
Abstract
Purpose:
Hereditary hemorrhagic telangiectasia (HHT) is under-recognized, and specialized referral centers are limited, specifically in Belgium. The primary aim of this study was to describe the clinical characteristics of our patients with HHT, in particular the prevalence of vascular malformations (VMs). Secondary objective was to describe associations between patient’s current work-up and anemia.
Methods:
We performed a retrospective analysis of 118 patients treated at our center between 2015 and 2024. We analyzed documented VM and correlation with genetic work-up, prevalence of epistaxis, and referral of patients to specialist ENT services. We studied the correlation between anemia and clinical parameters such as documented digestive telangiectasia.
Results:
The main results confirmed the dominance of epistaxis among the documented symptoms. Anemia was correlated with age and presence of documented digestive telangiectasia. Only 55% of patients with epistaxis were referred to an otorhinolaryngologist. The majority of patients (65%) had documented multisystem involvement of at least 2 organs, and the majority of important VMs (lung and liver) were documented properly. However, liver malformations remain not documented in 41% of cases and lung malformations in 31% of cases. In our cohort, 61% of patients underwent genetic analysis (80% ENG and/or ACVRL1). However, there was no significant association between genetic and multisystem work-up or specific phenotype.
Conclusions:
We highlighted the importance of epistaxis as a cardinal symptom in the management of HHT. We also highlighted strengths and gaps that could lead to future improvements, such as systematic referral of patients with epistaxis to ENT and improved coordination of care. Further longitudinal studies would allow us to assess the impact of improved pathways to improve patient care nationwide.
Introduction
Hereditary hemorrhagic telangiectasia (HHT) or Rendu-Osler-Weber disease is an autosomal-dominant genetic disorder. This disease is caused by pathogenic variants in ENG (endoglin), ACVRL1 (ALK1), SMAD4 (MADH4), GDF2 (BMP9-10), or RASA1 genes, leading to the dysregulation of angiogenesis.1-3 The first 3 genes account for 90% of documented cases.1-3 The disease is characterized by multisystem involvement, presenting with capillary dilatations, known as telangiectasias affecting the skin (face, lips, fingers, toes, etc) and mucous membranes of the nose and oral cavity/oropharynx as well as the digestive tract. It is associated with arteriovenous malformations (AVMs) in various organs (lungs, liver, central nervous system) and is the cause of numerous complications that contribute to the morbidity of the disease. The most common manifestation is epistaxis, which affects more than 90% of the affected population and usually occurs before the age of 20 years.3,4 Repeated epistaxis can lead to iron deficiency and chronic anemia, which can have a significant impact on quality of life. 5 Diagnosis is difficult due to the rarity of the disease (prevalence is estimated to be between 1/5000 and 1/8000 in Europe, with regional variations 6 ), the variable clinical presentation depending on age, and the lack of awareness of the disease among physicians. As a result, there is an average delay of around 20 years between the onset of symptoms and diagnosis. 4 Clinical diagnosis is based on the Curaçao criteria7,8 (Table 1). There is currently no curative treatment. Only symptomatic treatments are available.2,3,6 The management of HHT necessitates a multidisciplinary approach to address its diverse manifestations8-10; however, gaps in care coordination and documentation remain prevalent. 11
Curaçao Criteria (Faughnan et al 8 ).

Abbreviations: AVMs, arteriovenous malformations; HHT, hereditary hemorrhagic telangiectasia.
At present, there is a lack of reference centers in Belgium and more specifically in Wallonia, unlike our French, German, Dutch, and Swiss neighbors.12,13 In this context, the European Network of Reference for Rare and Multisystem Vascular Diseases (VASCERN) proposes the creation of a patient registry. 13 This is an essential tool for centralizing clinical data and information on rare vascular diseases. By collecting clinical data, the registry aims to improve research, diagnosis, and patient care.
The primary objective of this study was to characterize the clinical profile of patients with HHT disease treated in our center, providing an overview of the affected population and its management within our institution. Specifically, we aimed to determine the prevalence of vascular malformations (VMs) in these patients and to evaluate the completeness of their diagnostic work-up in relation to international guidelines. As a secondary objective, we investigated potential associations between VMs, anemia, and iron supplementation, given their role as markers of disease severity. This analysis was essential in assessing the appropriateness of the diagnostic tests performed and their impact on both disease severity and patient quality of life. Furthermore, by comparing international guidelines with our institutional practices, we sought to contextualize our findings within a broader clinical framework, identifying potential areas for improvement in patient care.
Material and Methods
This is a retrospective study including a population of 118 patients with HHT treated at the CHU of Liège between January 1, 2015, and November 30, 2024. Inclusion criteria were all patients with a confirmed clinical diagnosis of HHT according to the Curaçao criteria (Table 1). Exclusion criteria were limited to patients with an unconfirmed diagnosis. Data were extracted and anonymized directly from the medical records with the approval of the Ethics Committee of the University Hospital of Liège (Ref.: 2024/581). The collected data included demographic characteristics such as vital status (alive or deceased), age (current or at time of death), and sex (male-to-female ratio). Clinical parameters comprised the most recent recorded hemoglobin level, the presence or absence of organ damage (pulmonary, cerebral, hepatic, gastrointestinal, or cutaneous), the availability of relevant data, and the occurrence of epistaxis. Additionally, information was gathered on iron supplementation (oral or intravenous), regular ENT follow-ups, genetic counseling, genetic analysis, and the identification of the specific gene and mutation. Organ damage was graded according to standardized criteria based on the examinations performed and the details documented in the patients’ medical records.
For pulmonary involvement, the presence or absence of pulmonary VMs was determined using a contrast-enhanced chest computed tomography (CT) scan, which was regarded as the reference diagnostic modality. 8 Patients with a negative contrast-enhanced chest CT were classified as having no pulmonary involvement. Cerebral involvement was assessed by detecting VMs or, more rarely, a cerebral abscess, which could occasionally be the initial manifestation leading to the diagnosis.10,11 Contrast-enhanced brain MRI was the gold standard to rule out involvement, though in some cases contrast-enhanced CT scan could also reveal abnormalities. Patients with a negative contrast-enhanced brain MRI were considered as having no cerebral involvement.
For liver involvement, the exclusion of hepatic VMs was mainly based on Doppler ultrasound, which served as the first-line diagnostic tool. In certain cases, liver CT or MRI was used to confirm the absence of vascular abnormalities. For gastrointestinal involvement, gastrocolonoscopy served as the main investigation for assessing damage to the digestive tract. In some cases, video capsule endoscopy was employed for a more detailed examination to confirm the absence of lesions. Descriptive analysis provided an overview of the clinical characteristics of the patient population. Chi2, Pearson and Spearman correlation tests were applied where appropriate to examine the relationships between clinical characteristics and severity parameters. Independent values were compared using the Mann-Whitney U test or the t-test, depending on the distribution of the variables and the homogeneity of variances. The results are presented in tables and figures to illustrate the key findings.
Results
Demographic analysis of the 118 subjects showed a slightly-male-dominated population (56.78% males and 43.22% females), with a median age of 54.5 years, a 25th percentile of 38.2 years and a 75th percentile of 66 years (Table 2). The prevalence of epistaxis was 88.68% (n = 106), which is slightly lower than reported in the literature. Epistaxis was more common in older subjects (median age: 56 years with epistaxis vs 37 years without epistaxis, P = .0142). Among the patients, 49.5% (n = 105) required iron supplementation and had significantly-lower hemoglobin levels than those who did not (median hemoglobin: 12.4 g/dL with iron supplementation vs 14.2 g/dL without iron supplementation, P = .00011). On the other hand, hemoglobin levels tended to decrease with age (P = .0000577 with a Spearman correlation coefficient of −0.378). Despite these observations, only 55.3% of patients with epistaxis were referred to an otorhinolaryngologist for evaluation and management. Genetic analysis was performed in 61% of patients. The majority of (likely) pathogenic variants were identified in the ENG and ACVRL1 genes (54% and 24%, respectively, with 3% carrying a combination of these 2 mutations). Mutations in SMAD4 (MADH4) were less common (3% of cases). In 14% of cases, genetic analysis did not identify any pathogenic variants.
Results Extracted From the Database.
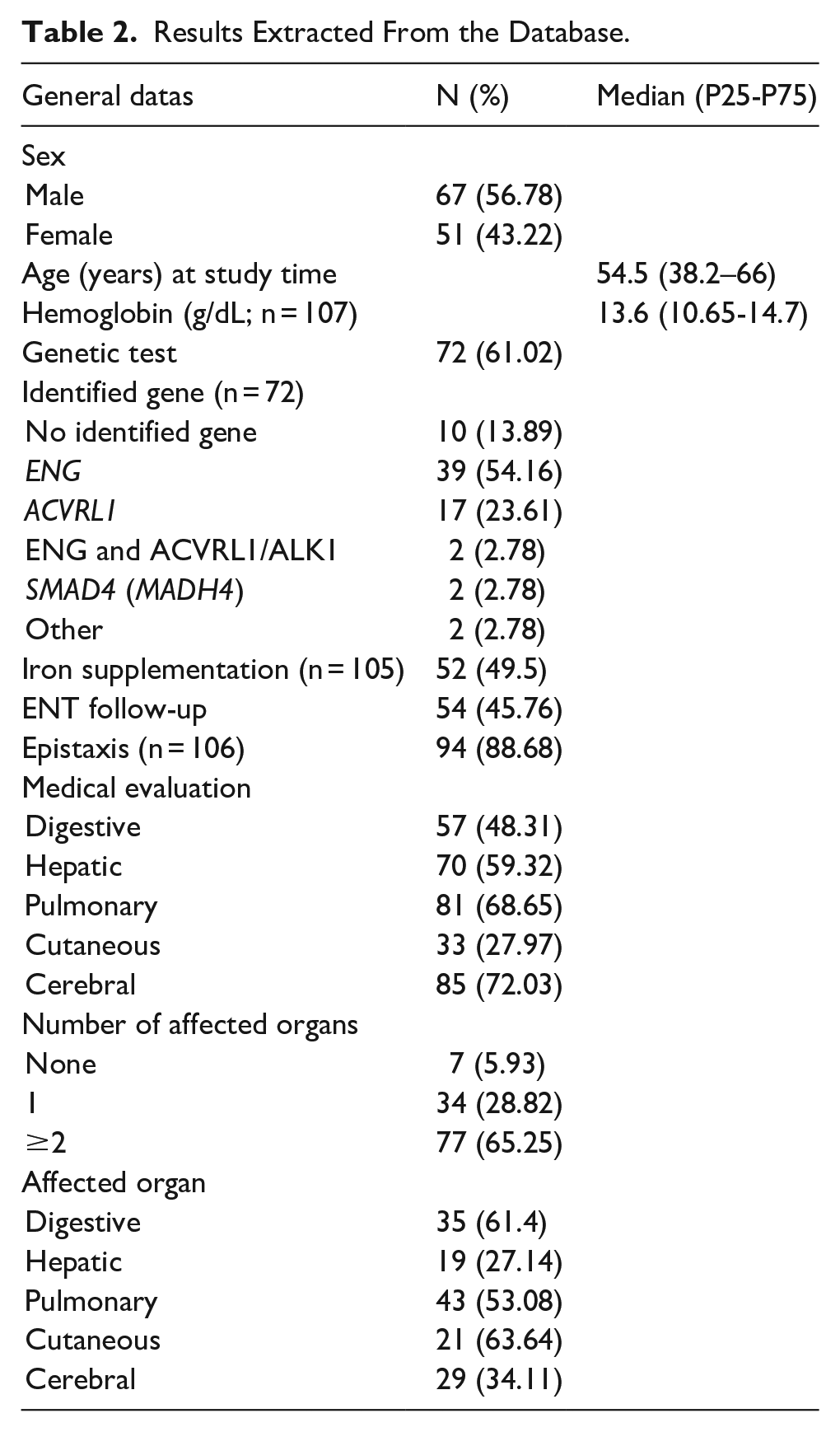
The population of patients who underwent genetic analysis was significantly-younger than those who did not (median age of 47.5 vs 63 years, P = .00022). We were not able to demonstrate an association between having received genetic testing and having received a multisystem work-up. We also identified gaps in documentation (Figure 1), with pulmonary assessments missing in 31% of cases, liver examinations in 41%, brain examinations in 28%, and gastrointestinal evaluations in 52%.
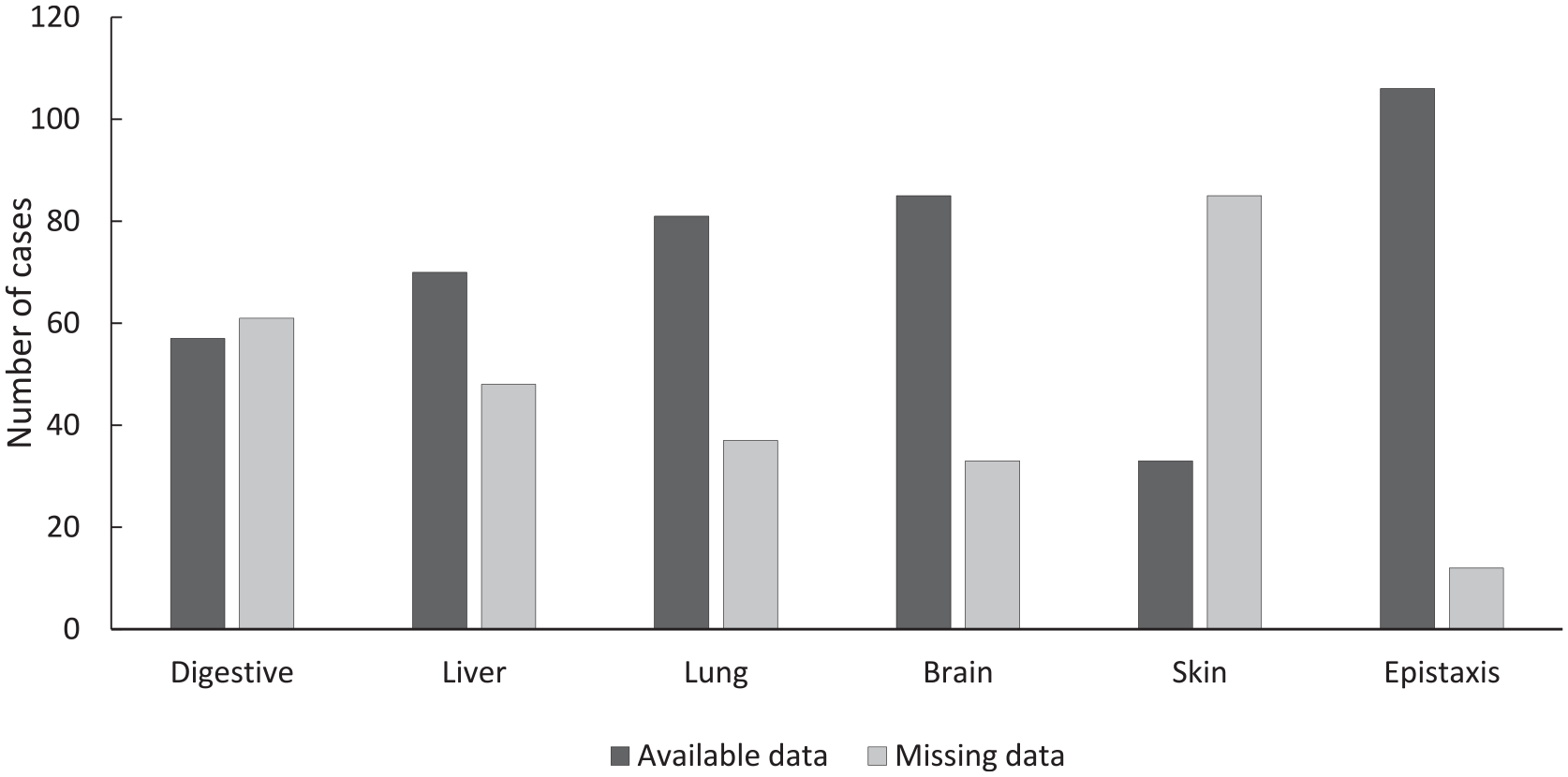
Ratios between missing and known data by organ.
Sixty-five percent of our subjects had documented multisystem involvement, with at least 2 organs affected. Of the 69% (N = 81) of subjects examined for lung disease, 53% (N = 43) showed involvement. Of the 59% (N = 70) of subjects tested in the liver, 27% (N = 19) were affected. Of the 72% (N = 85) of subjects examined for brain damage, 34% (N = 29) were affected, and of the 48% (N = 57) of subjects examined for gastrointestinal damage, 61% (N = 35) were affected (Figure 2). It should also be noted that subjects with documented gastrointestinal malformations had significantly-lower hemoglobin levels (median level of 10.45 g/dL) than those without malformations (median level of 14.15 g/dL, P = .00011; Figure 3). These malformations were also more prevalent with increasing age (median age: 55 years without digestive malformations vs 66 years with digestive malformation(s), P = .00082).
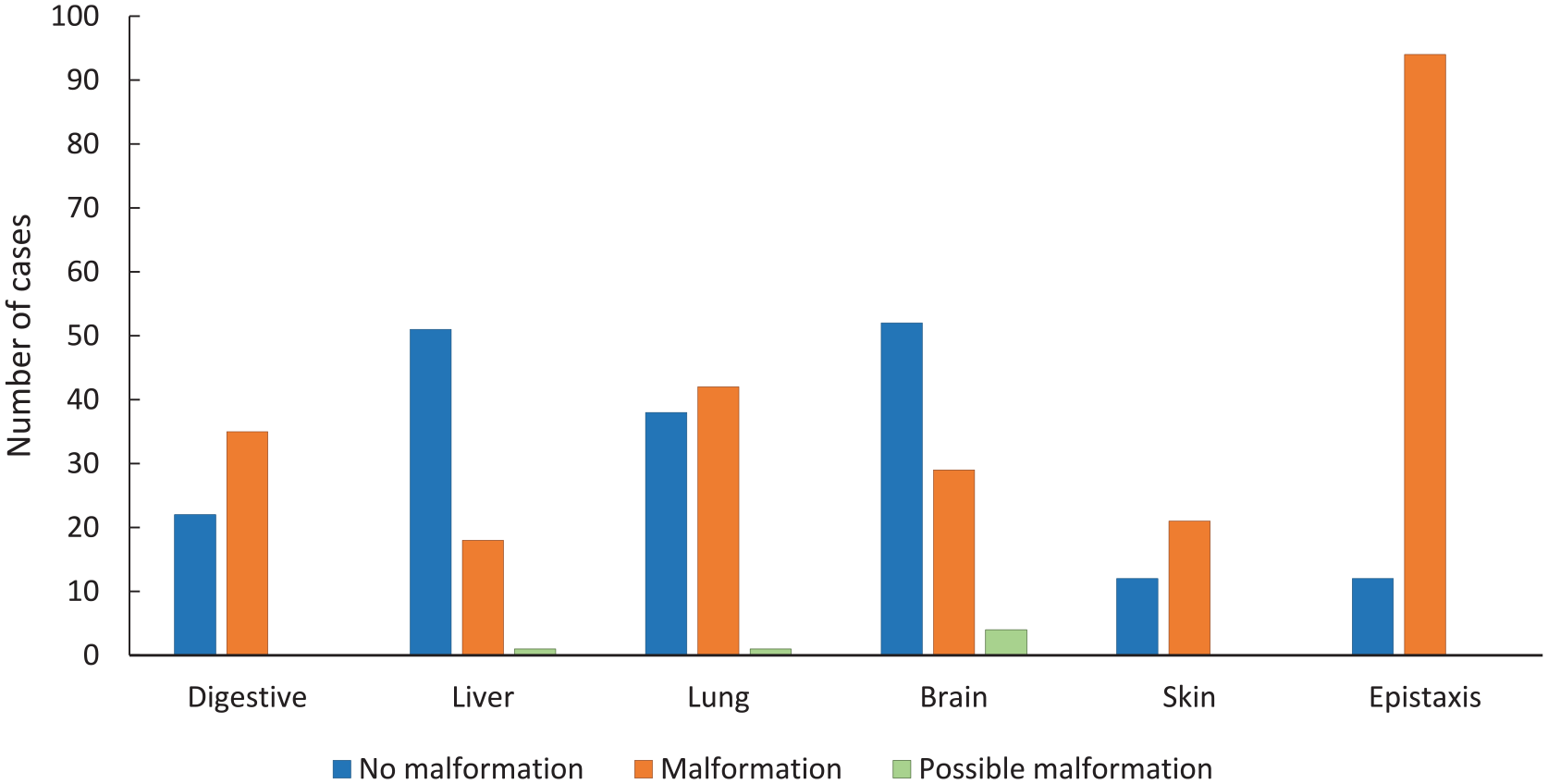
Prevalence of arteriovenous malformations.
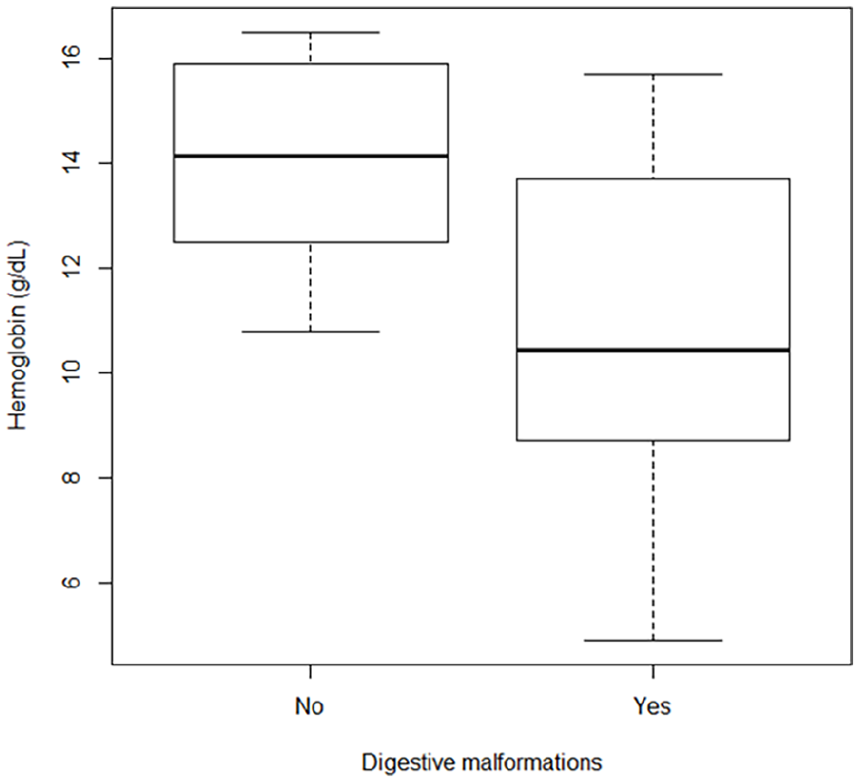
Distribution of hemoglobin levels according to the presence of digestive malformations. Subjects with documented gastrointestinal malformations had significantly-lower hemoglobin levels (median level of 10.45 g/dL) than those without malformations (median level of 14.15 g/dL, P = .00011).
Discussion
Epistaxis was the key symptom of HHT. One of the main strengths of this study is the detailed clinical and genetic profiling of a relatively-large cohort of patients with HHT, which provided a comprehensive picture of the disease as it presents in our institution. Our study highlighted gaps in clinical documentation and areas for improvement in the management of HHT, particularly in the completeness of diagnostic work-ups, which could inform future practice and guideline development.
However, there are several limitations to this study. The retrospective nature of the analysis, with reliance on medical records, introduces the possibility of incomplete data. Additionally, the study was conducted at a single center, limiting the generalizability of the findings to other settings. The sample size, though relatively large, may still not fully capture the spectrum of HHT cases, particularly rare manifestations of the disease. Lastly, while genetic analysis was performed in 61% of patients, this may not represent a fully-representative sample of the population.
Despite the high prevalence of epistaxis, patients were not systematically referred to ENT as recommended by international guidelines. 7 Epistaxis affects the quality of life of patients,5,14 in particular leading to chronic anemia. Its management is very specific. 15 Currently, only half of patients presenting with epistaxis were correctly referred. There is a high risk that patients and their caregivers were not informed about the appropriate management. Trauma should be avoided wherever possible. Nonabsorbable nasal packing should be avoided, and the systematic use of topical moisturizers, oral tranexamic acid, and absorbable tamponades should be recommended as first-line treatment.16,17 If packing were ever necessary, absorbable nasal packing should be preferred and should be accompanied by antibiotic therapy.16-18 These initial steps are followed by ablative therapies, combined or not with systemic anti-angiogenic agents, and finally radio-surgical interventions aimed at hemostasis in the most refractory cases. 18 The analysis of the management of epistaxis could be the subject of a separate study. The lack of an internal protocol for the management of patients presenting with epistaxis is also a failure to follow guidelines. This delays management and risks increasing morbidity. 19 It is therefore up to us to establish this protocol and educate our colleagues on the proper management of epistaxis in these patients. Our findings align with previous research on the prevalence of epistaxis and iron deficiency in HHT, as the high prevalence of epistaxis in our cohort (88.68%, Table 2) was consistent with other studies, which report epistaxis in 88% to 100% of HHT patients.20,21
Genetic consultations also provide an opportunity to integrate patients into multidisciplinary care. However, the lack of guidelines also hinders their effectiveness.
Although genetic analysis is emphasized in recommendations,
7
the Curaçao criteria do not consider pathogenic variant identification as essential for diagnosis. Sixty-one percent of patients underwent genetic analysis, but the relationship between phenotype-genotype correlation remains unclear. Studies have consistently reported
We assume that adapting treatment to genetic data would optimize multidisciplinary management.
While guidelines 7 recommend systematic investigation of the lungs and liver, analysis of patient records revealed a significant lack of documentation (eg, more than 30% of lung data were missing). This deviation from the recommendations suggests that the tests were not being performed systematically. This can lead to a delay in diagnosis or even a failure to diagnose, with potentially-fatal complications. It is therefore important to systematically screen for organ damage and record it in the patient’s medical record. 22 The association between gastrointestinal malformations and lower hemoglobin levels observed in our study has also been previously reported. 23 Gastrointestinal involvement, including bleeding from telangiectasia in the digestive tract, is well-documented as a source of significant anemia in HHT patients. The age-related increase in the prevalence of digestive malformations observed in our study is consistent with other studies suggesting that gastrointestinal involvement increases after age 50 years. 23 We have also found that 72% of our patients underwent brain imaging to check for AVM with 34% of them coming back positive.
In terms of anemia management, international recommendations 8 include the use of iron supplementation (per os or intravenous) and erythrocyte concentrate transfusion, depending on the severity of anemia, while searching for other potential causes if iron supplementation proves inadequate. 24 As the study was limited to measuring hemoglobin levels and whether or not iron supplementation was given, it was not possible to say whether this recommendation was followed. There is a risk that anemia may be treated inappropriately due to a lack of evidence and data.
Based on the above, the following suggestions were made to improve patient management:
Develop a standardized stepwise protocol for first-line treatment of epistaxis, as mentioned in the guidelines. 8
Implement a systematic screening protocol for liver and lung damage in patients diagnosed with HHT.
Increase referrals to ENT specialists in HHT and include geneticists and systemic care specialists to form specialized multidisciplinary teams.
Fill gaps in documentation and tailor care to genetic mutations.
The results of the study also highlight a number of strengths in current care practices. More than half of our patients receive lung and liver tests. We also have sufficient data to correlate clinical variables such as hemoglobin levels with digestive malformations. Statistically-significant trends have been identified between age and prevalence of epistaxis, digestive malformations, and severity of anemia. Our findings underscore the importance of comprehensive screening for multisystem involvement in the management of HHT and the need for adherence to international guidelines to ensure that all patients receive the most thorough and timely care possible.
Conclusion
In conclusion, our study provides valuable insights into the clinical characteristics and management of HHT in Belgium, identifying gaps in documentation and adherence to diagnostic guidelines. By analyzing a large dataset of HHT cases, we established key correlations and highlighted shortcomings in patient management, particularly insufficient ENT referrals, inadequate AVM documentation, and suboptimal integration of genetic results. Addressing these issues through a structured care protocol could enhance treatment adaptation and coordination. Future research should prioritize longitudinal studies to assess the long-term impact of optimized care pathways on disease progression and patient outcomes.
Footnotes
Ethical Considerations
The study was approved by the University Hospital of Liège Ethical committee under the approval number 2024/581, and all procedures were conducted in accordance with ethical guidelines.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.