certain small fraction was assumed to be ‘relatively fixed’ in the body, presumably in the form of bone bicarbonate and as a constituent of larger molecules with slow turnover. Compartment I included organs with high vascular perfusion (heart, liver, kidneys, intestinal tract, etc.), while Compartment II was represented by muscle, skin, and fat with a lower blood flow rate. (C39) Stubbs and Marshall (1993) modified this model slightly by defining a Compartment III, corresponding to the ‘fixed’ fraction and having a flow back to Compartment I with a half-time of 1000 h in accordance with the assumption for carbon metabolism in Publication 30 (ICRP, 1981). This half-time of 1000 h may not be sufficient to account for carbon deposited in the bone compartments with slow metabolic turnover, as there is experimental evidence for a much longer turnover time. (C40) The biokinetic model adopted here (Fig. C.1) is based on the models mentioned above with the following modifications. Compartment III has been further divided into three compartments, one (Compartment 3) being assumed to represent uptake in large molecules having a slow turnover with a biological half-time of 1000 h. The other compartments are assumed to represent bone. In the present model, bone has been divided into trabecular bone (Compartment 4) and cortical (compact) bone (Compartment 5), from which the activity is lost at a rate of 0.18 per year (half-time 3.9 years) and 0.03 per year (half-time 23 years), respectively (ICRP, 1995). Eighty percent of the bone mass is assumed to be cortical bone and 20% is assumed to be trabecular bone (ICRP, 1995). The rate of inflow to Compartments 3, 4, and 5 is chosen so that realistic carbonate/bicarbonate pool sizes are reached during a lifetime. This means that the inflow rate constants to the bone compartments have been set to twice the steady-state value calculated to give a carbonate/bicarbonate pool in the bone of 300 g. The model is shown in Fig. C.1, and the tables in this subsection show values used for the transfer constants. The numerical values for transfer constants not taken from Publication 70 (ICRP, 1995) or calculated from steady-state conditions are taken from Winchell et al. (1970). Biokinetic data for l-[methyl-11C]-methionine. This biokinetic model is not applicable for 14C. Biokinetic model for carbon dioxide/bicarbonate. The model presented is valid for adults. The transfer constants are given as h−1. Compartment 1 represents organs with high vascular perfusion (heart, liver, kidneys, intestinal tract, etc.), and Compartment 2 represents tissues with a lower blood flow rate (muscle, skin, and fat). Compartment model used to describe the kinetics of iodine (Leggett, 2010).



C.10.2. References for 14C-labelled urea including carbon dioxide/bicarbonate
C.11. 15O-water
C.11.1. Biokinetic model
(C41) Water labelled with 15O is widely used to evaluate regional cerebral blood flow using PET, and has been proposed for blood flow measurement in other organs and tissues. Early biokinetic models based on equilibrium tracer distribution in body water are inaccurate for use with 15O-water. On account of the short physical half-life of 15O (2.04 min), uniform radionuclide concentration in body water is not attained; consequently, such models underestimate dose values for this substance. (C42) A more satisfactory method of kinetic modelling is based on organ blood flow rates. Using this model, the concentration of 15O-water in a given organ is derived by convolution of the arterial blood concentration (arterial input function) and the transit time function (impulse response) of the organ. The latter is given by exp[- (F/Vd + λ)t], where F (ml min−1 g−1) is the organ blood flow, Vd (ml g tissue−1/ml × ml blood−1) is its relative water distribution space, and λ (min−1) is the radioactive decay constant of 15O. Thus, following intravenous administration of 15O-water and measurement of the arterial blood concentration, a retention equation can be derived for organs for which values of F and Vd are known. (C43) In practice, a measured amount of 15O-water activity is injected via a forearm vein, and the arterial blood concentration is monitored continuously from the other forearm. Residence time (min) in a given organ is calculated as the product of the areas under the curves of the arterial input function (min ml−1 per administered MBq) and the organ transit time function (min), multiplied by the total blood flow to the organ (ml min−1). The latter is given by F × M, where M is the mass (g) of the organ. Table C.22 lists organ residence times that lead to organ doses equal to the mean values that have been estimated using the blood flow model at four different centres (Berridge et al., 1991; Herscovich et al., 1993; Brihaye et al., 1995; Eichling et al., 1997). Direct measurements of the retention in some organs by PET (Smith et al., 1994) have shown good agreement with the model for brain, heart, liver, and spleen. Absorbed doses for l-[methyl-11C]-methionine. The physical half-life of 11C is 20.4 min. The urinary bladder wall contributes 56% of the effective dose.

C.11.2. References for 15O-water
C.12. 18F-labelled amino acids (generic model)
C.12.1. Biokinetic model
(C44) The generic biokinetic model for 18F-labelled amino acids is the same as the generic biokinetic model for 11C-labelled amino acids (see Section C.3.1). It is based on the following references: Coenen et al., 1989; Cottrall et al., 1973; Inoue et al., 1998; ICRP, 1987, 2008; Schmidt et al., 1997; Shoup et al., 1999; Stenhouse and Baxter, 1977; Stenström et al., 1996; Taylor, 2000; Taylor et al., 1973; Wester et al., 1999a, 1999b. Biokinetic data for 11C-labelled thymidine.
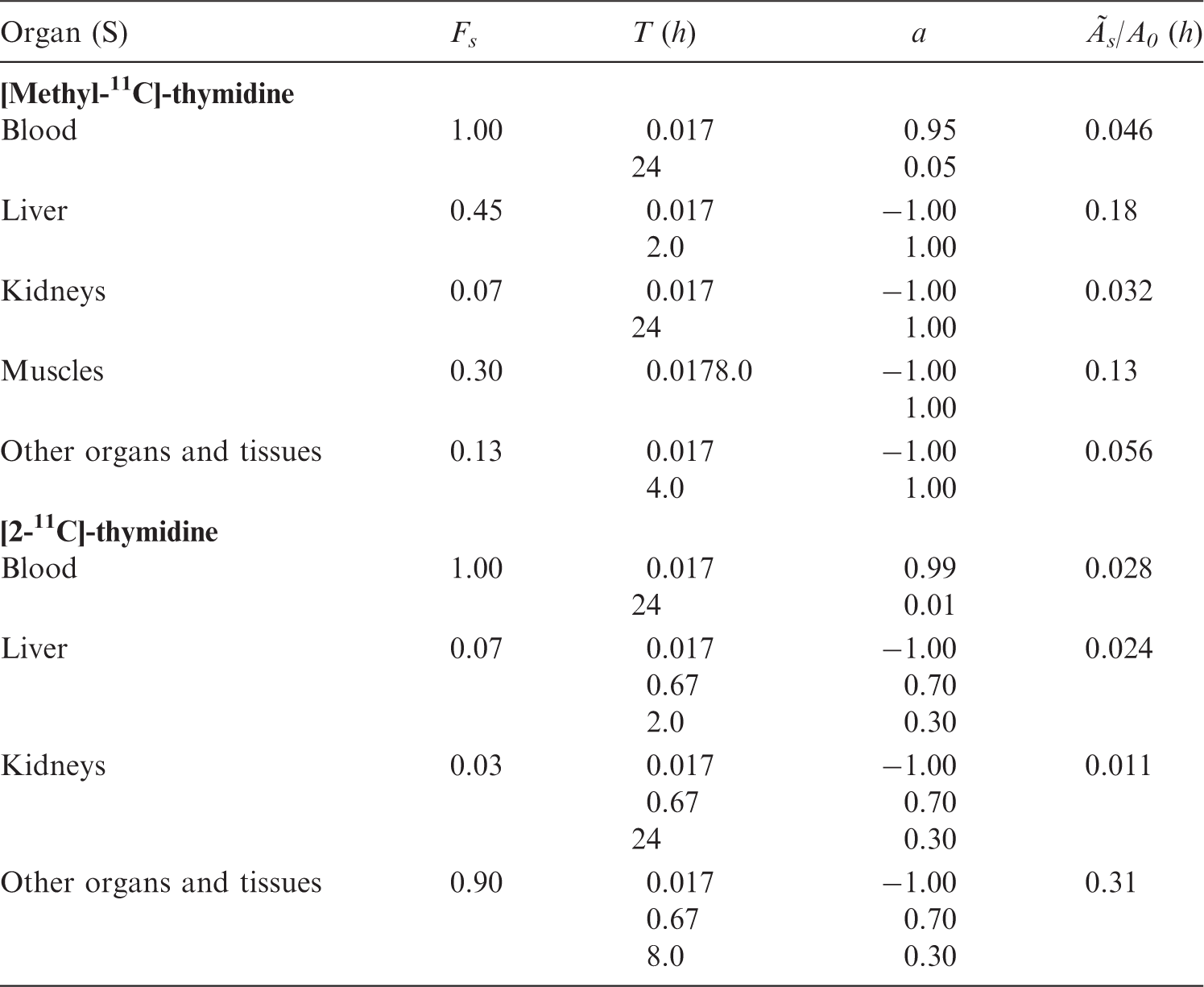
C.12.2. References for 18F-labelled amino acids (generic model)
C.13. 18F-labelled brain receptor substances (generic model)
C.13.1. Biokinetic model
(C45) A large number of radiopharmaceuticals labelled with 18F and 123I have been developed for PET and single photon emission computer tomographic (SPECT) studies of different types of receptor in the human brain. For many of these substances, the available biokinetic data are insufficient to construct realistic compound-specific biokinetic models for the calculation of absorbed dose to persons undergoing an investigation. Therefore, a generic model for radionuclide-labelled brain receptor substances that would predict the internal radiation dose with sufficient accuracy for general radiation protection purposes has been developed. (C46) A generic model for 11C-labelled brain receptor substances has been published previously (Nosslin et al., 2003). A review of the literature has identified biokinetic and dosimetric data for five 18F-labelled and 15 123I-labelled compounds considered to be potential substances for the clinical imaging of brain receptors (e.g. acetylcholinesterase receptors, benzodiazepine receptors, dopamine receptors, dopamine transporters, and serotonin receptors). These data indicate that despite fairly large differences in chemical structure, the patterns of uptake in the human brain, and other tissues for which information is available, appear to be sufficiently similar to justify a generic model for each radionuclide. For details, the reader is referred to Booij et al. (1998a, 1998b), Boundy et al. (1995), Deterding et al. (2001), Gründer et al. (2001, 2003), Kauppinen et al. (2003), Kuikka et al. (1994), Mitterhauser et al. (2004), Mozley et al. (1995, 1996), Taylor (2000), van de Wiele et al. (1999), Verhoeff et al. (1993a, 1993b), Versijpt et al. (2000), Votaw et al. (1995), Volkow et al. (1995), and Waterhouse et al. (2003). (C47) For some compounds, the published data on the dosimetry of 18F- and 123I-labelled receptor radiopharmaceuticals were derived from PET and SPECT studies in humans, and for other compounds, the biokinetic models were derived, at least in part, from studies of biodistribution in experimental animals. (C48) The generic model proposed for 18F-labelled substances assumes that fractions of 0.07, 0.08, 0.05, 0.02, 0.02, and 0.002 of the administered activity are distributed instantaneously to the brain, liver, lungs, kidneys, stomach wall, and thyroid, respectively, from where they are excreted with a biological half-time of 10 h. The remaining activity is assumed to be distributed uniformly throughout the rest of the body, and eliminated with a biological half-time of 10 h. A biological half-time of 10 h means that approximately 98% of the 18F will decay in the tissue of interest. It is assumed that of the activity entering the liver, 30% is eliminated via the gallbladder and the remainder would pass directly into the small intestine. A total of 90% of the administered activity is assumed to be excreted in the urine and 10% via the gastrointestinal tract. Absorbed doses for 11C-labelled thymidine. The physical half-life of 11C is 20.4 min.

C.13.2. References for 18F-labelled brain receptor substances (generic model)
C.14. 18F-choline
C.14.1. Biokinetic model
(C49) Choline uptake is increased in cancerous tissues because the high metabolic rates of tumour cells require choline for the synthesis of phospholipids. For example, choline kinase is overexpressed in prostate cancer cells (Ramirez de Molina et al., 2002; Ackerstaff et al., 2003), thus making choline a suitable indicator for early and differential diagnosis of prostate cancer. PET with radiolabelled choline is therefore used for diagnosis of malignant and recurrent tumours, and metastases in prostate cancer patients (DeGrado et al., 2002; Schmid et al., 2005; Kwee et al., 2006; Steiner et al., 2009). Correct evaluation of the patient dose and optimisation of the imaging protocols imply knowledge of the biodistribution and kinetics of the administered compounds. The biokinetics of 18F-choline (18F-FCH) in four prostate cancer patients were investigated in a study conducted in the frame of the European Collaborative project MADEIRA (Hoeschen et al., 2010; Uusijärvi et al., 2010). Six new patients were later included in the study. In these investigations, biodistribution and excretion data were collected for up to 4 h after injection of the radiopharmaceutical (Janzen et al., 2010; Giussani et al., 2012; Tavola et al., 2012). Previous human studies with 11C- or 18F-choline were limited up to 1 h after administration (Roivainen et al., 2000; DeGrado et al., 2002; Schmid et al., 2005; Kwee et al., 2006; Steiner et al., 2009; Sutinen et al., 2004). (C50) The biokinetics of 11C-choline and 18F-choline differ due to the different interactions of carbon and fluorine in the organism. A paper with dosimetric data on 11C-choline has been published by Tolvanen et al. (2010), and the biokinetics and dosimetry of 11C-choline will be subject to further evaluation by ICRP. Biokinetic data for 11C-labelled substances (realistic maximum).

C.14.2. References for 18F-choline
C.15. 2-[18F]-fluoro-2-deoxy-d-glucose (FDG)
C.15.1. Biokinetic model
(C51) 18F-fluoro-2-deoxy-d-glucose (FDG) is a glucose analogue used in the characterisation of glucose metabolism for diagnosis or follow-up of cancer diseases, and for investigation of myocardial and cerebral glucose metabolism. Following intravenous administration, most of the radiopharmaceutical is cleared rapidly from the circulation with a half-time of less than 1 min as it mixes within a large distribution space, although there are longer-term components with half-times of up to 1.5 h. Data from Hays et al. (2002) together with data obtained by Deloar et al. (1998) are used in this biokinetic model for the dose assessments to patients administered with 18F-FDG. These data confirm the assumption in Publication 53 (ICRP, 1987) of uptake of 0.04 in the heart wall, while uptake in the brain seems to be higher (0.07–0.1) than was given in the Publication 53 model (0.06). (C52) Additionally, there is an indication of significant uptake in the liver and the lungs. For the liver, uptake values of approximately 0.05 were derived by Deloar et al. (1998) and Meija et al. (1991). The model of Hays and Segall (1999) predicts greater uptake in the liver but it decreases rapidly to similar values. For uptake in the lungs, results range from 0.009 (Meija et al., 1991) to 0.029 (Deloar et al., 1998). Here again, the model by Hays and Segall (2002) indicates greater uptake followed by a rapid decrease. There are indications that there is a slight increase in activity in heart and brain, and a steep decrease in activity in lungs and liver (Meija et al., 1991; Hays and Segall, 1999). It is assumed that all activity is excreted by urine. (C53) The following biokinetic model has been derived (ICRP, 2008) based on this information. There is initial uptake of 18F-FDG in heart (0.04), brain (0.08), liver (0.05), lungs (0.03), and all other tissues (0.80). Retention in the specified source organs is considered to be infinite (without consideration of a delayed uptake). A fraction of 0.3 of the activity in other organs and tissues is considered to be excreted by urine with biological half-times of 12 min (25%) and 1.5 h (75%), according to the kidney–bladder model. Absorbed doses for 11C-labelled substances (realistic maximum). The physical half-life of 11C is 20.4 min. The urinary bladder wall contributes 79% of the effective dose.

C.15.2. References for 18F-fluoro-2-deoxy-d-glucose
C.16. O-(2-[18F]-fluoroethyl)-l-tyrosine (FET)
C.16.1. Biokinetic model
(C54) O-(2-[18F]-fluoroethyl)-L-tyrosine ([18F]FET) is actively taken up in tumour cells via amino acid transport system L, but is neither incorporated into proteins nor readily degraded, resulting in high intracellular concentrations of this imaging agent. Reflecting the increased amino acid transport capacity of tumour cells, [18F]FET is useful in PET brain tumour imaging because 18F-FDG, commonly used in PET tumour imaging, is relatively insensitive for detecting tumours in the brain due to the high levels of glycolytic metabolism in the normal cortex and, to a lesser extent, in white matter. (C55) The currently available information about the biokinetics, organ, and tissue distribution of [18F]FET in humans or animals is limited to the first 3–5 h after intravenous injection. Studies in mice and humans after administration of [18F]FET (Heiss et al., 1999; Wester et al., 1999; Pauleit et al., 2003; Tang et al., 2003; Abe et al., 2006; Langen et al., 2006) showed that the activity was removed rapidly from the blood plasma. Clinical PET studies with [18F]FET, performed in seven patients (Pauleit et al., 2003) and four normal men (Abe et al., 2006), showed that uptake of the radiopharmaceutical in the tissues studied was maximal within 0.25 h, and then decreased mono-exponentially with a biological half-time of between 8 and 12 h. The removal of the radioactivity from the blood plasma appeared to be bi-exponential with biological half-times of <0.05 h (40%) and ≈14 h (60%) (Pauleit et al., 2003). Approximately 25% of the administered substance was excreted in the urine in 5 h, suggesting an elimination half-time of ∼14 h (Pauleit et al., 2003; Langen et al., 2006). (C56) Estimates of radiation dose to human tissues after injection of [18F]FET based on biokinetic data for mice were published by Taylor (2000) and Tang et al. (2003), and an estimate based on clinical PET studies was reported by Pauleit et al. (2003). In general, these agree relatively well with each other. In order to take account of the human data published by Abe et al. (2006), all the available biokinetic data have been re-analysed, and a cautious biokinetic model for [18F]FET was developed (see below). (C57) It is assumed that [18F]FET entering the systemic circulation is distributed rapidly into the organs and tissues and then eliminated with a biological half-time of 14 h. The fractional uptake in the various organs is shown in the first two columns of Table C.32. It is also assumed that 99% of the 18F from [18F]FET is excreted through the urinary bladder with a biological half-time of 14 h; the remaining 1% of the administered activity is assumed to be eliminated via the small intestine and faeces. The selection of 14 h as the elimination half-time means that ∼99% of the administered activity decays in the body. (C58) The effective dose is nearly the same as that calculated by Pauleit et al. (2003) for [18F]FET (1.65E–02 mSv MBq−1). However, the dose to the urinary bladder is higher than that calculated by Pauleit et al. (2003) (6.0E-02 mGy MBq−1) because a longer bladder voiding interval (3.5 h) is assumed here compared with that assumed by Pauleit et al. (2003) (2 h). Biokinetic data for 11C-raclopride. Activity in liver is excreted according to the Publication 53 (ICRP, 1987) gallbladder model (90% passes the gallbladder). Activity in the small intestine wall and half of the activity in other tissues with the long half-time goes directly to the gastrointestinal tract contents. Remaining activity with finite biological half-time is excreted via the urinary bladder.

C.16.2. References for 2-[18F]-fluoroethyl-l-tyrosine
C.17. [18F]-fluoro-l-DOPA
C.17.1. Biokinetic model
(C59) The amino acid analogue 6-fluoro-l-dopa (L-DOPA) is taken up rapidly in the human brain and transformed into the important catecholamine neurotransmitter, dopamine (Luxen et al., 1992; Pauwels et al., 1994). After labelling with the positron-emitting radioisotope 18F, the resulting 18F-DOPA can be used for the scintigraphic investigation of normal and pathological dopamine metabolism in the human brain and tumours (Luxen et al., 1992; Meyer et al., 1995; Heiss et al., 1996), and for the quantitative assessment of dopaminergic function in Parkinson's disease and other conditions. (C60) Studies in normal human volunteers and dogs after administration of 18F-DOPA have shown that the activity is more or less uniformly distributed throughout the body tissues, and is removed by a bi-exponential process with biological half-times of approximately 12 h (67–94%) and 1.7–3.9 h (6–33%) (Harvey et al., 1985; Boyes et al., 1986). Both these half-times appear to be age-dependent (Harvey et al., 1985). 18F is excreted through the kidneys: 50% with a half-time of 0.7 h and 50% with a half-time of approximately 12 h (Harvey et al., 1985). (C61) On the basis of the biokinetic data given by Harvey et al. (1985) and Dhawan et al. (1996), the biokinetic model for 18F-DOPA illustrated in Table C.34 was developed. This model assumes that 100% of 18F is distributed homogeneously in the body and eliminated through the kidneys with biological half-times of 1 h (50%) and 12 h (50%). This model was, in spite of observations cited above, assumed to be independent of age. (C62) Human studies have shown that the uptake of 18F-DOPA in the striatum and cerebellum can be increased approximately two-fold by administration of the amino decarboxylase inhibitor, carbidopa (Melega et al., 1990; Hoffman et al., 1992; Brown et al., 1998). Absorbed doses after intravenous administration of 11C-raclopride. The physical half-life of 11C is 20.4 min.
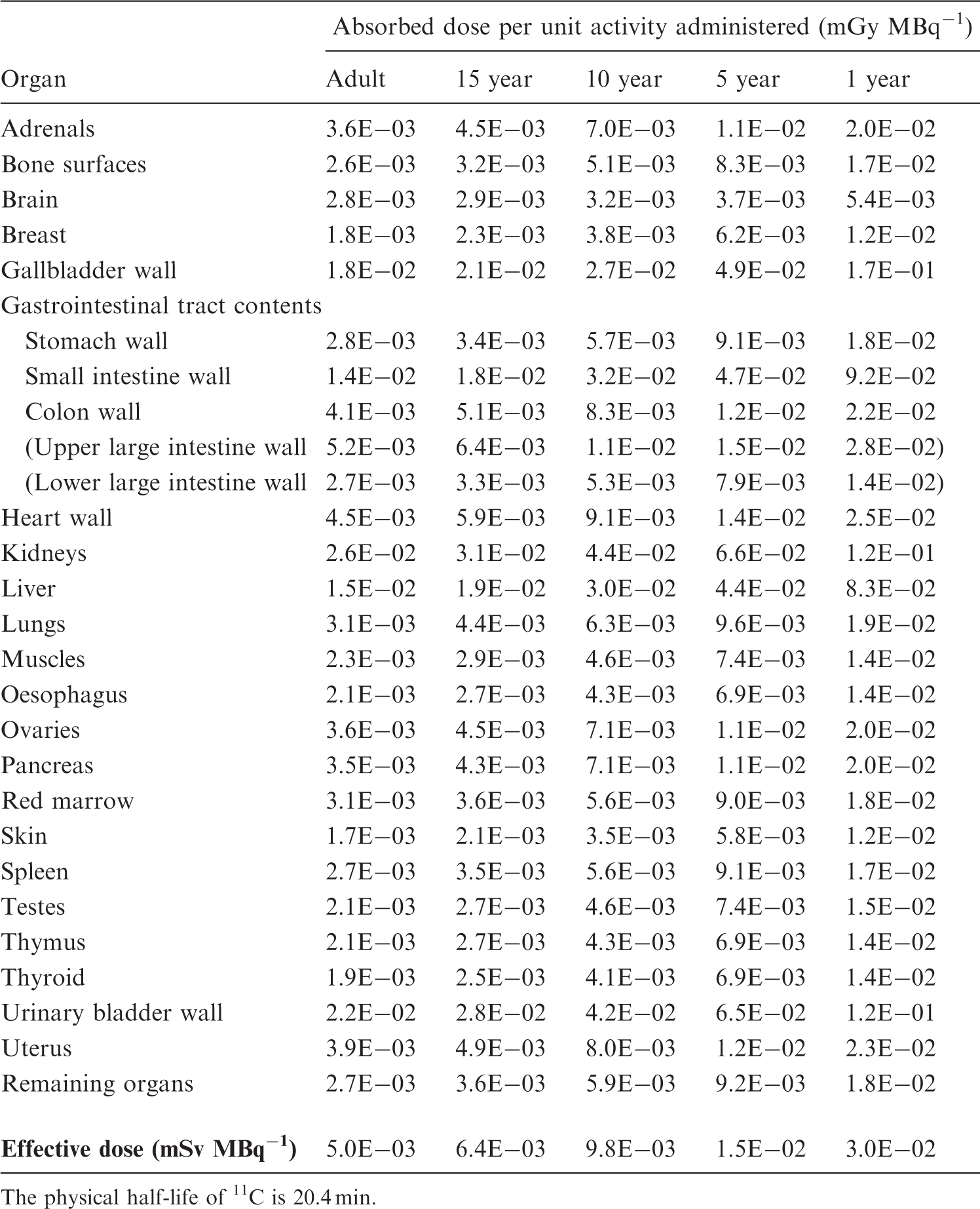
C.17.2. References for [18F]-fluoro-l-DOPA
C.18. 18F-fluoride
C.18.1. Biokinetic model
(C63) Considering the high uptake in mineral bone, and given that the skeleton model has been improved considerably since Publication 53 (ICRP, 1987) was published, ICRP has re-reviewed the literature and proposes a new biokinetic model and a new dose table for this substance. (C64) 18F-fluoride is a highly effective bone-seeking PET tracer used for the detection of skeletal abnormalities (Fair et al., 2010). The uptake mechanism of 18F-fluoride resembles that of 99mTc-methylene diphosphonate, but has better pharmacokinetic characteristics, including faster blood clearance and two-fold higher uptake in bone. Uptake of 18F-fluoride reflects blood flow and bone remodelling. The proposed biokinetic model is mainly based on the compartment model of Blake et al. (2001) and Park-Holohan et al. (2001). Additional information was extracted from Hawkins et al. (1992) and Doot et al. (2010). The Task Group has agreed on the following simple model: with a 15-min uptake half-time, the fraction of the administered activity in the skeleton is estimated to be 60%. Retention on the bone surface is considered to be infinitive. The immediate uptake of 40% of the administered activity in other organs and tissues is assumed to be retained with biological half-times of 15 min (75%) and 13 h (25%). Biokinetic data for 14C-neutral fat and free fatty acids.

C.18.2. References for 18F-fluoride
C.19. 3′-deoxy-[18F]-3′-fluorothymidine (FLT)
C.19.1. Biokinetic model
(C65) The antiviral nucleoside 3′-deoxy-[18F]-3′-fluorothymidine ([18F]FLT) is an analogue of the natural nucleoside thymidine, an essential component of the DNA molecule. The 18F-labelled compound can be applied for the scintigraphic visualisation of cell proliferation in tumours and other tissues using PET, and thus for the monitoring of cancer therapy using cytotoxic drugs or radiation (Grierson et al., 1995; Shields et al., 1996; Choi et al., 2003; Cobden et al., 2003; Vesselle et al., 2003; Buchmann et al., 2004). (C66) Non-radioactive FLT was developed for the treatment of human immunodeficiency virus infection (Shinazi et al., 1990). Clinical studies (Flexner et al., 1994) showed that the administration of ∼0.5 mmol FLT day−1 led to unacceptable toxicity in six out of 10 patients. At the biochemical level, FLT inhibits DNA synthesis and leads to apoptotic cell death. In-vitro tests suggest that FLT exhibits a relatively strong mutagenicity. It is, however, assumed that the single administration of 400 MBq and ∼0.01 µmol [18F]FLT would not cause any demonstrable chemical toxicity or mutagenic effect. (C67) Studies in humans (Cobden et al., 2003; Vesselle et al., 2003; Buchmann et al., 2004), dogs, and monkeys (Shinazi et al., 1990) suggested that the substance was not metabolised extensively in vivo, but was excreted largely unchanged, mainly in the urine (Muzi et al., 2005). Shortly after injection, [18F]FLT is taken up by rapidly proliferating cells, especially in bone marrow and tumours (Vesselle et al., 2003; Buchmann et al., 2004). Studies in dogs suggested that during the first hour after injection, the standardised uptake value in the bone marrow reached 4.2, and significant uptake in the kidneys and urinary bladder were seen (Shields et al., 2002). (C68) Clinical PET studies with [18F]FLT have indicated that the activity is distributed relatively rapidly throughout the body and then taken up by the body tissues. The maximum uptake in kidneys, liver, and bone marrow was found within 5 min of injection. The following standardised uptake values were calculated: bone marrow, 10; liver, 4–8; kidneys, 2–6; spleen, 2–4; and lungs, 0.5–1.2 (Vesselle et al., 2003; Buchmann et al., 2004). Little information is available regarding the elimination rates from the tissues. Vesselle et al. (2003) reported that the maximum uptake in the kidneys occurred within 1.5 min, and that thereafter, 80% of the radioactivity was eliminated with a biological half-time of 0.05 h, and the remainder was lost with a longer half-time. A clinical PET study suggested that 20% of the injected [18F]FLT accumulated in the urinary bladder within 1.5 h (Vesselle et al., 2003). (C69) Vesselle et al. (2003) calculated radiation doses for men and women following intravenous injection of [18F]FLT. On the basis of the limited human data mentioned above, the following cautious biokinetic model was developed. It is assumed that immediately after intravenous injection of [18F]FLT, the uptake in the liver, bone marrow, kidneys, and spleen is 14, 10, 8, and 0.6% of the injected activity, respectively. Of the activity deposited in the kidneys, it is assumed that 75% is eliminated with a biological half-time of 0.05 h. As no good human data are available for the retention times in the various organs and tissues, a biological half-time of 24 h is assumed for all tissues. It is further assumed that 15% of the total radioactivity would be eliminated in the urine. Absorbed doses for 14C-neutral fat and free fatty acids. The physical half-life of 14C is 5730 years.

C.19.2. References for 3′-deoxy-[18F]-3′-fluorothymidine
C.20. 51Cr-labelled ethylenediaminetetraacetic acid (EDTA)
C.20.1. Biokinetic model
(C70) Intravenous administration of chromium ethylenediaminetetraacetic acid (EDTA) results in initial distribution in the extracellular fluid, and the substance is excreted exclusively by the renal system according to the model for GFR substances and the kidney–bladder model [see Sections A.6 and A.5, respectively, in Publication 53 (ICRP, 1987)] (Bröchner-Mortensen et al., 1969; Chantler et al., 1969; O’Reilly et al., 1979). (C71) In the normal case, total body retention is described by a bi-exponential function with half-times of 100 min (0.99) and 7 days (0.01). The fraction excreted by the kidneys equals 1, and the renal transit time is 5 min. (C72) For the abnormal case, it is assumed that the retention half-time of the major component is 1000 min, and the renal transit time is increased to 20 min. (C73) 51Cr-EDTA administered orally is only absorbed minimally (i.e. approximately l–5%) from the gastrointestinal tract in the normal case. In conditions of abnormal gut permeability, absorption of this substance is increased significantly (Bjarnason et al., 1983), followed by rapid clearance of the absorbed fraction from extracellular fluid by glomerular filtration. The proportion of the administered activity appearing in the urine is an indication of the degree of permeability of the gut wall. (C74) For absorbed dose calculations, the gastrointestinal tract model [Section A.3 in Publication 53 (ICRP, 1987)] was applied. In cases with increased gut permeability, the absorbed activity is excreted more rapidly than the activity remaining in the intestines. Thus, absorbed doses in abnormal cases are lower than in normal cases and, for this reason, no separate absorbed dose values are presented for these cases. Biokinetic data for 14C-labelled urea.

C.20.2. References for 51Cr-ethylenediaminetetraacetic acid
C.21. 67Ga-citrate
C.21.1. Biokinetic model
(C75) The biokinetic model given in MIRD Dose Estimate Report No. 2 (MIRD, 1973), which is based on human data, is adopted here without change. In children, the bone uptake is predominantly in the metaphyseal growth zones; this is discussed in Section A.6, Paragraph A25. (C76) The activity excreted via faeces (0.09) is assumed to have entered the bowel in the small intestine. The mean residence times in the gut are those of the standard gastrointestinal tract model [see Section A.3 in Publication 53 (ICRP, 1987)]. Absorbed doses for 14C-labelled urea. The physical half-life of 14C is 5730 years.

C.21.2. References for 67Ga-citrate
C.22. 68Ga-labelled ethylenediaminetetraacetic acid (EDTA)
C.22.1. Biokinetic model
(C77) This generator-produced positron-emitting substance is used in PET studies. After intravenous administration and initial distribution in the extracellular fluid, the substance is excreted exclusively by the renal system according to the model for GFR substances and the kidney–bladder model [see Sections A.6 and A.5, respectively, in Publication 53 (ICRP, 1987)]. Assuming normal renal function, total body retention can be described by a bi-exponential function, with component half-times of 100 min (0.99) and 7 days (0.01). The fraction excreted by the kidneys is 1.0, and the renal transit time is 5 min. Biokinetic data for 15O-water. For adults, the ratio between cumulated activity in the gonads and that in the total body is proportional to the ratio of gonad weight and total body weight. For children, the same assumption is made.

c.22.2. Reference for 68Ga-labelled ethylenediaminetetraacetic acid
C.23. 75Se-labelled amino acids (generic model)
C.23.1. Biokinetic model
(C78) The generic biokinetic model for 75Se-labelled amino acids is the same as the generic biokinetic model for 11C-labelled amino acids (see Section C.3.1). For details, please see Bergmann et al. (1995), Coenen et al. (1989), Cottrall et al. (1973), Deloar et al. (1998), ICRP (1987, 2008), Inoue et al. (1998), Schmidt et al. (1997), Shoup et al. (1999), Stenhouse and Baxter (1977), Stenstrüm et al. (1996), Taylor (2000), and Taylor et al. (1973). Absorbed doses for 15O-water. The physical half-life of 15O is 2.04 min.

C.23.2. References for 75Se-labelled amino acids (generic model)
C.24. 75Se-labelled bile acid
C.24.1. Biokinetic model
(C79) Tauroselcholic acid (SeHCAT) is a bile acid analogue used to study various aspects of the enterohepatic circulation. Following oral administration, approximately 95% of the bile acid is absorbed in normal humans (Heaton, 1976), mainly by the terminal ileum, during each enterohepatic cycle, and is almost entirely confined to the lumen of the biliary ducts, gut, and liver (Nyhlin et al., 1983). SeHCAT first appears in the gallbladder, on average, 73 min after oral administration (Jazrawi et al., 1984), and the substance undergoes enterohepatic circulation approximately five times each day (Merrick et al., 1985). The distribution of the bile acid pool in the fasting state and postprandially was measured by Jazrawi et al. (1984), and was 30%, 62%, and 8%, on average, in gallbladder, small intestine, and liver, respectively. Whole-body retention data from normal subjects (Nyhlin et al., 1983) showed that 97–100% of the bile acid was excreted with a half-time of 2.6 days and that, in most cases, a small component of approximately 3% was slowly eliminated with a mean half-time of 62 days. (C80) Based on the above data, the biokinetic model for the normal case assumes that a fraction (0.97) of orally administered 75SeHCAT circulates within the enterohepatic system, and that a fraction (0.95) of this is absorbed by the terminal ileum during each cycle. The mean transit time through the small intestine prior to absorption is assumed to be 3 h and, on the basis of bile acid pool distribution, the transit times through liver and gallbladder are 0.4 and 1.4 h, respectively. These conditions lead to a total body retention half-time of 2.7 days. The small fraction of the substance transferred to the large intestine on each cycle is excreted according to the gastrointestinal tract model. The residual fraction (0.03) of the administered substance is assumed to be distributed uniformly in the total body and retained with a half-time of 62 days. (C81) In most clinical investigations for which this substance is used (e.g. Crohn’s disease), the effects of impaired ileal absorption and shorter gastrointestinal transit time tend to reduce the dose commitment compared with the normal case. However, in patients with severe cholestatic jaundice, the liver dose has been estimated to be approximately 100 times the normal value (Soundy et al., 1982). Biokinetic data for 18F-labelled amino acids (generic model).

C.24.2. References for 75Se-labelled bile acid
C.25. 82Rb-chloride
C.25.1. Biokinetic model
(C82) 82Rb-chloride is a clinical PET perfusion tracer. Myocardial perfusion imaging with 82Rb may be performed under both rest and stress conditions. The biokinetics and absorbed doses of organs have been reported to differ between stress and rest studies. The differences between stress and rest are, in almost all cases, so small that, considering the overall uncertainties, only one set of biokinetic model and dose coefficients is discussed here. ICRP (1998) developed dosimetry for this agent, assuming a model based on relative blood flow to various tissues as a proportion of cardiac output. It was noted that this model might be conservative for some organs. The effective dose equivalent (He) [using weighting factors from Publication 26 (ICRP, 1977)] was 4.8 × 10−3 mSv MBq−1 in adults. (C83) The radiation dosimetry for this agent has also been evaluated by several authors, with considerable variation in the results. Ryan et al. (1986) collected data in two human subjects over approximately 10 min post injection using an Anger camera configured to image the 511-keV photons; they presented cumulated activity values for a limited number of organs. The data of Ryan et al. (1986) were used as the basis for the dose estimates presented in NUREG/CR-6345 (Stabin et al., 1996), which suggested an He of 1.2 × 10−3 mSv MBq−1 (for adults). (C84) There was concern in the nuclear medicine community about the significant difference between these two dose estimates. Stabin (2010) performed an analysis of the two models, and introduced a third model, by Leggett et al. (1996), which proposed cumulated activity values for most organs of the body based on a blood flow model. The results of this model were in good agreement with those of Ryan et al. (1986) for organs that they presented. The effective dose for this model was 1.7 × 10−3 mSv MBq−1 using tissue weighting factors from Publication 60 (ICRP, 1991). (C85) In 2011, cumulated activity values were measured in patients under rest and stress conditions (Senthamizhchelvan et al., 2011) and reported for many organs. The measured values varied from approximately 0.2 to 1.5 times the values suggested by Leggett et al.’s (1996) model. After evaluation of these publications, the Task Group decided to use the data of Senthamizhchelvan et al. (2011) for organs for which contours were well defined and activity content could be quantified experimentally at various time points, and where there were large differences in the estimates between the two publications (e.g. for thyroid, heart contents, and heart wall). For bone and kidneys (accumulated and moving through to the urinary bladder), Leggett et al.'s (1996) cumulated activity data were used. The average between Senthamizhchelvan et al. (2011) and Leggett et al. (1996) was used for red marrow, stomach wall, intestine, spleen, adrenals, and other organs. The final value of effective dose using these combined values was 1.1 × 10−3 mSv MBq−1 (for adults, rest or stress). Absorbed doses for 18F-labelled amino acids (generic model). The physical half-life of 18F is 1.83 h.

C.25.2. References for 82Rb-chloride
C.26. 99mTc-apcitide
C.26.1. Biokinetic model
(C86) Apcitide is a peptide that binds to the glycoprotein IIb/IIIa receptor on the surface of activated platelets; a major component of active thrombus formation. Apcitide is used for the detection and localisation of acute venous thrombosis in the lower extremities (Taillefer et al., 2000). A biokinetic model with distribution in circulating blood and an effective half-life equal to the physical half-life is assumed. Biokinetic data for 18F-labelled brain receptor substances (generic model).

C.26.2. Reference for 99mTc-apcitide
C.27. 99mTc-labelled large colloids
C.27.1. Biokinetic model
(C87) These are defined in Section A.8 in Publication 53 (ICRP, 1987) for two types of 99mTc-labelled colloids:
large colloids (100–1000 nm) – sulphur colloid, tin colloid, micro-aggregated albumin, and phytate; and small colloids (<100 nm) – mini-/micro-aggregated albumin and antimony sulphide colloid. Absorbed doses for 18F-labelled brain receptor substances (generic model). The physical half-life of 18F is 1.83 h.
Due to the short physical half-life of 99mTc, it is assumed that no redistribution or excretion occurs.

C.27.2. Reference for 99mTc-labelled large colloids
C.28. 99mTc-labelled small colloids (intratumoural injection)
C.28.1. Biokinetic model
(C88) The typical procedure is to inject approximately 20 MBq 99mTc-colloid immediately adjacent to the breast tumour that is later to be removed. The patient is investigated with a gamma camera 4 h after injection and then operated on for the removal of the tumour very shortly afterwards. If no uptake of 99mTc in the lymph nodes is seen on the scan, the tumour and the site(s) of injection of the radioactivity are removed surgically. If lymph node uptake of activity is found, a more radical operation is performed. For more detailed information, the reader is referred to Bronskill (1983), Eshima et al. (2000), and Wilhelm et al. (1999). (C89) In either situation, the injected 99mTc-colloid is removed in its entirety by approximately 6 h after injection (this may be extended to 18 h in some circumstances). The only significant absorbed dose of radiation is that to surrounding tissues, mainly lung, as a result of irradiation from the local deposit of radionuclide in the breast during the few hours of exposure. This dose is generally considered to be very small. (C90) Current ICRP dosimetric models do not permit calculations of dose from breast as a source organ, and because the doses are likely to be very small, the Task Group does not consider it necessary to develop a new dosimetric model in which breast is treated as a source organ. (C91) Leakage of radionuclide from the injection site into the systemic circulation is not considered likely; should it happen, such leakage would be covered by the existing 99mTc-colloid model. Biokinetic data for 18F-choline.

C.28.2. References for 99mTc-labelled small colloids (intratumoural injection)
C.29. 99mTc-dimercaptosuccinic acid (DMSA)
C.29.1. Biokinetic model
(C92) Intravenous injection of 99mTc-dimercaptosuccinic acid (DMSA) gives rise to an initial distribution in the extracellular fluid. Of material entering the extracellular fluid, half is deposited in the renal cortex, where it is retained for a long time. A further fraction is temporarily retained in liver and spleen. Excretion is exclusively via the kidneys. For details, the reader is referred to Arnold et al. (1975), Elliott et al. (1976), Enlander et al. (1974), and Handmaker et al. (1975). (C93) Total body retention is described by tri-exponential functions. A fraction of 0.5 is taken up in the renal cortex, with an uptake half-time of 1 h, and is assumed to be retained permanently. Fractions of 0.1 and 0.01 are taken up in liver and spleen, respectively, with a half-time of 1 h, and eliminated with half-times of 2 h (0.5) and 1.8 days (0.5). Absorbed doses for 18F-choline. The physical half-life of 18F is 1.83 h.
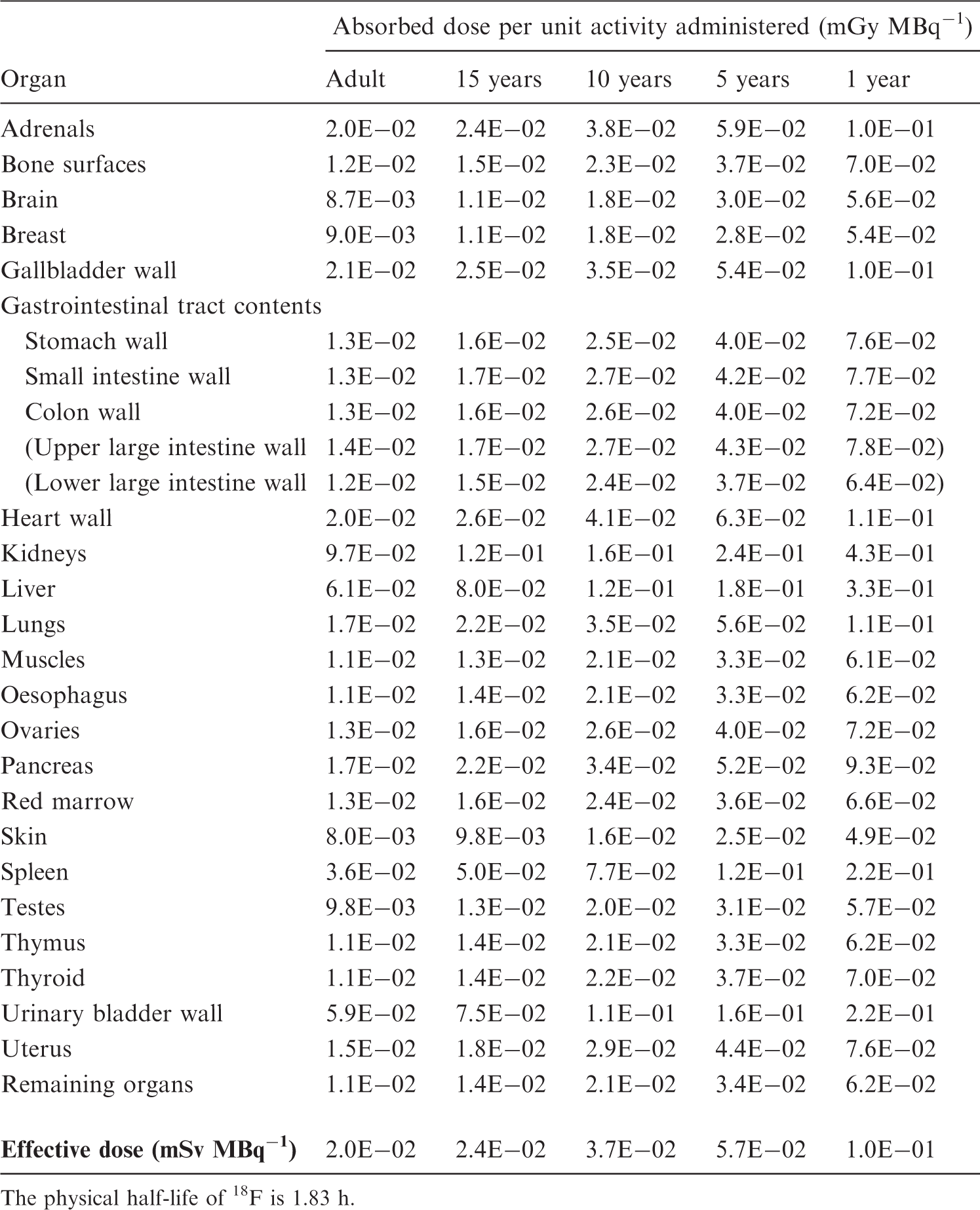
C.29.2. References for 99mTc-dimercaptosuccinic acid
C.30. 99mTc-diethylenetriaminepentaacetic acid (DTPA)
C.30.1. Biokinetic model
(C94) Intravenous administration of Tc-diethylenetriaminepentaacetic acid (DTPA) gives rise to an initial distribution in the extracellular fluid. Following this initial distribution phase, the substance is excreted exclusively by the renal system according to the model for GFR substances and the kidney–bladder model [see Sections A.6 and A.5, respectively, in Publication 53 (ICRP, 1987)] (Klopper et al., 1972; McAfee et al., 1979; O’Reilly et al., 1979). (C95) In the normal case, total body retention is described by a bi-exponential function with component half-times of 100 min (0.99) and 7 days (0.01). The fraction excreted by the kidneys is 1.0 (1.0), and the renal transit time is 5 min. (C96) For the abnormal case, it is assumed that the retention half-time of the major component is 1000 min, and the renal transit time is increased to 20 min. Biokinetic data for 18F-fluoro-2-deoxy-d-glucose.

C.30.2. References for 99mTc-diethylenetriaminepentaacetic acid
C.31. 99mTc-ethylenedicysteine (EC)
C.31.1. Biokinetic model
(C97) 99mTc-ethylenedicysteine (EC) is used for renal studies. The biokinetic behaviour of the substance is very similar to that of Hippuran (sodium orthoiodohippurate) with a half-time in the total body of 25 min (ICRP, 1987). The cumulated amount found in urine at different times is as follows: 40 min, 70%; 60 min, 80%; and 90 min, 95% (Surma et al., 1994; Surma, 1998a, 1998b; Liniecki, 1998). The clearance is approximately 70% of that of Hippuran. Absorbed doses for 18F-fluoro-2-deoxy-d-glucose. The physical half-life of 18F is 1.83 h.

C.31.2. References for 99mTc-ethylenedicysteine
C.32. 99mTc-ethylenedicysteine diester (ECD, Neurolite)
C.32.1. Biokinetic model
(C98) N,N’-1,2-ethylenediylbis-l-cysteinediethylester, (ECD) (Neurolite), labelled with 99mTc, is a neutral lipophilic complex that crosses the intact blood–brain barrier rapidly and is retained in the brain for a long time, making it possible to perform detailed tomographic studies of regional cerebral blood flow. (C99) Kinetic data from humans (Holman et al., 1989; Vallabhajosula et al., 1989; Léveillé et al., 1992) have shown that the substance is cleared rapidly from the blood after intravenous injection. Uptake in the brain reaches a maximum of 4.9–6.5% within 1 min, and remains relatively constant over several hours. Early whole-body imaging also shows uptake in lungs, liver, kidneys, and thyroid. (C100) In the model, it is assumed that there is immediate cellular uptake of the substance in the brain (0.06), lungs (0.06), liver (0.20), kidneys (0.10), and thyroid (0.003). The activity in the brain is cleared bi-exponentially with half-times of 1 h (0.40) and 1.5 days (0.60). At 48 h, approximately 80% has been excreted in urine and 20% in faeces. Activity in the liver is assumed to be excreted through the intestines, partly via the gallbladder, according to the model described in Section A.9 in Publication 53 (ICRP, 1987). The rest of the activity is assumed to be excreted via the kidneys and urinary bladder. (C101) For children, the fractional uptake in the brain is higher due to the relative weight of the brain (Barthel et al., 1997). Biokinetic data for 2-[18F]-fluoroethyl-l-tyrosine.

C.32.2. References for 99mTc-ethylenedicysteine diester
C.33. 99mTc-furifosmin (Q12)
C.33.1. Biokinetic model
(C102) [Trans-1,2-bis(dihydro-2,2,5,5-tetramethyl-3(2H)furanone-4-methyleneimino ethane) bis tris(3-methoxy-1propyl)–phosphine] technetium (III)-99 m is a non-reducible complex of Tc(III). 99mTc-furifosmin is prepared from a freeze-dried kit (TechneCard) and is used for studies of myocardial perfusion. (C103) 99mTc-furifosmin accumulates in viable myocardial tissue in proportion to regional blood flow in a manner similar to thallous chloride. After intravenous injection, the substance is cleared rapidly from the blood (<5% left by 20 min) and taken up predominantly in muscular tissues (including heart), liver, and kidneys. Biodistribution is generally similar to that of 99mTc-methyl oxy-isobutyl-isonitrile (MIBI, Cardiolite) (ICRP, 1991) and 99mTc-tetrofosmin (Myoview; ICRP, 1998), but there are some differences which have a bearing on diagnostic technique. 99mTc-furifosmin shows a heart uptake of 1.2–2.4%. It is cleared rapidly from the liver (<6.5% left by 1 h), and lung uptake is low (4%). More than 50% of the substance has entered excretory pathways by 24 h and the faecal:urinary excretion ratio is 60:40. (C104) It is assumed that the fractions of the substance taken up by the liver and kidneys are excreted in faeces and urine, respectively. When the substance is injected in conjunction with an exercise stress test, there is little change in heart uptake, and biodistribution is similar to that observed at rest. The initial rate of urinary clearance is lower than at rest, and the same faecal:urinary excretion ratio is assumed. (C105) The quantitative figures for uptake and excretion in man, presented in Table C.66, are based on the rest and exercise studies of Rossetti et al. (1994) with supplementary information from Gerson et al. (1994). Substance excreted by the hepatobiliary system is assumed to leave the body via the intestinal tract according to the ICRP model for the gastrointestinal tract (ICRP, 1979). (C106) The biodistribution and excretion data used to derive this model were based on a small number of subjects (seven at rest and three after exercise), with the result that significant differences between rest and exercise data could not be established. The difference between the biokinetic data tables presented for rest and exercise studies is therefore largely based on experience of models for similar 99mTc-labelled myocardial perfusion imaging agents such as MIBI and tetrofosmin. Absorbed doses for 2-[18F]-fluoroethyl-l-tyrosine. The physical half-life of 18F is 1.83 h.

C.33.2. References for 99mTc-furifosmin
C.34. 99mTc-labelled human immunoglobulin (HIG)
C.34.1. Biokinetic model
(C107) Labelled non-specific (polyclonal) human immunoglobulin of IgG-type (HIG) is used to localise and image focal sites of infection and inflammation in the body. Increased vascular permeability and trapping of HIG by inflammatory cells is believed to play a role in the accumulation of radioactivity in the foci. (C108) Native IgG has a half-time in the body of approximately 23 days (Solomon et al., 1963). After labelling with 99mTc or 111In, a much shorter half-time of the radioactivity in the body is observed, probably due to some alteration of the protein during the labelling procedure and to partial dissociation of the label from the carrier protein. (C109) After intravenous injection, the initial distribution is determined by the blood content of the organs. The early whole-body image therefore shows the heart, major blood vessels, lungs, liver, spleen, kidneys, mucosae of the nose and vagina, and the external genitalia. There is also early activity in the bladder. Blood activity falls slowly due to the combined effect of distribution of HIG into extravascular spaces, uptake and excretion via the kidneys of some dissociated label, and some active uptake in the liver. As a consequence, delayed images (at 24 h) are dominated by activity in liver and kidneys, while activity in other blood-rich organs has decreased. There is no visible uptake in the bone marrow, and activity in the intestines is only seen occasionally. The urine contains 27–50% of administered activity after 24 h. For detailed information, see Buscombe et al. (1990), Corstens and Claessens (1992), Datz et al. (1995), Hovi et al. (1993), Kinne et al. (1993), Saptogino et al. (1991), and Sciuk et al. (1991). (C110) The dosimetric model assumes an initial blood pool distribution, with organ blood content according to Publication 23 (ICRP, 1975). There is rapid uptake (T1/2 = 1 h) in the liver (5%) and kidneys (8%); the kidney contents are excreted in the urine with a half-time of 6 h. The rest of the blood activity falls with a half-time of 12 h because of distribution into other organs and tissues (50%) and direct excretion in the urine (37%). In view of the short physical half-life of the label, infinite half-time is assumed for activity remaining in the body. Biokinetic data for [18F]-fluoro-l-DOPA.

C.34.2. References for 99mTc-labelled human immunoglobulin
C.35. 99mTc-labelled hexamethylpropyleneamineoxine (HM-PAO)
C.35.1. Biokinetic model
(C111) The d,1 diastereoisomer of hexamethylpropyleneamineoxine (HM-PAO) labelled with 99mTc is a lipophilic complex that crosses the intact blood–brain barrier rapidly and is retained in the brain for a long time, making detailed tomographic studies of the regional cerebral blood flow possible. Quantitative studies in man have shown that the substance is rapidly cleared from the blood after intravenous injection. Uptake in the brain reaches a maximum of 4–6% within 1 min, and there is little loss of activity for the following 24 h. Early whole-body scans also show uptake in lungs, liver, gastrointestinal tract, kidneys, and thyroid. A great part of the activity is widely distributed throughout the body, particularly in muscle and soft tissue. Except for the brain, the activity leaves the organs and tissues within a few days by excretion in urine and faeces. At 48 h, approximately 40% has been excreted in urine and 15% in faeces. The reader is referred to Costa et al. (1986), Sharp et al. (1986), Soundy et al. (1990), and Vestergren et al. (1991) for further information. (C112) In the model, it is assumed that there is an immediate cellular uptake of the substance in the brain (0.05), lungs (0.10), liver (0.15), gastrointestinal tract (0.05), kidneys (0.09), and thyroid (0.008). The uptake in gastrointestinal walls (0.05) is assumed to be apportioned according to the relative weights of the walls. The activity of the labelled radiopharmaceutical is cleared from the brain mono-exponentially with a half-time of 4 days, while most of the other organs and tissues are cleared bi-exponentially. Activity in the liver is assumed to be excreted to the intestine, partly via the gallbladder, according to the model described in Section A.9 in Publication 53 (ICRP, 1987). (C113) For children, the fractional uptake in the brain is higher due to the higher relative weight of the brain. In some cases, uptake in the lacrimal gland has also been seen. Villanueva-Meyer et al. (1990) reported significant uptake in 15 of their 138 adult patients, resulting in an absorbed dose of 0.014 mGy MBq−1 to the lacrimal gland. This does not influence the effective dose value. Absorbed doses for [18F]-fluoro-l-DOPA. The physical half-life of 18F is 1.83 h. The urinary bladder wall contributes 51% of the effective dose.

C.35.2. References for 99mTc-labelled hexamethylpropyleneamineoxine
C.36. 99mTc-labelled iminodiacetic acid derivatives (HIDA, etc.)
C36.1. Biokinetic model
(C114) Radiopharmaceuticals belonging to this group are predominantly taken up in the liver and excreted, via the biliary tract, to the intestine. A minor fraction is excreted by the kidneys. The individual compounds are based on N-substitution of IDA, and are named according to type of substitute [e.g. BIDA, DISIDA (disofenin), EIDA, HIDA, PBIDA, PIPIDA, and mebrofenin]. (C115) The dosimetry model has been presented in Section A.9 in Publication 53 (ICRP, 1987). Calculations are performed for the normal case, and for three pathological conditions, namely parenchymal liver disease, cholecystitis with occlusion of the cystic duct, and occlusion of the common bile duct. The assumed magnitude and rate of the different flows in the model are evident from the FS and T values in Table C.72. The final excretion from the body follows the model for the gastrointestinal tract [Section A.3 in Publication 53 (ICRP, 1987)] and the kidney–bladder model [Section A.5 in Publication 53 (ICRP, 1987)]. In the atresia case, it is assumed that activity initially taken up in the liver is transported slowly (T1/2 = 8 days) back to blood for excretion by the kidneys. Biokinetic data for 18F-fluoride.

C36.2. Reference for 99mTc-labelled iminodiacetic acid derivatives
C.37. 99mTc-labelled macro-aggregated albumin (MAA)
C.37.1. Biokinetic model
(C116) The model for 99mTc-labelled macro-aggregated albumin (MAA) is the same as that used for iodine-labelled MAA [see p. 293 in Publication 53 (ICRP, 1987)], with the modification that released technetium is assumed to be excreted by the kidneys according to the model proposed for pertechnetate when a blocking agent has been given. Absorbed doses of 18F-fluoride. The physical half-life of 18F is 1.83 h.

C.37.2. Reference for 99mTc-labelled macro-aggregated albumin
C.38. 99mTc-labelled mercaptoacetyl triglycine (MAG3)
C.38.1. Biokinetic model
(C117) 99mTc-labelled mercaptoacetyl triglycine (MAG3) has been developed as a possible replacement for radioiodine-labelled Hippuran for investigation of renal function with improved imaging quality. (C118) In the normal case, following intravenous administration of MAG3, the substance is rapidly distributed in the extracellular fluid and excreted entirely by the renal system according to the kidney–bladder model. Total body retention is described by tri-exponential functions (Stabin et al., 1992). The renal transit time is assumed to be 4 min, as for Hippuran. The reader is referred to Bubeck et al. (1990), Jafri et al. (1988), and Taylor et al. (1986) for further information. (C119) When renal function is bilaterally impaired, it is assumed that the clearance rate of the substance is one-tenth of that for the normal case, that the renal transit time is increased to 20 min, and that a fraction of 0.04 is taken up in the liver. (C120) As an example of acute unilateral renal blockage, it is assumed that a fraction of 0.5 of the administered radiopharmaceutical is taken up by one kidney, slowly released to the blood with a half-time of 5 days, and subsequently excreted by the other kidney, which is assumed to function normally. Biokinetic data for 3′-deoxy-[18F]-3′-fluorothymidine.

C.38.2. References for 99mTc-labelled mercaptoacetyl triglycine
C.39. 99mTc-labelled non-absorbable markers
C.39.1. Biokinetic model
(C121) Substances labelled with technetium are used as non-absorbable markers in studies of the gastrointestinal tract. For absorbed dose calculations, a modified ICRP model for the gastrointestinal tract is used, as described in Section A.3 in Publication 53 (ICRP, 1987). The reader is referred to Chadhuri (1974), Fisher et al. (1976), and Meyer et al. (1976) for further information. Absorbed doses of 3′-deoxy-[18F]-3′-fluorothymidine. The physical half-life of 18F is 1.83 h.

C.39.2. References for 99mTc-labelled non-absorbable markers
C.40. 99mTc-labelled methoxy-isobutyl-isonitrile (MIBI, Sestamibi, Hexamibi)
C.40.1. Biokinetic model
(C122) Technetium-methyl oxy-isobutyl-isonitrile (MIBI, Sestamibi, Hexamibi) is a cationic complex prepared from a lyophilised kit (Cardiolite). It is used for studies of myocardial perfusion and cardiac ventricular function. (C123) 99mTc MIBI is accumulated in viable myocardial tissue in proportion to regional blood flow in a manner similar to thallous chloride. After intravenous injection, the substance is cleared rapidly from the blood and taken up predominantly in muscular tissues (including heart), liver, and kidneys, with a smaller amount in salivary glands and thyroid. Other organs and tissues show low uptake with a uniform distribution. When the substance is injected in conjunction with a stress test, there is a considerable increase of uptake in heart and skeletal muscles, with correspondingly lower uptake in all other organs and tissues. No redistribution takes place, and there is no evidence of any metabolism of the substance. The principal pathway for excretion is via the hepatobiliary system to the gastrointestinal tract, with some additional excretion via the kidneys. Results of animal studies do not indicate direct uptake and excretion via the gastrointestinal wall (Andersson et al., 1990). The major part of the injected substance is excreted within 48 h. (C124) The quantitative figures for uptake and excretion in man, presented in Table C.80, are based on reports by Wackers et al. (1988) and Leide et al. (1992). Substance excreted by the hepatobiliary system is assumed to leave the body via the intestinal tract according to the Publication 30 gastrointestinal tract model (ICRP, 1979). The kidney–bladder model presented in Publication 53 (ICRP, 1987) is used for substance excreted in urine. It is further assumed that all substance leaving organs and tissues with half-times stated in Table C.80 is excreted by the liver (75%) and kidneys (25%). Biokinetic data for 51Cr-ethylenediaminetetraacetic acid.

C.40.2. References for 99mTc-labelled methoxy-isobutyl-isonitrile
C.41. 99mTc-labelled monoclonal tumour-associated antibodies
C.41.1. Biokinetic model
(C125) Radiolabelled monoclonal antibodies against antigenic substances within or on the surface of malignant cells are used in medical research and for diagnosis and treatment of cancer. The antibody is an immunoglobulin, usually IgG1 or IgG2a, and is used either as the intact molecule (molecular weight 150 kDa) or as fragments F(ab’)2 (100 kDa) and F(ab’) (50 kDa). Antibodies against a large number of tumour-associated antigens have been produced and investigated, but only a few are in regular use as commercial products for diagnostic purposes. (C126) There is great variation in production with regard to type of antigen, type of cells used (mouse, goat, human, etc.), and possible genetic modification (chimeric, humanised). There is also variation in the mode of application of the product with regard to amount of substance administered, possible pre-treatment with unlabelled antibody or other modifying substances, route of administration (intravenous injection or infusion, subcutaneous or intraperitoneal injection, etc.), type of radionuclide used as label, and method of labelling. (C127) In spite of these variations, certain common features in the behaviour of the antibodies can be distinguished. Directly after intravenous injection, the highest activity is seen in organs with high vascular perfusion, such as liver, spleen, bone marrow, and kidneys. Organ uptake is mainly a function of molecular size, with the intact molecule showing uptake mainly in liver and bone marrow, while smaller fragments concentrate to a greater degree in the kidneys. Also, the rate of degradation and elimination is mainly a function of molecular size, being more rapid with smaller fragments (Bischof Delaloye and Delaloye, 1995; Britton and Granowska, 1987; Fishman et al., 1989; ICRP, 1987). (C128) Based on these general properties, a set of models can be defined, assuming principal uptake in the above-mentioned organs and even distribution of the remainder in the rest of the body. The quantitative data for uptake and elimination have been defined after an extensive survey of published reports, and are to be looked upon as typical values for the intact antibody and ‘large’ and ‘small’ fragments. (C129) The antibodies and fragments are metabolised within the body. The technetium thus set free is assumed to be handled by the body according to the biokinetic model for pertechnetate. The contribution from released technetium can be calculated as:
Absorbed doses for 51Cr-ethylenediaminetetraacetic acid. The physical half-life of 51Cr is 27.7 days. The urinary bladder wall contributes up to 60% of the effective dose.


C.41.2. References for 99mTc-labelled monoclonal tumour-associated antibodies
C.42. 99mTc-Pertechnegas
C.42.1. Biokinetic model
(C130) Pertechnegas is a 99mTc-labelled sodium chloride aerosol that is soluble because it lacks the carbon coating of Technegas, and has a median particle diameter of 167 nm (Lloyd et al., 1995). Pertechnegas is thus a modified form of Technegas, produced by heating 99mTc pertechnetate in argon containing 3% oxygen. Following inhalation, Pertechnegas shows lung clearance properties similar to those of a 99mTc-pertechnetate aerosol. Approximately 75% of inhaled Pertechnegas is lost from the lungs with a half-time of 9–11 min in both smokers and non-smokers. The remainder of the material appears to leave the lungs with a half-time of 2–3 h (Isawa et al., 1996; Kotzerke et al., 1996). The material leaving the lungs is assumed to enter the blood as pertechnetate. (C131) The biokinetic model for Pertechnegas assumes that 75% of the total inhaled activity is lost from the lungs with a half-time of 10 min; the remaining 25% leaves the lungs with a half-time of 160 min. All of the activity leaving the lungs is assumed to be absorbed to blood and to behave as intravenously injected 99mTc-pertechnetate. Biokinetic data for 67Ga-citrate.

C.42.2. References for 99mTc-Pertechnegas
C.43. 99mTc-pertechnetate
C.43.1. Biokinetic model
(C132) The MIRD Dose Estimate Report No. 8 (MIRD, 1976) presents two sets of biological parameters based on a compartmental model and constructed using data from measurements on different groups of subjects. The two groups are possibly related to different levels of physical activity (resting and non-resting). For most organs and tissues, the difference in absorbed dose per unit administered activity between the two groups is small (less than a factor of two). (C133) The following model is based on mean values for the parameters in the two MIRD groups. Data published by Dayton et al. (1969) on renal clearance, by Beasley et al. (1966) on distribution in humans, and by Andros et al. (1965) have also been used. All published studies have demonstrated early active uptake in the thyroid, salivary glands, and stomach, and delayed uptake in the colon. The remaining fraction of administered activity is assumed to be distributed uniformly throughout all other organs and tissues (except brain). Elimination is by way of the kidneys and intestines. (C134) Pre-treatment with blocking agents such as perchlorate or iodide inhibits active uptake and diminishes whole-body retention (Coffey et al., 1984). The model for this case therefore assumes uniform distribution and a higher rate of renal excretion than in the standard model set out above. (C135) For oral administration, the fractional absorption is taken to be 0.8 (ICRP, 1980). Absorbed doses for 67Ga-citrate. The physical half-life of 67Ga is 3.26 days.

C.43.2. References for 99mTc-pertechnetate
C.44. 99mTc-labelled phosphates and phosphonates
C.44.1. Biokinetic model
(C136) This group of radiopharmaceuticals includes phosphates, such as pyrophosphate and polyphosphate, and phosphonates such as methylene diphosphonate (medronate), hydroxymethylene diphosphonate (oxidronate), hydroxyethylidene diphosphonate, ethane-hydroxy diphosphonate, dicarboxypropane diphosphonate, imido diphosphate, and similar compounds used for bone imaging. The biokinetic behaviour of these substances is sufficiently similar to justify the use of a common biokinetic model. (C137) The main uptake is in bone, with further small uptake in kidneys, and excretion is via the renal system. On the basis of the references given below (Ackerhalt et al., 1974; Krishnamurthy et al., 1975; Makler and Charkes, 1980; Rudd et al., 1977; Subramanian et al., 1975), it is assumed that a fraction of 0.5 of the injected activity is taken up by bone with a half-time of 15 min, and retained there with half-times of 2 h (0.3) and 3 days (0.7). In children, uptake is predominantly in the metaphyseal growth zones; this question is discussed in Section A.6, paragraph A25. Kidney uptake is set at 0.02 with retention identical to that of the total body, having half-times (with fractional retention) of 0.5 h (0.3), 2 h (0.3), and 3 days (0.4). (C138) In pathological cases, there may be higher uptake and/or longer retention in bone, especially in kidney diseases. The 24-h total body retention, which normally amounts to 30%, has been reported as 40% in osteomalacia, 50% in primary hyperparathyroidism, 60% in Paget’s disease, and 90% in renal osteodystrophia (Fogelman et al., 1978). For absorbed dose calculations in pathological cases, average bone uptake of 70% is assumed, with no excretion. Biokinetic data for 68Ga-labelled ethylenediaminetetraacetic acid.

C.44.2. References for 99mTc-labelled phosphates and phosphonates
C.45. 99mTc-labelled erythrocytes
C.45.1. Biokinetic model
(C139) Erythrocytes can be labelled with 99mTc in vitro, in vivo, or with a combined in-vitro/in-vivo method (Callahan et al., 1982). Several studies have demonstrated some elution of technetium from the circulating cells, with half-times of 40 and 80 h after in-vitro or in-vivo labelling, respectively. The exact mechanism of elution and the fate of the eluted technetium is not known, but approximately 15% of the activity is excreted in the urine during the first day (Porter et al., 1983). The reader is referred to Dahlström et al. (1979), Larson et al. (1978), and Ryo and Pinsky (1976) for further information. (C140) In the model chosen, the activity is assumed to be distributed in the blood, being removed with a half-time of 60 h by excretion via the kidneys. No specific uptake in any organ or tissue is assumed. The model assumes 100% efficiency in labelling of the erythrocytes. In the case of incomplete labelling, the separate contribution from free pertechnetate has to be taken into account. Absorbed doses for 68Ga-labelled ethylenediaminetetraacetic acid. The physical half-life of 68Ga is 68.06 min.

C.45.2. References for 99mTc-labelled erythrocytes
C.46. 99mTc-Technegas
C.46.1. Biokinetic model
(C141) Technegas is used for ventilation lung scintigraphy. It is an aerosol incorporating 99mTc atoms that is prepared by evaporating sodium 99mTc-pertechnetate in normal saline to dryness in a graphite crucible. The crucible is then heated at 2500℃ for 15 s in an atmosphere of pure argon (Burch et al., 1986). Technegas appears to consist of 99mTc atoms attached to carbon particles with a median diameter of 140–160 nm (Strong and Agnew, 1989; Lloyd et al., 1995; Isawa et al., 1996). Following inhalation, Technegas shows good penetration to the lung periphery, and the material is deposited on the lung parenchyma, where it is retained with a half-time that is long compared with the physical half-life of 99mTc (Burch et al., 1986; Isawa et al., 1991). The observed deposition of Technegas in the bronchial airways is approximately 5% (Lloyd et al., 1995), and the biological retention in the pulmonary tissue amounts to 85% at 24 h (Isawa et al., 1991). (C142) The biokinetic model for Technegas assumes that 95% of the inhaled material is deposited in the lungs, with 5% in the main bronchial airways. The inhaled material is assumed to be lost from the pulmonary tissue with a biological half-time of 4 days. The material deposited in the bronchii is assumed to be elevated by the ciliary escalator and swallowed. The material absorbed from the gastrointestinal tract is assumed to behave as orally administered 99mTc-pertechnetate (ICRP, 1987). Biokinetic data for 75Se-labelled amino acids (generic model). For [75Se]-selenomethionine, the compound-specific data (ICRP, 1987) should be used.

C.46.2. References for 99mTc-Technegas
C.47. 99mTc-labelled tetrofosmin
C.47.1. Biokinetic model
(C143) Technetium-99m-1,2-bis[bis(2-ethoxyethyl)phosphino]ethane is a lipophilic technetium phosphine dioxo cation {[99mTc-(tetrofosmin)2 O2]+} prepared from a freeze-dried kit (Myoview). It is used for studies of myocardial perfusion. (C144) 99mTc-tetrofosmin accumulates in viable myocardial tissue in proportion to regional blood flow in a manner similar to thallous chloride. After intravenous injection, the substance is cleared rapidly from the blood (<5% left by 10 min) and taken up predominantly in muscular tissues (including heart), liver, kidneys, and salivary glands, with a smaller amount in the thyroid. Biodistribution is generally similar to that of 99mTc-MIBI (Cardiolite) [Publication 62 (ICRP, 1991)], but there are some differences that have a bearing on diagnostic technique. 99mTc-tetrofosmin shows heart uptake of 1.2%, and is cleared very rapidly from the liver (<4.5% left by 1 h) and lungs. More than 80% of the substance is excreted in 48 h, and the faecal:urinary excretion ratio is 54:46. When the substance is injected in conjunction with an exercise stress test, there is a considerable increase in uptake in skeletal muscle but little change in heart uptake. Initial rates of urinary and faecal clearance are lower than at rest, and the faecal:urinary excretion ratio is 46:54. (C145) The quantitative figures for uptake and excretion in man, presented in Table C.94, are based on reports by Smith et al. (1992) and Higley et al. (1993). Substance excreted by the hepatobiliary system is assumed to leave the body via the intestinal tract according to the Publication 30 gastrointestinal tract model (ICRP, 1979). The kidney–bladder model presented in Publication 53 (ICRP, 1987) is used for substance excreted in urine. Absorbed doses for 75Se-labelled amino acids (generic model). The physical half-life of 75Se is 119.8 days. For [75Se]-selenomethionine, the compound-specific data (ICRP, 1987) should be used.

C.47.2. References for 99mTc-labelled tetrofosmin
C.48. 99mTc-labelled leukocytes
C.48.1. Biokinetic model
(C146) The same model is used as for indium-labelled leukocytes [see p. 255 in Publication 53 (ICRP, 1987)], with the exception that, in view of the short physical half-life, the retention half-times are set to infinity. For further information, the reader is referred to Hanna et al. (1984), Kelbaek et al. (1985), and Schroth et al. (1981). Biokinetic data for 75Se-labelled bile acid.

C.48.2. References for 99mTc-labelled leukocytes
C.49. 111In-labelled human immunoglobulin (HIG)
C.49.1. Biokinetic model
(C147) 111In-labelled human immunoglobulin (HIG) behaves in principle in the same way as described in the biokinetic model for technetium-labelled HIG, i.e. an initial blood pool distribution, followed by some active accumulation in liver and kidneys, some additional uptake in spleen, and some direct excretion of radioactivity in the urine. The model has been constructed based on data in the literature (Buijs et al., 1990; Claessens et al., 1994; Datz et al., 1995; Fishman et al., 1988; Morrel et al., 1989; Oyen et al., 1990). Compared with the technetium-labelled substance, blood clearance is slower (T1/2 = 24 h for the main part), liver and spleen uptake is somewhat higher, and kidney uptake and urinary excretion are somewhat lower. Early excretion in urine is set to 20%, and it is assumed that there is slow excretion of remaining activity in the body with the same half-time as found for indium in ionic form (i.e. 70 days). Absorbed doses for 75Se-labelled bile acid. The physical half-life of 75Se is 119.8 days.

C.49.2. References for 111In-labelled human immunoglobulin
C.50. 111In-labelled monoclonal tumour-associated antibodies
C.50.1. Biokinetic model
(C148) The models for indium-labelled monoclonal tumour-associated antibodies and fragments are the same as those used for the corresponding technetium-labelled substances, with the modification that released indium is handled by the body according to the model for ionic indium (Bischof Delaloye and Delaloye, 1995; Britton and Granowska, 1987; Fishman et al., 1989; ICRP, 1987). This biokinetic model is not intended to apply to therapeutic use of the substance. Biokinetic data for 82Rb-chloride.

C.50.2. References for 111In-labelled monoclonal tumour-associated antibodies
C.51. 111In-labelled octreotide
C.51.1. Biokinetic model
(C149) 111In-DTPA- (C150) The biokinetic model is based on scintigraphic studies of a total of 24 humans by Krenning et al. (1992), Forssell Aronsson et al. (1995, 1999), and Leide-Svegborn et al. (1996). Tissue samples have been analysed by Forssell Aronsson et al. (1995). These studies have demonstrated uptake in liver, spleen, kidneys, and thyroid. In some patients, there may also be uptake in the pituitary gland. For further information, the reader is referred to Bajc et al. (1994), Krenning et al. (1993), and Stabin et al. (1997). There is wide variation in uptake values between the subjects. The main route of excretion is via the kidneys, and less than 2% is excreted in faeces. Although some degradation seems to occur, the great majority of activity excreted in urine is still peptide bound, even after 48 h. The biokinetic data come from patients with carcinoid tumours and neuroendocrine tumours in the gastrointestinal tract. Uptake in tumour tissue present in any given organ may therefore be included in the published organ uptake values. (C151) Intravenously injected 111In-DTPA- (C152) An observed excretion of 85% via urine after 24 h fits well with the model proposed. Absorbed doses for 82Rb-chloride. The physical half-life of 82Rb is 1.27 min.

C.51.2. References for 111In-labelled octreotide
C.52. 123I, 124I, 125I, and 131I-iodide
C.52.1. Biokinetic model
(C153) The kinetic behaviour of iodide has been studied extensively, and several biokinetic models have been suggested (Riggs, 1952; Berman et al., 1968; MIRD, 1975; Smith, 1988; Zanzonico, 2000; Johansson et al., 2003; Leggett, 2010). These may be described either as compartment models or in terms of fractional uptakes and half-times. The model presented in Publication 53 (ICRP, 1987) is a highly simplified kinetic model presenting the absorbed dose for different thyroid uptakes, expressed as the fractional distribution of iodide in the thyroid, Fs, in the range 0.05–0.55. ICRP has published another model to be used for members of the public, including occupational exposure to radioiodine, in Publications 56 and 67 (ICRP, 1990, 1993), and this model is also a greatly simplified model, expressed as half-times and fractional distributions. In addition to these models, Publication 88 (ICRP, 2001) presents a more complex compartment model for the biokinetics of iodine in a pregnant mother and the fetus, with the aim of calculating the dose to the embryo and fetus. (C154) The biokinetic model adopted for the present report was developed by Leggett (2010). It is described as a compartment model including inorganic iodide as well as organically bound iodine released to the body tissues following discharge from the thyroid. For a more comprehensive discussion and description of the model, the reader is referred to the original paper (Leggett, 2010). Applying the transfer coefficients proposed by Leggett (Table C.104) results in 26% uptake of 131I in the thyroid 24 h after administration. The same 24-h uptake is obtained with the Publication 53 model if a fractional distribution in the thyroid, Fs, of 0.33 is used (ICRP, 1987). The adopted model is also well in accordance with the ICRP model for occupational exposure (ICRP, 1990), where it is assumed that 0.3 of iodine entering the blood is taken up by the thyroid. (C155) The Leggett model, as presented in Fig. C.2, has, for the purpose of dose calculations, been adjusted in order to be congruent with other radiopharmaceutical models. The modifications are included in Table C.104.
For the biokinetics of the gastrointestinal tract, the HAT model (ICRP, 2006) has been used. The resulting cumulated activities in the different parts of the colon have, for dose calculations thereafter, been redistributed to ULI and LLI, as defined by the old gastrointestinal tract model (ICRP, 1979). An organ ‘kidneys 3’ has been introduced for the transfer of iodide to the urinary bladder. The mean residence time in ‘kidneys 3’ is 5 min, which is in accordance with other models for radiopharmaceutical dosimetry (ICRP, 1998). (C156) The uptake of iodide in the normal thyroid depends on the dietary intake of stable iodine (Stanbury et al., 1954; Zvonova, 1989) which varies in different regions of the world. In order to take this variation into account, the Task Group has chosen to include dose data for typical ‘low’ and ‘high’ thyroid uptake of iodide. This has been accomplished by adjusting the transfer coefficient for thyroid uptake (i.e. from blood to thyroid), aiming at 24-h uptake of 16% and 36%, respectively, for 131I. For the Publication 53 model (ICRP, 1987), this is obtained if Fs is set to 0.20 and 0.45, respectively. All other transfer factors are independent of the uptake in thyroid, including the release of organically bound iodine from the thyroid.
Blocked thyroid
(C157) In order to obtain the cumulated activities in different organs when the thyroid has been blocked, the transfer between the compartments representing inorganic iodide in the thyroid to the compartment for organically bound iodine is set to zero. The dose to the thyroid in this case is thus only a result of uptake of iodide and irradiation from other parts of the body. Incomplete blockage will, however, increase the radiation dose to the thyroid; one may use dose data for the thyroid for different uptakes to get some idea of the magnitude of this dose.
Age dependence
(C158) There is no significant age dependence of uptake in the thyroid after the first few days after birth. However, for organically bound iodine, the biological half-time of thyroid to blood shows distinct age dependence. The half-time in adults is 90 days, and 65, 50, 30, and 15 days for 15, 10, 5, and 1 year olds, respectively (Leggett, 2015). In general, the turnover rate of organically bound iodine, also between other compartments, may be assumed to be faster (Leggett, 2015) with a transfer coefficient approximately 50% larger for the youngest ages. Biokinetic data for 99mTc-apcitide.

C.52.2. References for 123I, 124I, 125I, and 131I
C.53. 123I-labelled fatty acids
C.53.1. Biokinetic model
(C159) Free fatty acids are major energy sources for the myocardium, and iodine-labelled free fatty acids are used to study the energy metabolism of the heart. Long-chain fatty acids are taken up rapidly by the heart and metabolised by β-oxidation (Tamaki et al., 2000). The first iodine-labelled free fatty acids that were developed had the disadvantage of excessive release of radioiodide. This was overcome by the introduction of 123I-para-iodophenyl pentadecanoic acid (123I-IPPA), a terminally phenylated straight-chain fatty acid, where the iodine was substituted in the phenyl group (Machulla et al., 1980; Reske et al., 1982; Reske, 1985; Dudzcak et al., 1986). (C160) The rapid clearance of 123I-IPPA from the myocardium is, however, a problem when tomography (SPECT) is performed. This problem has been overcome by the introduction of a methyl group on the 3-carbon of the fatty acid. 3-Methyl-branched fatty acids are metabolised in the peroxisomes by initial α-oxidation followed by peroxisomal β-oxidation, a process that is slower than mitochondrial β-oxidation (Casteels et al., 2003). This principle was first used by Knapp et al. (1986) by the introduction of β-methyl-p-(123I)-iodophenylpentadecanoic acid (123I-BMIPP) [see references in Knapp and Kropp (1995)]. (C161) After intravenous injection, 123I-IPPA and 123I-BMIPP are cleared rapidly from the blood (biological half-time 2.5–3.0 min) (Knapp et al., 1995) due to fast uptake in various organs and tissues (Torizuka et al., 1991; Yoshizumi et al., 2000). Whole-body pictures shortly after the injection (Torizuka et al., 1991; Sloof et al., 1997; Caveliers et al., 1998; Yoshizumi et al., 2000) show a concentration of activity in liver and heart, and uniform distribution in the rest of the body. (C162) After uptake, only some 123I-IPPA and 123I-BMIPP will be metabolised immediately to water-soluble low-molecular-weight products. 123I-IPPA is, to a large extent, metabolised like long-chain fatty acids by rapid mitochondrial β-oxidation, resulting in p-(123I)-iodobenzoic acid which is excreted in a conjugated form in the urine. The metabolism of 123I-BMIPP is slower than that of 123I-IPPA due to the methyl group on the β-carbon. The end product is p-(123I)-iodophenyl acetic acid, which is also excreted as a conjugate in the urine. In either case, no release of free iodine has been detected. The initially unmetabolised part of 123I-IPPA and 123I-BMIPP will become incorporated into the fat stores in the body, which have slow turnover thus causing considerably delayed metabolism. (C163) Time–activity curves for the heart and liver indicate bi-exponential elimination of 123I-BMIPP (Torizuka et al., 1991; De Geeter et al., 1998). Out of these curves, initial uptake has been calculated to be 5.0–5.7% of the activity administered (excluding blood activity) in the heart and 13–14% in the liver. For 123I-BMIPP, the biological half-time of the fast phase is approximately 1 h, and that of the slow phase is approximately 2 days. The fast phase corresponds to a fraction of 0.43 of uptake in the heart. In the liver, the fast-eliminated fraction is 0.33–0.36. The final metabolite is excreted via the kidneys and urinary bladder. After 16 h, 15% (Dudzcak et al., 1986) has been excreted, and this increases to 22.6% after 24 h (Torizuka et al., 1991). There are no data for 123I-BMIPP covering longer time periods, but from studies on labelled fatty acids (Gunnarsson et al., 2003), one must assume uptake into body fat and, consequently, slow turnover of part of the administered activity. (C164) The biokinetic model for 123I-BMIPP adopted here assumes initial uptake of 6% of the administered activity in the heart and 14% in the liver. The rest is assumed to be distributed uniformly in the remaining organs and tissues. From the heart, 40% is excreted with a biological half-time of 1 h and 60% with a half-time of 48 h. For the liver, the fractions are 30% and 70%, respectively. Elimination from the rest of the body is assumed to be bi-exponential, with a fast phase with a half-time of 48 h and a slow phase with a half-time exceeding 100 h (Gunnarsson et al., 2003). (C165) The faster phase corresponds to the combined fast and slow phases of the heart and liver, and represents the turnover of a more dynamic fat pool of the body. The slow phase represents the turnover of the rest of the body fat. The size of the latter long-lasting pool is taken to be 20% of the administered activity; a high value according to data in the literature (Gunnarsson et al., 2003). (C166) For 123I-IPPA, there are no data suitable for dose estimations. Initial uptake in the heart, liver, and other organs and tissues is assumed to be the same as for 123I-BMIPP. The first-phase elimination from heart and liver, however, should be much faster as β-oxidation is not inhibited. The model assumes a half-time that is five times lower (i.e. a biological half-time of 10 min for the initial fast phase). For the slow phase of the heart and liver, and for elimination from the rest of the body, the same figures are used as in the 123I-BMIPP model. Note that the models are intended for 123I only. Absorbed doses for 99mTc-apcitide. The physical half-life of 99mTc is 6.01 h.
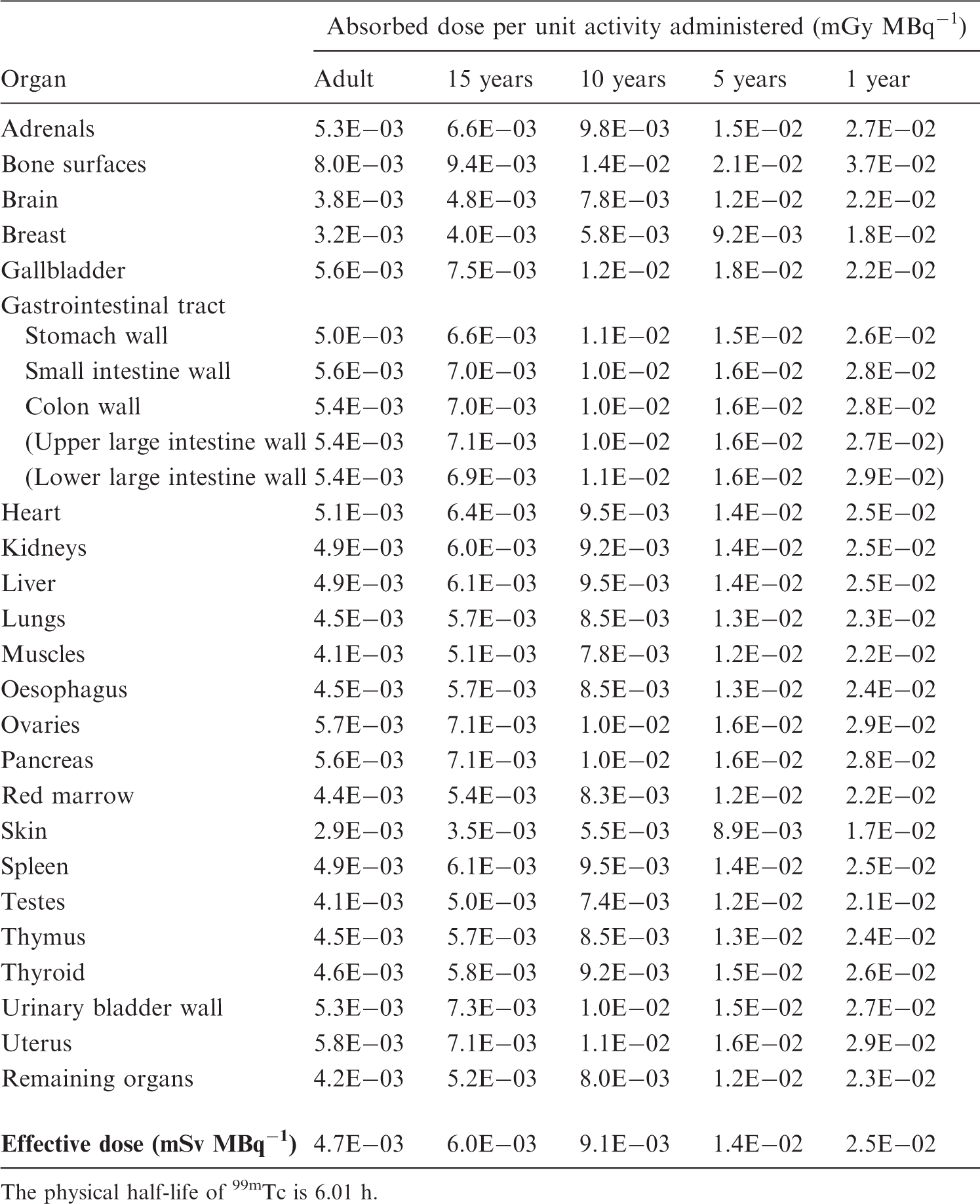
C.53.2. References for 123I-labelled fatty acids
C.54. 123I-labelled brain receptor substances (generic model)
C.54.1. Biokinetic model
(C167) A large number of radiopharmaceuticals labelled with 18F and 123I have been developed for PET and SPECT studies of different types of receptor in the human brain. For many of these substances, the available biokinetic data are insufficient to construct realistic compound-specific biokinetic models for the calculation of absorbed dose to persons undergoing an investigation. Therefore, a generic model for radionuclide-labelled brain receptor substances that would predict the internal radiation dose with sufficient accuracy for general radiation protection purposes has been developed. (C168) A generic model for 11C-labelled brain receptor substances has been published (Nosslin et al., 2003). A review of the literature has identified biokinetic and dosimetric data for five 18F-labelled and 15 123I-labelled compounds considered to be potential substances for the clinical imaging of brain receptors (e.g. acetylcholinesterase receptors, benzodiazepine receptors, dopamine receptors, dopamine transporters, and serotonin receptors). These data indicate that despite fairly large differences in chemical structure, the patterns of uptake in the human brain, and other tissues for which information are available, appear to be sufficiently similar to justify a generic model for each radionuclide. (C169) For some compounds, the published data on the dosimetry of 18F- and 123I-labelled receptor radiopharmaceuticals were derived from PET and SPECT studies in humans, and for other compounds, the biokinetic models were derived, at least in part, from studies of biodistribution in experimental animals. (C170) For the 123I model, it is assumed that fractions of 0.06 and 0.003 of the administered activity are distributed instantaneously to brain and thyroid, respectively. The activity is excreted from these tissues with a biological half-time of 100 h (i.e. more than 99% of 123I will decay in situ). It is also assumed that 0.20 of the administered activity is distributed instantaneously to the lungs and then excreted with a biological half-time of 8 h. Fractions of 0.20 and 0.03 are assumed to be deposited in the liver and the kidneys, and are excreted bi-exponentially with biological half-times of 8 h (50%) and 100 h (50%), respectively. Thirty percent of the activity uptake in liver is eliminated via the gallbladder, and the remainder of the liver uptake is passed directly into the small intestine. It is assumed that 75% of the administered 123I is excreted in the urine and 25% via the gastrointestinal tract. Biokinetic data for 99mTc-labelled large colloids.

C.54.2. References for 123I-labelled brain receptor substances (generic model)
C.55. 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)- N-(3-fluoropropyl) nortropane (FP-CIT, β-CIT-FP, ioflupane)
C.55.1. Biokinetic model
(C171) 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)-N-(3-fluoropropyl) nortropane (otherwise referred to as 123I-FP-CIT, 123I-ß-CIT-FP, or 123I-ioflupane) is a radioiodinated cocaine analogue that binds with high affinity and selectivity to dopamine transporters, which solely exists in the presynaptic terminals of dopaminergic neurons. By introducing an agent that binds to the dopamine transporters, a quantitative measure and spatial distribution of the transporters can be obtained (Booij et al., 1997; Colloby et al., 2004; Walker et al., 2004; Tolosa et al., 2007). 123I-FP-CIT thus probes dopaminergic cell loss. It is used to detect and distinguish states of degeneration of presynaptic dopaminergic neurons in the striatum (caudate nucleus and putamen) from essential tremor in the diagnosis and staging of neurodegenerative disorders (e.g. Parkinson’s disease, Alzheimer's disease, and dementia with Lewy bodies). Thyroid blocking via oral administration of potassium iodide is recommended to minimise unnecessary excessive uptake of radioiodine. (C172) The kinetics of 123I-FP-CIT are relatively fast and allow adequate striatal uptake for imaging 3 h post injection. Booij et al. (1998) studied uptake and retention of 123I-FP-CIT in six male and six female healthy volunteers (mean age 40 years) during a 48-h period using conjugate view scanning and analysis of blood and urine samples. Sydoff et al. (2013) performed a similar study in 10 male patients (mean age 70 years) using planar and combined planar and SPECT/CT imaging over a 48-h period, and blood and urine were also sampled. After uptake, the activity was mainly concentrated in liver, lungs, and brain (Abi-Dargham, 1997; Booij et al., 1998; Tavola et al., 2012; Sydoff et al., 2013). In the brain, the activity is concentrated in the striatum, which could have 10–15 times higher concentrations than the average for the whole brain. Activity is also seen in the spleen, gastrointestinal tract, and urinary bladder. Sixty percent of the administered activity is assumed to be excreted via urine and 14% through faeces (Boiij et al., 1998). The reader is referred to Booij et al. (1997, 2007) and Lim et al. (2009) for further information. (C173) The biokinetic model for 123I-FP-CIT adopted here assumes initial uptake of 31% of the administered activity in the liver, 11% in the lungs, and 4% in the brain. The rest is assumed to be distributed uniformly in the remaining organs and tissues. For all organs and tissues, 80% is assumed to be excreted with a biological half-time of 58 h, and 20% with a half-time of 1.6 h. It is further assumed that 60% of the injected activity is excreted to the urine, and 40% is excreted to the gastrointestinal tract for all organs and tissues. Activity in the liver is excreted according to the Publication 53 gallbladder model (ICRP, 1987), where 30% is eliminated via the gallbladder and the remainder passes directly into the small intestine. (C174) Absorbed dose calculations have been undertaken by Booij et al. (1998) and Sydoff et al. (2013). Both papers show very similar organ doses for the organs with highest uptake and for effective doses. These values do not differ significantly from the values presented here. Note that the models are intended for 123I-labelled substance only. Absorbed doses for 99mTc-labelled large colloids. The physical half-life of 99mTc is 6.01 h.

C.55.2. References for 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)-N-(3-fluoropropyl) nortropane
C.56. 123I-labelled monoclonal tumour-associated antibodies
C.56.1. Biokinetic model
(C175) The models for iodine-labelled antibodies and fragments are the same as those used for the corresponding technetium-labelled substances (see Section C.41.1), with the modification that released iodine is assumed to be handled by the body according to the model proposed for iodine with blocking of uptake in the thyroid (ICRP, 1987, p. 275). The reader is referred to Bischof Delaloye and Delaloye (1995), Britton and Granowska (1987), and Fishman et al. (1989) for further information. This biokinetic model is not intended to apply to therapeutic use of the substance. Biokinetic data for 99mTc-labelled small colloids (intratumoural injection).

C.56.2. References for 123I-labelled monoclonal tumour-associated antibodies
C.57. 131I-labelled monoclonal tumour-associated antibodies
C.57.1. Biokinetic model
(C176) The models for iodine-labelled antibodies and fragments are the same as those used for the corresponding technetium-labelled substances (see Section C.41.1), with the modification that released iodine is assumed to be handled by the body according to the model proposed for iodine with blocking of uptake in the thyroid (ICRP, 1987, p. 275). The reader is referred to Bischof Delaloye and Delaloye (1995), Britton and Granowska (1987), and Fishman et al. (1989) for further information. This biokinetic model is not intended to apply to therapeutic use of the substance. Absorbed doses for 99mTc-labelled small colloids (intratumoural injection). The physical half-life of 99mTc is 6.01 h. In the model, it is assumed that no leakage occurs. Dose to the remaining breast has been assumed to be equal to the dose to the lungs.

C.57.2. References for 131I-labelled monoclonal tumour-associated antibodies
C.58. 127Xe gas
C.58.1. Biokinetic model
(C177) Radioactive xenon can be administered as a gas by inhalation or as xenon dissolved in saline through intravenous injection. Xenon gas is inhaled as a single breath or it can be contained in a closed spirometer system from which the patient is rebreathing for 2–60 min, usually 5 or 10 min. There are also other techniques that can be regarded as combinations of the methods described. (C178) The MIRD model (Atkins et al., 1980), which is partly based on Ackery and Goddard (1975), Goddard and Ackery (1975), and Susskind et al. (1977), is adopted here. The total body retention of xenon has been described as the sum of four exponential functions associated with xenon retention in the lungs (air and tissue), lean body mass, and fat (two fat components). For the purpose of absorbed dose calculations, it is assumed that xenon not present in the lungs is distributed uniformly throughout the rest of the body. The rate of uptake in the rest of the body during breath holding and rebreathing is assumed to be the same as the elimination rate observed after discontinuing xenon administration. Biokinetic data for 99mTc-dimercaptosuccinic acid.

C.58.2. References for 127Xe gas
C.59. 133Xe gas
C.59.1. Biokinetic model
(C179) Radioactive xenon can be administered as a gas by inhalation or as xenon dissolved in saline through intravenous injection. Xenon gas is inhaled as a single breath or it can be contained in a closed spirometer system from which the patient is rebreathing for 2–60 min, usually 5 or 10 min. There are also other techniques that can be regarded as combinations of the methods described. (C180) The MIRD model (Atkins et al., 1980), which is partly based on Ackery and Goddard (1975), Goddard and Ackery (1975), and Susskind et al. (1977), is adopted here. The total body retention of xenon has been described as the sum of four exponential functions associated with xenon retention in the lungs (air and tissue), lean body mass, and fat (two fat components). For the purpose of absorbed dose calculations, it is assumed that xenon not present in the lungs is distributed uniformly throughout the rest of the body. The rate of uptake in the rest of the body during breath holding and rebreathing is assumed to be the same as the elimination rate observed after discontinuing xenon administration. Absorbed doses for 99mTc-dimercaptosuccinic acid. The physical half-life of 99mTc is 6.01 h.

C.59.2. References for 133Xe gas
C.60. 201Tl ion
C.60.1. Biokinetic model
(C181) Intravenously injected ionic monovalent thallium leaves the blood rapidly by uptake into the cells of all organs and tissues. The distribution is largely determined by the magnitude of the regional blood flow, and is therefore dependent on the degree of physical activity. Compared with the situation at rest, which is considered in the present model, uptake in muscles increases two- to three-fold during exercise, with a corresponding reduction in other tissues. (C182) The organ uptake data in the model are based on the reports by Samson et al. (1978), Atkins et al. (1977), and Chen et al. (1983). Bartlett et al. (1984) showed that 80% of thallium is excreted via the gastrointestinal tract and 20% by the renal tract. The whole-body retention curve can be represented by a bi-exponential function, with half-times of 7 days for 63% of the injected activity and 28 days for 37% of the injected activity (Chen et al., 1983). It is assumed here that all organs and tissues have similar retention kinetics, with the exception of the heart which shows more rapid initial clearance (Freeman et al., 1986). (C183) The uptake of thallium ions in the testes has been studied extensively. Direct organ measurements at autopsy in two cases (Samson et al., 1978) showed 0.11–0.12%, while Hosain and Hosain (1981) and Gupta et al. (1981) derived values of 0.8–1.0% from gamma camera images of the testicular–scrotal region. More recent studies, however, have indicated lower uptake (Rao et al., 1995; Nettleton et al., 2004; Thomas et al., 2005). Thomas et al. (2005) measured testicular uptake in 28 individuals using a collimation method, so that the testes were shielded from body background with lead during imaging. However, activity in the scrotum could still influence the measurements. Based on data from Thomas et al. (2005) and Krahwinkel et al. (1988), uptake in the testes of 0.3% has been adopted. Biokinetic data for 99mTc-diethylenetriaminepentaacetic acid.

C.60.2. References for 201Tl ion
Absorbed doses for 99mTc-diethylenetriaminepentaacetic acid.

The physical half-life of 99mTc is 6.01 h.
The urinary bladder wall contributes up to 57% of the effective dose.
Biokinetic data for 99mTc-ethylenedicysteine.
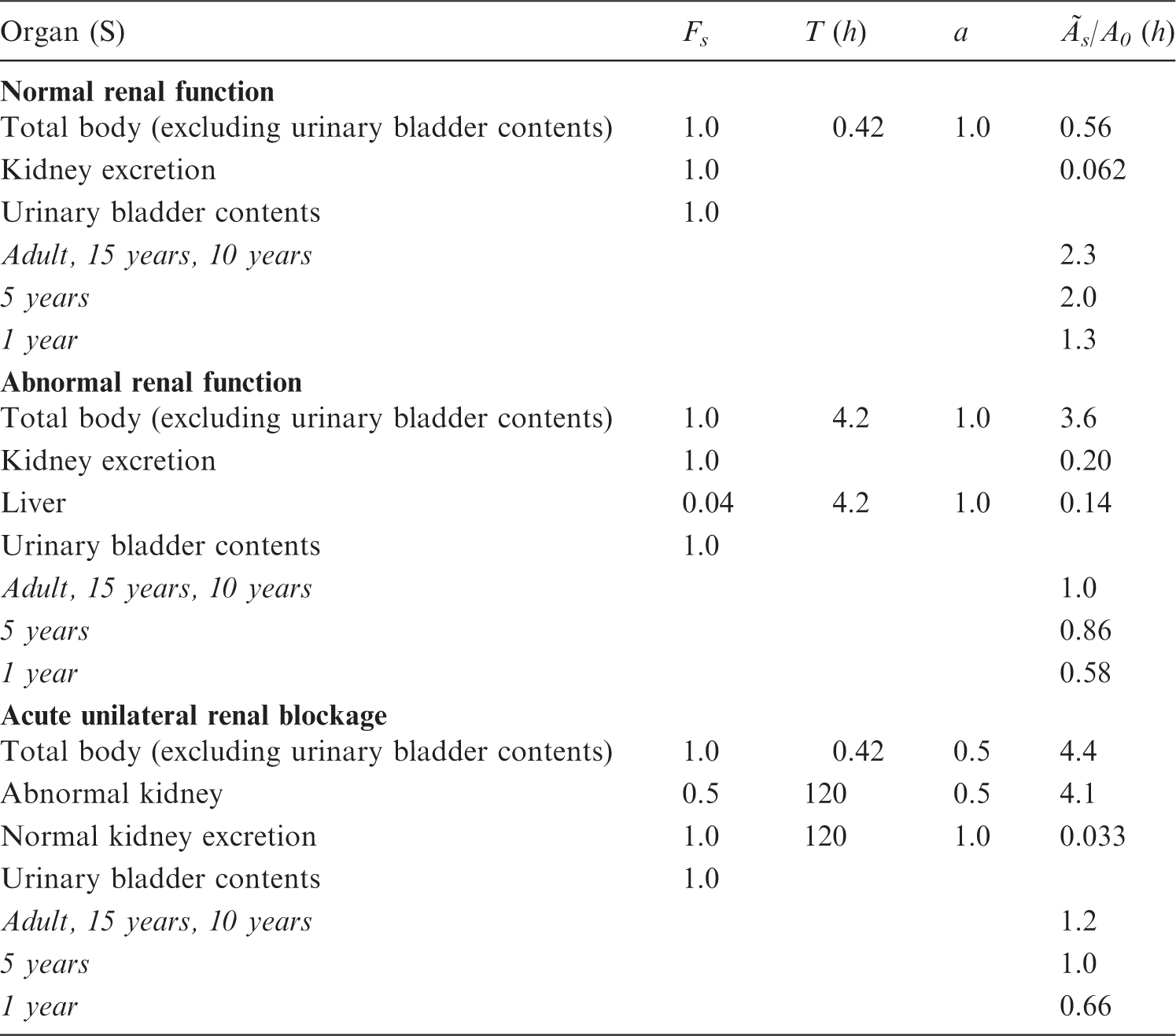
Absorbed doses for 99mTc-ethylenedicysteine.

The physical half-life of 99mTc is 6.01 h.
The urinary bladder wall contributes 76% of the effective dose.
Biokinetic data for 99mTc-ethylenedicysteine diester.

Absorbed doses for 99mTc-ethylenedicysteine diester.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-furifosmin.

Absorbed doses for 99mTc-furifosmin.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled human immunoglobulin.
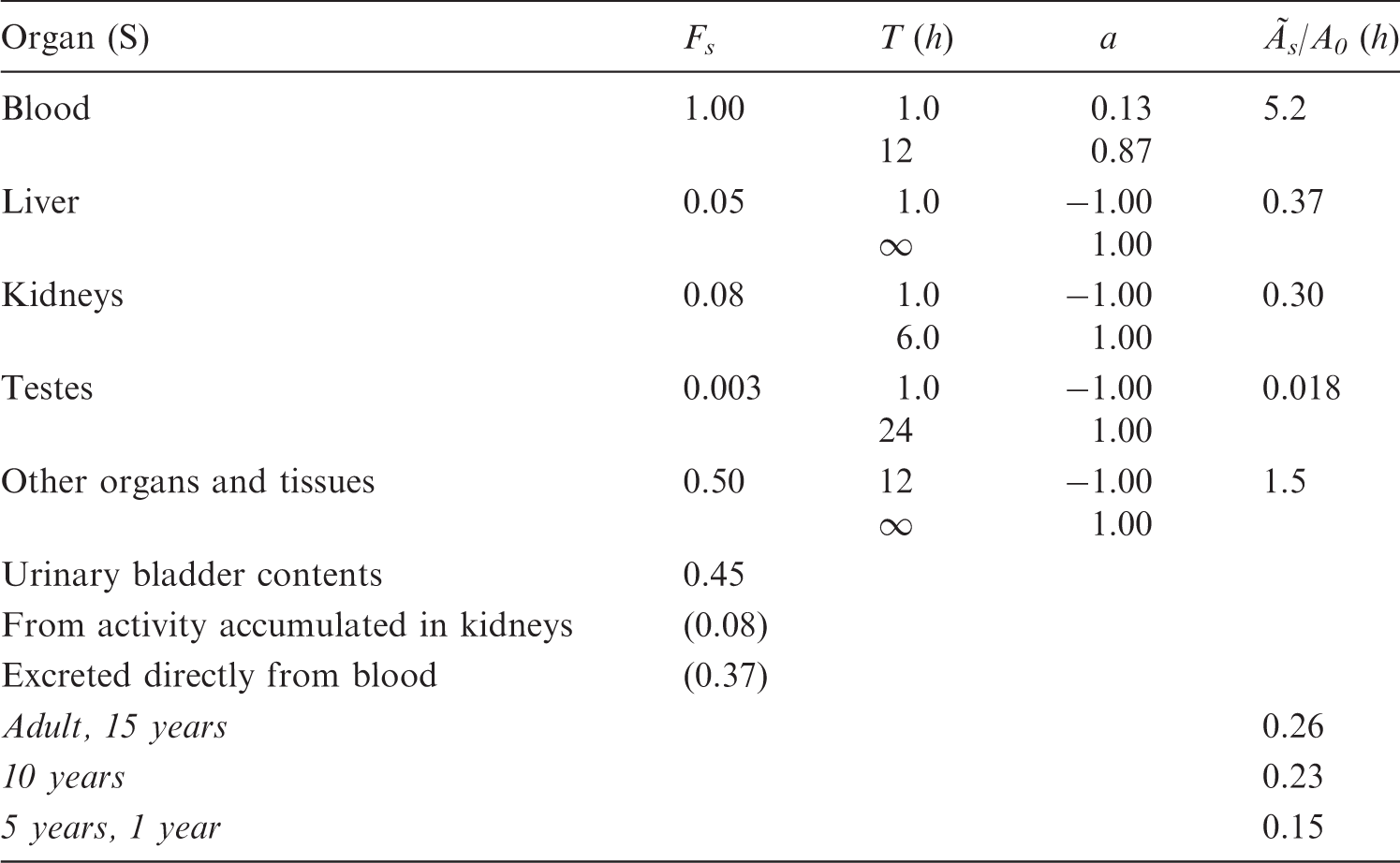
Absorbed doses for 99mTc-labelled human immunoglobulin.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled hexamethylpropyleneamineoxine.

Absorbed doses for 99mTc-labelled hexamethylpropyleneamineoxine.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled iminodiacetic acid derivatives.

Absorbed doses for 99mTc-labelled iminodiacetic acid derivatives.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled macro-aggregated albumin.

Absorbed doses for 99mTc-labelled macro-aggregated albumin.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled mercaptoacetyl triglycine.

Absorbed doses for 99mTc-labelled mercaptoacetyl triglycine.

The physical half-life of 99mTc is 6.01 h.
The urinary bladder wall contributes up to 80% of the effective dose.
Biokinetic data for 99mTc-labelled non-absorbable markers.

Absorbed doses for 99mTc-labelled non-absorbable markers.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled methoxy-isobutyl-isonitrile.

Absorbed doses for 99mTc-labelled methoxy-isobutyl-isonitrile.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled monoclonal tumour-associated antibodies.
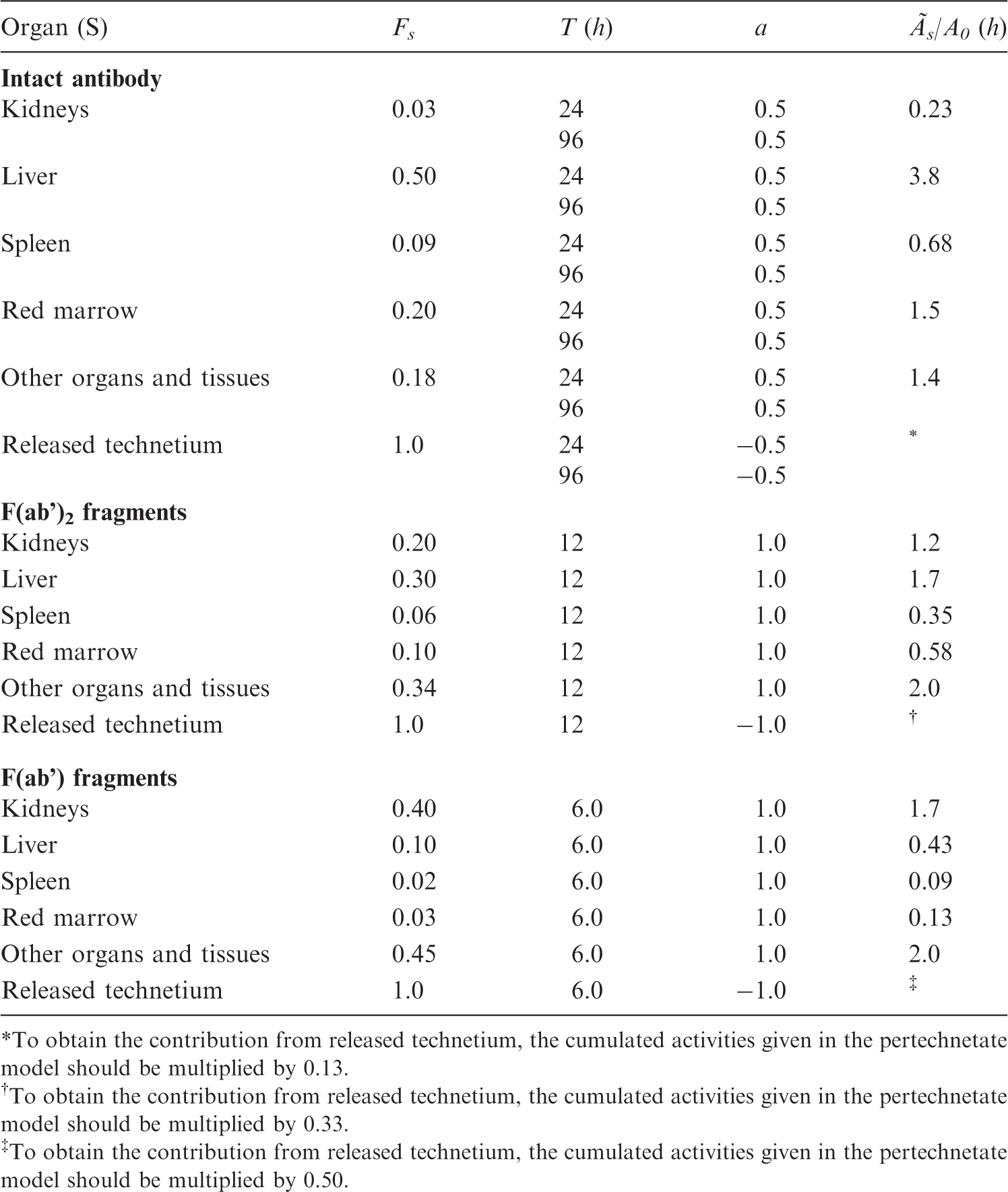
To obtain the contribution from released technetium, the cumulated activities given in the pertechnetate model should be multiplied by 0.13.
To obtain the contribution from released technetium, the cumulated activities given in the pertechnetate model should be multiplied by 0.33.
To obtain the contribution from released technetium, the cumulated activities given in the pertechnetate model should be multiplied by 0.50.
Absorbed doses for 99mTc-labelled monoclonal tumour-associated antibodies.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-Pertechnegas.

Absorbed doses for 99mTc-Pertechnegas.
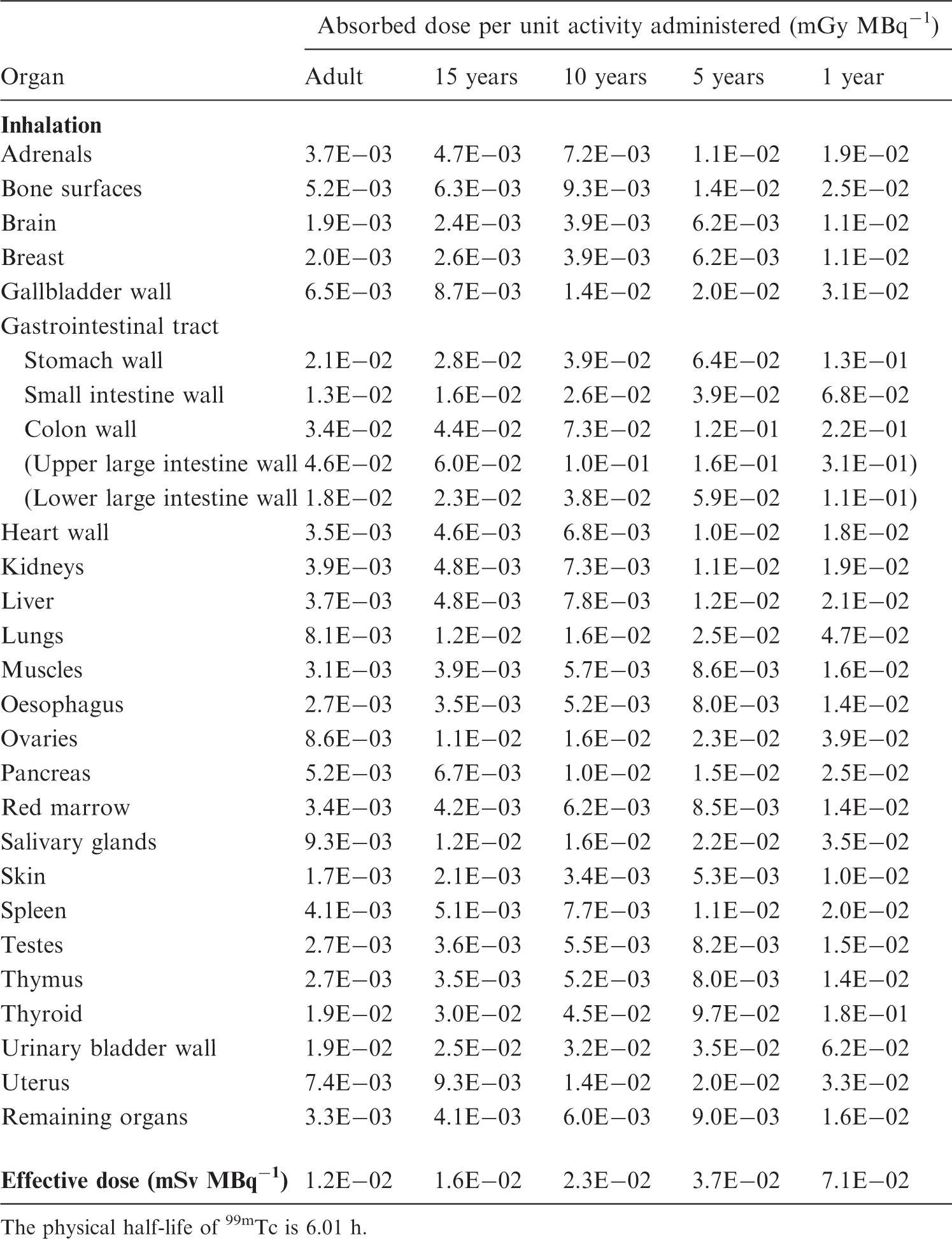
The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-pertechnetate.

Absorbed doses for 99mTc-pertechnetate.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled phosphates and phosphonates.

Absorbed doses for 99mTc-labelled phosphates and phosphonates.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled erythrocytes.

Absorbed doses for 99mTc-labelled erythrocytes.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-Technegas.

Absorbed doses for 99mTc-Technegas.
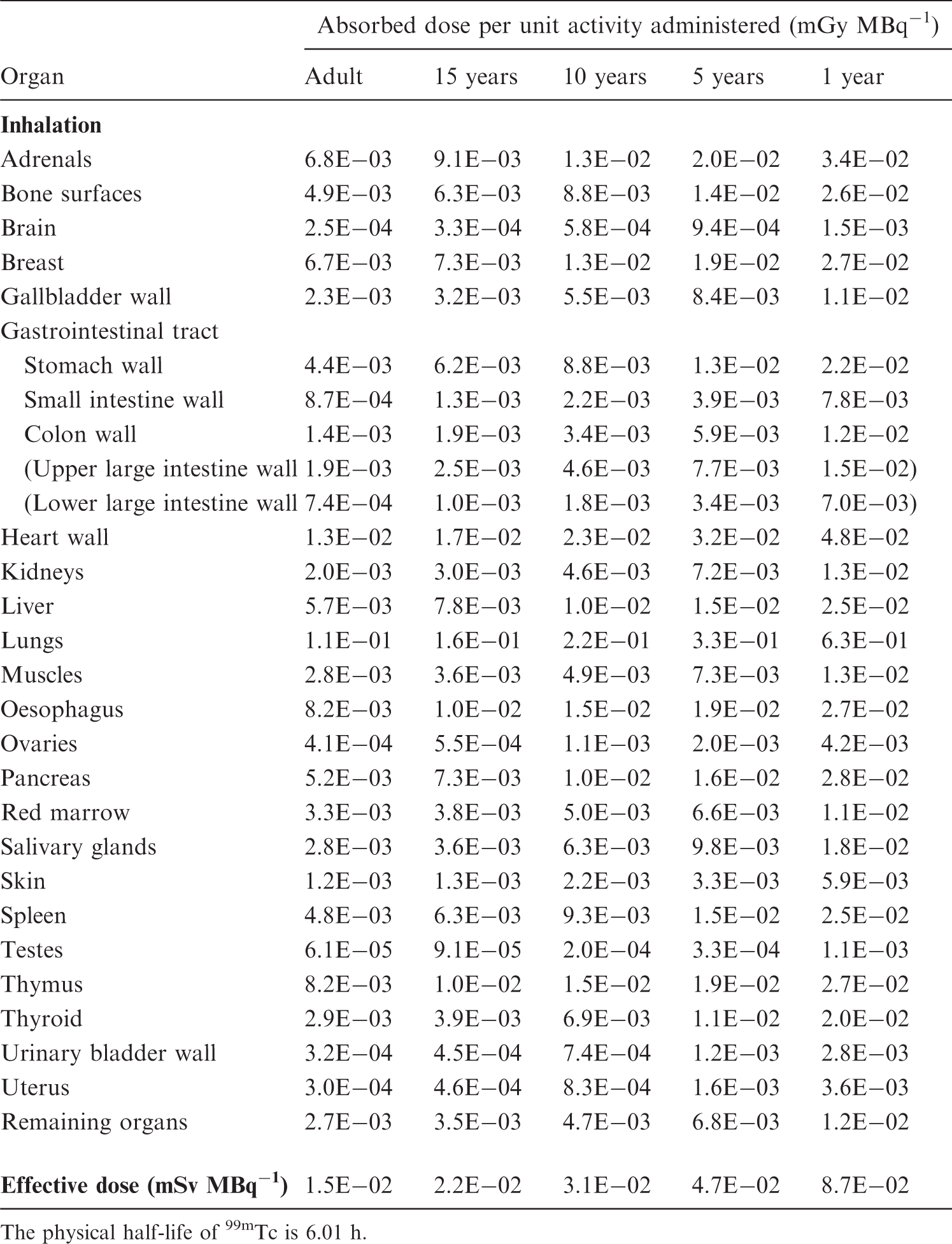
The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled tetrofosmin.

Absorbed doses for 99mTc-labelled tetrofosmin.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 99mTc-labelled leukocytes.
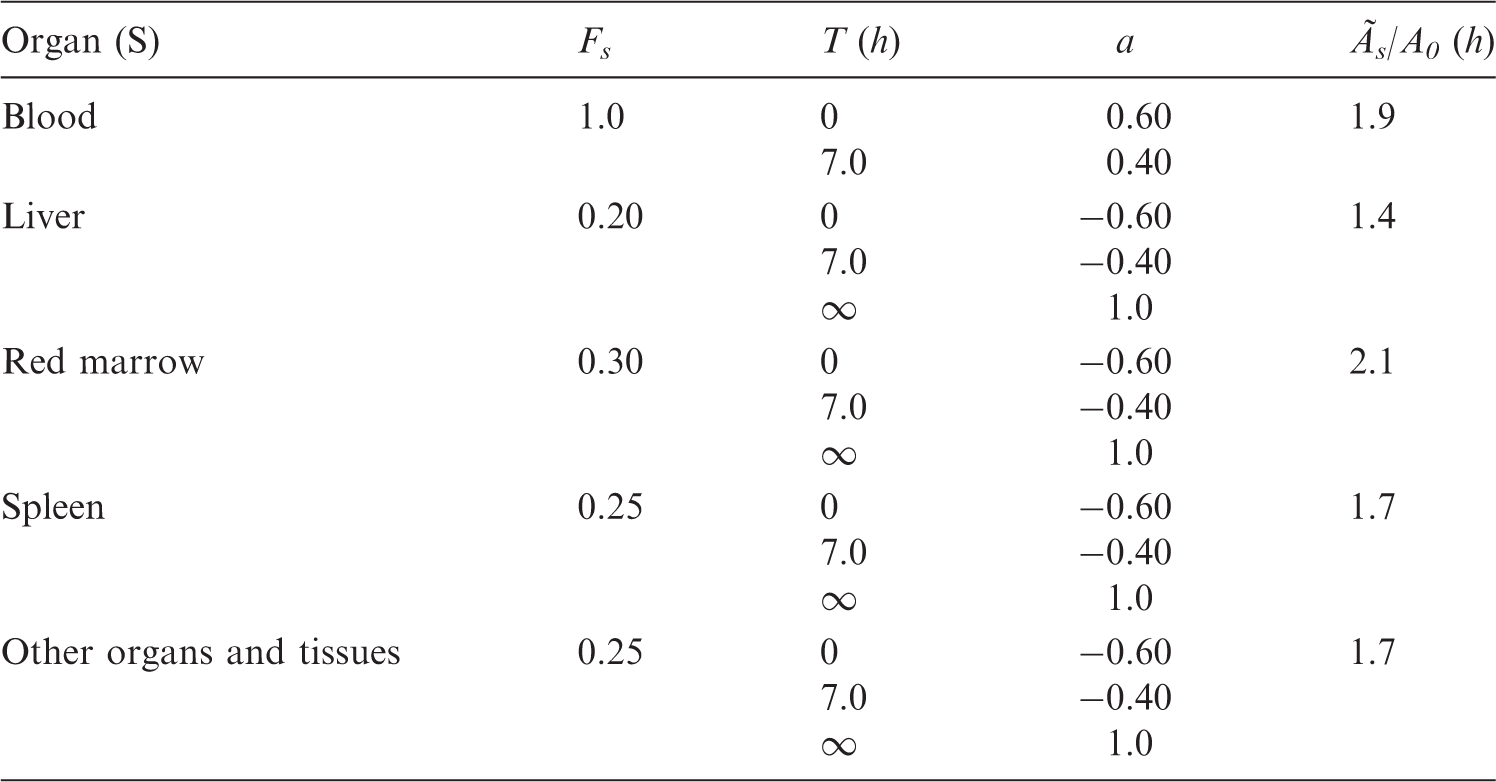
Absorbed doses for 99mTc-labelled leukocytes.

The physical half-life of 99mTc is 6.01 h.
Biokinetic data for 111In-labelled human immunoglobulin.

Absorbed doses for 111In-labelled human immunoglobulin.

The physical half-life of 111In is 67.9 h.
Biokinetic data for 111In-labelled monoclonal tumour-associated antibodies.

To obtain the contribution from released indium, the cumulated activities given in the model for ionic indium (ICRP, 1987) should be multiplied by 0.58.
To obtain the contribution from released indium, the cumulated activities given in the model for ionic indium (ICRP, 1987) should be multiplied by 0.88.
To obtain the contribution from released indium, the cumulated activities given in the model for ionic indium (ICRP, 1987) should be multiplied by 0.92.
Absorbed doses for 111In-labelled monoclonal tumour-associated antibodies.

The physical half-life of 111In is 67.9 h.
Biokinetic data for 111In-labelled octreotide.

Absorbed doses for 111In-labelled octreotide.

The physical half-life of 111In is 67.9 h.
Transfer coefficients (h−1) for the compartment model.

See text for explanation of organs not illustrated in Fig. C.2.
Biokinetic data for 123I, 124I, 125I, and 131I.

Ãs/A0, time-integrated activity (cumulated activity) in organ S for 1 year.
Absorbed doses for 123I-iodide.

The physical half-life of 123I is 13.2 h.
Absorbed doses for 124I-iodide.

The physical half-life of 124I is 4.18 days.
Absorbed doses for 125I-iodide.

The physical half-life of 125I is 59.4 days.
Absorbed doses for 131I-iodide.

The physical half-life of 131I is 8.04 days.
Biokinetic data for 123I-labelled fatty acids [beta-methyl-p-(123I)-iodophenylpentadecanoic acid].

No free iodide is released.
This biokinetic model is intended for 123I alone.
Biokinetic data for 123I-labelled fatty acids (123I-para-iodophenyl pentadecanoic acid).

No free iodide is released.
This biokinetic model is intended for 123I alone.
Absorbed doses for 123I-labelled fatty acids [beta-methyl-p-(123I)-iodophenylpentadecanoic acid].

The physical half-life of 123I is 13.2 h.
Absorbed doses for 123I-labelled fatty acids (123I-para-iodophenyl pentadecanoic acid).

The physical half-life of 123I is 13.2 h.
Biokinetic data for 123I-labelled brain receptor substances (generic model).

Absorbed doses for 123I-labelled brain receptor substances (generic model).

The physical half-life of 123I is 13.2 h.
Biokinetic data for 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)-N-(3-fluoropropyl) nortropane.

No free iodide is released.
This biokinetic model is intended for 123I-labelled substance only.
Absorbed doses for 123I-labelled 2ß-carbomethoxy 3ß-(4-iodophenyl)-N-(3-fluoropropyl) nortropane.
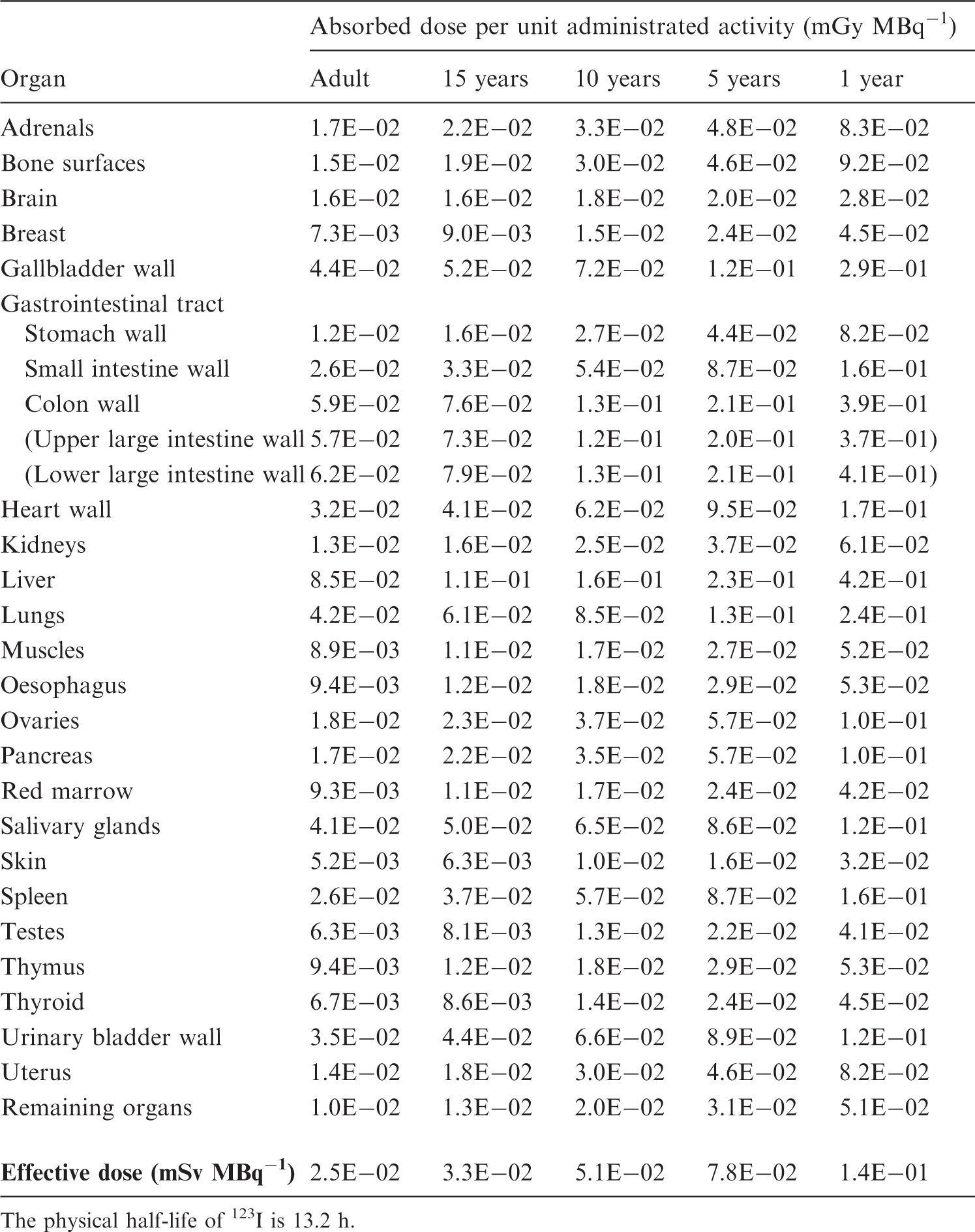
The physical half-life of 123I is 13.2 h.
Biokinetic data for 123I-labelled monoclonal tumour-associated antibodies.

To obtain the contribution from released 123I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.24.
To obtain the contribution from released 123I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.52.
To obtain the contribution from released 123I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.69.
Absorbed doses for 123I-labelled monoclonal tumour-associated antibodies.

The physical half-life of 123I is 13.2 h.
Biokinetic data for 131I-labelled monoclonal tumour-associated antibodies.

To obtain the contribution from released 131I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.78.
To obtain the contribution from released 131I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.94.
To obtain the contribution from released 131I, the cumulated activity given in the model for iodide with blocked thyroid should be multiplied by 0.97.
Absorbed doses for 131I-labelled monoclonal antibodies.

The physical half-life of 131I is 8.04 days.
Biokinetic data for 127Xe gas.

Absorbed doses for 127Xe gas.

The physical half-life of 127Xe is 36.4 days.
Biokinetic data for 133Xe gas.
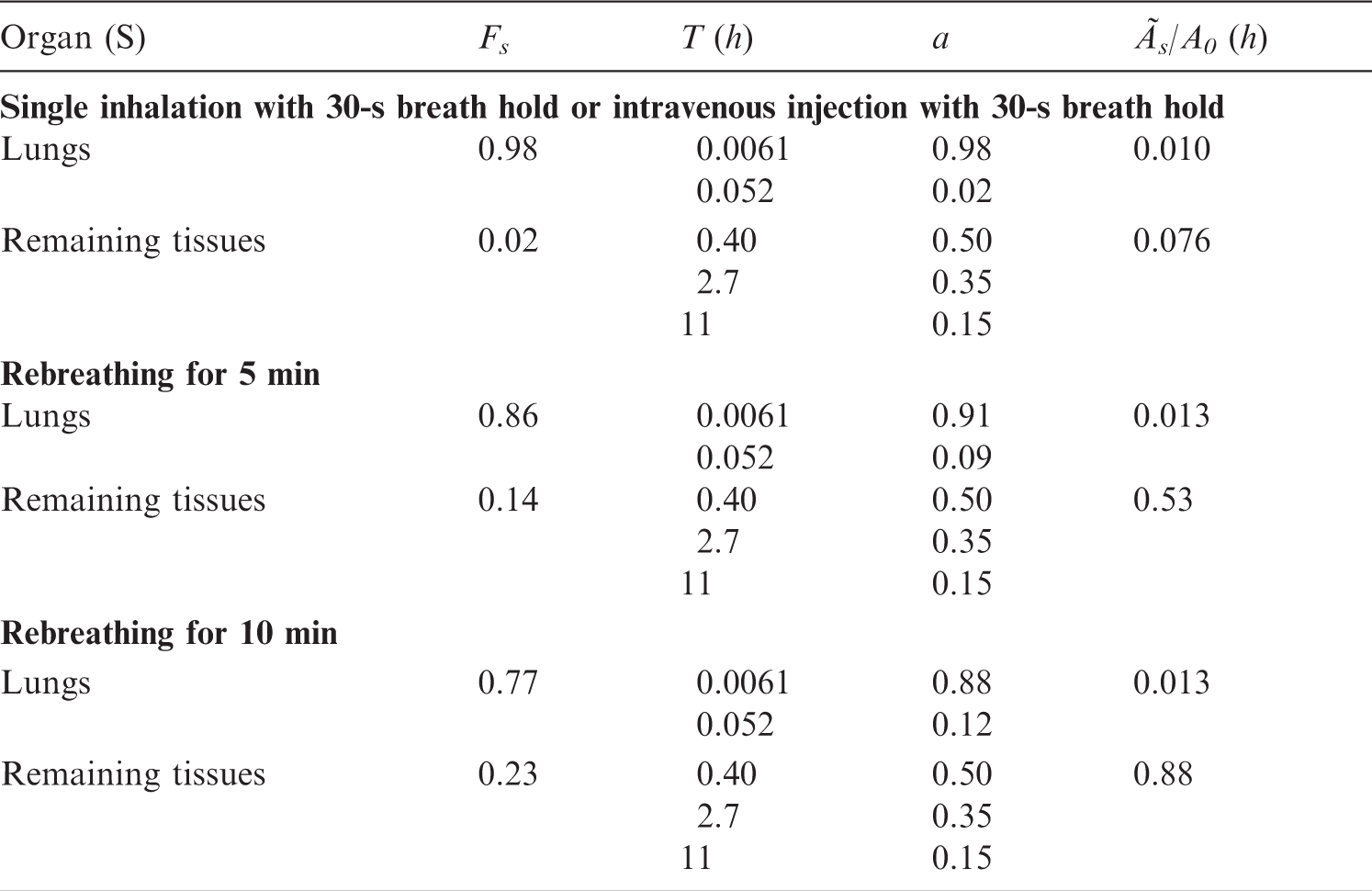
Absorbed doses for 133Xe gas.

The physical half-life of 133Xe is 5.25 days.
Biokinetic data for 201Tl ion.

Absorbed doses for 201Tl ion.

The physical half-life of 201Tl is 3.05 days.
Annex D. Recommendations on Breast-Feeding Interruptions
D.1. Introduction
(D1) As many radiopharmaceuticals are secreted in breast milk, it is safest to assume that, unless there are data to the contrary, some radioactive compound will be found in the breast milk when a radiopharmaceutical is administered to a lactating female. Consideration should be given to postponing the procedure. If the procedure is performed, the child should not be breast fed until the radiopharmaceutical is no longer secreted in an amount estimated to give an effective dose >1 mSv to the child. It is therefore recommended that the actions shown in Table D.1 should be taken for various radiopharmaceuticals (Ahlgren et al., 1985; Castronovo et al., 2000; Evans et al., 1993; Johnston et al., 1996; McCauley and Mackie, 2002; Mountford and Coakley, 1989; Rose et al., 1990; Rubow et al., 1994; Stabin and Breitz, 2000; Tobin and Schneider, 1976). Recommendations on breast-feeding interruptions after a nuclear medicine investigation. MDP, methylene diphosphonate. Interruption not essential. Interruption not essential for most of the 99mTc-labelled compounds, under the circumstance that no free pertechnetate exists in the radiopharmaceutical. An interruption of 4 h during which one meal is discarded can be advised to be on the safe side. Interruption for 3 weeks (504 h) at least. However, difficult to maintain the milk supply → cessation. For all substances labelled with 123I (except iodo-hippurate), interruption for >3 weeks is recommended due to the risk of contamination of other iodine isotopes. ¶For 11C, 13N, and 15O-labelled substances, interruption not essential due to short physical half-life.
