
Abstract
Congenital arhinia or nasal absence is a rare condition, with only less than 100 cases published in the literature to date. It is a rare condition that causes respiratory distress during the neonatal period. Although stabilization of the airway is the priority, management is not clearly defined, given the rarity of the malformation. We report a case of arhinia in a female newborn and briefly review the literature.
Introduction
Congenital arhinia is an extremely rare disease, with only fewer than 100 cases reported in the literature to date. 1 It is defined as the absence of the external nose, nasal cavities, and the olfactory apparatus. It represents a challenge for pediatric, maxillofacial, and plastic surgeons.
Arhinia may predispose the patient to respiratory and feeding problems at birth, requiring early intervention.2,3 The most severe presentation is called Bosma Arhinia Microphthalmia Syndrome and includes arhinia, microphthalmia, and hypogonadotropic hypogonadism; it was first described by Bosma in 1981. 4
Case Report
We report the case of a female newborn from a second-degree consanguineous marriage, a 38-year-old mother, and siblings in good health. The pregnancy was moderately followed with 4 prenatal consultations by a city gynecologist and was complicated by hydramnios. Morphological ultrasound was not performed. The diagnosis of a nasal defect was made on the third-trimester ultrasound data. The newborn was born at term, and delivery was by cold cesarean section for a tricicatricial uterus. The adaptation to extra uterine life was average with an APGAR score of 6/8/9 with a weight of 3650 g. She presented immediate respiratory distress that required hospitalization in the neonatal intensive care unit. Examination showed facial dysmorphia with 2 frontal humps, hypertelorism, arhinia, low implanted hair, low implanted ears in the form of horns, retrognathism, and a short neck (Figure 1).
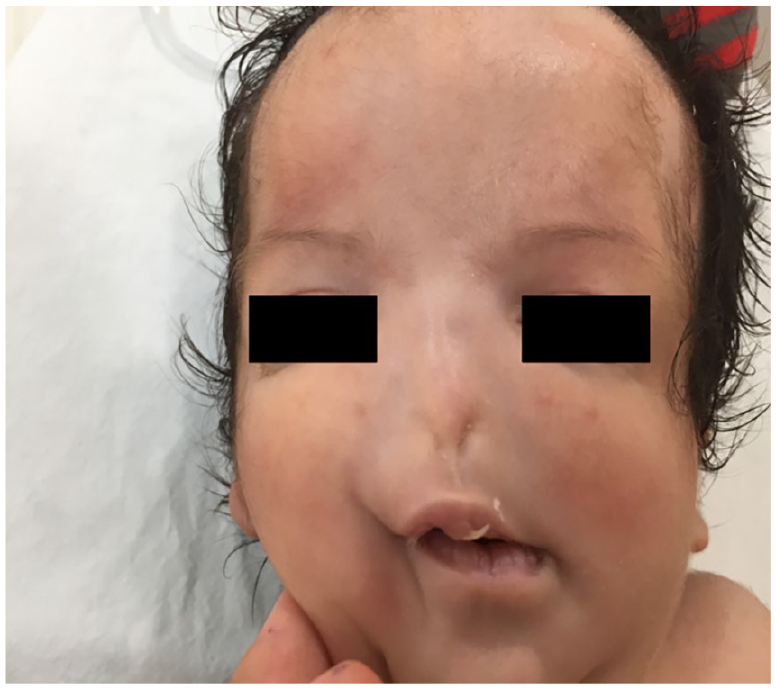
Facial dysmorphia with 2 frontal humps, hypertelorism, arhinia, low implanted hair, and low implanted ears.
She was conditioned with the placement of a Mayo cannula and oxygen therapy through a hood enclosure, and he was perfused. An emergency CT scan of the facial mass showed agenesis of the external nose, hypoplasia of the nasal bone, hypoplasia of both piriform sinuses, total agenesis of the vomer, total agenesis of the perpendicular lamina, and agenesis of the ethmoidal labyrinth (Figure 2).

CT scan of the facial mass.
The evolution was favorable, and the newborn was weaned from oxygen on day 3 of life, the introduction of tube feeding was well tolerated. She was breathing spontaneously by mouth without a Mayo tube. The team of maxillofacial surgeons advised considering a surgical procedure around school age. Therefore, a malformation assessment was carried out, abdominal, renal, and cardiac ultrasounds were normal. The transfontanellar ultrasound showed a slight triventricular dilatation and for a better exploration of the cerebral parenchyma, a cerebral magnetic resonance imaging (MRI) showed agenesis of the olfactory bulbs and congenital stenosis of the Magnum foramen. An ophthalmological examination concluded that the nasal lacrimal canal was absent and that there was no iridal or retinal coloboma. The ENT examination showed no reaction to loud sounds with the absence of acoustic otoemission on both sides. He was taken out at 24 days of age. The newborn was seen in the outpatient clinic at 37 days of age and was fed by tulip feeding. Death occurred at 42 days of age at home due to asphyxial respiratory distress.
Discussion
The pathogenesis of congenital arhinia is not well understood. Embryologically, the nose forms between the third and 10th week of gestation. Arhinia is due to lack of development of the medial and lateral nasal processes, insufficient resorption of the nasal epithelial plug during the 13th to 15th week of gestation, or premature fusion of the medial nasal prominences. A neural crest cell migration defect has also been proposed in the pathogenesis of arhinia. 5
In most cases, congenital arhinia occurs sporadically, however, familial cases have been reported. 6 Furthermore, there is no genetic mutation associated with arhinia, nevertheless, an aberration of chromosome 9. 7 Kaminker et al 8 described a case of arhinia in a mosaic trisomy 21. Treacher Collins syndrome has been reported in some cases.9,10 It should be noted that the prenatal history is often uneventful, 2 however, gestational diabetes and hydramnios were also reported. 11
Arhinia is often diagnosed after birth. However, a prenatal diagnosis is possible as early as the 23rd gestational week by fetal MRI. 3 Ultrasound screening helps reveal a flat fetal profile.9,12
This malformation is usually associated with other midface and orbital malformations. 1 Orbital manifestations include microphthalmia or anophthalmia, choroidal malformations, colobomas, cataracts, and hypertelorism.13 -16 The absence of nasolacrimal ducts is another feature that can cause visual impairment due to the overproduction of tears.1,16
Other abnormalities that have also been observed include cleft palate, umbilical hernia, hypospadias, syndactyly, and abnormalities of the central nervous system.2,14 Auricular anomalies sometimes accompany arhinia. 2
The facial mass scan determines the thickness of the atretic plate, choanal atresia, brain abnormalities, and the extent of microphthalmia and is an important tool in planning surgical correction.
Treatment of arhinia usually involves the treatment of airway obstruction and the management of feeding difficulties. Because newborns breathe compulsorily through the nose, the simultaneous needs of sucking and breathing result in respiratory distress. Inhaling and exhaling through the oral passage alone can cause chest retraction, further exacerbating this distress. Temporary measures, such as oral airways and orogastric feeding are effective. Most cases require channeling of the nasal passage or tracheostomy depending on the severity of neonatal respiratory distress.13,17,18
Due to the rarity of the disease, there is no best surgical treatment. A wide variety of surgical interventions have been proposed, including the placement of a nasal prosthesis 19 or nasal reconstruction with maxillary osteotomy and local flaps.9,10
Conclusion
Congenital arhinia is one of the rare craniofacial malformations that can cause severe respiratory distress at birth due to obstruction of the upper airways. Our patient is the first case of congenital arhinia in Tunisia. Contrary to the belief that newborns should breathe through the nose, our patient adapted well to breathing through the mouth.
Footnotes
Availability of Data and Materials
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Statement of Informed Consent
Written informed consent was obtained from the parent of the patient(s) for their anonymized information to be published in this article.