
Abstract
Keywords
Introduction
Congenital sensorineural hearing loss may occur in association with defects in pigmentation. This depigmentation can be found in areas that are naturally colored by melanin, like the skin, iris, and hair. These conditions, collectively termed auditory pigmentary disorders (APDs), represent a group of extremely heterogeneous hereditary diseases and include Waardenburg syndrome with its various types, Tietz syndrome, Clouston syndrome, and piebaldism, among others. APDs are part of neurocristopathies—a group of congenital multisystem disorders caused by altered development of neural crest cells and multipotent progenitors that differentiate into various cell lineages, including peripheral nervous system glial cells and melanocytes. 1 As a result, patients with APDs can develop a wide range of abnormalities affecting their skin, hair, eyes, and the auditory system. The constellation of clinical features that typically constitute an APD is seldom encountered in routine clinical practice; however, the presentation might be suggestive enough to ask for genetic tests for confirmation. The present case series illustrates the clinical features and work-up of 3 children diagnosed with APD. It emphasizes the fact that congenital pigmentary disorders might coexist with congenital sensorineural hearing loss leading to the non-development of verbal speech, and a syndromic disorder should be suspected and evaluated in such cases.
Case Series
Patient# 1
A 2-year-old boy presented with hearing impairment since birth leading to non-development of verbal speech. On examination, the child had hypoplastic depigmented (blue) irises, dystopia canthorum with W-index > 1.95 (please refer to the Discussion section for details), hypertrichosis of medial eyebrows (synophrys), dystrophia of lacrimal puncta, and a broad nasal root. He had no abnormality of the upper limbs. His 28-year-old father also had similar features. He had perceptive hearing loss with no speech development, along with depigmentation of the eyes (heterochromia irides) and scalp, and also facial hair (early greying) (Figure 1). Both the child and his father showed normal external auditory canal and tympanic membrane and had bilateral severe-to-profound hearing loss (no identifiable peak V till 95 dBnHL) on brainstem-evoked response audiometry (BERA). High-resolution computed tomography (HRCT) of the temporal bones carried out on the child showed no inner ear abnormalities. The constellation of the clinical features suggested Waardenburg syndrome type 1. This was confirmed by whole-genome sequencing which showed a novel mutation (c.9959-5T > g) in the Pax3 (paired box 3) gene in both the father and his son.
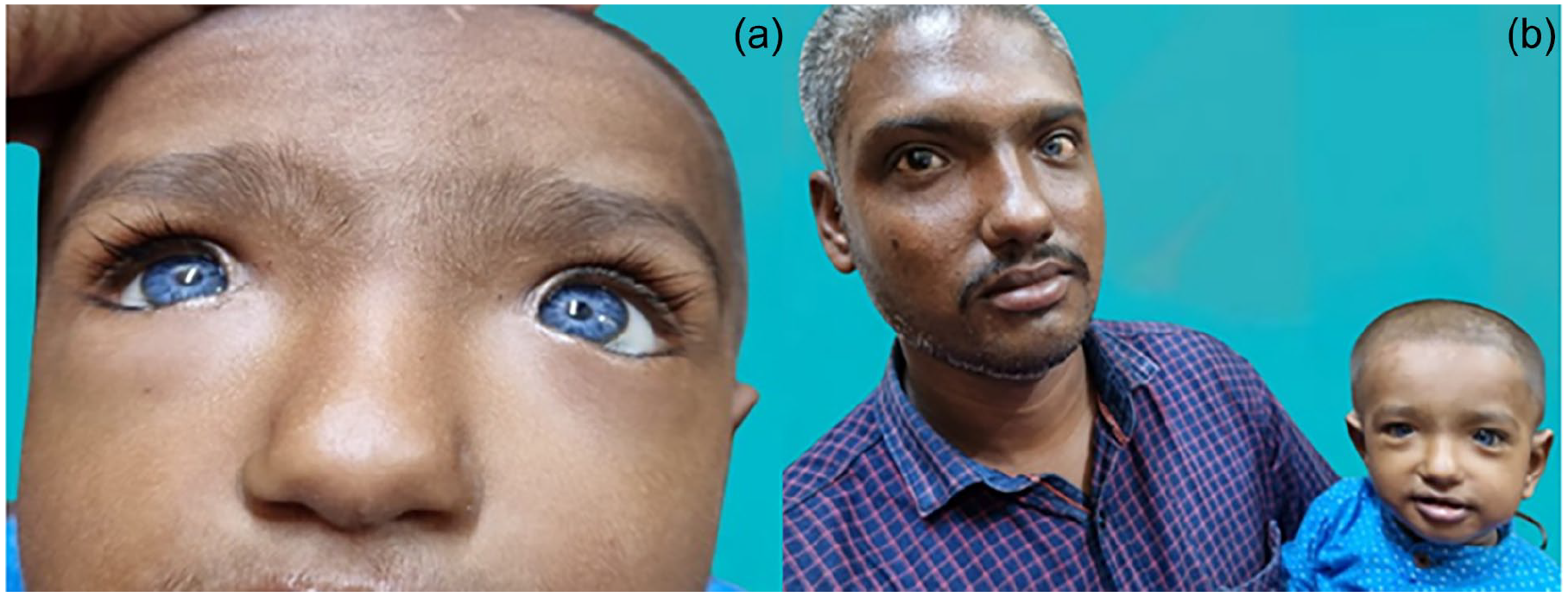
This 2-year-old child (patient 1) showed hypoplastic blue irides and dystopia canthorum (a). His 28-year-old father also showed dystopia canthorum and heterochromia, along with early graying of the scalp and facial hairs (b).
Patient# 2
A 1-year-old male child presented with congenital depigmentation of eyes, skin, and hair. The areas of depigmentation gradually progressed to involve his face, limbs, and trunk as patches. He had blue irises and white hair on his forehead (Figure 2). His parents had consanguineous marriages (first cousins). On examination, his tympanic membrane and external auditory canal were normal. Features such as poliosis, hypoplastic blue irides with brilliant red reflex, hypopigmentation of scalp hair, and congenital leukoderma over the limbs and thorax were noted (Figure 3). His developmental milestones were within normal limits. BERA showed bilateral severe-to-profound hearing loss (no identifiable peak V at 95 dBnHL). HRCT of the temporal bones showed no inner ear abnormalities and normal vestibular aqueducts. There was no clinico-radiologic evidence of Hirschsprung disease. There was no depigmentation of the retinal vessels on fundoscopy, and no abnormality of the upper limbs. The clinical diagnosis suggested Waardenburg syndrome type 2. However, on genetic analysis, the child was found to be an index case of Clouston syndrome with G6PD (glucose-6-phosphate dehydrogenase) gene carrier (Figure 4).
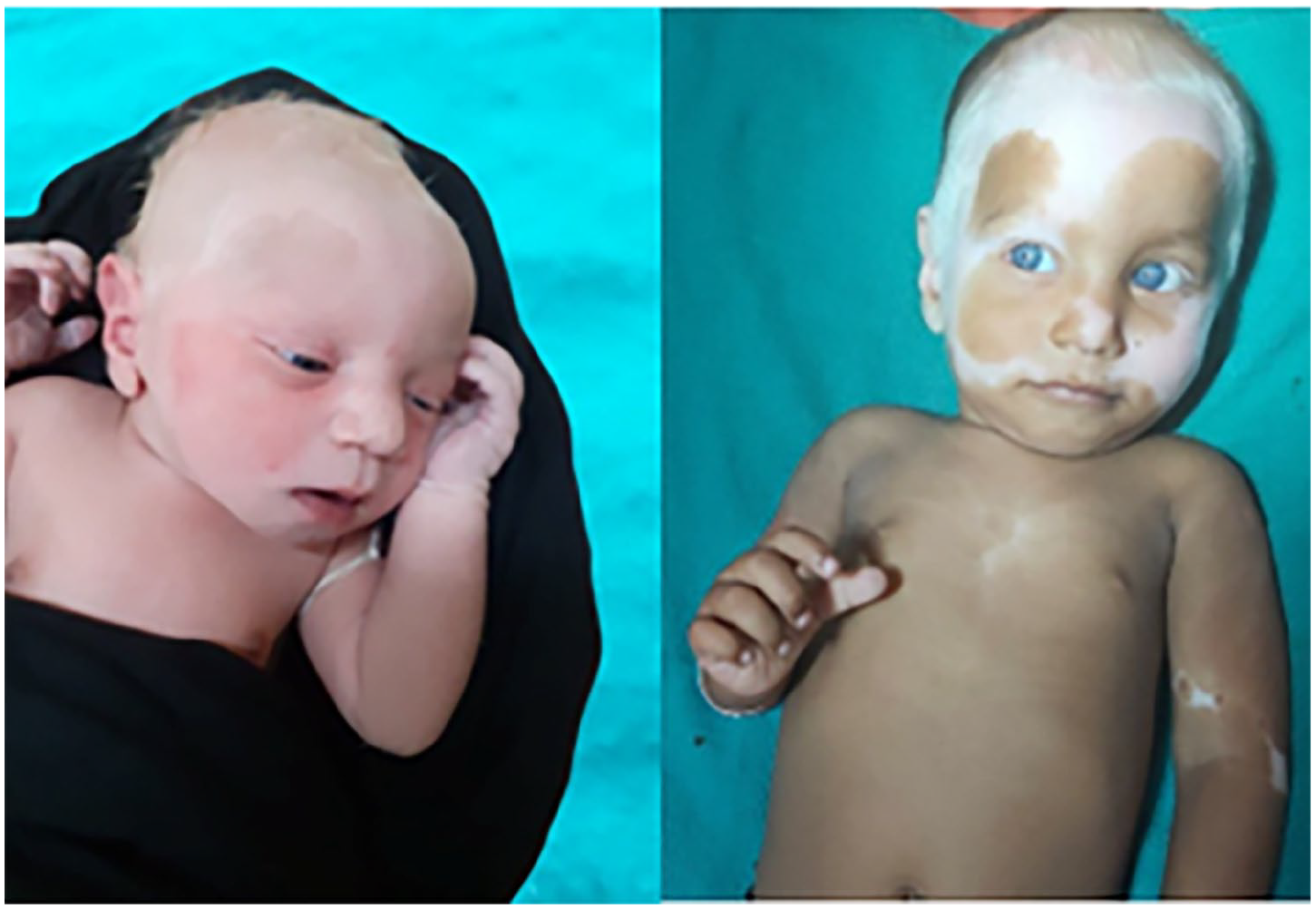
This 1-year-old child (patient 2) had 100% oculocutaneous albinism at birth (left), and hypopigmented skin patches with hypoplastic blue irides (right).
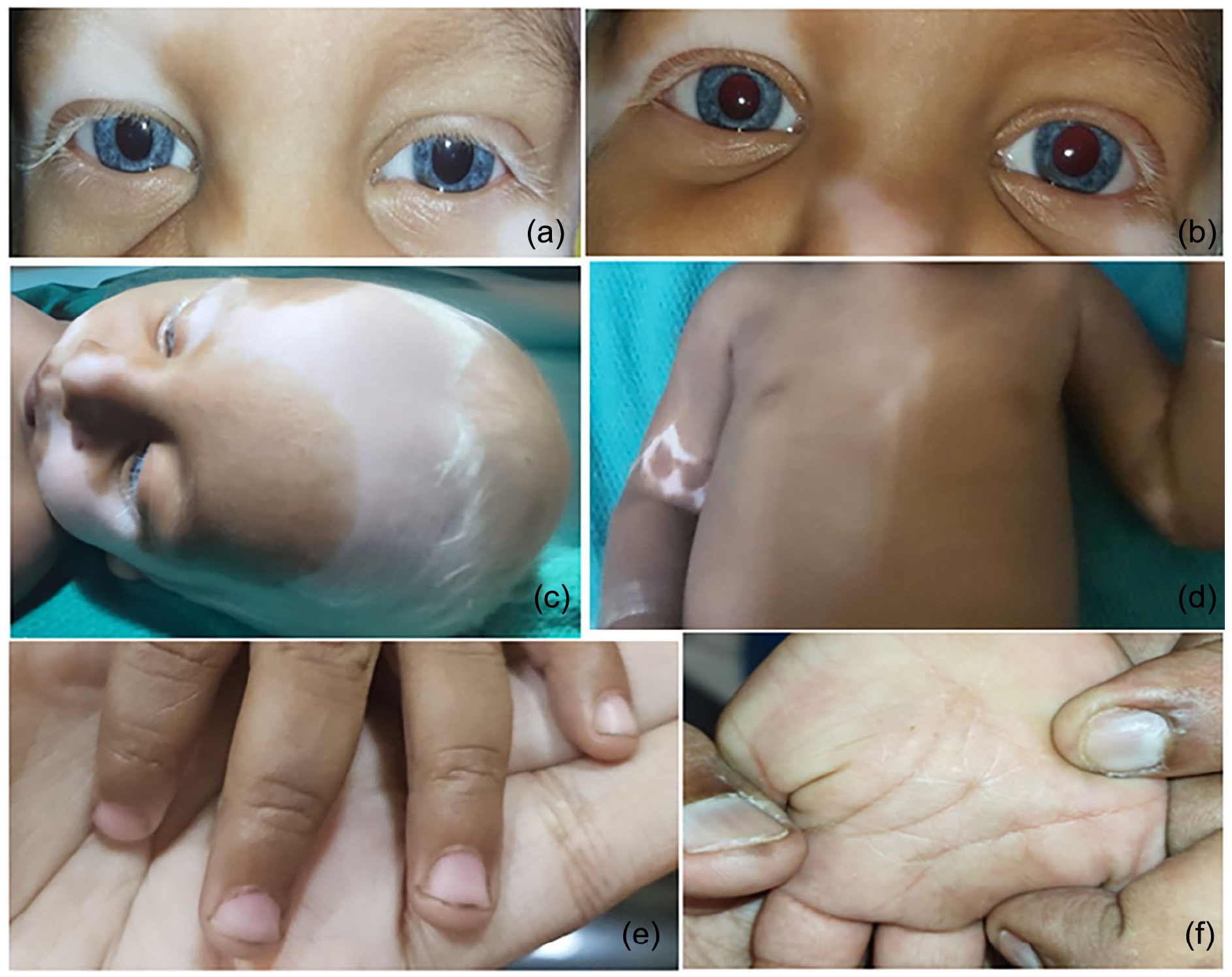
The child (patient 2) showed poliosis with W-index < 1.95 (a), hypoplastic blue irides with a brilliant red reflex (b), hypopigmented scalp hair (c), congenital leukoderma over limbs and thorax (d), absence of nail dystrophy (e), and absence of palmar keratoderma (f).
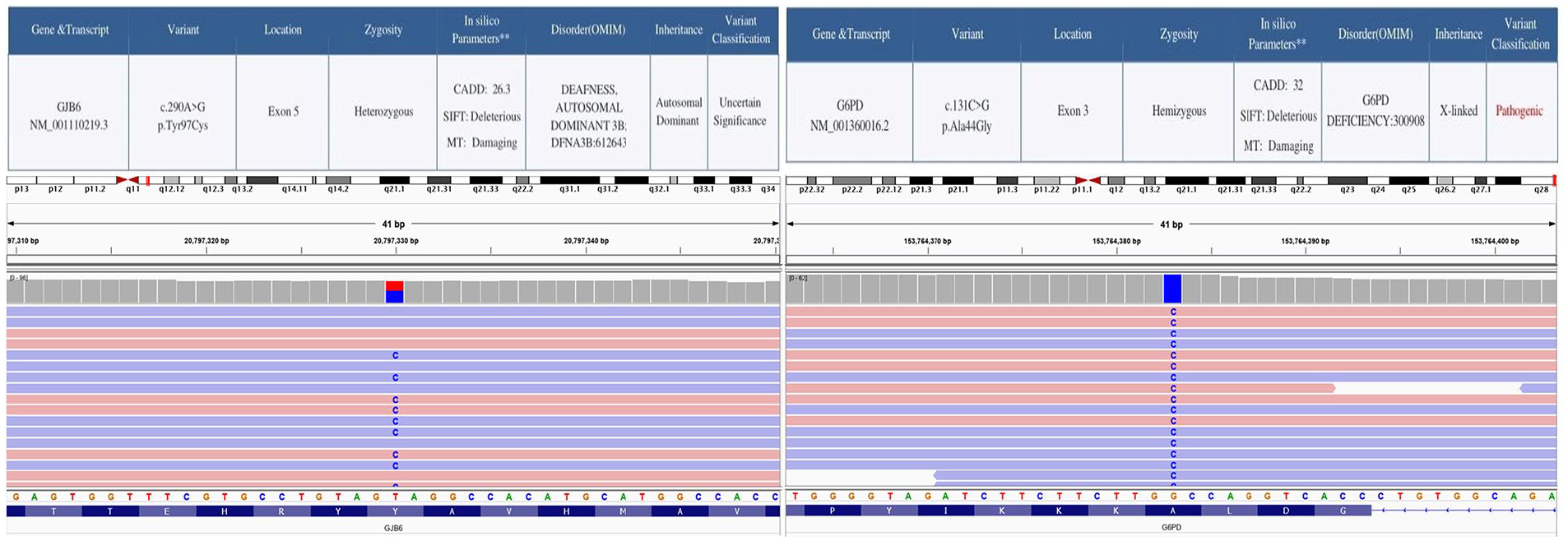
The child (patient 2) showed mutations in the GJB6 gene causing Clouston syndrome. He was also found to be a G6PD carrier.
Patient# 3
A 14-month-old male child presented with impaired hearing since birth that led to non-development of verbal speech. He suffered from global developmental delay and had a history of multiple seizure episodes, head banging, and frequent oral ulcers and boils. Routine examination revealed a normal tympanic membrane and external auditory canal. There were bilateral hypoplastic blue irides, W-index < 1.95, and absence of white forelock, ruling out Waardenburg syndrome type 1 (Figure 5). BERA revealed bilateral moderately severe-to-severe hearing loss with no identifiable peak V in both ears till 95 dBnHL. Arterial blood gas examination showed metabolic acidosis. The child was already on levetiracetam at the presentation. HRCT of the temporal bones was unremarkable with normal vestibular aqueducts. The child had been suffering from Hirschsprung disease (Figure 6), which excluded Waardenburg syndrome type 2. There was depigmentation of the retinal vessels on fundoscopy (Figure 6). The child had no loss in visual acuity, a normal echocardiogram, and no abnormalities in his upper limbs.
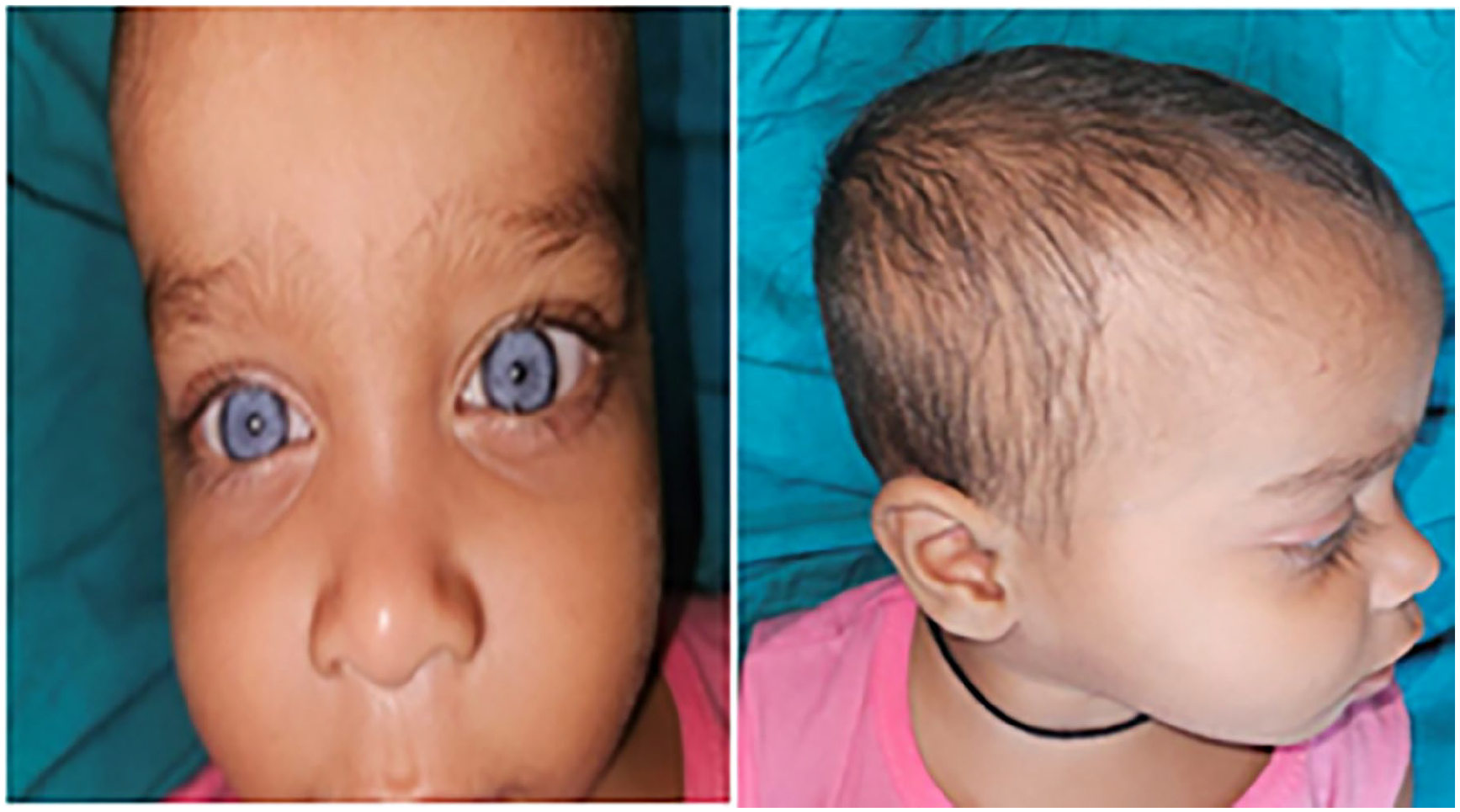
This child (patient 3) showed bilateral hypoplastic irides with a W index < 1.95 (left) and the absence of a white forelock (right).
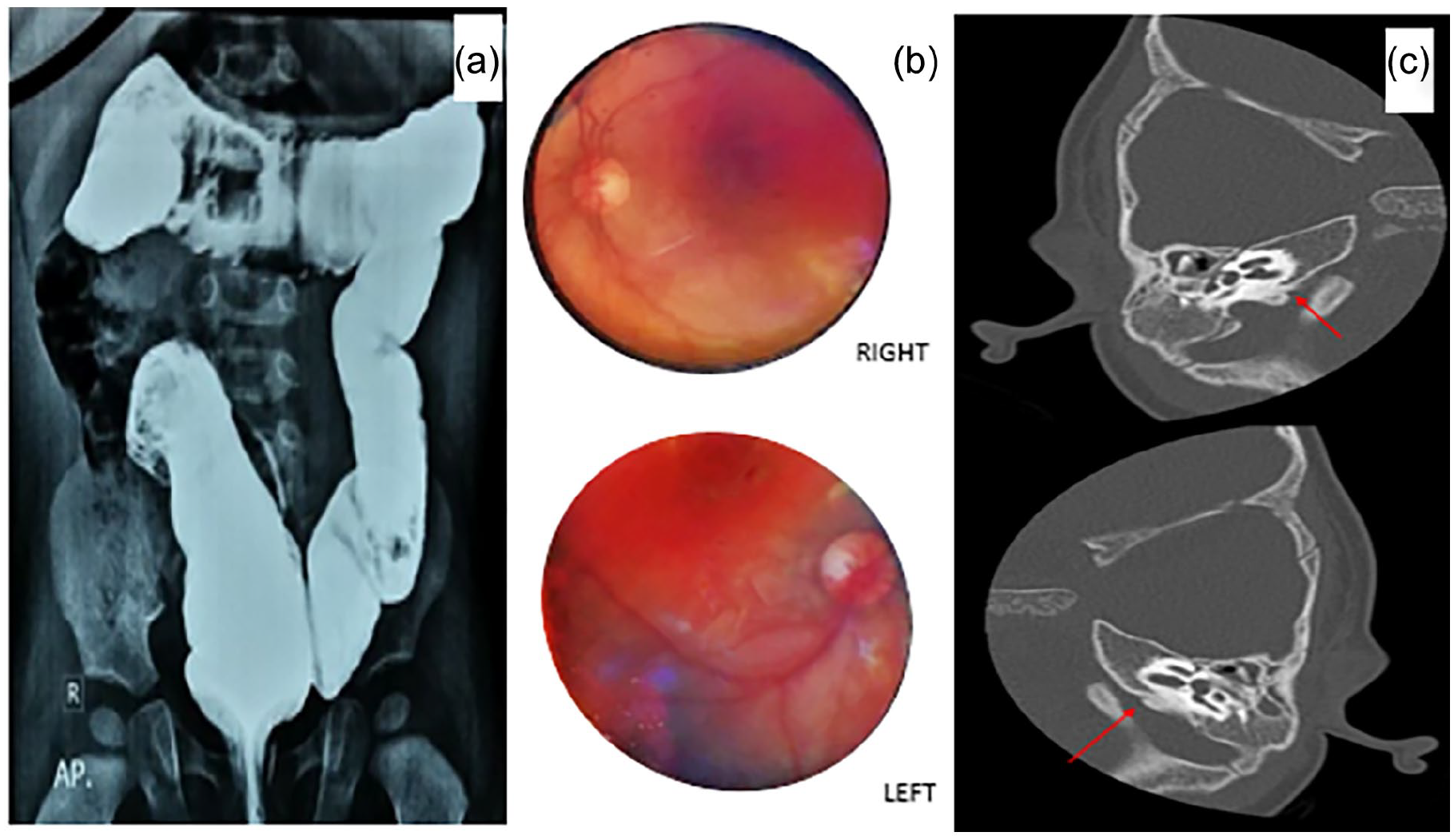
This child (patient 3) showed features of Hirschsprung disease on barium enema (a), depigmentation of retinal vessels on fundoscopy (b), and normal vestibular aqueducts on HRCT of temporal bones (red arrows) (c).
The clinical features here favored Waardenburg syndrome type 4 (Waardenburg-Shah syndrome), which was confirmed on genetic analysis. The child had compound heterozygous variants such as Waardenburg syndrome type 4a showing mutation in EDNRB (endothelin receptor type B) gene, Phelan-McDermid syndrome with mutation in the SHANK3 (SH3 and multiple ankyrin repeat domains 3) gene (PMS-SHANK3-related), Delpire-McNeill syndrome with mutation in the SLC12A2 (Solute Carrier Family 12 Member 2) gene, and PRORP (Protein Only RNase P Catalytic Subunit) gene mutation denoting Combined Oxidative Phosphorylation deficiency 54 (Figure 7).

The child (patient 3) showed compound mutations in the genome that suggested a compound heterozygous variant of Waardenburg type 4 (Waardenburg-Shah) syndrome.
All the children discussed above were planned for cochlear implantation after an appropriate hearing aid trial. At the time of writing this report, the first and the third child had been receiving speech therapy following a successful implant and were reported to have satisfactory outcomes regarding auditory and verbal rehabilitation. The second child had just undergone a cochlear implant and was waiting to switch on the device.
Discussion
The association between congenital hearing loss and de-/hypopigmentation is intriguing and is yet to be fully explained. The melanocytes in the stria vascularis of the cochlea are derived from the neural crest cells during the development of the otocyst/otic vesicle (the embryonic inner ear sensory organ). Animal studies have shown that these melanocytes are not associated with the function of pigmentation; rather, they play a pivotal role in ionic homeostasis in the membranous labyrinth fluids (K+ transport in the endolymph), responsible for normal hearing.2,3 Thus, lack of melanocytes in the stria vascularis may lead to sensorineural hearing loss without any features of depigmentation, and vice versa, albinism may not always be associated with congenitally impaired hearing. The role of the strial melanocytes in generating the endocochlear potential, and the resultant sensorineural hearing loss in the absence of this function, is regulated by a group of genes (Mitf [melanocyte inducing transcription factor], c-kit [receptor tyrosine kinase proto-oncogene], Steel, etc.). 2 However, whether there appear to be separate sets of gene regulations as connecting links that control both the strial melanocytes and those responsible for oculocutaneous pigmentation is debatable and is a matter of ongoing research. Several studies do link these 2 phenotypes by the selective contributions of the melanocytes, like the association of sensorineural hearing loss with alopecia areata and vitiligo, with newer animal studies coming up attributing the loss of Pax3 gene (that causes Waardenburg syndrome in humans) to the reduction of strial melanocytes.4-6 The association of congenital sensorineural hearing loss with oculocutaneous pigment disorders was first explored systematically by the Dutch geneticist and ophthalmologist, Petrus Johannes Waardenburg, in 1951, after whom, the Waardenburg syndrome was named. 7
Waardenburg syndrome is characterized by the loss of pigmentary cells in the skin, hair, and eyes, along with the stria vascularis of the cochlea. It affects about 1 in 42 000 population; however, its association with hearing loss and non-development of verbal speech is not common (0.9%-2.8% of the patients). 8 Waardenburg syndrome is ubiquitous and affects people of all races regardless of sex, with a normal life expectancy. The pathophysiology of this syndromic disorder involves aberrant dispersion of melanocytes during embryogenesis, leading to patchy areas of depigmentation. 8 Based on the clinical characteristics, Waardenburg syndrome can be of 4 types (Table 1).9,10
The 4 Types of Waardenburg Syndrome (WS), Their Etiology, and Clinical Features.

The W-index is a biometric scale used to diagnose Waardenburg syndrome type 1 and identify dystopia canthorum in patients with this syndrome. The W-index is based on the 3 measurements of ocular distance (in mm) obtained from patients with Waardenburg syndrome: inner canthal distance (a); interpupillary distance (b); and outer canthal distance (c). The formula stands as W-index = X + Y + a/b, where X = [2a−(0.2119c + 3.909)/c], and Y = [2a−(0.2479b + 3.909)/b]. 9 Dystopia canthorum, one of the major criteria for the diagnosis of Waardenburg syndrome type 1, is said to be present when the W-index is >1.95. 9 However, there may be variations to this value based on geographic regions and ethnicities. 11
Also known as hidrotic ectodermal dysplasia, Clouston syndrome is an uncommon autosomal dominant disorder of ectodermal dysplasia caused by mutations in the connexin gene and is characterized by hyperkeratotic palms and soles, dystrophic nails, and global hypotrichosis. 12 Connexins, also known as gap junction proteins, are a family of structurally related transmembrane proteins that facilitate direct communication between cells and are in charge of the transport of ions and chemicals among the neighboring cells. 12 Owing to the similarities, Clouston syndrome is occasionally confused with Waardenburg syndrome. Although Clouston syndrome does not affect the eyes, ears, or limbs like Waardenburg syndrome, both conditions present with abnormalities of the skin, hair, and nails. The scalp hair in Clouston syndrome is lighter in color, sparse, and patchy. The condition progressively worsens and culminates in complete hair loss by adolescence. 13 Unlike Waardenburg syndrome, Clouston syndrome features characteristic abnormalities in the nails, like severe deformity, spontaneous detachment, poor development, unusual thickness, and discoloration; also, the skin on the palms and soles is thickened, and there may be areas of darker pigmentation. 14 While Waardenburg syndrome is caused by mutations in genes like Pax3, Mitf, and Sox10 (SRY-box transcription factor 10), Clouston syndrome (which has an autosomal dominant inheritance) is specifically caused by mutations in the GJB6 (gap junction protein beta 6) gene, as seen in the second child in this series.8,12
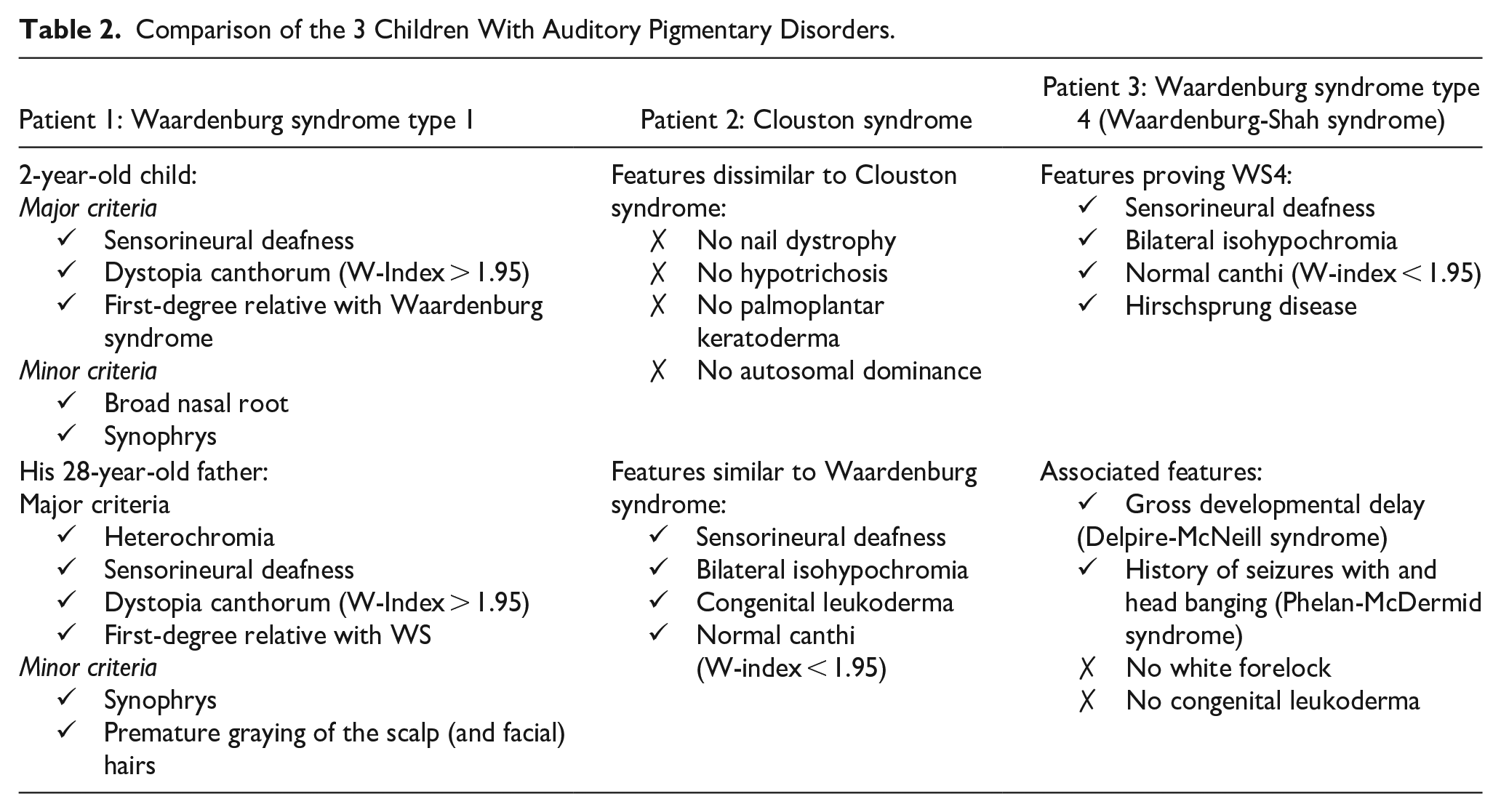
The clinical features of the 3 children with APDs discussed here are elaborated and compared in a tabular format (Table 2). The first child diagnosed with Waardenburg syndrome type 1 had typical features of bilateral profound sensorineural hearing loss with hypoplastic blue irides and dystopia canthorum. The second child presented with atypical and sporadic features of Clouston syndrome where the phenotype did not match the classic triad of hypotrichosis, nail dystrophy, and palmar keratoderma; however, there was severe congenital hearing loss leading to non-development of verbal speech. Mutation-proved Clouston syndrome is extremely rare among the Asian-Indians. 12 And as per the authors’ knowledge, Clouston syndrome with congenital hearing loss sans the skin and nail findings are yet to be documented in the literature. A case-based search in the PubMed/MEDLINE database on Clouston syndrome with congenital hearing loss revealed a 2013 report by Sugiura et al. 15 that described a 24-year-old Japanese woman with Clouston syndrome who had mild prelingual bilateral sensorineural hearing loss in addition to the classic triad of nail dystrophy, hypotrichosis, and palmoplantar keratoderma. Another report in 2018 by Khatter et al. 12 discussed a large family with a novel GJB6 mutation causing Clouston syndrome without palmoplantar keratoderma and congenital sensorineural hearing loss. In a 2014 report, Clouston syndrome sans hearing loss was described in a patient who developed eccrine syringofibroadenomas. 16 These reports assert that sensorineural hearing loss, while not a cardinal feature, can occasionally be associated with Clouston syndrome. Nevertheless, the prevalence of congenital hearing loss in this condition appears to be low based on the limited volume of published literature. More research is needed to fully characterize the range of clinical manifestations associated with Clouston syndrome.
Comparison of the 3 Children With Auditory Pigmentary Disorders.
The third child had a sporadic presentation of an autosomal recessive form of Waardenburg syndrome type 4 (aka Waardenburg-Shah syndrome). Characterized by sensorineural hearing loss, hypoplastic blue iris, and Hirschsprung disease, it is the least common type of Waardenburg syndrome and is therefore exceedingly rare.17-19 PMS-SHANK3-related disorder is associated with several developmental delays involving speech production, neuromuscular affections, and facial dysmorphism. 20 Most children tend to develop low intelligence quotient in the future and may have gait disorders (ataxia), poor motor skills, sleep deprivation, and epilepsy. According to the Phelan-McDermid Syndrome Foundation, almost 1200 children with this syndrome are on record. 20 Delpire-McNeill syndrome with SCL12A2 mutation causes mild-to-severe neurodevelopmental disorders and global developmental delay with sensorineural hearing loss as described in a case series by Alisdair McNeill. 21 Bi-allelic variants in the PRORP gene have recently been associated with a newly defined syndrome—combined oxidative phosphorylation deficiency 54 (COXPD54)—including a phenotypic spectrum of Perrault syndrome and leukodystrophy. 22 Perrault syndrome is a rare genetic disorder affecting both males and females, characterized primarily by sensorineural hearing loss; male subjects generally retain normal fertility. 22 The third child of the present series matched the clinical features of this syndrome (history of seizures, head banging, global developmental delay, non-development of verbal speech), along with the characteristic genetic mutations that clinched the diagnosis. Hence, this is the first report of a patient with a compound heterozygous variant of Waardenburg-Shah syndrome, consisting of Waardenburg syndrome type 4a, Phelan-McDermid syndrome, Delpire-McNeill syndrome, and PRORP mutation denoting COXPD54.
Cochlear implantation remains the mainstay of treatment for the sensorineural hearing loss that is associated with Waardenburg syndrome. 23 Special attention is needed in these children with syndromic disorders because there may be associated inner ear malformations in about 17% to 18% of cases, most importantly absence of posterior semicircular canal, malformation in the internal acoustic meatus, cochlear hypoplasia, enlarged vestibular aqueduct, and Mondini deformity.23-25 However, with proper individualized preoperative planning and parental counseling, the potential for auditory outcome following cochlear implantation in children with Waardenburg syndrome is comparable to those with non-syndromic pre-lingual sensorineural hearing loss.23,26 Fortunately, none of the children in the present series had inner ear abnormalities, and the outcomes of cochlear implantation and post-implant auditory and speech rehabilitation in them hold promise.
To conclude, this case series illustrates 3 children with rare genetic disorders/syndromes characterized by congenital oculocutaneous albinism/hypopigmentation, sensorineural hearing loss, and non-development of verbal speech. The striking features of depigmentation in the melanin-containing areas of the body could be the first presentation before the parents are aware of the associated hearing and speech problems. For the otolaryngologists-in-practice, congenital oculocutaneous hypopigmentation in a child, either as the first presentation or as a referral from a pediatric medicine colleague, should arouse suspicion for other coexisting congenital stigmata, like perceptive hearing loss and subsequent non-development of verbal speech. APDs are seldom encountered in routine otolaryngology practice. Therefore, a high index of suspicion is needed for these syndromic disorders characterized by the tandem of perceptive hearing loss and poor speech, which might go unnoticed if not specifically searched for. Early identification of these syndromes should help alleviate the quality of life of the affected children who may be offered appropriate auditory rehabilitation and speech therapy at the earliest. To execute the management plan for these children, a multidisciplinary team of otolaryngologists, dermatologists, psychiatrists, ophthalmologists, pediatricians, rheumatologists, and genetic analysts is essential.
Footnotes
Data Availability Statement
The dataset and all relevant information disclosed in this manuscript are available with the first author and can be reproduced on demand, as and when necessary.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Standards Compliance
The paper is in compliance with the Ethical Standards of national and international guidelines on human experimentation, as laid down in the Helsinki Declaration of 1964, as revised in 2013 at Fortaleza, Brazil.
Ethical Approval
Being a case series, the Scientific Technical Advisory Committee and Institutional Ethics Committee have exempted this paper from formal ethical approval. However, as stated above, the authors have ensured that the content of the manuscript and the patient care decisions duly followed the concerned ethical standards of international agreements.
Consent for Publication
Due informed written consents were obtained from the parents of all the children mentioned in the article for their images and other clinical details for publication/reproduction in a medical journal. The documents have been submitted along with the manuscript.