
Abstract
GAPO syndrome is an exceptionally-rare autosomal recessive disorder characterized by growth retardation, alopecia, pseudoanodontia, and optic abnormalities, with fewer than 60 cases reported globally. We present the first documented case in Syria, highlighting novel otolaryngological and radiological findings that expand the clinical spectrum of this syndrome. A 27-year-old male presented with chronic right-sided otalgia, unilateral conductive hearing loss, and persistent sinonasal symptoms. Examination revealed hallmark features of GAPO syndrome, including craniofacial anomalies, external auditory canal stenosis, and pseudoanodontia. Computed tomography demonstrated total aplasia of the paranasal sinuses and mastoid air cells—findings not previously reported in GAPO syndrome. Audiological evaluation revealed moderate conductive hearing loss attributed to external auditory canal stenosis and eustachian tube dysfunction, contrasting with the predominantly-sensorineural hearing loss reported in earlier cases. Additionally, unique ophthalmic findings, including peripheral congenital cataracts and a myelinated retinal nerve fiber layer, were observed. This case underscores the importance of comprehensive evaluations, including advanced imaging and audiological assessments, in identifying subtle or atypical manifestations of GAPO syndrome. It also highlights challenges in airway management due to craniofacial anomalies. The findings emphasize the necessity for a multidisciplinary approach to optimize care and improve outcomes in patients with GAPO. Further research is needed to clarify genotype-phenotype correlations and refine diagnostic criteria.
Introduction
GAPO syndrome is a rare genetic disorder caused by recessive mutations in the ANTXR1 gene. 1 The name of the syndrome (GAPO) is based on its main features, which include growth retardation (G), alopecia (A), pseudoanodontia (P), and optic atrophy (O). 2 It is believed to have an incidence of approximately one in every 1,000,000 live births and does not display any discernible gender, ethnic, or geographic predispositions. 3 Patients with GAPO syndrome may present with various physical abnormalities, including frontal bossing, underdeveloped midface, absence of eyebrows and eyelashes, prominent scalp veins, increased intracranial pressure, micrognathia, protruding lips and auricles, depressed nasal bridge, altered sweating ability, redundant skin, hyperextensible joints, rough thickened fingernails, umbilical hernia, delayed bone growth, hepatomegaly, and underdevelopment of the genitalia and mammary glands. Those patients reported to have shortened life span, which is thought to be caused by generalized interstitial fibrosis and atherosclerosis.4,5 Our report presents the first documented case of GAPO syndrome in Syria, which is characterized by unilateral conductive hearing loss and other ophthalmic manifestations and novel finding in computed tomography (CT) scan that have not been reported before, to our best knowledge.
Case Presentation
A 27-year-old male patient of Syrian origin, who was diagnosed with GAPO syndrome, was admitted to our ENT department due to persistent right-sided ear pain, which started a month ago, accompanied by a small amount of yellowish, sticky, odorless ear discharge, in addition to right sided hearing loss that started several months ago. There was no history of dizziness, recurrent sinus infections, facial trauma, or endoscopic sinus surgeries. The patient reported that he had been suffering from headache, fullness of the face, breathy voice, blocked nose, and snoring, along with hyposmia since childhood, so that he underwent an adenoidectomy that improved the symptoms for less than 6 months. His parents were first-degree cousins (consanguineous parents), and his perinatal events were normal. He stated that his sister had the same clinical features and had died in her 16s because of cardiac problems, while the rest of the family history was unremarkable. Right ear examination revealed chronic otitis externa, which was treated with topical antibiotics and steroids, in addition to stenosis in the external auditory canal impeding the view of the tympanic membrane, while the left ear examination revealed external auditory canal stenosis. In intranasal examination, we found severe bilateral septal deviation with pale nasal mucosa. His physical examination showed that his psychomotor development and intellectual status were age-appropriate, and he exhibited several characteristic clinical features of GAPO syndrome, including frontal bossing, micrognathia, prematurely-aged face, absent eyebrows and eyelashes, total alopecia, protruding thick lips, buphthalmos, depressed nasal bridge, failure of tooth eruption (pseudoanodontia) (Figure 1), high-arched palate, and increased skin laxity. His cardiovascular, respiratory, and gastrointestinal system examination were within normal limits. The results of the CXR, echocardiogram, and ECG examination were all normal. By optimizing auditory function, pure tone audiometry revealed moderate right-sided conductive hearing loss (CHL) across all frequencies. His ophthalmic examination revealed that he had peripheral congenital cataract, severe astigmatism, myelinated retinal nerve fiber layer, supraorbital ridges hypoplasia, and floppy eyelid syndrome. Head and neck CT showed bilateral external auditory canal stenosis, bilateral total mastoid air cells aplasia, heavy septal deviation, total aplasia of the paranasal sinuses, and pseudoanodontia (Figure 2). Biochemical investigations were within normal limits, except for a slight increase in LH and FSH.
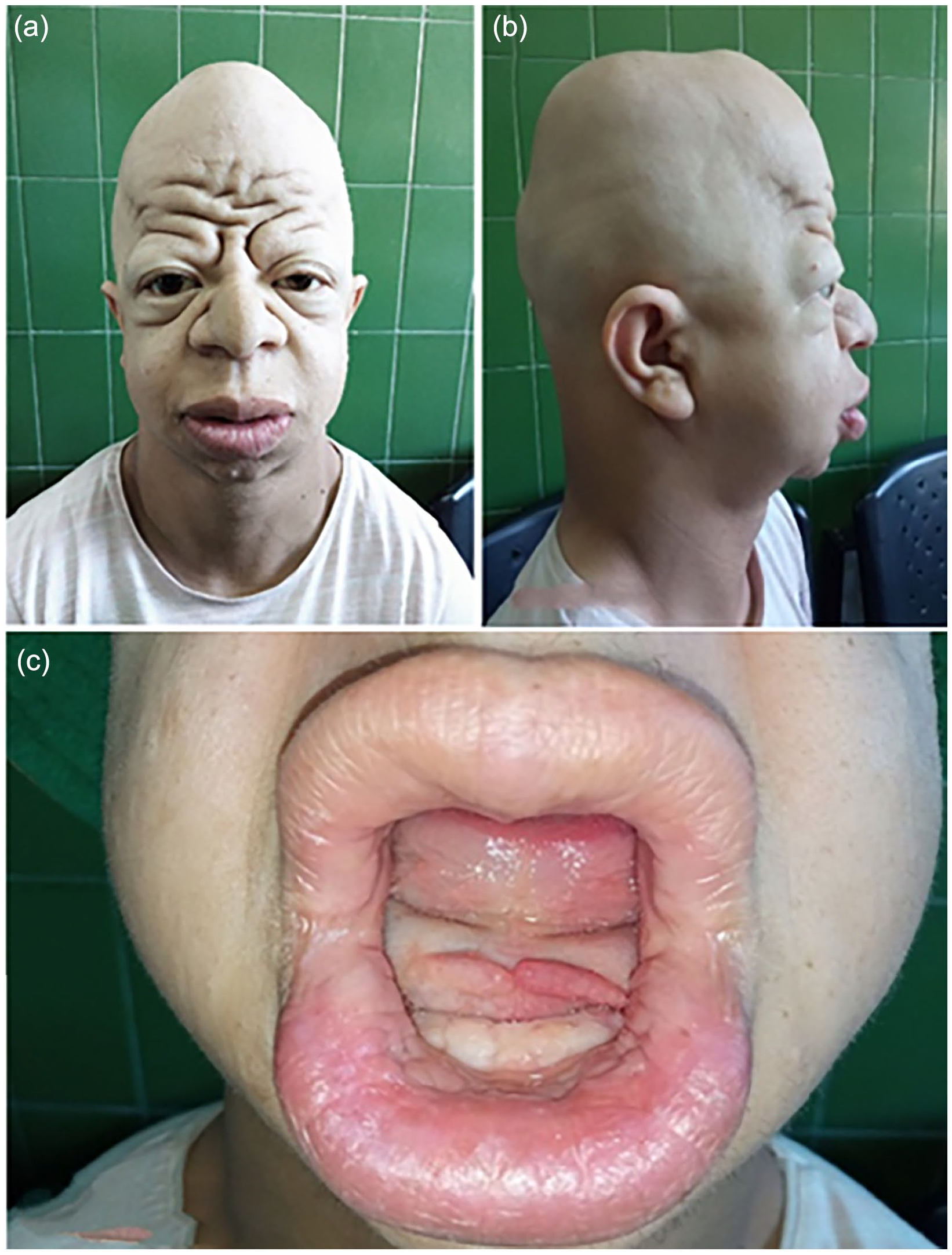
Frontal and lateral photographs of our patient. (a, b) frontal bossing, micrognathia, absent eyebrows and eyelashes, and total alopecia. (b) Buphthalmos and depressed nasal bridge. (c) Failure of tooth eruption and protruding thick lips.
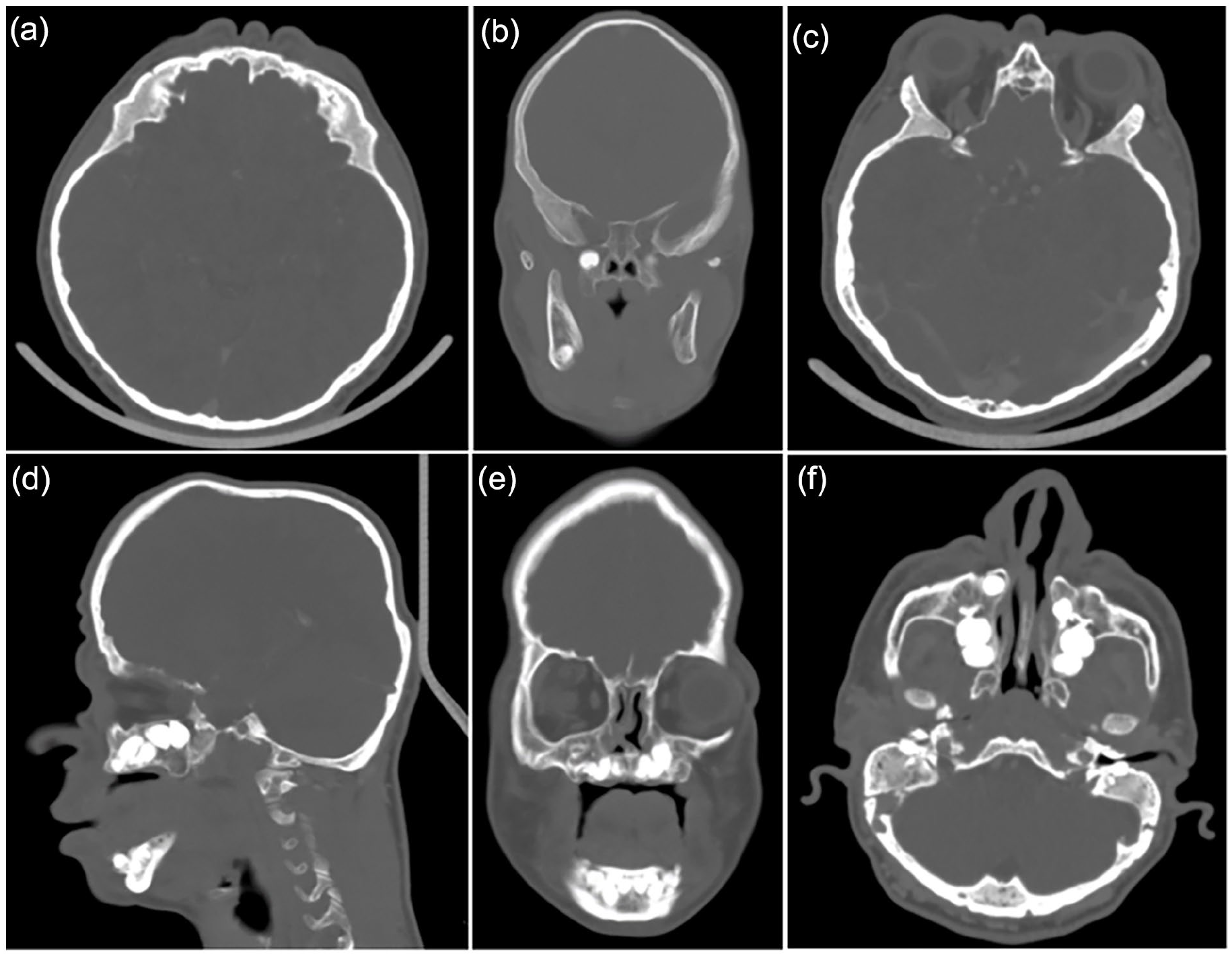
Head coronal, sagittal, and frontal CT scan showing (a) frontal sinus aplasia, (b) sphenoid sinus aplasia, (c) total ethmoid sinus aplasia, (d) pseudoanodontia and sphenoid sinus aplasia, (e) pseudoanodontia and nasal septum deviation, (f) left external auditory canal stenosis, bilateral total mastoid air cells, and maxillary sinus aplasia.
Discussion
GAPO syndrome is an exceptionally-rare genetic disorder characterized by a distinctive clinical presentation. Since its initial description by Andersson and Pindborg in 1947, only around 53 cases have been documented in the medical literature. The condition was officially termed GAPO syndrome by Tipton and Gorlin in 1984.6,7 To date, this represents the first reported case in Syria, with the majority of previously-identified cases originating from Brazil, Turkey, and more recently, Egypt.8,9 In our case, as well as in the majority of reported cases, there is no evidence of intellectual disability.8,10 The patient exhibits all the hallmark features of the syndrome, with the exception of the fourth criterion, “optic atrophy.” This particular feature is considered variable, as it has been documented in only about one-third of cases. Consequently, it has been proposed that the “O” in the syndrome’s acronym should represent ocular findings more broadly, rather than specifically referring to optic atrophy.11,12 The patient presents with severe breathing difficulties, characterized by bilateral nasal obstruction, mouth breathing, rhinolalia clausa, snoring, and obstructive sleep apnea. These symptoms are likely attributed to facial anomalies, macroglossia, micrognathia, and nasopharyngeal abnormalities, as confirmed through flexible fiberoptic laryngoscopy. Such structural issues also compromise middle ear ventilation, resulting in bilateral otitis media with effusion. This pattern aligns with a previously-reported case where an adenoidectomy was performed during infancy to enhance breathing and middle ear ventilation. However, in our patient, dyspnea recurred a few months postoperatively. Currently, the patient is scheduled for septoplasty in our department to address these persistent issues. 13 The literature describes two additional cases of middle ear disorders in individuals with this syndrome. The first involved a mild bilateral CHL 14 while the second concerned complicated, untreated otitis media. 5 Our patient, however, represents the first reported case of unilateral CHL caused by chronic otitis externa and external auditory canal stenosis, compounded by eustachian tube dysfunction. Other reported cases of hearing loss predominantly involved bilateral sensorineural hearing loss (SNHL), which may result from mutations in gamma- and beta-actin genes. These mutations could disrupt connexin 30 function and contribute to extracellular connective tissue matrix accumulation characteristic of this syndrome.12,15 Additionally, the patient’s high Mallampati score and associated anatomical deformities suggest potential risks during general anesthesia, including challenges with ventilation and intubation. However, our patient, like others reported in the literature, has successfully undergone multiple surgical procedures without complications.5,15 Notably, our patient exhibits a myelinated retinal nerve fiber layer, a feature first documented in two Turkish children with GAPO syndrome in 2013. Furthermore, he presents with severe astigmatism and peripheral congenital cataracts, two findings that have not been previously reported in association with this syndrome. 16 To the best of our knowledge, this case represents only the third reported occurrence of complete paranasal sinus aplasia in the medical literature. 17 Furthermore, it is the first documented instance of GAPO syndrome associated with the total absence of all paranasal sinuses combined with complete mastoid air cell aplasia. In contrast, a prior study involving three Brazilian patients noted hypoplasia of the sphenoid sinuses and mastoid sclerosis, with frontal sinus agenesis identified in only two cases. 8 While individuals with paranasal sinus hypoplasia or aplasia are typically asymptomatic and diagnosed incidentally, our patient experienced facial fullness and chronic recurrent headaches, consistent with findings described by H. Korkmaz and M. Korkmaz. 17
This case underscores novel clinical and radiological findings in GAPO syndrome, shedding light on its phenotypic variability and emphasizing the necessity of multidisciplinary management. While the diagnosis of GAPO syndrome primarily relies on hallmark features—such as growth retardation, alopecia, pseudoanodontia, and ocular abnormalities—this case highlights the value of comprehensive evaluations, including advanced imaging, to identify subtle or previously-unreported manifestations. One of the most notable findings in this case is total aplasia of the paranasal sinuses and mastoid air cells, a rare structural anomaly that adds a new dimension to the craniofacial abnormalities associated with GAPO syndrome. Functionally, these anomalies may result in impaired sinus ventilation and drainage, contributing to chronic headaches, facial pressure, and recurrent upper respiratory symptoms. Such findings emphasize the importance of detailed radiological assessments in patients with GAPO presenting with sinonasal complaints, as these anatomical variants may necessitate tailored medical or surgical interventions. Additionally, the rarity of these findings raises the question of whether similar anomalies are underreported due to the limited use of imaging in previous GAPO cases. This case also expands the spectrum of auditory involvement in GAPO syndrome by identifying unilateral CHL, attributed to chronic otitis externa and external auditory canal stenosis. While bilateral SNHL has been previously reported, this case suggests that external auditory canal abnormalities and eustachian tube dysfunction could contribute to CHL in GAPO syndrome. The observed canal stenosis may be linked to the generalized connective tissue dysplasia characteristic of the syndrome. These findings reinforce the importance of routine audiological evaluations, as early identification and management of hearing loss can significantly enhance quality of life. The ophthalmic findings in this patient, including peripheral congenital cataract, severe astigmatism, and myelinated retinal nerve fibers, expand the recognized spectrum of ocular involvement in GAPO syndrome. These observations suggest that the “O” in GAPO might more accurately represent “ocular findings” rather than solely “optic atrophy.” The presence of myelinated retinal nerve fibers—a feature typically associated with benign developmental anomalies—raises intriguing questions about the genetic or developmental mechanisms affected in GAPO syndrome. Future research should explore whether mutations in ANTXR1 play a direct or indirect role in retinal development. From a genetic perspective, ANTXR1 mutations are central to GAPO syndrome. This gene encodes a type I transmembrane protein initially identified as a receptor for anthrax toxin and Seneca Valley virus. While its physiological roles remain incompletely understood, ANTXR1 is known to be involved in craniofacial mesenchyme and vascular development. 18 Given its low expression in adult tissues and its upregulation in pathological states, 18 future genetic studies could investigate whether ANTXR1 mutations contribute to the ocular or craniofacial phenotypes observed in GAPO syndrome. Whole-genome sequencing may help establish genotype-phenotype correlations, providing deeper insights into the syndrome’s pathophysiology. The craniofacial and oropharyngeal abnormalities observed in this patient, including micrognathia and a high Mallampati score, raise concerns about potential airway challenges during anesthesia. While prior reports suggest that patients with GAPO often tolerate surgeries without complications, this case highlights the importance of preoperative airway assessments. Such evaluations are particularly critical for procedures such as septoplasty or other surgical interventions, where advanced airway techniques may be necessary to mitigate intraoperative risks. Finally, this case contributes to the ongoing discussion about systemic manifestations of GAPO syndrome. Although systemic fibrosis has been well-documented in the syndrome, often manifesting as interstitial fibrosis or vascular atherosclerosis, this patient exhibited normal cardiovascular findings and mild endocrine abnormalities, including elevated LH and FSH levels. These findings suggest significant variability in the severity of fibrotic and endocrine manifestations. Long-term follow-up studies may clarify whether endocrine dysfunctions are secondary to organ fibrosis or represent independent features of the syndrome. This case broadens the clinical understanding of GAPO syndrome by identifying previously-undocumented phenotypic traits, including total aplasia of the paranasal sinuses, unilateral CHL, and unique ophthalmic findings. These findings highlight the importance of comprehensive evaluations and individualized management strategies in patients with GAPO. Future research should focus on unraveling the mechanisms underlying these diverse manifestations, refining diagnostic criteria, and exploring potential therapeutic targets. Early recognition and a multidisciplinary approach remain crucial for optimizing patient outcomes and improving quality of life.
Conclusion
This case report highlights the extensive phenotypic variability and the unique otolaryngological and craniofacial features of GAPO syndrome, offering significant insights into its clinical presentation and management. The documented unilateral CHL, external auditory canal stenosis, and total aplasia of the paranasal sinuses and mastoid air cells underscore the importance of thorough audiological and radiological evaluations in patients with GAPO syndrome, especially when sinonasal or auditory symptoms are present. These findings suggest that previously-underrecognized structural anomalies may contribute to the functional impairments seen in these patients, such as chronic sinonasal infections, hearing loss, and upper airway obstruction. The novel ophthalmic manifestations and craniofacial abnormalities identified in this case further expand the known clinical spectrum of the syndrome, emphasizing the need for individualized diagnostic and therapeutic strategies. The findings also raise important considerations for otolaryngologists regarding surgical planning, airway management, and long-term multidisciplinary care. Future research and genetic analyses may clarify the underlying mechanisms driving these diverse manifestations, aiding in the refinement of diagnostic criteria and the development of targeted interventions. Early recognition and comprehensive care are pivotal in improving the quality of life for individuals with GAPO syndrome.
Footnotes
Data Availability
The data that support the findings of this study are available from the author or corresponding author upon reasonable request.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Our institution does not require ethics approval for reporting individual cases. Written informed consent was obtained from the patient for publication of this article.